Fra-1 promotes growth and survival in RAS-transformed thyroid cells by controlling cyclin A transcription (original) (raw)
Abstract
Fra-1 is frequently overexpressed in epithelial cancers and implicated in invasiveness. We previously showed that Fra-1 plays crucial roles in RAS transformation in rat thyroid cells and mouse fibroblasts. Here, we report a novel role for Fra-1 as a regulator of mitotic progression in _RAS_-transformed thyroid cells. Fra-1 expression and phosphorylation are regulated during the cell cycle, peaking at G2/M. Knockdown of Fra-1 caused a proliferative block and apoptosis. Although most Fra-1-knockdown cells accumulated in G2, a fraction of cells entering M-phase underwent abortive cell division and exhibited hallmarks of genomic instability (micronuclei, lagging chromosomes and anaphase bridges). Furthermore, we established a link between Fra-1 and the cell-cycle machinery by identifying cyclin A as a novel transcriptional target of Fra-1. During the cell cycle, Fra-1 was recruited to the cyclin A gene (ccna2) promoter, binding to previously unidentified AP-1 sites and the CRE. Fra-1 also induced the expression of JunB, which in turn interacts with the cyclin A promoter. Hence, Fra-1 induction is important in thyroid tumorigenesis, critically regulating cyclin expression and cell-cycle progression.
Keywords: AP-1, cyclin A, Fra-1, RAS transformation, thyroid
Introduction
RAS oncogene activation is frequently associated with different types of tumors originating from the follicular epithelium of the thyroid gland. In the highly frequent papillary thyroid carcinoma (PTC), RAS mutations are mutually exclusive with BRAF point mutations and RET/PTC oncogenic rearrangements (Kimura et al, 2003), providing genetic evidence for the role of the RET/PTC–RAS–BRAF signaling cascade in thyroid cancer (Melillo et al, 2005).
The rat FRTL-5 cell line, which recapitulates the fully differentiated thyroid phenotype, represents a model system for studying the thyroid-specific gene expression and cell growth (Kimura et al, 2001). RAS transformation of FRTL-5 cells disrupts thyroid-specific gene expression and mitogenic pathways by multiple mechanisms, affecting the activity of thyroid-specific transcription factors (Avvedimento et al, 1991; Francis-Lang et al, 1992; Missero et al, 2000). A major consequence of the oncogene-driven thyroid cell dedifferentiation is the uncoupling of cell growth control from the thyroid-specific mitogen TSH, both in vitro (Fusco et al, 1987) and in human anaplastic thyroid carcinomas.
AP-1 is a dimeric transcription factor composed of members of the Jun and Fos proto-oncogene families, as well as some ATF and Maf family proteins (Shaulian and Karin, 2002; Hess et al, 2004). In addition to its role in proliferation, apoptosis and differentiation in normal cells, AP-1 is a critical target during neoplastic transformation and is implicated in the complex transcriptional programs that control many hallmarks of cancer (Eferl and Wagner, 2003).
By investigating the changes in AP-1 composition associated with RAS transformation, we previously reported the upregulation of the Fos family component Fra-1, both in mouse NIH3T3 fibroblasts and in rat FRTL-5 thyrocytes (Mechta et al, 1997; Vallone et al, 1997). Fra-1 accumulation is involved in oncogenesis in both cell systems, as ectopic Fra-1 (and c-Jun) expression results in oncogenic transformation in NIH3T3 fibroblasts (Mechta et al, 1997), whereas Fra-1 inhibition attenuates the neoplastic phenotype in the FRTL-5K-Ras thyroid cell line (Vallone et al, 1997). In FRTL-5K-Ras cells, the effect of RAS on Fra-1 accumulation is mediated by both Fra-1-dependent transcriptional autoregulation and post-translational stabilization of the protein. The increased protein stability is due to Fra-1 phosphorylation by the MEK/ERK pathway (Casalino et al, 2003), and protection from proteasomal degradation in human colon carcinoma cells (Vial et al, 2003).
The role of Fra-1 in cell motility and invasiveness, originally described in mouse adenocarcinoma cells (Kustikova et al, 1998), and further characterized in detail in human breast cancer cell lines (Belguise et al, 2005), involves several Fra-1 target genes implicated in extracellular matrix degradation (uPA, uPAR, PAI-1, MMP-3, MMP-9 and TIMP-1). The effect of Fra-1 on tumor cell motility is characterized by a Fra-1-dependent decrease in stress fibers and focal adhesions in human colon carcinoma cell lines (Vial et al, 2003; Pollock et al, 2005). Conversely, the invasion suppressor E-cadherin acts as a negative regulator of Fra-1 in human epidermoid carcinoma cells (Andersen et al, 2005). Compared to other Fos-family members, a determinant role for Fra-1 in human tumor progression is strongly suggested from its overexpression in a variety of cell lines and clinical samples, representing a wide range of epithelial tumors (Milde-Langosch, 2005).
However, in contrast to the consolidated evidence concerning Fra-1 function in tumor cell motility and invasion, little is known about the role of Fra-1 in tumor cell proliferation. In contrast to the individual Jun family members, for which specific roles in cell-cycle progression have been established from analysis of knockout (c-jun, junB, junD) mouse embryonic fibroblasts (MEFs), the absence of cell-autonomous proliferation defects in MEFs lacking distinct Fos family members suggests a possible functional redundancy between Fos proteins in normal cell-cycle control (Brown et al, 1998). Regarding Fra-1, neither its ectopic expression (Jochum et al, 2000) nor conditional inactivation (Eferl et al, 2004) affects osteoblast proliferation, consistent with the previously reported normal proliferation of _fra-1-_null primary fibroblasts. As Fra-1 is essential for placental vascularization in mice (Schreiber et al, 2000), dispensable in most adult tissues (Eferl et al, 2004) and re-expressed in tumors, we decided to investigate the role of Fra-1 in tumor cell proliferation.
Here, we present evidence that Fra-1 is essential for the growth and survival of RAS-transformed thyroid cells. We observed Fra-1 accumulation and increased phosphorylation during the G2/M phase, followed by degradation in early G1. Fra-1 knockdown led to decreased proliferation, cell-cycle arrest in G2 and cell death associated with mitotic defects. These effects may be linked to the combined influence of cyclin E upregulation and cyclin A downregulation. Among various cell-cycle regulators affected by Fra-1 inhibition, we identify the cyclin A2 gene (ccna2) as a novel transcriptional target of Fra-1 both in vitro and in intact cells. Fra-1-dependent induction of JunB, which is also directly implicated in cyclin A transcription, suggests that Fra-1 regulates the ccna2 promoter by both direct and indirect mechanisms in RAS-transformed thyroid cells.
Results
Variations in Fra-1 levels during cell-cycle progression
To investigate a potential link between Fra-1 expression and cell-cycle progression in FRTL-5K-Ras cells, we monitored its level in cycling cells, by 2D flow cytometry of cells stained simultaneously with propidium iodide and anti-Fra-1-FITC. We observed a significant increase (about ninefold) in Fra-1-FITC fluorescence intensity in cells with a double DNA content (G2/M fraction) compared to G0/G1 cells (Figure 1A).
Figure 1.
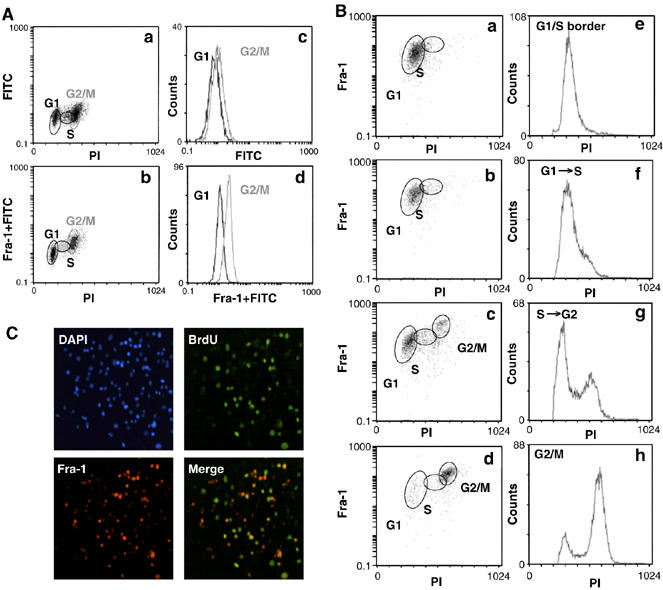
Cell-cycle-dependent regulation of Fra-1 expression in FRTL-5K-Ras cells. (A, B) 2D FACS analysis of Fra-1 level/DNA content in FRTL-5K-Ras cells. (A) Biparametric flow cytometry analysis of asynchronously growing FRTL-5K-Ras cells. (B) FRTL-5K-Ras cells were arrested by thymidine block and released for 4, 8 or 10 h, to enrich for the G1 to S, S to G2 and G2/M cell fractions. (C) Indirect IF of asynchronous FRTL-5K-Ras cells. BrdU-labeled cells were co-stained with the α-BrdU-FITC or an α-Fra-1 antibody, followed by a Texas Red-conjugated secondary antibody. Nuclei were marked by DAPI staining.
To confirm this finding, we analyzed synchronized FRTL-5K-Ras cell cultures isolated at different times following release from a thymidine block at G1/S. Gating of results from 2D-FACS analysis (Figure 1B) of cell fractions showed that, although all three populations (G1, S and G2/M) were positive for Fra-1 expression, the average signal was considerably higher in the cell subset with double (4_n_) DNA content. These results showed that Fra-1 was predominantly accumulating during the G2- and/or M-phases of the cell cycle.
We further substantiated the unequal distribution of Fra-1 expression by BrdU labeling and double immunofluorescence (IF) staining of asynchronously growing FRTL-5K-Ras cells (Figure 1C), showing that the Fra-1 signal was restricted to only a fraction of the total cell population. Merge of the BrdU- and Fra-1-specific immunostaining images revealed the lack of coincidence between the two signals; only a fraction of the Fra-1-expressing cells were BrdU positive, whereas not all BrdU-positive cells expressed Fra-1. These results suggest that the Fra-1 level was fluctuating within S-phase itself.
We further investigated cell-cycle-dependent Fra-1 expression and modification by Western blotting (WB) analysis of FRTL-5K-Ras cell extracts, at different time points following release from a double thymidine block. The G2- and M-phase-enriched cell fractions were obtained by nocodazole/thymidine-combined treatment, or alternatively, mitotic cells were isolated by mechanical shake-off from untreated cultures. Alignment between cell-cycle distribution and immunoblotting data (Figure 2A) revealed that Fra-1 concentration was very low in cells arrested at the G1/S boundary, but increased during synchronous progression toward the G2 phase. Notably, Fra-1 accumulation began in the late S-phase and peaked during the G2/M transition; Fra-1 levels increased in both the adherent G2-enriched cell population and in floating mitotic cells. In addition to the quantitative variations, we observed cell-cycle-dependent modifications of the Fra-1 protein, revealed by multiple species with different electrophoretic mobilities. The G2/M phase was characterized by slower migrating modified forms of Fra-1, whereas early-G1 cells were characterized by the predominantly faster moving isoforms. Fra-1 modifications were not caused by the drugs used for cell synchronization, as comparable Fra-1 changes were detected in mitotic cells mechanically separated from asynchronous cell cultures (Figure 2A). To test if these differences of Fra-1 electrophoretic mobility are due to phosphorylation, cell extracts were treated in vitro with alkaline phosphatase before WB. As shown in Figure 2B, the treatment resulted in a single, faster migrating form, suggesting that the cell-cycle-dependent modifications of Fra-1 are indeed due to phosphorylation (Figure 2B).
Figure 2.
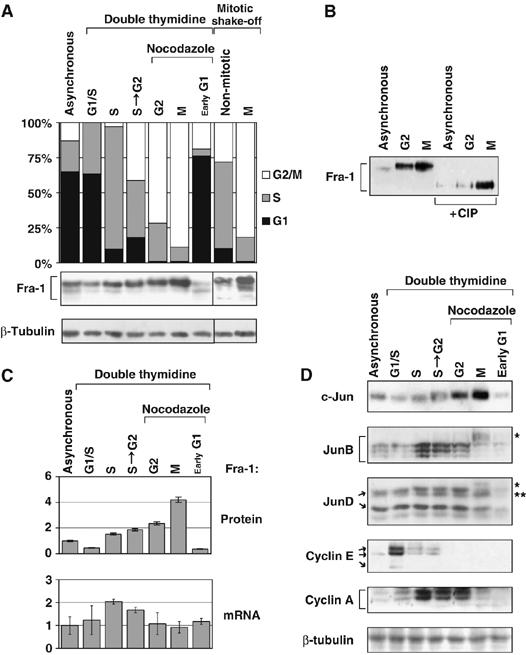
Expression of Fra-1 and Jun-family partners in synchronized FRTL-5K-Ras cells. FRTL-5K-Ras cells were released from a double-thymidine block and allowed to proceed synchronously through the S-phase (4 h) and toward the G2 phase (S to G2: 8 h). Mitotic cells (M) were mechanically separated by shake-off from nocodazole-arrested (11 h) cells, or from interphase cells in asynchronous growth. (A) Diagram: cell-cycle profile of indicated cell samples. Immunoblotting: the bracket shows the major Fra-1 isoforms, ranging between 33 and 43 kDa. (B) WB analysis of Fra-1 after in vitro dephosphorylation by ChIP. (C) Time course of Fra-1 protein and mRNA expression in synchronized cell samples. Upper diagram: Fra-1 protein accumulation, determined by densitometric scanning of (A) Lower diagram: Fra-1 transcript level, determined by RT–QPCR analysis. The diagram indicates the transcript fold change after normalization. (D) WB analysis of c-Jun, JunB and JunD, along with cyclins E and A. Brackets indicate the three major JunB isoforms (38–40 kDa). The JunD isoforms (41 and 37 kDa) and cyclin E isoforms (50 and 42 kDa) are shown by arrows. Single asterisks indicate the M-phase modified isoforms of JunB and JunD; double asterisk indicates the c-Jun protein detected by cross-reaction with the anti-JunD antibody.
Comparison between the variation of Fra-1 protein and mRNA levels revealed that the small fluctuation of the Fra-1 transcript undergoing a small (less than two-fold) variation during cell cycle did not correlate with the strong change of protein level (over ten-fold), thus suggesting that the major determinant of Fra-1 accumulation in mitotic cells occurs at the post-translational level (Figure 2C).
We also analyzed the expression of the three Jun-family partners, along with cyclins E and A, in thymidine/nocodazole-synchronized FRTL-5K-Ras cell cultures (Figure 2D). c-Jun protein levels increased concomitant with Fra-1 in the G2/M cell fraction, exhibiting maximum accumulation in mitotic cells. In contrast, the marked degradation of JunB was associated with electrophoretic shift of the protein, coinciding with the peak of Fra-1 and c-Jun accumulation in mitotic cells. JunD, in turn, exhibited modest variations during progression from the S- toward G2-phase, and displayed an electrophoretic retardation in mitotic cells, followed by downregulation in early G1. A peak of cyclin E expression coincided with the low levels of Fra-1 at the G1/S transition. Cyclin A accumulation correlated with both Fra-1 and JunB expression in cells proceeding from G1/S to G2, but the decline of cyclin A in mitotic cells coincided with both JunB downregulation and accumulation of the modified Fra-1 isoform. In summary, these results confirmed that Fra-1 was regulated during cell cycle in FRTL-5K-Ras cells; Fra-1 accumulation coincided with electrophoretic retardation of the protein in G2/M phase, followed by decrease in the early G1 phase. Whereas in G2 Fra-1 was associated with significant levels of the three Jun family partners, in mitotic cells, Fra-1 expression coincided with the peak of c-Jun and downregulation of JunB (and partially JunD). These results suggest a possible interplay between different Fra-1-containing heterodimers during cell-cycle progression. Finally, quantitative real-time PCR (RT-QPCR) analysis revealed that the increase in c-Jun, during the G2 to M transition, and the increase in JunB during S-phase correlated with increased mRNA levels, whereas the mitotic downregulation of both JunB and JunD did not reflect variations in the transcript levels (data not shown).
Inhibition of Fra-1 synthesis resulted in a proliferative block in G2-phase and increased apoptosis in FRTL-5K-Ras cells
We silenced Fra-1 expression by stable transfection of FRTL-5K-Ras cells with shRNA expression vectors targeting different portions of Fra-1 mRNA. By screening various pools, we isolated two cell clones (shFra-1/cl2 and cl7), with drastically reduced Fra-1 expression, as detected by IF (Figure 3A), WB and RT–QPCR (Figure 3B) analysis. Fra-1 was almost completely suppressed in the shFra-1/cl2 cell line, whereas the shFra-1/cl7 exhibited intermediate silencing compared to control cell lines. Variations at the protein level paralleled the variations in the fra-1 transcript (Figure 3B).
Figure 3.
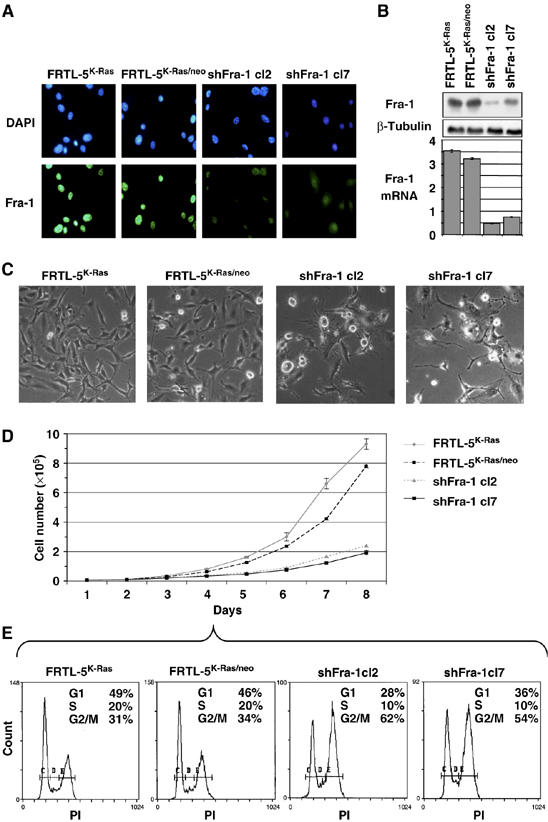
Morphology and proliferation of Fra-1 RNAi-knockdown cell clones. Cell lines expressing Fra-1-specific shRNAs were generated by stable transfection and characterization of individual cell clones. (A) Indirect IF: controls and shFra-1 cells were labeled with α-Fra-1 and FITC-coupled secondary antibodies. Nuclei were marked by DAPI staining. (B) Immunoblotting and RT–QPCR analysis of Fra-1 expression in controls and knockdown cell clones. (C) Phase-contrast images of semi-confluent cells. (D, E) Proliferation rates and cell-cycle profiles. Both for FACS analysis and cell count, cell lines were plated at the same density. (D) Cell counts were determined daily on triplicate samples and average values are shown. (E) Flow cytometry profiles of exponentially growing cell lines, FACS-analyzed at day 4, after propidium iodide labeling.
Phase-contrast microscopy showed that both knockdown cell clones exhibited a rather rounded cell shape compared to controls, with many apoptotic bodies and floating dead cells (Figure 3C).
We performed cell proliferation analysis (Figure 3D) and observed a strongly reduced proliferation rate in Fra-1-knockdown cells (doubling time 35±3 h) compared to the control or parental cells (FRTL-5K-Ras/neo and FRTL-5K-Ras; doubling time 18±3 h), suggesting that the Fra-1 expression is critical for proliferation and/or survival of RAS-transformed thyroid cells. We determined the cell-cycle distribution in the two knockdown cell clones during the exponential phase of asynchronous growth (day 4 after cell seeding; Figure 3E). Flow cytometry analysis showed an increased G2/M cell fraction, which was more evident in the clone with the lowest amount of Fra-1 (cl2), linked to a decreased G1 fraction (Figure 3E). Thus, the slower proliferation rate might be consequent to the inability to proceed through the G2/M transition in the absence of Fra-1.
The apoptotic bodies and floating dead cells (Figure 3C) in subconfluent cultures of knockdown cell clones suggested that apoptotic cell death contributes to the reduced cell count associated with decreased Fra-1. Therefore, we analyzed shFra-1/cl2 at a later stage of culture (day 7 after plating), when alterations in proliferation and cell-cycle distribution were more evident (Figure 4A). Flow cytometry showed that control cells (FRTL-5K-Ras and FRTL-5K-Ras/neo) accumulated predominantly in G0/G1 (65—70%) with only a small S-phase cell fraction (16%) at confluence. In contrast, the Fra-1-knockdown cells exhibited, in addition to the modified G2/M and G0/G1 subpopulations, a significant increase of the sub-G1 fraction (7%), likely representing a subpopulation undergoing cell death.
Figure 4.
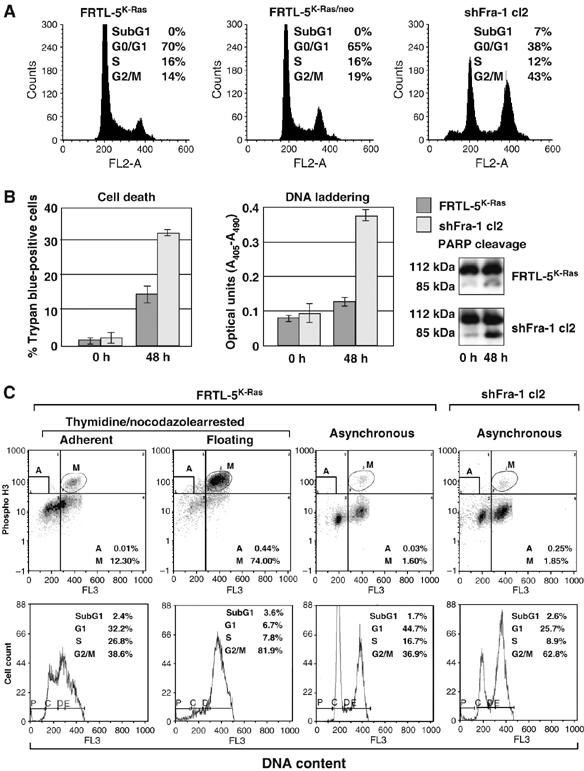
Spontaneous apoptosis and growth arrest in Fra-1-knockdown cell clones. (A) Flow cytometry (FACS) analysis of the sub-G1 fraction with <2_n_ DNA content. The same cell samples described in Figure 3D (day 7 after cell plating) were fixed and analyzed. (B) Apoptotic cell death was monitored in subconfluent cultures, at 0 versus 48 h after cell plating. Left panel: percentage of Trypan blue-positive dead cells. Center panel: DNA laddering, quantitated by ELISA detection of DNA fragmentation. Right panel: immunoblot analysis of poly-ADP-ribose-polymerase (PARP) cleavage. (C) Quantitation of mitotic cell fraction (M) by biparametric FACS analysis of phospho-histone H3-positive cells with 4_n_ DNA content. Apoptotic cells (<2_n_ DNA content) exhibiting strong phospho-histone H3 staining are boxed in (A). Lower diagrams: monoparametric flow cytometry, showing cell-cycle distribution.
To confirm the apoptotic cell death, we analyzed cytoplasmic DNA fragmentation and proteolytic cleavage of poly (ADP-ribose) polymerase (PARP), as markers of caspase cascade activation. We observed a significant increase in Trypan blue-positive cells, a fourfold increase in DNA laddering and extensive PARP cleavage in shFra-1/cl2 cells compared to parental cells (Figure 4B). Thus, the reduced expression of Fra-1 caused a proliferation defect, resulting from both cell accumulation in G2/M and caspase-dependent apoptosis.
The increased number of cells with a 4_n_ DNA content could result from a block at the G2/M checkpoint, or involve cells undergoing mitosis. To determine the proportion of cells in M phase, we performed 2D flow cytometry analysis on asynchronously growing cultures labeled with an anti-phospho-Ser-H3, to identify the subset of mitotic cells containing high amounts of Ser10-phosphorylated isoforms of histone H3 (P-H3) (Figure 4C). We gated the cell distribution data by defining the mitotic cells (M) (highly P-H3-positive cells with 4_n_ DNA content, upper right quadrant) and the fraction of apoptotic cells (A) (P-H3-positive with <2_n_ DNA content, upper left quadrant). As a positive control, we used floating FRTL-5K-Ras cells collected by mitotic shake-off of nocodazole-treated cultures, as a prominent proportion of these mitotic cells give a strong signal for the phospho-histone H3 (74%). The results showed a similar percent of P-H3-positive cells in both the parental and knockdown lines (1.6 versus 1.85%). Therefore, the percentage of mitotic cells cannot account for the increased G2/M fraction in shFra-1 (63%) compared to parental cells (37%), indicating that the major alteration in the cell-cycle profile in Fra-1-knockdown clones results from the accumulation of cells in G2 before initiation of cell division. However, we also found that about 10% of apoptotic shFra-1 cells displayed a high content of phosphorylated histone H3.
To test the possibility that defective mitosis might trigger the apoptotic program in a small fraction of cells proceeding from the G2/M boundary, we analyzed the number and morphology of mitotic shFra-1/cl2 and FRTL-5K-Ras cells by IF and DAPI staining. From statistical analysis (Figure 5A), we found a comparable fraction of mitotic cells (defined on the basis of both chromosome condensation and high P-H3 content) between FRTL-5K-Ras (7.1%) and shFra-1/cl2 (8.0%), confirming the flow cytometry results. However, the overall number of P-H3-positive cells was slightly higher in the knockdown clone (10.8%), owing to the apoptotic subpopulation of P-H3-positive cells (2.8%). Moreover, P-H3 staining revealed a significant difference between the FRTL-5K-Ras and shFra-1/cl2 cell distribution in mitotic stages. Pro/metaphases and ana/telophases represented 4.7 and 2.4%, respectively, of total P-H3-positive cells in the parental cell population, whereas we detected less anaphases (only 0.9%) in knockdown cells. In addition, mitosis appeared frequently disorganized in shFra-1/cl2 cells, as shown in representative micrographs (Figure 5B and C), where phospho-H3-associated green fluorescence was not uniformly distributed but condensed in spots in anaphases and telophases (Figure 5Bf–h). A large amount of mitotic cells contained misaligned chromosomal material, represented by DAPI-positive structures left behind at the metaphase plate (Figure 5Cd). As a consequence, lagging chromosomes (Figure 5Bh and Figure 5Ce–i) were observed during anaphase, often in combination with additional abnormalities like condensed chromatin bridges (Figure 5Cg and h), whereas nuclei with bridges of decondensed chromatin (Figure 5Cj–l) and micronuclei (Figure 5Cm–o) were frequent in interphase. The incidence of abnormalities was higher in shFra-1 cells than in control cells (chromatin bridges: 5.3 versus 0.8%; micronucleation: 9.1 versus 1.7%). To test if these chromosomal segregation defects might be linked to alterations in mitotic spindle, we analyzed the microtubule bundles and poles, by β- and γ-tubulin immunostaining, in FRTL-5K-Ras and Fra-1-knockdown cells. In interphase FRTL-5K-Ras cell nuclei, γ-tubulin staining usually revealed two centrosomes colocalizing at one pole (Figure 5Da). Most mitotic FRTL-5K-Ras cells exhibited normal, mostly biconical, spindles, with the number of centrosome-associated γ-tubulin spots equal to the number of spindle poles (Figure 5D, panels b and c). Most Fra-1-knockdown cells also had a couple of interphasic centrosomes and bipolar spindles (Figure 5Dd and e); however, γ- and β-tubulin staining revealed a high incidence of centrosome duplication (Figure 5Dg) and a more heterogeneous mitotic apparatus, often with additional centrosomes (Figure 5Dh and i). Moreover, alteration of spindle polarity was also detected with a significant incidence, as a consequence of centrosome amplification: mitotic cells with multipolar spindle originating by supernumerary centrosomes (Figure 5Df). Therefore, cells with reduced Fra-1 expression appear less efficient in assembling normal spindles and correctly segregating the bulk of DNA. To confirm that mitotic disasters were triggering apoptosis, we stained shFra-1 cells with both phospho-histone H3 and cleaved caspase-3. The nuclei were counterstained with DAPI. We found that some shFra-1 mitotic cells and post-mitotic cells with fragmented nuclei, identified by both DAPI and phosphohistone H3 staining, were also positive for caspase-3 activation, indicating the onset of apoptotic cell death.
Figure 5.
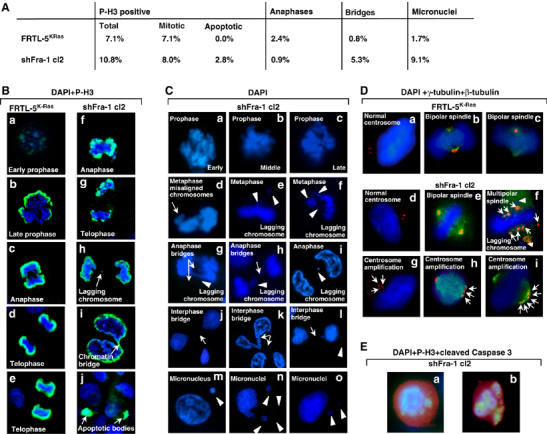
Mitotic defects in shFra-1 cells. Indirect IF analysis of mitosis in exponentially growing FRTL-5K-Ras and shFra-1/cl2 cells. (A) Statistical analysis of distribution within mitotic stages and frequency of mitotic defects in FRTL-5K-Ras and shFra-1/cl2 cells. (B–D) Cells were stained with anti-phospho-histone H3 (green) (B), anti-γ-tubulin (red) and anti-β-tubulin (green) (D) or, anti-phospho-histone H3 (green)/anti-cleaved caspase 3 (red) (E). Nuclei were counterstained with DAPI. Representative images of different mitotic stages are indicated in individual panels. (B) Arrows indicate lagging chromosome (panel h), chromatin bridge (panel i) and apoptotic bodies (panel j). (C) Morphology of DAPI-stained nuclei. Arrows indicate misaligned chromosomes (panel d), anaphase bridges (panels g and h); interphase bridges (panels h and j–l); arrowheads point to lagging chromosomes (panels e–i) and micronuclei (panels l–o). (D) γ- and β-tubulin staining of mitotic spindles. Arrows indicate supernumerary centrosomes (panels f–i); arrowheads in panel f point to lagging chromosomes. (E) Cells with PH3-stained condensed chromatin positive for activated caspase 3: cells with mitotic (a) or fragmented post-mitotic (b) nuclei.
In summary, we conclude that the increased apoptosis in the presence of decreased Fra-1 (Figure 4) results from the increased incidence of mitotic catastrophe events, likely consequent to genomic instability, suggested by the increased incidence of chromosomal abnormalities and defects in spindle formation in Fra-1-knockdown cells (Figure 5).
Fra-1 inhibition affects the expression of multiple cell-cycle regulators
To investigate the molecular basis of the cell-cycle alteration caused by Fra-1 inhibition, we analyzed several cell-cycle regulators, both in the Fra-1-knockdown clones (shFra-1/cl2 and cl7) and in a pool of non-transformed FRTL-5-derived cells ectopically expressing a Fra-1 FLAG fusion protein (FRTL-5/Fra-1FLAG; Figure 6A). The expression of cyclin D1, cyclin E and cyclin A was significantly influenced by the changes in Fra-1 levels (Figure 6B), whereas cyclins B1 and D3 were essentially unaffected (not shown). In particular, both cyclin D1 and cyclin A expression correlated positively with the Fra-1 levels, being downregulated in the silenced cell clones and upregulated in the FRTL-5/Fra-1FLAG cells. In contrast, cyclin E expression was characterized by an opposite response to Fra-1 levels. In addition, we analyzed the expression of cdk inhibitors and found that p16INK4A, p21CIP/WAF and p19ARF were essentially unaffected by Fra-1 inhibition (not shown), whereas the p27KIP level was significantly decreased by Fra-1 downregulation but unaffected by the ectopically expressed Fra-1 (Figure 6B).
Figure 6.
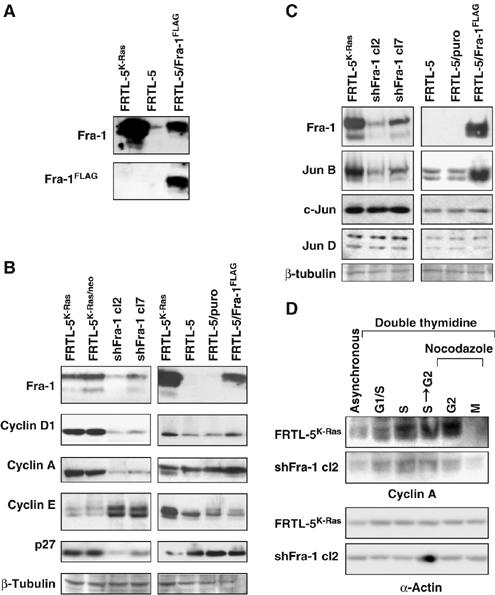
Expression of cell-cycle regulators and Jun proteins in Fra-1-knockdown and -overexpressing cell lines. Immunoblotting analysis of total extracts from exponentially growing (A–C) or synchronized (D) FRTL-5K-Ras, Fra-1-knockdown clones (cl2 and cl7) and FRTL-5/Fra−1FLAG cells. (A) Comparison between the endogenous Fra-1 and the ectopic Fra-1-FLAG expression levels. (B, C) Correlation between Fra-1 expression and levels of different cyclins (cyclin D1, A and E) and CDKI (p27Kip1) (B) and levels of c-Jun, JunB and JunD (C). (D) Immunoblotting analysis of kinetics of cyclin A accumulation in synchronously growing FRTL-5K-Ras and shFra-1/cl2 cells. Equal loading was verified by β-tubulin or α-actin detection.
To determine whether the observed modifications were due only to the decreased Fra-1, or to additional changes of AP-1 composition, we also investigated the level of Fra-1 dimerization partners, c-Jun, JunB and JunD. Immunoblotting results showed that, whereas c-Jun and JunD expression levels were unaffected, JunB levels were diminished by Fra-1 downregulation, correlating with the extent of residual Fra-1 expression in the two knockdown cell clones (Figure 6C). Fra-1 was also sufficient to trigger JunB accumulation independently from RAS expression, as shown from the analysis of FRTL-5/Fra-1FLAG cells, in which the ectopic Fra-1 specifically induced the accumulation of JunB, but not of c-Jun or JunD proteins (Figure 6C).
Finally, we confirmed the differences of expression of cell-cycle regulators in thymidine–nocodazole-synchronized cell cultures. In particular, the time-course analysis of cyclin A in synchronized Fra-1-knockdown cells (sh-Fra-1/cl2) confirmed the effect of Fra-1 downregulation and showed that the compromised cyclin A expression was more evident in the S–G2 and G2 cell fractions (Figure 6D), thus suggesting that Fra-1 might be required to sustain the expression of cyclin A in G2.
Fra-1 directly regulates cyclin A transcription and is recruited on the ccna2 promoter in a cell-cycle-dependent manner
As cyclin A inhibition is known to cause cell-cycle arrest in G2 (Pagano et al, 1992), and because of the evidence for the role of AP-1 in regulating cyclin A transcription (see Discussion), we studied the ccna2 gene (encoding the ubiquitously expressed cyclin A2 gene product) as a potential link between Fra-1 expression and cell-cycle control in FRTL-5K-Ras cells. Downregulation of ccna2 mRNA in both knockdown cell clones (more evident in shFra-1/cl2 compared to cl7; Figure 7A) suggested that cyclin A might be controlled by Fra-1 at the transcriptional level. To test the Fra-1 dependence of the ccna2 promoter, we analyzed the activity of a murine ccna2 promoter-luciferase reporter construct (Andrecht et al, 2002) in shFra-1/cl2, FRTL-5K-Ras and FRTL-5 cells. The ccna2 promoter was about fourfold less active in the cl2 cells compared to the FRTL-5K-Ras cell line (Figure 7B).
Figure 7.
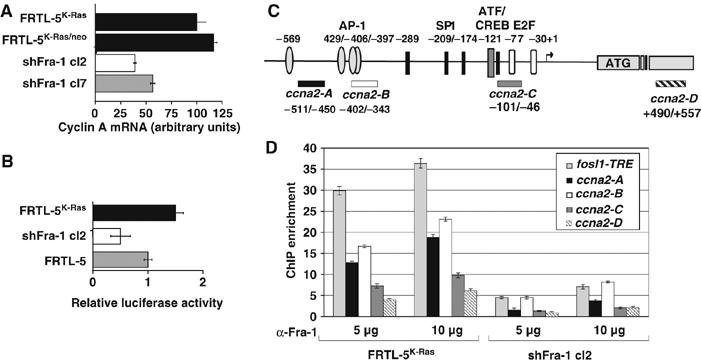
Functional and biochemical interaction of Fra-1 with the cyclin A2 gene (ccna2). (A) Cyclin A2 mRNA expression. RNA isolated from Fra-1-knockdown and control cell lines was reverse-transcribed. RT–QPCR analysis was performed in triplicate with two independent RNA preparations. (B) Activity of the (−795/+95) _ccna2_-luciferase reporter construct, transfected into Fra-1-knockdown (cl2), FRTL-5 and FRTL-5K-Ras cells. The diagram represents the results from two independent experiments on triplicate samples. (C) Scheme of the rat ccna2 promoter region. The arrow shows the transcription start site, based on the alignment to the human cyclin A promoter. The different boxes indicate the previously characterized binding sites for E2F, ATF/CREB and Sp1 transcription factors. In addition, four potential AP-1 binding sites, predicted by computer analysis (TRANSFAC), are indicated. The bars below the diagram represent the amplicons used for ChIP experiments. (D) ChIP analysis of the recruitment of Fra-1 to the ccna2 promoter in FRTL-5K-Ras versus shFra-1/cl2 cells. Immunoprecipitated chromatin was subjected to RT-QPCR with the ccna2 promoter-specific primers, along with the _fosl1_TRE primers. Fold enrichment represents the recovery of chromatin-associated DNA fragments immunoprecipitated by the Fra-1-specific antibody compared to controls. Error bars indicate s.d. between two independent experiments performed in triplicate. Different bar colors correspond to distinct amplicons, as indicated.
The effect of the Fra-1 expression levels on _ccna2_-luciferase reporter activity suggested that Fra-1 might bind directly to the ccna2 promoter. Alternatively, as JunB was reported to be a positive regulator of the ccna2 promoter, Fra-1 might affect the ccna2 transcription by an indirect mechanism, involving the Fra-1-dependent increase of the endogenous JunB (Figure 6D), which could in turn transactivate the ccna2 promoter with a different dimerization partner, such as ATF2 (Andrecht et al, 2002). To test the direct binding of Fra-1 to the ccna2 promoter in intact FRTL-5K-Ras cells, we performed chromatin immunoprecipitation (ChIP) and analyzed the products by using four primer sets encompassing distinct amplicons in the ccna2 promoter region (ccna2 A–D; Figure 7C). As a positive control, we utilized the rat fosl1 TRE fragment, containing the Fra-1 autoregulatory site of the fosl1 first intron (Casalino et al, 2003). Amplification of the _fosl1_TRE-containing fragment resulted in the highest fold-enrichment compared to the four ccna2 promoter fragments, which gave different amounts of PCR products: _ccna2_-B>_ccna2_-A>_ccna2_-C>_ccna2_-D (Figure 7D). In Fra-1-knockdown cells, the recovery was roughly fivefold lower for each fragment, but the ratio between the four amplicons was similar, confirming the specificity of the ChIP assay. In summary, our results suggest that Fra-1 regulates the cyclin A2 promoter and is directly recruited to the ccna2 upstream region in FRTL-5K-Ras cells (Figure 7B and D).
The cell-cycle-dependent expression of Fra-1 and Jun proteins in FRTL-5K-Ras cells (Figure 2) suggested that the association of Fra-1-Jun dimers with target promoters might fluctuate during cell-cycle progression. Therefore, we investigated the cell-cycle-dependent recruitment of Fra-1 to the ccna2 promoter (_ccna2_-B amplicon) in synchronized FRTL-5K-Ras cells (Figure 8A), and found that the Fra-1/ccna2 chromatin association was strongly increased in the cell population entering the G2-phase, compared to cells in G1/S and S-phase. Mitotic cells, expressing the highest Fra-1 content (Figure 2), displayed background levels of Fra-1 association with both the fosl1 and ccna2 regions, probably because of the inaccessibility of condensed mitotic chromosomes to transcription factors, and/or reduced DNA-binding activity of mitotically modified Fra-1 isoforms (Figure 8A). To investigate the role of Jun family heterodimeric partners in mediating the Fra-1 interaction with the ccna2 promoter, we performed sequential ChIP assays, using anti-Fra-1 for the first ChIP (Figure 8A) followed by a second immunoprecipitation with antibodies against individual Jun proteins (Figure 8B). These experiments revealed that the recruitment of Jun proteins to the ccna2 promoter was also cell-cycle dependent and characterized by different kinetics of chromatin association for c-Jun, JunB and JunD. At the G1/S transition, c-Jun was the major Fra-1 partner associated with the ccna2 promoter. JunD and JunB increased in cells proceeding along the S-phase, and JunB became the predominant Fra-1 partner associated with the _ccna_2 promoter in both S- and G2-phase (Figure 8B). In summary, these data show a cell-cycle-dependent interplay between distinct Jun proteins interacting with cyclin A2 upstream region (Figure 8C). JunB represents the major Jun protein recruited to the ccna2 promoter along with Fra-1, during the time period of ccna2 transcription, spanning from S- to late G2-phase. Finally, the comparison between the kinetics of cyclin A mRNA expression and Fra-1/ccna2 chromatin association during the cell cycle revealed partial overlap between the peak of Fra-1 recruitment to the ccna2 promoter in G2 and the peak of the cyclin A transcript level in the S to G2 transition, suggesting that the Fra-1-mediated control might be implicated in the late stage of cyclin A expression (Figure 8A and D).
Figure 8.
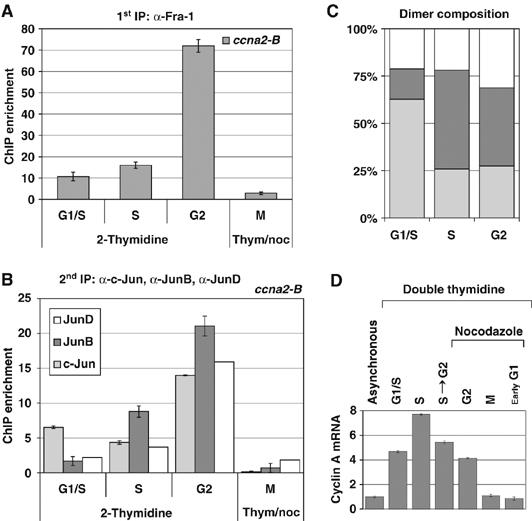
Cell-cycle-dependent recruitment of Fra-1 to the ccna2 promoter (_ccna2_-B amplicon) in synchronized FRTL-5K-Ras cells. (A–C) Sequential ChIP analysis of Fra-1 recruitment on the ccna2 promoter in synchronized FRTL-5K-Ras cells and its association with c-Jun, JunB and JunD during cell-cycle progression. Cell synchronization was performed as in Figure 2. Results are expressed as ChIP enrichment and error bars are indicated between triplicate samples. (A) First ChIP was carried out using the anti-Fra-1 antibody and RT–QPCR was performed with the _ccna2_-B primers. (B) Re-ChIP analysis: Fra-1-bound chromatin was subjected to a second round of immunoprecipitation, using anti-c-Jun, -JunB or -JunD antibodies and analyzed by RT–QPCR. (C) Relative proportion of individual Jun proteins detected in association with Fra-1 on the ccna2 promoter (B) expressed as percentage of the total amount of Fra-1-IP chromatin in synchronized cells. (D) RT–QPCR analysis of cyclinA mRNA level in synchronized FRTL-5K-Ras cells. The diagram shows the transcript fold change, after normalization with the internal control.
Fra-1-dependent transactivation and in vitro binding to the ccna2 promoter
The results of the ChIP analysis prompted us to investigate the direct role of Fra-1 in direct control of the ccna2 promoter by cotransfection analysis of a mouse _ccna2_-luciferase reporter in the FRTL-5 cell line. The results showed that the _ccna2_-luciferase reporter was transactivated by the c-Jun∼Fra-1 tethered heterodimer (Supplementary Figure S1). Comparison between two promoter deletions indicated that Fra-1-responsive element(s) were localized within an upstream region (−512 to −238) encompassing both _ccna2_-A and _ccna2_-B amplicons. This region includes four AP-1-like sites (at −569, −429, −406 and −397; Figure 7C). As the site-directed mutation of the element at −569, representing a canonical AP-1 sequence in the _ccna2_-luciferase construct, did not affect the Fra-1 responsiveness of the ccna2 promoter, we performed the in vitro binding analysis on three variant AP-1 sites (at −429, −406 and −397) within the same region (Figure 7C). Gel retardation and antibody supershift assays showed that AP-1 complexes, containing Fra-1 along with c-Jun, JunB and JunD, interacted with the three closely located variant sites and the binding activity was strongly decreased in the Fra-1-knockdown cell clones (Supplementary Figure S2). In addition, the in vitro analysis of the proximal CRE revealed that although the major complex contained ATF1 (and to a lesser extent CREB and ATF2), a minor contribution of Fra-1 could also be detected.
In summary, these data show that the Fra-1 recruitment on the ccna2 promoter (Figure 7) is mediated by Fra-1 binding to previously unidentified upstream TRE-like elements, at positions −429, −406 and −397, in addition to the previously characterized promoter-proximal ATF site. Moreover, the identification of JunB binding to both the TREs and the CRE shows that Fra-1 impacts the ccna2 promoter both by direct binding and by control of JunB expression.
Discussion
In this study, we show that Fra-1 expression is regulated during the cell cycle and is essential for the proliferation and survival of RAS-transformed thyroid cells, via mechanisms involving Fra-1-dependent regulation of the cyclin A promoter.
The Fra-1 species accumulating in the G2 and M fractions in synchronized FRTL-5K-Ras cells exhibit an electrophoretic shift, resulting from increased phosphorylation (Figure 2B). MEK-dependent phosphorylation is essential for Fra-1 stability in RAS-transformed thyroid and colon carcinoma cells, in which high levels of ERK activity protect Fra-1 from proteasome-dependent degradation (Casalino et al, 2003; Vial and Marshall, 2003). However, the slowest migrating Fra-1 isoform in the G2 and M cell fractions does not correlate with increase of active phosphorylated ERKs (data not shown), suggesting that the G2/M-specific modification might depend on a Fra-1 kinase different from ERK and active during the G2- and M-phase. Interestingly, the peak of Fra-1 accumulation coincides with the M-phase-associated degradation of JunB (Figure 2D), caused by JunB phosphorylation by the Cdc2–cyclin B complex (Bakiri et al, 2000).
The increased expression in the G2 and M fractions suggests a role for Fra-1 in the G2 to M transition. Accordingly, the Fra-1-knockdown cell clones exhibit a strong impairment of G2 progression (Figure 3E). A similar effect on cell-cycle distribution has been recently described in a human breast cancer cell line, in which the ⩾4_n_ cell fraction accumulating in the absence of Fra-1 was found to include not only G2/M cells but also G1-arrested cells with polyploid DNA content (Belguise et al, 2005).
In addition to the cell-cycle arrest in G2, we found that Fra-1 inhibition in _RAS_-transformed thyroid cells triggers apoptosis (Figure 4), associated with increased frequency of mitotic abnormalities (Figure 5). Overrepresentation of disorganized metaphases and anaphases, along with chromosomal bridges and micronuclei, suggests that loss of genetic material occurs as a consequence of mechanisms related to delayed metaphase to anaphase transition (activation of G2/M check point by misaligned or unattached chromosomes) and mitotic spindle defects. Potentially lethal alterations in gene dosage (mitotic catastrophe) might be responsible for the increased rate of apoptosis in Fra-1-knockdown cells. Interestingly, the same features of genomic instability (misaligned chromosomes, bridging and micronuclei) are induced by acute expression of RAS, via the MAPK pathway, in the PCCL3 rat thyroid cell line (Saavedra et al, 2000). We speculate that, among the targets induced by the chronic expression of RAS and implicated in the addiction to the oncogene in neoplastically transformed thyroid cell lines and tumors, Fra-1 might be required for preventing RAS-induced large-scale genomic abnormalities incompatible with tumor cell survival.
On the basis of the counteracting effects of cyclin E and cyclin A on the DNA replication machinery, we postulate that the abnormalities detected in the absence of Fra-1 are due to the unbalanced expression of cyclins E and A (Figure 6). Cyclin E–cdk2 plays a central role in licensing DNA replication origins by regulating the temporal window for the assembly of pre-replication complexes (preRC), mainly by phosphorylation-dependent stabilization of Cdc6 (Mailand and Diffley, 2005), whereas cyclin A–cdk2 is implicated in restricting the initiation of replication to only once per cell cycle, through its dual role in triggering DNA synthesis and preventing preRC assembly (Coverley et al, 2002). Therefore, we suggest that chromatin non-disjunction events are due to DNA reduplication, resulting from the combined effect of cyclin E overexpression and cyclin A downregulation, in the absence of Fra-1. In addition, the severe abnormalities in mitotic spindles observed in Fra-1-knockdown cells might be related to the essential control of centrosome duplication by cdk2–cyclin A activity (Meraldi et al, 1999).
We found that the ccna2 gene transcription is compromised in the absence of Fra-1 and that Fra-1 interacts directly with the ccna2 promoter (Figure 7), both in intact cells and in vitro, in a cell-cycle-dependent manner (Figure 8). Although we have found all three Jun proteins interact with the ccna2 promoter, the peak of chromatin-associated Fra-1 in G2 coincided with maximum recruitment of JunB and no chromatin-bound Fra-1 could be detected in mitotic cells. These results agree with previous reports describing no association between any of the Jun and Fos proteins and condensed chromatin, during mitosis (Lallemand et al, 1997).
To our knowledge, this is the first report showing the biochemical and functional interaction between endogenous Fra-1 and the cyclin A2 promoter. Several lines of evidence indicate that overexpression of Fos family members can induce cyclin A in distinct cell types. Ectopic c-Fos can accelerate cell-cycle progression and induce cyclin A synthesis in osteoblasts, but not in NIH3T3 fibroblasts (Sunters et al, 2004), whereas the c-Jun∼Fra-2 tethered heterodimer can transactivate the ccna2 promoter in NIH3T3 cells (Bakiri et al, 2002), suggesting a role for the cellular context in the regulation of cyclin A promoter by specific AP-1 heterodimers.
Our in vitro binding analysis in RAS-transformed thyroid cells showed that Fra-1 could interact with three previously uncharacterized non-consensus TREs from the ccna2 promoter. The low-affinity binding detected in vitro, in contrast with the chromatin association in intact cells, suggests the possible cooperation between the three TREs in mediating the in vivo binding of Fra-1 to the ccna2 promoter.
The ccna2 CRE site was characterized as a cell- and stimulus-specific target of multiple CREB/ATF family members, such as CREB1, ATF1, ATF2 and ATF4, in response to various extracellular signals (Yoshizumi et al, 1997; Shimizu et al, 1998; Beier et al, 2000). In addition, the analysis of serum-dependent induction of cyclin A in vascular smooth muscle cells (VSMC) showed that c-Fos binds to the ccna2 CRE (Sylvester et al, 1998). As both in VSMC and in our RAS-transformed cell line, both CREB/ATF1- and c-Fos- or Fra-1-containing complexes were found to interact with the ccna2 CRE, and given the lack of dimerization between CREB/ATF and Fos family proteins (Newman and Keating, 2003), the CREB/ATF-containing dimers and c-Fos/Fra-1-containing complexes might compete for binding to the ccna2 CRE. Here, we also show that, in addition to direct DNA binding, the effect of Fra-1 on the ccna2 CRE might be ascribed to Fra-1-dependent upregulation of JunB, previously characterized as a transactivator of the ccna2 CRE in mouse fibroblasts (Andrecht et al, 2002). A mechanism of Fra-1-dependent transcriptional induction of JunB has been suggested by the identification of junB among the genes upregulated by Fra-1 in a malignant glioma cell line, in which the ectopic Fra-1 strongly increases the tumorigenic phenotype (Debinski and Gibo, 2005).
In addition to cyclin A, we found good correlation between Fra-1 and cyclin D1 expression, in agreement with recent findings supporting the role of Fra-1 as a positive regulator of cyclin D1 transcription and G1/S transition in lung epithelial cells (Burch et al, 2004). However, the absence of a block in G1, in our Fra-1-knockdown cell lines, suggests that other cell-cycle regulators, such as cyclin D3, can compensate the decreased expression of cyclin D1, in agreement with similar observations made in Fra-1-silenced breast cancer cells (Belguise et al, 2005).
Our findings raise the question of the role of Fra-1 in cell-cycle progression in normal cells. Our results on Fra-1 silencing in RAS-transformed thyroid cells are in apparent contrast with the normal proliferation rates of Fra-1-null (_fosl1_−/−) mouse cells, including immortalized MEFs, ES cells and primary osteoblasts (Schreiber et al, 2000; Eferl et al, 2004). Accordingly, we did not detect Fra-1 on the ccna2 promoter by ChIP analysis using primary mouse osteoblasts (data not shown). Fra-1 deficiency in normal cells might not result in decreased proliferation rate, possibly because of counterbalancing alterations of cell-cycle distribution, as described for junB−/− fibroblasts (Andrecht et al, 2002). Alternatively, Fra-1 could be dispensable for asynchronous growth in normal cells, because of redundancy with other Fos proteins, similar to the compensation between c-Fos and FosB, revealed by the double knockout c-fos−/− fosB−/− MEFs exhibiting impaired proliferation and cyclin D1 expression (Brown et al, 1998). The unessential function of Fra-1 in most normal cell types, in contrast with its consolidated roles in neoplastic transformation, suggests that Fra-1 might represent an important element in the cellular addiction to oncogenic pathways. Therefore, it will be important to test the in vivo significance of our findings, by investigating the effect of RAS oncogenes in the recently described fra-1 conditional knockout mouse model (Eferl et al, 2004).
In summary, our results show that in addition to its function as a master switch of tumor cell motility and invasion, Fra-1 plays an important role in tumor cell proliferation in response to the RAS oncogene. The identification of ccna2 as a novel Fra-1 transcriptional target is relevant for oncogenesis, because of the frequent overexpression of cyclin A in tumors (Yam et al, 2002). Cyclin A is a target of adhesion-dependent signals and its adhesion-independent expression is sufficient to trigger anchorage-independent cell growth (Guadagno et al, 1993; Kang and Krauss, 1996). Because of Fra-1 involvement in anchorage-independent growth of a variety of tumor cell lines, it will be important to understand whether the ccna2 gene product plays a role in mediating anchorage independence in response to Fra-1 upregulation. As cyclin A has been recently characterized as a c-Jun target gene necessary for anchorage-independent growth in Rat1 cells (Katabami et al, 2005), cyclin A induction might represent a crucial aspect of the cooperation between Jun and Fra-1 proteins in tumorigenesis.
Materials and methods
RNAi constructs targeting Fra-1 were derived from pRNA-H1.1/Neo vector (GenScript). Antibodies were obtained from Santa Cruz, Cell Signalling and Sigma. The cyclin A-luciferase reporter and pBabe Fra-1 FLAG retroviral vectors have been described (Andrecht et al, 2002; Bakiri et al, 2002). Transfections were performed by electroporation (Gene Pulser, Bio-Rad) or lipofection (Fugene 6, Roche). Cell synchronization was obtained by double thymidine and/or nocodazole block, and flow cytometry was performed by Epics XL (Coulter) or FACScalibur (Beckman) flow cytometers. For fluorescence microscopy, cells were fixed/permeabilized in cold methanol/acetone or 1% PFA and 0.2% Triton X-100/PBS. Cell divisions were analyzed under a Zeiss Axioplan 2-imaging upright microscope equipped with the Zeiss Apo-Tome system. RT-PCR was performed by the ABI Prism 7900 Sequence Detection System (Applied Biosystems). Apoptosis was measured by photometric enzyme-immunoassay using the Cell Death Detection ELISAPLUS kit (Roche). Rat thyroid cells propagation, immunoblotting and EMSA were performed as described (Casalino et al, 2003). Detailed protocols are described in Supplementary data.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary data
Acknowledgments
We thank Marco Pontoglio, Franck Toledo and Massimo Santoro for helpful discussions and Marina Schorpp-Kistner for luciferase constructs. We thank Chaouki Miled and Andreas Reimann for assistance with ChIP experiments, Pasquale Barba for advice with FACS and Maria Terracciano for technical support. We acknowledge CNR, INSERM, EMBO, the Association for International Cancer Research (AICR), FEBS and the EU-TM Program for financial support to LC. This article is dedicated to the memory of Graziella Persico.
References
- Andersen H, Mejlvang J, Mahmood S, Gromova I, Gromov P, Lukanidin E, Kriajevska M, Mellon JK, Tulchinsky E (2005) Immediate and delayed effects of e-cadherin inhibition on gene regulation and cell motility in human epidermoid carcinoma cells. Mol Cell Biol 25: 9138–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrecht S, Kolbus A, Hartenstein B, Angel P, Schorpp-Kistner M (2002) Cell cycle promoting activity of JunB through cyclin A activation. J Biol Chem 277: 35961–35968 [DOI] [PubMed] [Google Scholar]
- Avvedimento VE, Musti AM, Ueffing M, Obici S, Gallo A, Sanchez M, DeBrasi D, Gottesman ME (1991) Reversible inhibition of a thyroid-specific trans-acting factor by Ras. Genes Dev 5: 22–28 [DOI] [PubMed] [Google Scholar]
- Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M (2000) Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J 19: 2056–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakiri L, Matsuo K, Wisniewska M, Wagner EF, Yaniv M (2002) Promoter specificity and biological activity of tethered AP-1 dimers. Mol Cell Biol 22: 4952–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier F, Taylor AC, LuValle P (2000) Activating transcription factor 2 is necessary for maximal activity and serum induction of the cyclin A promoter in chondrocytes. J Biol Chem 275: 12948–12953 [DOI] [PubMed] [Google Scholar]
- Belguise K, Kersual N, Galtier F, Chalbos D (2005) FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene 24: 1434–1444 [DOI] [PubMed] [Google Scholar]
- Brown JR, Nigh E, Lee RJ, Ye H, Thompson MA, Saudou F, Pestell RG, Greenberg ME (1998) Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol 18: 5609–5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch PM, Yuan Z, Loonen A, Heintz NH (2004) An extracellular signal-regulated kinase 1- and 2-dependent program of chromatin trafficking of c-Fos and Fra-1 is required for cyclin D1 expression during cell cycle reentry. Mol Cell Biol 24: 4696–4709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L, De Cesare D, Verde P (2003) Accumulation of Fra-1 in ras-transformed cells depends on both transcriptional autoregulation and MEK-dependent posttranslational stabilization. Mol Cell Biol 23: 4401–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coverley D, Laman H, Laskey RA (2002) Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat Cell Biol 4: 523–528 [DOI] [PubMed] [Google Scholar]
- Debinski W, Gibo DM (2005) Fos-related antigen 1 modulates malignant features of glioma cells. Mol Cancer Res 3: 237–249 [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF (2003) AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 3: 859–868 [DOI] [PubMed] [Google Scholar]
- Eferl R, Hoebertz A, Schilling AF, Rath M, Karreth F, Kenner L, Amling M, Wagner EF (2004) The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J 23: 2789–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis-Lang H, Zannini M, De Felice M, Berlingieri MT, Fusco A, Di Lauro R (1992) Multiple mechanisms of interference between transformation and differentiation in thyroid cells. Mol Cell Biol 12: 5793–5800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco A, Berlingieri MT, Di Fiore PP, Portella G, Grieco M, Vecchio G (1987) One- and two-step transformations of rat thyroid epithelial cells by retroviral oncogenes. Mol Cell Biol 7: 3365–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadagno TM, Ohtsubo M, Roberts JM, Assoian RK (1993) A link between cyclin A expression and adhesion-dependent cell cycle progression. Science 262: 1572–1575 [DOI] [PubMed] [Google Scholar]
- Hess J, Angel P, Schorpp-Kistner M (2004) AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 117: 5965–5973 [DOI] [PubMed] [Google Scholar]
- Jochum W, David JP, Elliott C, Wutz A, Plenk H Jr, Matsuo K, Wagner EF (2000) Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat Med 6: 980–984 [DOI] [PubMed] [Google Scholar]
- Kang JS, Krauss RS (1996) Ras induces anchorage-independent growth by subverting multiple adhesion-regulated cell cycle events. Mol Cell Biol 16: 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katabami M, Donninger H, Hommura F, Leaner VD, Kinoshita I, Chick JF, Birrer MJ (2005) Cyclin A is a c-Jun target gene and is necessary for c-Jun-induced anchorage-independent growth in RAT1a cells. J Biol Chem 280: 16728–16738 [DOI] [PubMed] [Google Scholar]
- Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA (2003) High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC–RAS–BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 63: 1454–1457 [PubMed] [Google Scholar]
- Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP (2001) Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev 22: 631–656 [DOI] [PubMed] [Google Scholar]
- Kustikova O, Kramerov D, Grigorian M, Berezin V, Bock E, Lukanidin E, Tulchinsky E (1998) Fra-1 induces morphological transformation and increases in vitro invasiveness and motility of epithelioid adenocarcinoma cells. Mol Cell Biol 18: 7095–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand D, Spyrou G, Yaniv M, Pfarr CM (1997) Variations in Jun and Fos protein expression and AP-1 activity in cycling, resting and stimulated fibroblasts. Oncogene 14: 819–830 [DOI] [PubMed] [Google Scholar]
- Mailand N, Diffley JF (2005) CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 122: 915–926 [DOI] [PubMed] [Google Scholar]
- Mechta F, Lallemand D, Pfarr CM, Yaniv M (1997) Transformation by ras modifies AP1 composition and activity. Oncogene 14: 837–847 [DOI] [PubMed] [Google Scholar]
- Melillo RM, Castellone MD, Guarino V, De Falco V, Cirafici AM, Salvatore G, Caiazzo F, Basolo F, Giannini R, Kruhoffer M, Orntoft T, Fusco A, Santoro M (2005) The RET/PTC–RAS–BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J Clin Invest 115: 1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA (1999) Centrosome duplication in mammalian somatic cells requires E2F and Cdk2–cyclin A. Nat Cell Biol 1: 88–93 [DOI] [PubMed] [Google Scholar]
- Milde-Langosch K (2005) The Fos family of transcription factors and their role in tumourigenesis. Eur J Cancer 41: 2449–2461 [DOI] [PubMed] [Google Scholar]
- Missero C, Pirro MT, Di Lauro R (2000) Multiple ras downstream pathways mediate functional repression of the homeobox gene product TTF-1. Mol Cell Biol 20: 2783–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JR, Keating AE (2003) Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science 300: 2097–2101 [DOI] [PubMed] [Google Scholar]
- Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G (1992) Cyclin A is required at two points in the human cell cycle. EMBO J 11: 961–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock CB, Shirasawa S, Sasazuki T, Kolch W, Dhillon AS (2005) Oncogenic K-RAS is required to maintain changes in cytoskeletal organization, adhesion, and motility in colon cancer cells. Cancer Res 65: 1244–1250 [DOI] [PubMed] [Google Scholar]
- Saavedra HI, Knauf JA, Shirokawa JM, Wang J, Ouyang B, Elisei R, Stambrook PJ, Fagin JA (2000) The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene 19: 3948–3954 [DOI] [PubMed] [Google Scholar]
- Schreiber M, Wang ZQ, Jochum W, Fetka I, Elliott C, Wagner EF (2000) Placental vascularisation requires the AP-1 component fra1. Development 127: 4937–4948 [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M (2002) AP-1 as a regulator of cell life and death. Nat Cell Biol 4: E131–E136 [DOI] [PubMed] [Google Scholar]
- Shimizu M, Nomura Y, Suzuki H, Ichikawa E, Takeuchi A, Suzuki M, Nakamura T, Nakajima T, Oda K (1998) Activation of the rat cyclin A promoter by ATF2 and Jun family members and its suppression by ATF4. Exp Cell Res 239: 93–103 [DOI] [PubMed] [Google Scholar]
- Sunters A, Thomas DP, Yeudall WA, Grigoriadis AE (2004) Accelerated cell cycle progression in osteoblasts overexpressing the c-fos proto-oncogene: induction of cyclin A and enhanced CDK2 activity. J Biol Chem 279: 9882–9891 [DOI] [PubMed] [Google Scholar]
- Sylvester AM, Chen D, Krasinski K, Andres V (1998) Role of c-fos and E2F in the induction of cyclin A transcription and vascular smooth muscle cell proliferation. J Clin Invest 101: 940–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallone D, Battista S, Pierantoni GM, Fedele M, Casalino L, Santoro M, Viglietto G, Fusco A, Verde P (1997) Neoplastic transformation of rat thyroid cells requires the junB and fra-1 gene induction which is dependent on the HMGI-C gene product. EMBO J 16: 5310–5321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial E, Marshall CJ (2003) Elevated ERK–MAP kinase activity protects the FOS family member FRA-1 against proteasomal degradation in colon carcinoma cells. J Cell Sci 116: 4957–4963 [DOI] [PubMed] [Google Scholar]
- Vial E, Sahai E, Marshall CJ (2003) ERK–MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 4: 67–79 [DOI] [PubMed] [Google Scholar]
- Yam CH, Fung TK, Poon RY (2002) Cyclin A in cell cycle control and cancer. Cell Mol Life Sci 59: 1317–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizumi M, Wang H, Hsieh CM, Sibinga NE, Perrella MA, Lee ME (1997) Down-regulation of the cyclin A promoter by transforming growth factor-beta1 is associated with a reduction in phosphorylated activating transcription factor-1 and cyclic AMP-responsive element-binding protein. J Biol Chem 272: 22259–22264 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary data