Cytokinesis in yeast meiosis depends on the regulated removal of Ssp1p from the prospore membrane (original) (raw)
Abstract
Intracellular budding is a developmentally regulated type of cell division common to many fungi and protists. In Saccaromyces cerevisiae, intracellular budding requires the de novo assembly of membranes, the prospore membranes (PSMs) and occurs during spore formation in meiosis. Ssp1p is a sporulation-specific protein that has previously been shown to localize to secretory vesicles and to recruit the leading edge protein coat (LEP coat) proteins to the opening of the PSM. Here, we show that Ssp1p is a multidomain protein with distinct domains important for PI(4,5)P2 binding, binding to secretory vesicles and inhibition of vesicle fusion, interaction with LEP coat components and that it is subject to sumoylation and degradation. We found non-essential roles for Ssp1p on the level of vesicle transport and an essential function of Ssp1p to regulate the opening of the PSM. Together, our results indicate that Ssp1p has a domain architecture that resembles to some extent the septin class of proteins, and that the regulated removal of Ssp1p from the PSM is the major step underlying cytokinesis in yeast sporulation.
Keywords: cytokinesis, intracellular budding, septins, sporulation, yeast
Introduction
In the bakers' yeast Saccharomyces cerevisiae, vegetative cell division is accompanied by formation of a bud, which is connected to the mother by the bud neck. Subsequent division processes involve polarized growth of the plasma membrane as well as sequestration of cytoplasmic contents including organelles and half of the nucleus through this connection into the daughter. A completely different morphogenetic program, called sporulation, is performed during the meiotic cell division. This time, the four meiotic progeny, the spores, are constructed entirely in the cytoplasm of the mother cell (Moens and Rapport, 1971; Peterson et al, 1972; Zickler and Olson, 1975). This type of cell division is not restricted to meiosis, but occurs in many fungal species (e.g. Ashbya gossippii and Coccidioides immitis) (Wendland and Walther, 2005) as an alternative mode of vegetative cell division, and is also common in protists (e.g_. Toxoplasma gondii_) (Shaw et al, 2000; Morrissette and Sibley, 2002). It is termed intracellular budding or endodyogeny. Interestingly, many of these species are pathogens, and in some species intracellular budding is specifically associated with their pathogenic form (e.g. Coccidioides immitis) (Miyaji et al, 1985; Nemecek et al, 2006). During intracellular budding, the nuclear divisions (one, two or several) are uncoupled from the physical cell division process and the nuclei become enwrapped at the end of their divisions by new membranous compartments, one per nucleus. This process then leads to physical separation of the new cells from the mother cytoplasm. In yeasts, these membranes are termed prospore membranes (PSMs, S. cerevisiae) or forespore membranes (Schizosaccharomyces pombe) (Shimoda, 2004; Neiman, 2005) and encompass compartments that initially resemble flattened pouches. They become assembled at the spindle pole bodies (SPBs) early in meiosis II (Okamoto and Iino, 1982; Davidow and Byers, 1984; Knop and Strasser, 2000). With progression through meiosis II, the four membranes grow out through the cytoplasm around lobes of the nucleus. Throughout this process, the SPBs are connecting the PSMs with the nuclear envelope to ensure the faithful inheritance of the genomes into the newly formed compartments. Simultaneously, cytoplasmic content such as secretory organelles, or mitochondria that associate during meiosis II with the nuclear envelope (Gorsich and Shaw, 2004), become enwrapped by the PSMs. At the end of the meiotic divisions, each of the four new nuclei is formed through fission of the nuclear envelope and subsequently fully engulfed by one PSM. The process of spore formation then proceeds by closure of the PSM. This generates two membrane bilayers on top of each other. The intervening space is then filled up with different layers of macromolecular compounds that together constitute the spore wall (Briza et al, 1988; Coluccio et al, 2004).
Two protein structures specific for PSM formation have been described (for reviews, see Moreno-Borchart and Knop, 2003; Neiman, 2005). The meiotic plaque at the cytoplasmic face of the SPB is required for initiation of membrane formation (Knop and Strasser, 2000). It substitutes the mitotic outer plaque of the SPB and consists of three essential components (Mpc54p, Mpc70p and Spo74p) and one non-essential protein (Ady4p) (Knop and Strasser, 2000; Bajgier et al, 2001; Nickas et al, 2003). In the absence of meiotic plaques, precursors of the PSM cannot be delivered to the SPBs and remain as clusters in the cytoplasm. The precursors are characterized by their content of the proteins Ssp1p/Spo3p, Ady3p and Don1p, and some of them were also found to contain membrane markers, the t-SNAREs Sso1p and Sso2p (Knop and Strasser, 2000; Moreno-Borchart et al, 2001). Another essential structure associated with PSMs is a coat that covers the leading edge of the growing membrane, termed the LEP coat. It is built of Ssp1p, Ady3p and Don1p during initiation of PSM formation (Knop and Strasser, 2000; Moreno-Borchart et al, 2001; Nickas and Neiman, 2002). Whereas Don1p and Ady3p are not essential for PSM and spore formation, deletion of Ssp1p completely abolishes the formation of spores, and no LEP coat can be found in these mutants. Initiation of PSM formation at the SPBs was unaltered, but it acquired irregular shapes and often formed tubular structures that tightly enwrapped nuclear fragments. Also, minicompartments encircled by PSM-like membranes were visible. From these results it was concluded that the LEP coat functions in maintaining the opening during PSM assembly (Moreno-Borchart et al, 2001).
It is an open question how the shape of the spores is regulated or how equal and efficient sequestration of membranous material to the four PSMs is achieved and controlled. Furthermore, the mechanism of closure of PSMs during meiotic cytokinesis is not known. These questions are particularly puzzling because actin and microtubules are not required for any of these steps (Gordon et al, 2006; Taxis et al, 2006). Thus, new mechanisms that compensate for actin-mediated polar transport can be expected.
Here, we report on the role of Ssp1p during PSM formation and meiotic cytokinesis. We found that Ssp1p plays a role early in the process, in regulation of the dynamics of precursors of the PSM. Late in the process, during cytokinesis at the end of meiosis II, active removal of Ssp1p from the PSM substitutes for the need of a contractile activity, such as an acto-myosin ring. Detailed biochemical and genetic analysis of Ssp1p revealed a certain degree of functional conservation of this protein with septins by several criteria: phosphoinositide (PIP) binding, mediated by an N-terminal cluster of basic amino-acid residues, SUMO (Smt3p) modification near the C-terminus, different protein–protein interaction domains and the overall domain architecture. Only the classical signature of septins, the GTPase domain, could not be identified. Together, our work describes how the different steps of spore plasma membrane de novo biogenesis are organized, as well as the role Ssp1p plays within these processes.
Results
Ssp1p mediates clustering of exocytic vesicles
Ssp1p is expressed exclusively during meiosis (Moreno-Borchart et al, 2001). In order to gain insight into potential activities of Ssp1p to regulate housekeeping machinery present in mitotic and meiotic cell division processes, we expressed Ssp1p in mitotic cells under the control of the strong inducible GAL1 promoter. This revealed that Ssp1p is toxic and prevents vegetative growth of the cells (Figure 1A). In these cells, Ssp1p localized to the plasma membrane with a preference to areas of membrane growth (buds of dividing cells; Figure 1B). Additionally, granulose structures near or inside the buds were visible in all cells (arrows in Figure 1B). Electron micrographs of Ssp1p-overexpressing cells revealed a 5–8-fold accumulation of secretory vesicles in the area of the emerging bud in small budded cells. Furthermore, the accumulated vesicles appeared to be smaller in size (35–60 nm compared with approximately 70 nm in the control cells) and a striking package of the vesicles into clusters was apparent (Figure 1C). In order to analyze the functioning of the secretory pathway in the Ssp1p overexpression strain, we investigated the biosynthesis of various secretory marker proteins in these cells (Avaro et al, 2002). This revealed no specific defects on the transport of these proteins through the secretory pathway (ER to late Golgi and vacuolar sorting, data not shown). This result and the accumulation of exocytotic vesicles in the bud therefore points to a defect late in secretion. To address this further, we analyzed the dynamics of vesicle delivery and fusion with the plasma membrane using a Sec2p-GFP fusion. Sec2p is a guanidine exchange factor for the Rab-like protein Sec4p that localizes predominantly to secretory vesicles at the bud tip or at the bud neck (Ortiz et al, 2002). In control cells Sec2p-GFP was hardly visible on mobile vesicles moving towards the bud tip or bud neck. In Ssp1p-overexpressing cells, however, bright and mobile Sec2p-GFP structures were seen that moved toward the bud. Figure 1D shows some frames derived from movies made from control and Ssp1p-overexpressing cells (movie showing more cells provided as Supplementary Movies S1 and S2). The increased brightness of the vesicles further suggests that not single but clusters of mobile structures are transported in the Ssp1p overexpression strain. This is consistent with the observation of clustered vesicles by electron microscopy (EM) (Figure 1C) and may point to a function of Ssp1p in mediating the formation of vesicle clusters.
Figure 1.
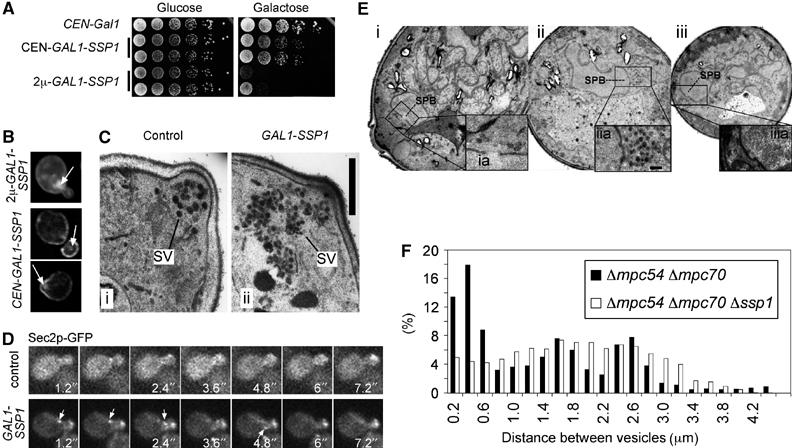
Ssp1p promotes secretory vesicle cluster formation in mitosis and meiosis. (A) Overexpression of Ssp1p is toxic for vegetative cell growth. SSP1 was expressed under the control of the GAL1 promoter from a low-copy (CEN, pKS89) or high-copy plasmid (2μ, pKS116) in cells of strain ESM356-1. Serial dilutions of cells containing the indicated plasmids were spotted on either glucose- or galactose/raffinose-containing plates and photographs were taken following incubation at 30°C after 2 days (glucose) or 3 days (galactose). (B) Localization of Ssp1p upon medium (CEN) or strong (2μ) overexpression following 3 h of induction of the GAL1 promoter using immunofluorescence microscopy (strains of (A)). Arrows point to Ssp1p-stained aggregates in the areas of the bud/budneck. (C) Visualization of secretory vesicles in wild-type cells and cells expressing GAL1_-SSP1 from a chromosomal location. Cells of strains ESM356-1 (i) and YKS207-14 (ii; strain ESM356-1 containing several copies of GAL1-SSP1 integrated in the URA3 locus) were grown in the presence of galactose for 3 h and processed for electron microscopy. SV, secretory vesicles. Bar, 400 nm. (D) Dynamics of secretory vesicles in small budded cells visualized using a Sec2p-GFP fusion (strain YMF178). Frames from a control cell (plasmid without SSP1) and a cell expressing SSP1 from the GAL1 promoter (plasmid pKS89) are shown. Arrows indicate a Sec2p-GFP containing cluster that moves toward the bud (see Supplementary Movies S1 and S2). (E) Analysis of 65–70 nm vesicle distributions in cells in meiosis II. OsO4-fixed and EPON-embedded cells of a Δ_mpc54 Δ_mpc70_ strain (YKS65; i) or a Δ_mpc54_ Δ_mpc70_ Δ_ssp1_ strain (YKS135; ii and iii) were used for this experiment. Magnifications from areas around the SPBs are shown. (F) Histogram of all distances between observed vesicles in cells (_n_=8) of each of the two strains. Coordinates of secretory vesicles were recorded manually using Metamorph™ software.
When we overexpressed Ssp1p in vegetative cells, we found that low-level expression from a low-copy number plasmid using a weakened GAL1 promoter (GALS) had no effect in wild-type cells, but it was lethal in cells that lack either one of the two t-SNARE genes (SSO1 and SSO2) (Supplementary Figure S1). These proteins are required for vesicle fusion with the plasma membrane. We furthermore noticed that this weak overexpression of Ssp1p led to a significant reduction of the restrictive temperature of temperature-sensitive sec4 and sec2 mutants (data not shown), another two proteins that function also during vesicle fusion at the plasma membrane. This indicates that Ssp1p has a dominant-negative function on the level of the core machinery that encompasses vesicle fusion with the plasma membrane.
To investigate the vesicle clustering function of Ssp1p in meiosis, we compared the distribution of secretory vesicles (65–70 nm) in the Δ_mpc54_ Δ_mpc70_ mutant (where PSM assembly is blocked and precursor structures are accumulated in the cells; Knop and Strasser, 2000) with the situation in the Δ_ssp1_ Δ_mpc54_ Δ_mpc70_ mutant. We used EM (Figure 1E) and recorded the position of visible 65–70 nm vesicles from cells as depicted in the figure. The histograms of the distances between the vesicles revealed that the additional deletion of SSP1 in the Δ_mpc54_ Δ_mpc70_ mutant leads to a more uniform scattering of vesicles throughout the cells, whereas in the Δ_mpc54_ Δ_mpc70_ mutant, an increased frequency of short distances between the vesicles is apparent (Figure 1F). This result fits well with our previous report that meiotic Δ_ssp1_ Δ_mpc54_ Δ_mpc70_ cells showed an even distribution of the t-SNARES Sso1p and Sso2p throughout the cytoplasm instead of the dot-like staining pattern typical for precursor membranes in the Δ_mpc54_ Δ_mpc70_ strain (Moreno-Borchart et al, 2001).
Together, these findings suggest that Ssp1p has a function in clustering vesicles and that it is able to block specifically the fusion of vesicles with the plasma membrane.
Ssp1p binds to PI(4,5)P2 at the plasma membrane
The observed localization of Ssp1p to membranes (Figure 1) indicates that Ssp1p interacts with lipids or specifically localized proteins, or both. To address this further, we tested the binding of Ssp1p to serial dilutions of all biologically relevant PIP species, as well as other lipids, spotted on membranes using a previously described overlay assay (Kanai et al, 2001). We found that the N-terminal fragment of Ssp1p (amino acids (aa) 1–269) as well as the full-length protein (not shown) did bind to PIPs (Figure 2A), but not to other phospholipids (data not shown). Ssp1p showed highest affinity to PI(4,5)P2.
Figure 2.
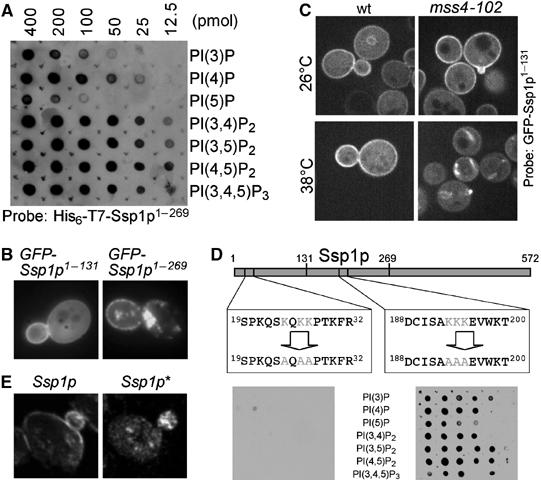
Protein–protein and protein–lipid interactions of Ssp1p. (A) Purified N-terminally 6HisT7-tagged Ssp1p (aa 1–269) was used to probe a nitrocellulose membrane containing spots of serial dilutions of the indicated lipids. Binding of Ssp1p was detected using a specific antibody that recognizes the T7 tag (Novagen). No signal was detected using an unrelated 6HisT7-tagged protein (data not shown). (B) Localization of GFP-Ssp1p1–131 and GFP-Ssp1p1–269 (in strain ESM356). (C) Localization of GFP-Ssp1p1–131 in WT (strain SEY6210) and the mss4-102 mutant (strain AAY202) at 26°C and following a shift to 38°C for 20 min. The pictures show sections acquired from the center of the cells using a spinning disc confocal microscope (Perkin-Elmer). (D) PIP binding of Ssp1p depends on a cluster of positively charged residues close to the N-terminus of the protein and not on the net charge of the protein. Experimental setup as for (A), but using equal amounts of a purified Ssp1p or Ssp1p* fragment governing aa 1–269 as a bait. (E) Plasma membrane binding but not binding to the structures inside or close to the bud is dependent on PIP binding of Ssp1p. Localization of full-length Ssp1p and the PIP binding-deficient mutant Ssp1p* following overexpression from a CEN-GAL1 plasmid in vegetative cells (of strain ESM356). Immunofluorescence microscopy was performed and pictures were taken using a confocal microscope.
Overexpression of GFP-tagged Ssp1p fragments revealed that the first 131 aa are sufficient to localize to the plasma membrane (Figure 2B). In contrast to the full-length protein and the fragment spanning aa 1–269 this construct failed to localize to vesicles inside the bud and it no longer enriched in the area of the bud (Figure 2B). This indicates that different domains of the proteins mediate plasma membrane binding and binding to vesicles and that the PI(4,5)P2 binding domain resides in the N-terminal 131 aa.
In order to investigate whether plasma membrane binding of GFP-Ssp1p1–131 depends on PI(4,5)P2 in vivo, we used a temperature-sensitive mss4-102 mutant, which is conditionally defective in the only PI4P-5-kinase (Audhya and Emr, 2002; Stefan et al, 2002). In this mutant, GFP-Ssp1p1–131 disappeared from the plasma membrane and localized to cytoplasmic structures within 10–20 min after a shift to the restrictive temperature (Figure 2C). A similar result was previously also obtained for the PI(4,5)P2-specific PH domain of PLCδ (Stefan et al, 2002).
Direct binding of proteins to PIPs usually involves clusters of negatively charged amino-acid residues. We inspected the amino-acid sequence of the N-terminal domain (aa 1–131) of Ssp1p and found one such cluster. To test the involvement of these residues (K24, K26 and K27) in PIP binding, we substituted them with alanine. This mutant of Ssp1p (called Ssp1p*) was no longer able to bind to PIPs using the spot blot method. In contrast, a mutant with three Lys → Ala substitutions in the central domain of the protein exhibited unchanged lipid-binding properties (Figure 2D). This result clearly demonstrates that the N-terminal basic cluster of residues is mediating PIP binding and not the net charge of the protein (pI=5.6).
In order to address the function of PI(4,5)P2 binding of Ssp1p in vivo, we tested whether overexpression of Ssp1p* is still toxic for the cells. This was still the case (data not shown), however, the cellular localization of Ssp1* was changed compared with the WT protein. The protein was no longer able to bind to the plasma membrane, whereas it still stained the structures present in the bud of dividing cells (Figure 2E). This confirms the previous notion that a separate function of Ssp1p mediates vesicle localization, independent of the PIP-binding capability of the N-terminal domain.
In order to get further insight into the domain architecture of Ssp1p, we investigated the subclones of Ssp1p for toxic effects upon mitotic overexpression and two-hybrid interaction with its known binding partner Ady3p (Moreno-Borchart et al, 2001) (Figure 3). This analysis revealed that the N-terminal half (aa 1–269) of Ssp1p mediates self-interaction, whereas the C-terminus (aa 217–572) binds to Ady3p. Toxicity of Ssp1p requires aa 1–160. Together, our results demonstrate that distinct domains within Ssp1p mediate membrane binding, toxicity and protein–protein interaction.
Figure 3.
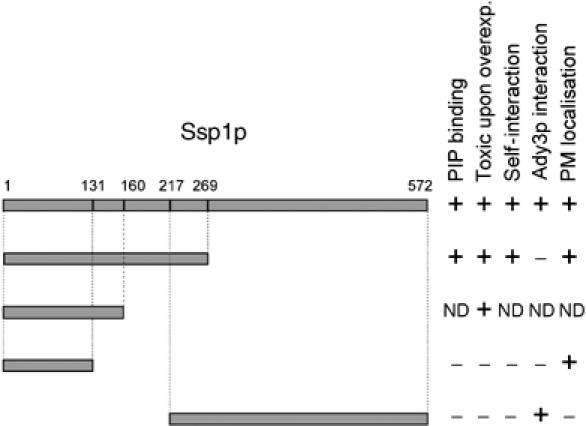
Subcloning of the different domains of Ssp1p. PIP binding was assayed using purified proteins and the blot technique of Figure 2A. Toxic growth effects were assayed as described in the legend to Figure 1A (using 2μ-GAL1 plasmids). Self interaction and interaction with Ady3p were determined using the two-hybrid system. Localization to the plasma membrane was performed using live cell imaging of GFP fusions and untagged constructs and immunofluorescence microscopy for all subcloned fragments of Ssp1p with two methods (GFP fusions in living cells (Figure 2B) and immunofluorescence microscopy (Figure 2E)) (ND, not determined).
PI(4,5)P2 binding of Ssp1p influences the movement of precursor structures
Previously, it has been shown that PI(4,5)P2 is present at the plasma membrane of sporulating cells and at the membranes of the spores, as soon as they become visible (Nakanishi et al, 2004; Rudge et al, 2004). Using the GFP-PLCδPH fusion we confirmed, that in fact PI(4,5)P2 can be found at the plasma membrane also during earlier stages of sporulation, before the PSMs become assembled (during late stages of meiosis I) (data not shown). At this stage, Ssp1p localizes to 10–30 punctuate structures inside the cells, which are the precursors of the PSMs (Moreno-Borchart et al, 2001). We recently investigated in detail the assembly of PSMs from these precursor structures and found that actin-dependent as well as Brownian movements of precursors occur, and that actin-dependent transport is restricted to areas underneath the plasma membrane of the cell (Taxis et al, 2006). It could therefore be that PI(4,5)P2 binding of Ssp1p mediates interaction of precursors with the plasma membrane and thereby influences the movements of precursors. To test this idea, we used time-lapse microscopy. We addressed whether precursor movements are changed in the strain that expresses Ssp1p* as compared with WT. To follow precursor movements, we used Don1p-GFP as a specific marker (Knop and Strasser, 2000). Don1p colocalizes with Ssp1p to precursors and the LEP coat of the PSMs (Moreno-Borchart et al, 2001). With frame rates of 1 frame/∼4 s and projections of the entire cells, we found that the precursors of the Ssp1p*-expressing strain exhibit ∼10% faster movements as compared with WT (Figure 4A). This difference, although not large, is significant because the analysis is based on more than 10 000 single measurements per strain (using automated object tracking) and in three independent measurements (P<0.001 using _t_-test analysis). In order to have an internal control, we additionally measured the movements of Don1p-GFP precursors at the SPBs in cells in early phases of meiosis II in the same movies (as an internal control). The SPBs can easily be recognized by their brighter decoration with Don1p-GFP (Knop and Strasser, 2000) and their pairwise movements, which is caused by the short metaphase spindles that connect them (Taxis et al, 2006). This revealed that the observed movements of the SPBs in the WT and the Ssp1p* strains were exactly the same (0.4% difference). Using high frame rates (1 frame/∼0.2 s) and single plane live cell recording, we noticed a 22% faster movement of the Ssp1p* precursors as compared with the precursors in the Ssp1p strain (Figure 4A). In this case, we could not identify SPBs (due to missing spatial information).
Figure 4.
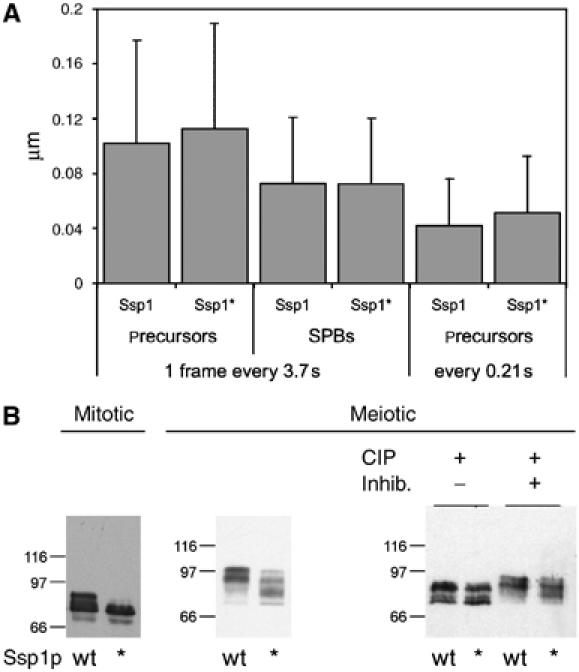
PIP binding of Ssp1p is required for fast precursor movements and phosphorylation of Ssp1p. (A) Altered dynamics of precursor structures (visualized using Don1p-GFP) in meiotic cells expressing Ssp1p* as compared with wild type Ssp1p. Movies were recorded following 5–6 h after induction of sporulation. Don1p-GFP movies were analyzed using the automated object tracking function of Metamorph™. Movements of about 200–300 individual precursor structures per strain were recorded over 50 frames. Two clones per strain were analyzed. One movement corresponds to the movement of a precursor structure from one frame to the next in the movie. Whole-cell projection (3.77 s/frame) and single-section (0.21 s/frame) recordings were used. The dynamics of LEP coats was analyzed in whole-cell projections (_n_=30 cells per strain) (strains: YKS65 containing pRS41H-SSP1 (pMM80) or SSP1*). Bars indicate standard deviations of the mean velocities. (B) Electrophoretic mobility of Ssp1p and Ssp1p* in extracts of mitotic and meiotic cells (mitotic cells: strain ESM356 containing p416-_GAL1_-SSP1 (pKS89) or SSP1*; meiotic cells: strains of (A)). For meiotic cells, alkaline phosphatase (CIP)-treated extracts without and with inhibitors (50 mM 3-glycero phosphate, 50 mM NaF) were analyzed as well.
Together, these results indicate that the PIP binding of Ssp1p reduces the movements of precursor structures. The observation of larger differences in faster movies is consistent with the idea that the movements are due to Brownian motion. Therefore, the difference between the movement of Ssp1p and Ssp1p* precursors may best be explained by weak interactions of Ssp1p with the plasma membrane, which inhibits Brownian movements.
Next, we performed immunoblotting with meiotic cell extracts from the cells used for the analysis shown in Figure 4A and also from vegetative cells with overexpressed Ssp1p and Ssp1p* proteins. As can be seen in Figure 4B, protein levels for Ssp1p and Ssp1p* were comparable; however, in both the mitotic and the meiotic cells, WT Ssp1p showed an additional band with reduced mobility on the gel. Also, Ssp1p and Ssp1p* from meiotic cells showed multiple bands with reduced electrophoretic mobility compared with the mitotically expressed Ssp1p species. To test whether these different mobilities were due to phosphorylation, we treated meiotic extracts with alkaline phosphatase. In mitotic extracts, this shifted the bands of Ssp1p to the same position as the bands of Ssp1p*; however, in the meiotic extracts, both proteins still showed different bands (Figure 4B). This suggests that impaired PI(4,5)P2 binding concomitantly leads to reduced phosphorylation of Ssp1p. Additionally, this experiment revealed the presence of another modification of Ssp1p.
Ssp1p is sumoylated
In order to address the nature of the Ssp1p modification, we tested whether meiotically expressed Ssp1p as well as ectopically expressed Ssp1p from vegetative cells is modified by the small ubiquitin-like protein SUMO (Smt3p in yeast; Johnson and Blobel, 1999). As shown in Figure 5A, Ssp1p from meiotic cells was indeed sumoylated and migrated approximately 20 kDa above the unmodified version. In vegetative cells, only very little sumoylation was visible, suggesting meiosis-specific regulation of Ssp1p sumoylation.
Figure 5.
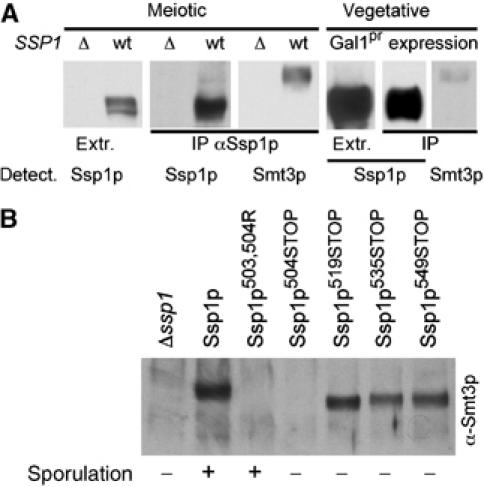
Ssp1p is modified by Smt3p/SUMO. (A) Smt3p/SUMO modification of Ssp1p in meiotic and vegetative cells (upon CEN-GAL1 expression). Immunoprecipitated Ssp1p (using anti-Ssp1p antibodies) was analyzed for Smt3p modification using specific antibodies (as indicated). Extr., crude cell extract. The bands shown in this figure are taken from the blot shown in Supplementary Figure S2B.) (B) Sumoylation of immunoprecipitated Ssp1p mutant proteins. Extracts of cells mostly in meiosis II (6 h after induction of sporulation) were used. Mutations are as indicated. For all strains equal amounts of Ssp1p were present in the immunoprecipitates (not shown). The ability of the different mutants to promote spore formation is qualitatively indicated below the blot. ‘–' indicates a sporulation efficiency <1%, ‘+' >20% (precise values are given for some strains in Figure 7A).
We also tested whether the other components of the LEP coat and of PSMs were sumoylated. For Ady3p and Don1p, we could not detect any Smt3p modification (data not shown). The septin Cdc3p, but not the meiosis-specific septin Spr3p (Fares et al, 1996), was sumoylated in meiotic cells, as reported for Cdc3p for mitotic cells (Johnson and Blobel, 1999) (Supplementary Figure S2A).
To investigate the requirement of assembled PSMs for the sumoylation of septins and Ssp1p, we used several different mutants defective for different steps of PSM biogenesis (Δ_mpc54_, Δ_spo14_, Δ_mso1_ and Δ_sma1_; Knop and Strasser, 2000; Rudge et al, 2004; Knop et al, 2005; Riedel et al, 2005). The pattern and abundance of sumoylated Spr3p, Cdc3p and Ssp1p was similar in all mutants (Supplementary Figures S2A and S2B). This suggests that Smt3p modification of the septins and Ssp1p is independent of PSM assembly.
Next we generated truncations of Ssp1p, starting with deletions from the C-terminus, in order to identify the lysine residues that are used for the covalent attachment of Smt3p. As shown in Figure 5B, deletion of 67 aa (Ssp1p504STOP) but not 52 aa (Ssp1p519STOP) from the C-terminus prevented Ssp1p from Smt3p modification. Using site-directed mutagenesis, we found that Lys503 and/or Lys504 were required for Smt3p modification (Figure 5B). These results indicate that specific residue(s) were used for the attachment of Smt3p.
When analyzing the functionality of C-terminal truncations of Ssp1p, we found that all truncations we constructed were no longer able to support spore formation (Figure 5B). Because some of these truncations were still modified by Smt3p and the Smt3p modification-impaired Ssp1p503R, 504A mutant was able to support spore formation (Figure 5B), the C-terminus of Ssp1p must exhibit an Smt3p-independent function, which is essential for the functioning of the protein.
Removal of Ssp1p is required for closure of the PSM
We analyzed protein levels of the C-terminally truncated variants of Ssp1p in meiotic time course experiments. As shown in Figure 6A, wild type Ssp1p is only transiently detected in sporulating cells approximately 6, 5 and 8 h after induction of sporulation. These time points correspond to the stages where most cells are undergoing meiosis II and PSM assembly. In contrast, the C-terminal truncations of Ssp1p exhibited higher stability, and the stability increased with increasing length of the truncations. The Smt3p modification-impaired Ssp1p mutant (K503, 504R) behaved as the wild type protein (data not shown), suggesting that the C-terminus of Ssp1p but not Smt3p modification is required for regulation of protein stability after progression through meiosis II.
Figure 6.
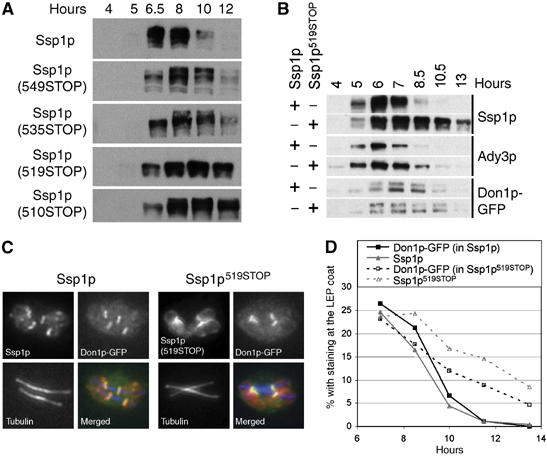
C-terminal truncation of Ssp1p (Ssp1p-ΔC) leads to impaired removal and the degradation of Ssp1p from the PSM. (A) Sporulation time-course experiments with strains expressing the C-terminally truncated Ssp1p variants (strain YKS95 with plasmid pMM80 (SSP1) or a derived plasmid containing the indicated mutation in SSP1). Cells from aliquots of the culture were analyzed. (B) Western blot analysis of Ady3p, tubulin, Ssp1p and Ssp1p519STOP in aliquots of cells from a time-course experiment (strains as in (A)). (C) Immunofluorescence localization of Ssp1p (length: 572 aa) and Ssp1p (519STOP) at the LEP coats. Ssp1p, Don1p-GFP (as a marker for the LEP coat) and tubulin were detected using specific antibodies as indicated (strains as in (A)). (D) Persistence of Ssp1p and Don1p-GFP at the LEP coats in synchronously sporulating cells expressing Ssp1p or Ssp1p519STOP. Hours: time after induction of sporulation. Aliquots of the cells were processed for immunofluorescence microscopy and the fraction of cells containing Ssp1p- and Don1p-labeled LEP coats was determined at each time point (_n_>200 cells/time point) (strains as in (A)).
We also analyzed protein levels of the other components of the LEP coat during sporulation in wild type cells and the _SSP1_519STOP mutant. Ady3p, that interacts with Ssp1p and requires Ssp1p for binding to the PSM (Moreno-Borchart et al, 2001), exhibited higher stability, although not as pronounced as the truncated Ssp1p. In contrast to this, Don1p, which interacts with Ady3p, was degraded with similar rates as in the wild type strain (Figure 6B). Using immunofluorescence microscopy, we found that the truncated Ssp1p exhibited similar localization to the LEP coat as the wild type Ssp1p protein (Figure 6C). Using immunofluorescence microscopy performed with cells from different time points during sporulation, we then studied the kinetics of LEP coat disassembly in the mutant and wild type strains. This revealed that the mutant exhibited a delay in disassembly of the LEP coats, as shown by anti-Ssp1p or anti-Don1p staining (Figure 6D). By analyzing spindle staining, we could not obtain any indication for a delay in cell-cycle progression in the Ssp1p mutant (data not shown).
Together, these data indicate that the C-terminus of Ssp1p mediates removal and/or degradation of Ssp1p and the entire LEP coat. This correlates with the ability of sporulating cells to undergo cytokinesis (closure of the PSMs) at the end of the meiotic divisions.
The enhanced stability of the LEP coat in the truncated Ssp1p mutants may be the only reason for the defect in cytokinesis in the C-terminally truncated Ssp1p mutants. Alternatively, Ssp1p could also exhibit regulatory function(s) necessary for cytokinesis that require the C-terminus of the protein. To test these possibilities, we made use of the temperature-sensitive ssp1 mutant (spo3-1) that was originally identified in a screen for mutants defective in meiosis and sporulation (Esposito and Esposito, 1969; Moens et al, 1974). We sequenced the spo3-1 allele of SSP1 and found that it contained a single point mutation that leads to a non-conservative amino-acid exchange (L225S). We then combined this mutation with a C-terminal truncation of Ssp1p (535STOP) and addressed the ability of these cells to form spores. We found that the additional L225S mutation restored spore formation of Ssp1p (535STOP) (Figure 7A). This is consistent with the idea that the L225S mutation destabilizes the Ssp1p protein. As a consequence, this either leads to a weakened LEP coat or an increased protein degradation rate of Ssp1p, which may compensate the regulated removal of the LEP coat and thereby facilitate the closure of the PSM in the C-terminally truncated Ssp1p mutant. Therefore, we tested whether the L225S mutation reverts the degradation rate of the C-terminally truncated Ssp1p mutant back to wild type of Ssp1p. As this was not the case (Figure 7B), it is suggested that removal of Ssp1p from the PSM and degradation of Ssp1p are not necessarily coupled processes, and that the impaired spore formation of C-terminally truncated Ssp1p mutants is not due to a prolonged presence of the molecule inside the cells. We also found that Don1p-GFP in spores was present in plasma membrane-associated aggregates in the Ssp1pL225S, 535STOP mutant, whereas it was diffusely localized to the cytoplasm in the WT and Ssp1pL225S (Supplementary Figure S3). This scaffolding and recruitment of Don1p-GFP indicates the continuous presence of Ssp1pL225S, 535STOP inside and not outside the spores. This suggests that Ssp1p degradation is mostly occurring after closure of the PSM inside the immature spores.
Figure 7.
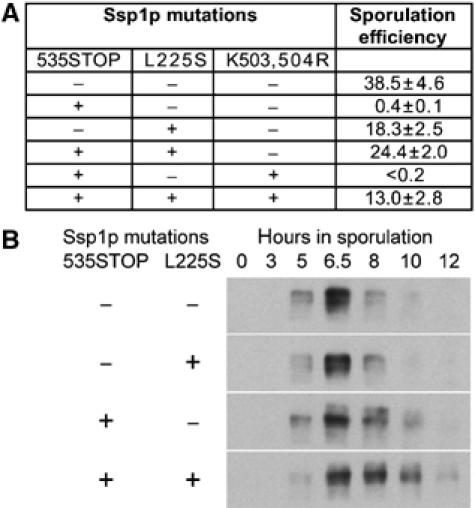
A destabilizing mutation of Ssp1p rescues the Ssp1p-ΔC defect in PSM closure. (A) Table showing the sporulation efficiency of various mutations in Ssp1p, alone or in combination (as indicated). A sporulation efficiency of 100% corresponds to a situation where all cells have formed four spores. Values were averaged from three independent experiments (strains as in Figure 6A). (B) Protein levels and stability of Ssp1p and the indicated mutants of Ssp1p during meiotic time-course experiments. Synchronized cultures of sporulating yeast cells were used and the level of Ssp1p was analyzed in aliquots of cultures using Western blotting. Ssp1p and its mutant alleles were expressed from low copy number (CEN) plasmids (pMM80 and derived plasmids) in a Δ_ssp1_ strain (YKS95).
Together, these results indicate that removal of Ssp1p from the LEP coat during cytokinesis in meiosis is a key step necessary for the closure of the PSM and the faithful formation of spores.
Discussion
We have shown that Ssp1p has different domains with different functions during PSM assembly. The N-terminal domain mediates binding of the protein to PI(4,5)P2, which is required for phosphorylation of the protein and has an influence on the movement of precursors of the PSM. Ssp1p is able to mediate vesicle cluster formation and exhibits activity that can abolish fusion of exocytic vesicles with the plasma membrane, when ectopically expressed in vegetative cells. We found that Ssp1p is modified by Smt3p/SUMO and that the C-terminus of Ssp1p is required for its removal from the PSM at a late stage of sporulation.
In a previous work, we showed that Ssp1p is required for formation and recruitment of proteins to the leading edge protein coat of the PSM, and that in the absence of Ssp1p, PSMs close in an uncontrolled and untimely manner (Moreno-Borchart et al, 2001). Our data are thus consistent with the idea that controlled removal of Ssp1p is required to promote closure of the PSMs during meiotic cytokinesis. Figure 8 summarizes our findings.
Figure 8.
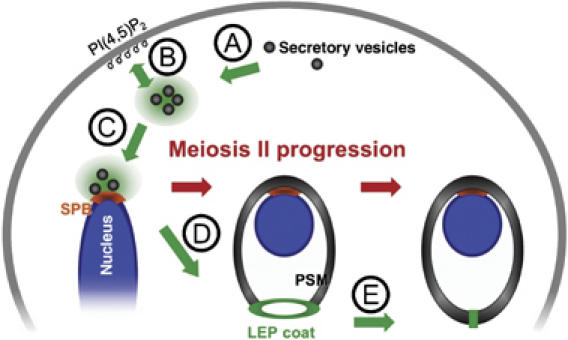
Model for the different functions of Ssp1p during sporulation. The cartoon illustrates the different steps of prospore membrane (PSM) biogenesis and the functions of Ssp1p within. The localization of Ssp1p is indicated in green. Steps during meiosis I until early in meiosis II: (A) Ssp1p appears to cluster secretory vesicles and (B) mediate their interactions with the plasma membrane in meiotic cells. (C) Ssp1p further mediates interaction of vesicles with the spindle pole body (SPB), most likely through the interaction of Ssp1p with Ady3p (Moreno-Borchart et al, 2001). Steps during meiosis II: (D) Vesicles at the SPB fuse and form a new compartment, the PSM. Ssp1p forms a coat at the opening of the PSM, together with Ady3p and Don1p, as soon as the PSM grows outwards beyond the region of the SPB (Moreno-Borchart et al, 2001). (E) Regulated closure of the PSM during cytokinesis requires the regulated removal of Ssp1p from the PSM. Associated with this process, shrinkage of the leading edge protein coat of the PSM (LEP coat) can be observed (Taxis et al, 2006). Ssp1p becomes degraded during or after the removal from the PSM. During growth, the PSM is anchored to the nuclear envelope via the SPB and each PSM grows around a nucleus. Upon cytokinesis, the PSM closes up. This gives rise to immature spores (prospores).
Role of Ssp1p during regulation of vesicle transport
One key difference between vegetative growth of a bud and PSM biogenesis during sporulation is that polarized delivery of membranes to sites of membrane growth is necessary only in the first case, because depolymerization of actin inhibits budding, but not formation, shaping or closure of the PSM during sporulation (Taxis et al, 2006). In meiosis, spores are formed within the lumen of the mother cell and sequestration and distribution of membranes to the four daughters are controlled by unknown mechanisms. Here, we report that Ssp1p can promote vesicle cluster formation. Previously, we reported that it interacts with Ady3p, a protein that can bind to the meiotic SPB where PSM assembly takes place via fusion of vesicles of homogenous size (Moreno-Borchart et al, 2001; Knop et al, 2005; Riedel et al, 2005). This puts Ssp1p in a position where it could act as a key regulator for control of membrane formation. Deletion of SSP1, however, does not abolish membrane formation per se, indicating that Ssp1p might function more as an inhibitor of untimely membrane fusion events, a hypothesis that is consistent with the inhibition of exocytotic vesicle fusion by Ssp1p in vegetative cells (Figure 1). To address this point, we tried to identify other molecular interactors of Ssp1p, in particular proteins that are components of secretory vesicles, but with standard procedures we could not identify any protein. This analysis is complicated by the fact that Ssp1p becomes nonfunctional upon fusion with a tag to either of its termini (data not shown).
The PI(4,5)P2 interaction domain of Ssp1p seems to promote binding of Ssp1p to the plasma membrane in meiotic cells. This is consistent with the finding that PI(4,5)P2 could indeed be detected at the PSM during this stage of meiosis (Rudge et al, 2004). We investigated whether PI(4,5)P2 can be detected also at the PSM, but found that the first detectable amounts of this lipid (using the GFP-PLCδPH sensor; Stefan et al, 2002) appear only after closure of the PSMs (data not shown). Binding of Ssp1p to secretory vesicles, which have been found to colocalize with precursor structures, might thus be mediated by a different interaction. This binding does not appear to be mediated by a phosphoinositide because a PIP binding-deficient mutant of Ssp1p did still bind to the vesicles. Also phosphorylation of Ssp1p appears to play no major role on the level of precursor structures because phosphorylation is dependent on binding of Ssp1p to PI(4,5)P2.
Our findings are consistent with the idea that Ssp1p mediates the occasional binding of precursor structures to the plasma membrane, and that this binding can cause reduced mobility of the precursors. A plasma membrane-localized kinase may then phosphorylate Ssp1p. Recently, we reported actin cable-based transport of precursors in sporulating cells. This transport mechanism is not essential and seems to occur solely to increase mobility of precursors (Taxis et al, 2006). As the actin cables were detected along the plasma membranes (Taxis et al, 2006), it might be that PI(4,5)P2 binding of Ssp1p serves to increase the chance for a precursor structure to be loaded on an actin cable.
Shaping and closing the PSM
In cells deleted for SSP1, the PSMs assemble and the membranes bend strongly toward the nuclear envelope in a manner that leads to complete enwrapping of nuclear structures without engulfed cytoplasmic content (Moens et al, 1974; Moreno-Borchart et al, 2001). Often many minicompartments that contain fractions of the nucleus are tightly enwrapped by PSMs, which indicates that Ssp1p is required to prevent PSMs from untimely closure. This suggests the existence of forces or mechanisms that counteract the Ssp1p-mediated opening of the PSM during assembly and that constantly try to close the membrane. Their existence is also suggested by the phenotype of the Δ_sma2_ mutation (Rabitsch et al, 2001), in which the PSM is no longer bent (P Maier and M Knop, manuscript in preparation). We therefore hypothesize that the LEP coat has the properties of a stiff ring that keeps the membrane open. Within such a model, closure of the PSM during cytokinesis could best be achieved by simple removal of the LEP coat through the removal of Ssp1p. We reported previously that the LEP coat of cells undergoing PSM closure and cytokinesis is constantly shrinking over a period of approximately 6 min (using Don1p-GFP as a marker) until no coat can be detected anymore. Thereafter, Don1p-GFP is specifically enriched in the lumen of the spores, but not in the remaining space of the cell, indicating physical separation of the spore cytoplasm and the lumen of the original cell (Taxis et al, 2006). This is consistent with a model where the amount of Ssp1p at the LEP coat determines the size of the opening of the PSM. Our molecular analysis indicates that the C-terminal domain of Ssp1p is required for efficient removal of the LEP coat during spore formation. The C-terminus of Ssp1p (aa 542–569) is predicted to form a coiled-coil domain. As the Smt3p modification occurs in direct vicinity of the C-terminus, it is tempting to speculate that sumoylation might serve as a means to fine-regulate LEP coat removal, for example through protection of Ssp1p removal/degradation in situations where a checkpoint is activated. We also investigated ubiquitination of Ssp1p, but could not obtain conclusive results, mostly owing to the lack of appropriate reagents that can be used with meiotic cells (many mutant alleles are not available in the SK1 background, and the fact that meiosis is temperature sensitive per se (Byers and Goetsch, 1982) excludes the use of many temperature-sensitive mutants as well).
When we analyzed the primary amino-acid sequence of Ssp1p, we noticed the presence of a destruction box (D-Box) and two KEN boxes. One KEN box did overlap with the Smt3p sites of Ssp1p (503-KKEN-506). KEN and D boxes have been shown to mediate the degradation of proteins via the anaphase-promoting complex (APC) (Harper et al, 2002; Peters, 2006). We performed a preliminary analysis of some of these motifs, but could not obtain indication that they are involved in degradation of Ssp1p. For the case of the motor protein Cin8p, it has been shown that these degradation signals are only active upon translocation of the molecule into the nucleus (Hildebrandt and Hoyt, 2001), where they become recognized for degradation via the APC. For Ssp1p, we have no indication for nuclear translocation, which could explain why these motifs are not involved in its degradation. However, further work will be needed to unravel the degradation pathway of Ssp1p and how it is linked to removal of the protein from the PSM.
Is Ssp1p a divergent septin?
Interestingly, Ssp1p exhibits several similarities to septins that play multiple roles at the bud neck in vegetatively dividing yeast. Septin have been demonstrated to bind to PIPs through their N-terminal domain, become Smt3p modified near to the C-terminus and have a C-terminal coiled-coil domain (Casamayor and Snyder, 2003). For septins, PIP binding is important but not completely essential for septum formation. In our case, it appears not to be required for binding to the PSMs, but for another (non-essential) step earlier in the process. Smt3p modification of septins was found to regulate septin ring stability in vegetative cells but not to affect cell viability (Johnson and Blobel, 1999). For Ssp1p, we cannot exclude slight effects of Smt3p modification on Ssp1p/LEP coat disassembly or degradation because the synchrony of sporulating cells is not high enough owing to technical limitations. This makes precise measurements of small kinetic differences very difficult. The coiled-coil region of septins was found to mediate inter-septin interactions, but also interactions with Bem4p (Casamayor and Snyder, 2003). Similarly, the corresponding region of Ssp1p could mediate protein–protein interactions necessary for removal/degradation of the LEP coat, and it would be interesting to identify the responsible machinery in order to address the question how this process is regulated with respect to the meiotic cell cycle.
One notable structural difference between Ssp1p and septin exists: Ssp1p lacks the characteristic signature of a GTPase domain. We tried to measure GTPase activity of purified Ssp1p, but did not obtain conclusive results, because we could not identify residues that abolished the weak GTPase activity we measured. Also, we tried to address the other prominent function of septins, which is filament formation. Using published protocols that work well with septins (Versele and Thorner, 2004), we could not detect formation of Ssp1p filaments in vitro. However, when we overexpressed truncated variants of Ssp1p in vegetative cells, we could see for some constructs that small circles are formed that float in the cytoplasm (data not shown). It is not clear whether these circles are associated with membranous compartments, but if not, this could indicate that Ssp1p can form filaments as well. Further biochemical work is required to solve this point.
Yeast possesses two septins that are specific for sporulation (Spr3p and Spr28p), and only a subset of the septins that are used during vegetative cell division is also expressed in sporulation (De Virgilio et al, 1996; Fares et al, 1996). Interestingly, the septins do not cover the leading edge of the PSM, they rather form parallel bar like assemblies, 2–3 per PSM, that run in parallel from the LEP coat to the rear of the PSM. Moreover, the deletions each of the sporulation-specific or the non-essential vegetative septin do not impair sporulation, making it unlikely that they function in an analoguous manner as during vegetative cell division. Gip1p, a regulatory subunit of the phosphatase Glc7p, is required for septin bar formation in meiosis. However, the Δ_gip1_ mutant is not impaired in closure of the PSM but in the deposition of the spore wall (Tachikawa et al, 2001). Thus, in meiosis, the septins are in a bad position with respect to their function as ‘septins', whereas Ssp1p is in an excellent position to function as a genuine ‘septin'.
Altogether, our results indicate that Ssp1p is a major player involved in PSM formation and postmeiotic cytokinesis. We think that Ssp1p should be considered as a functional relative but evolutionarily very divergent member of the septin family of proteins that has acquired a specialized function in sporulation/intracellular budding.
Materials and methods
Yeast strains, plasmids and materials and methods
The genotypes of strains and the plasmids used in this work are listed in Supplementary Table 1. Materials and methods are provided as Supplementary data.
Supplementary Material
Supplementary Table S1
Supplemental Figure Legends
Supplemental Online Materials and Methods and References
Supplementary Movies S1
Supplementary Movies S2
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Acknowledgments
We are grateful to N Belgareh-Touze and J-M Galan for investigation of the secretory processes in the Ssp1p overexpression strain. We thank the following: J Audhya and S Emr for PIP kinase mutants and the GFP-PLCδPH construct and C Hoege, S Jentsch and F Barr for antibodies. We thank A Habermann and M Matzner for help with electron microscopy. K Strasser is kindly acknowledged for additional help and for strain and plasmid constructions, and J Jaentti for providing yeast strains. R Esposito is kindly acknowledged for communication of the identity of the spo3-1 mutation with SSP1. We thank the staff of the advanced light microscopy facility (ALMF) of EMBL and Timo Zimmermann, Stefan Terjung and Jens Rietdorf for help with microscopes. We thank Applied Precision (DeltaVision) and Leica for continuous support of the ALMF. This work was supported in parts by the German Research Council (DFG), Grant KN498/2-2.
References
- Audhya A, Emr SD (2002) Stt4 PI 4-kinase localizes to the plasma membrane and functions in the Pkc1-mediated MAP kinase cascade. Dev Cell 2: 593–605 [DOI] [PubMed] [Google Scholar]
- Avaro S, Belgareh-Touze N, Sibella-Arguelles C, Volland C, Haguenauer-Tsapis R (2002) Mutants defective in secretory/vacuolar pathways in the EUROFAN collection of yeast disruptants. Yeast 19: 351–371 [DOI] [PubMed] [Google Scholar]
- Bajgier BK, Malzone M, Nickas M, Neiman AM (2001) SPO21 is required for meiosis-specific modification of the spindle pole body in yeast. Mol Biol Cell 12: 1611–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briza P, Ellinger A, Winkler G, Breitenbach M (1988) Chemical composition of the yeast ascospore wall. The second outer layer consists of chitosan. J Biol Chem 263: 11569–11574 [PubMed] [Google Scholar]
- Byers B, Goetsch L (1982) Reversible pachytene arrest of Saccharomyces cerevisiae at elevated temperature. Mol Gen Genet 187: 47–53 [DOI] [PubMed] [Google Scholar]
- Casamayor A, Snyder M (2003) Molecular dissection of a yeast septin: distinct domains are required for septin interaction, localization, and function. Mol Cell Biol 23: 2762–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccio A, Bogengruber E, Conrad MN, Dresser ME, Briza P, Neiman AM (2004) Morphogenetic pathway of spore wall assembly in Saccharomyces cerevisiae. Eukaryot Cell 3: 1464–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidow LS, Byers B (1984) Enhanced gene conversion and postmeiotic segregation in pachytene-arrested Saccharomyces cerevisiae. Genetics 106: 165–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Virgilio C, DeMarini DJ, Pringle JR (1996) SPR28, a sixth member of the septin gene family in Saccharomyces cerevisiae that is expressed specifically in sporulating cells. Microbiology 142: 2897–2905 [DOI] [PubMed] [Google Scholar]
- Esposito MS, Esposito RE (1969) The genetic control of sporulation in Saccharomyces. I. The isolation of temperature-sensitive sporulation-deficient mutants. Genetics 61: 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares H, Goetsch L, Pringle JR (1996) Identification of a developmentally regulated septin and involvement of the septins in spore formation in Saccharomyces cerevisiae. J Cell Biol 132: 399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon O, Taxis C, Keller PJ, Benjak A, Stelzer EH, Simchen G, Knop M (2006) Nud1p, the yeast homolog of Centriolin, regulates spindle pole body inheritance in meiosis. EMBO J 25: 3856–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsich SW, Shaw JM (2004) Importance of mitochondrial dynamics during meiosis and sporulation. Mol Biol Cell 15: 4369–4381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Burton JL, Solomon MJ (2002) The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev 16: 2179–2206 [DOI] [PubMed] [Google Scholar]
- Hildebrandt ER, Hoyt MA (2001) Cell cycle-dependent degradation of the Saccharomyces cerevisiae spindle motor Cin8p requires APC(Cdh1) and a bipartite destruction sequence. Mol Biol Cell 12: 3402–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ES, Blobel G (1999) Cell cycle-regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. J Cell Biol 147: 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB (2001) The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol 3: 675–678 [DOI] [PubMed] [Google Scholar]
- Knop M, Miller KJ, Mazza M, Feng D, Weber M, Keranen S, Jantti J (2005) Molecular interactions position Mso1p, a novel PTB domain homologue, in the interface of the exocyst complex and the exocytic SNARE machinery in yeast. Mol Biol Cell 16: 4543–4556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Strasser K (2000) Role of the spindle pole body of yeast in mediating assembly of the prospore membrane during meiosis. EMBO J 19: 3657–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaji M, Nishimura K, Ajello L (1985) Scanning electron microscope studies on the parasitic cycle of Coccidioides immitis. Mycopathologia 89: 51–57 [DOI] [PubMed] [Google Scholar]
- Moens PB, Esposito RE, Esposito MS (1974) Aberrant nuclear behavior at meiosis and anucleate spore formation by sporulation-deficient (SPO) mutants of Saccharomyces cerevisiae. Exp Cell Res 83: 166–174 [DOI] [PubMed] [Google Scholar]
- Moens PB, Rapport E (1971) Spindles, spindle plaques, and meiosis in the yeast Saccharomyces cerevisiae (Hansen). J Cell Biol 50: 344–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Borchart AC, Knop M (2003) Prospore membrane formation: how budding yeast gets shaped in meiosis. Microbiol Res 158: 83–90 [DOI] [PubMed] [Google Scholar]
- Moreno-Borchart AC, Strasser K, Finkbeiner MG, Shevchenko A, Knop M (2001) Prospore membrane formation linked to the leading edge protein (LEP) coat assembly. EMBO J 20: 6946–6957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissette NS, Sibley LD (2002) Cytoskeleton of apicomplexan parasites. Microbiol Mol Biol Rev 66: 21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, de los Santos P, Neiman AM (2004) Positive and negative regulation of a SNARE protein by control of intracellular localization. Mol Biol Cell 15: 1802–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman AM (2005) Ascospore formation in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev 69: 565–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemecek JC, Wuthrich M, Klein BS (2006) Global control of dimorphism and virulence in fungi. Science 312: 583–588 [DOI] [PubMed] [Google Scholar]
- Nickas ME, Neiman AM (2002) Ady3p links spindle pole body function to spore wall synthesis in Saccharomyces cerevisiae. Genetics 160: 1439–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickas ME, Schwartz C, Neiman AM (2003) Ady4p and Spo74p are components of the meiotic spindle pole body that promote growth of the prospore membrane in Saccharomyces cerevisiae. Eukaryot Cell 2: 431–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Iino T (1982) Genetic block of outer plaque morphogenesis at the second meiotic division in an hfd1-1 mutant of Saccharomyces cerevisiae. J Gen Microbiol 128: 1309–1317 [DOI] [PubMed] [Google Scholar]
- Ortiz D, Medkova M, Walch-Solimena C, Novick P (2002) Ypt32 recruits the Sec4p guanine nucleotide exchange factor, Sec2p, to secretory vesicles; evidence for a Rab cascade in yeast. J Cell Biol 157: 1005–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7: 644–656 [DOI] [PubMed] [Google Scholar]
- Peterson JB, Gray RH, Ris H (1972) Meiotic spindle plaques in Saccharomyces cerevisiae. J Cell Biol 53: 837–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabitsch KP, Toth A, Galova M, Schleiffer A, Schaffner G, Aigner E, Rupp C, Penkner AM, Moreno-Borchart AC, Primig M, Esposito RE, Klein F, Knop M, Nasmyth K (2001) A screen for genes required for meiosis and spore formation based on whole-genome expression. Curr Biol 11: 1001–1009 [DOI] [PubMed] [Google Scholar]
- Riedel CG, Mazza M, Maier P, Korner R, Knop M (2005) Differential requirement for phospholipase D/Spo14 and its novel interactor Sma1 for regulation of exocytotic vesicle fusion in yeast meiosis. J Biol Chem 280: 37846–37852 [DOI] [PubMed] [Google Scholar]
- Rudge SA, Sciorra VA, Iwamoto M, Zhou C, Strahl T, Morris AJ, Thorner J, Engebrecht J (2004) Roles of phosphoinositides and of Spo14p (phospholipase D)-generated phosphatidic acid during yeast sporulation. Mol Biol Cell 15: 207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MK, Compton HL, Roos DS, Tilney LG (2000) Microtubules, but not actin filaments, drive daughter cell budding and cell division in Toxoplasma gondii. J Cell Sci 113: 1241–1254 [DOI] [PubMed] [Google Scholar]
- Shimoda C (2004) Forespore membrane assembly in yeast: coordinating SPBs and membrane trafficking. J Cell Sci 117: 389–396 [DOI] [PubMed] [Google Scholar]
- Stefan CJ, Audhya A, Emr SD (2002) The yeast synaptojanin-like proteins control the cellular distribution of phosphatidylinositol (4,5)-bisphosphate. Mol Biol Cell 13: 542–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachikawa H, Bloecher A, Tatchell K, Neiman AM (2001) A Gip1p–Glc7p phosphatase complex regulates septin organization and spore wall formation. J Cell Biol 155: 797–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C, Maeder C, Reber S, Rathfelder N, Greger K, Miura K, Stelzer EHK, Knop M (2006) Dynamic organization and requirement of the actin cytoskeleton during meiosis and sporulation in budding yeast. Traffic 7: 1628–1642 [DOI] [PubMed] [Google Scholar]
- Versele M, Thorner J (2004) Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. J Cell Biol 164: 701–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland J, Walther A (2005) Ashbya gossypii: a model for fungal developmental biology. Nat Rev Microbiol 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Zickler D, Olson LW (1975) The synaptonemal complex and the spindle plaque during meiosis in yeast. Chromosoma 50: 1–23 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1
Supplemental Figure Legends
Supplemental Online Materials and Methods and References
Supplementary Movies S1
Supplementary Movies S2
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3