Specific interaction of HTLV tax protein and a human type IV neuronal intermediate filament protein (original) (raw)
Abstract
The human T-cell leukemia virus (HTLV) is associated with adult T cell leukemia and neurological disorders (TSP/HAM). The HTLV transcriptional transactivator, Tax, is known to exert its effect through protein–protein interaction with several transcription factors that activate genes in T cell proliferation. The pathogenic mechanism in the CNS is less defined. Using the yeast two-hybrid system, we have identified a specific Tax-binding protein as the neuronal specific intermediate filament protein, α-internexin. Tax binds to the domain corresponding to the rod region of α-internexin, which is essential for neurofilament assembly. The Tax domains involved in binding are separable from those involved in transactivation. TxBP-1/α-internexin and Tax are expressed in the cytoplasm and nucleus, respectively, when expressed alone, but in coexpressing cells, colocalization of both proteins was observed in a perinuclear, punctate distribution. This in vivo interaction also resulted in a dramatic reduction in Tax transactivation and the network formation by α-internexin. The specific interaction of Tax and a neuronal specific intermediate filament protein may provide a clue to the pathogenesis of TSP/HAM.
Human T-cell leukemia virus type 1 (HTLV-1) is the causative agent of adult T-cell leukemia (ATL) (1) and a degenerative neurological disorder known as tropical spastic paraparesis/HTLV-1 associated myelopathy (TSP/HAM) (2, 3). Like other complex retroviruses, HTLV-1 encodes both regulatory and structural gene products (4). The regulatory protein, Tax, is a 40-kDa nuclear protein that activates viral transcription through a 21-bp repeat enhancer element in the long terminal repeat (LTR) (5). In addition, Tax has been implicated in the transcriptional regulation of several cellular genes including interleukin 2 (IL-2) and the IL-2 receptor, both involved in T-cell proliferation; granulocyte-macrophage colony-stimulating factor (GM-CSF); vimentin, the cytoskeletal protein; the c-fos protooncogene; and the HIV LTR (6). Existing evidence suggests that Tax does not bind DNA directly (7, 8), but rather interacts with DNA-binding cellular proteins (9, 10). These include the CREB/ATF family proteins that bind to the CRE-like elements in the 21-bp enhancer directly (7, 8, 11, 12), those that bind to the NF-κB binding site, and the serum responsive element (13). Extracellular Tax also exerted a transactivation function and was shown to promote DNA-binding activity of the transcriptional factor NF-κB (14), and up-regulate the expression of IL-2Rα (15). The activation of cellular genes that promote T-cell proliferation by Tax may contribute to HTLV leukemogenesis (6). Transgenic mice expressing Tax from the HTLV promoter developed mesenchymal tumors and neurofibromas (16, 17).
HTLV has been shown to be associated with the neurological disease TSP/HAM (2, 3, 18), which is pathologically characterized by atrophy of the thoracic spinal cord with axonal degeneration and demyelination of the lateral and anterior spinal tracts. Although the existence of the neurological disorder has long been identified and extensive studies have been carried out, the mechanism of the CNS tissue damage in TSP/HAM remains virtually unknown. HTLV-1 isolates from patients with TSP/HAM and ATL generally show no distinguishing genetic features (2, 3, 19), although a Tax mutation associated with TSP/HAM was reported (20). Higher viral burden as well as immune responses (both humoral and cytotoxic) to Tax have been detected in TSP/HAM patients relative to ATL patients (21, 22). Using in situ hybridization, Moritoyo et al. (23) and Lehky et al. (24) detected Tax gene expression in infiltrating CD4+ T lymphocytes in areas of inflammation and white matter destruction in the spinal cord of TSP/HAM patients. Although many favor the hypothesis that the immune response against Tax or other viral proteins is implicated in the pathogenesis of TSP/HAM, a direct role of these viral proteins cannot be ruled out. However, because HTLV-1 does not infect neurons, any effect of the virus on the induction and/or progression of TSP/HAM must be mediated by an indirect mechanism, such as the ability of Tax to function as an extracellular molecule (15, 25). In a search for host cell proteins potentially pertinent to the neuropathogenesis of HTLV-1, we employed the yeast two-hybrid system to screen a human brain library to identify protein(s) that specifically interact with Tax. In this report, we describe the isolation and characterization of a neuronal specific cDNA that encodes such a protein [Tax binding protein-1 (TxBP-1)]. We further demonstrated that the TxBP-1-binding domain(s) of Tax is separable from those involved in transactivation, and that the Tax:TxBP-1 interaction altered the cellular localization and function of both proteins. TxBP-1 corresponds to the neuronal intermediate filament protein, α-internexin. These results are of particular interest because deregulated expression of two other neuronal intermediate filaments (NF-L and NF-H) have been shown to induce motor neuron disorders in the transgenic mouse model (26–28).
MATERIALS AND METHODS
Construction of Plasmids and Yeast Two-Hybrid Assay.
The construction of yeast GAL4 DNA-binding domain fusion expression vectors, pGBT9-Tax (wild type and mutants), pGBT9-Tat, pGBT9-Vpr, pGBT9-Nef, and GST-Tax have been described previously (12). The TxBP-1 amino-terminal deletion mutants: pGAD10 TxBP-1 Δ150, Δ300, Δ480, and Δ660 were constructed by cloning the PCR-amplified fragments into the _Bam_HI and _Eco_RI sites of pGAD10, which contains the GAL4 activation domain. The carboxyl-terminal deletion mutants were constructed by partial digestion of TxBP-1 cDNA with _Pst_I enzyme, followed by religation. GST-TxBP-1: the _Bam_HI-_Bgl_II fragment (1.5 kb) of pGAD10-TxBP-1 was directly cloned at the _Bam_HI site of the GST plasmid. The human α-internexin (from Alex Chu, Albert Einstein Medical College, Bronx, NY) was cloned into the _Eco_RI sites of pcDNA3 vector. A yeast two-hybrid expression library (CLONTECH) derived from human brain was screened as described previously (12).
Oligonucleotides Synthesized.
We have used the following oligonucleotides: TxBP-1 forward primers Δ150, 5′-GCTGTGGCTAAACCAGAC-3′; Δ300, 5′-GCCATCCGAGCCAGCCGA-3′; Δ480, 5′-GACCTGAGGAACACCAAG-3′; Δ660, 5′-AGTTATTTGCTCCCTCCT-3′; GAL4 activation domain forward and reverse primers, 5′-TACCACTACAATGGATG-3′ and 5′-GAGATGGTGCACGATGCA-3′; actin forward and reverse primers, 5′-TACATGGCTGGGGTGTTGAA-3′ and 5′-AAGAGAGGCATCCTCACCCT-3′.
Cell Lines and Media.
The human T-cell line, H9 (human T cells), and U937 (monocytic cell line) were maintained in RPMI 1640 medium and PC12 (rat pheochromocytoma cell line); NIH 3T3 and MEL (strain 745A); HeLa, NT2, and SW13 (vim-) cells were maintained in DMEM supplemented with 10% fetal bovine serum.
RNA Isolation, Reverse Transcription (RT), and PCR.
Total RNA was isolated from each of 5 × 106 MEL, H9, PC 12, NIH 3T3, and U937 cells, and the distribution of TxBP-1 was examined by a sensitive RNA PCR by using the internal forward and reverse primers 5′-TACATGGCTGGGGTGTTGAA-3′ and 5′-AAGAGAGGCATCCTCACCCT-3′ derived for β-actin gene and 5′-GCTGTGGCTAAACCAGAC-3′ and 5′-TTACATTTTTTGGCTGGA-3′ for TxBP-1.
The DNA products generated by RT-PCR (12) were analyzed by Southern blotting, by using the 32p end-labeled probes 5′-TACATGGCTGGGGTGTTGAA-3′ for β-actin and 5′GACCTGAGGAACACCAAG-3′ for TxBP-1.
Expression of GST-Tax and GST-TxBP-1 Fusion Proteins.
GST-Tax and GST-TxBP-1 recombinant plasmids were transformed into Escherichia coli strain DH5α. Overnight-grown cultures were diluted 1:10 with Luria–Bertani medium containing 100 μg/ml ampicillin and allowed to grow for 1 hr. After 1 mM IPTG was added, they continued to grow for an additional 3 hr. The bacterial cultures were harvested, washed with PBS, and lysed by sonication at 4°C in NETN (20 mM Tris, pH 8.0/100 mM NaCl/1 mM EDTA/0.5% Nonidet P-40) containing 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, and 2 μg/ml each of aprotinin, leupeptin, and pepstatin. The insoluble cell debris was pelleted by centrifugation at 10,000 rpm for 15 min at 4°C. The resultant supernatant was mixed with glutathione-Sepharose beads (Pharmacia) that were presoaked with NETN buffer containing 0.5% milk powder and rocked for 30 min at 4°C. Nonspecific proteins bound to beads were removed by washing the glutathione-Sepharose beads three times with NETN buffer and analyzed by SDS/PAGE.
In Vitro Binding Assay and Western Blot Analysis.
For the in vitro binding assay, TxBP-1 was cleaved from GST beads at 4°C with 2 μg of thrombin per μg of protein in 20 mM Tris⋅HCl (pH 8.0) containing 2.5 mM CaCl2, and the supernatant was dialyzed against the column buffer NETN buffer. A 20-μl (approximately 0.3 μg) aliquot of cleaved TxBP-1 was added to 25 μl of glutathione-bound GST-Tax fusion protein slurry and incubated at 4°C for 1 hr in 100 μl of column buffer. The beads were washed three times with 0.5 ml of column buffer at each time. Washed beads were suspended in SDS buffer and boiled for 5 min, and the beads were pelleted by centrifugation. The supernatant was analyzed by SDS/PAGE and were transferred onto nitrocellulose membrane according to standard protocols. Nitrocellulose membranes were incubated with the polyclonal antibodies raised in rabbits against rat α-internexin protein, and the affinity of antibody to the protein was detected with ECL Western blotting detection.
Transfections and Chloramphenicol Acetyltransferase (CAT) Assays.
HeLa and SW13 (vim-) cells were transfected with the DNAs, as shown in Fig. 4, by calcium phosphate method. pcDNA3 was used as filler DNA to equalize the amount of DNA for each transfection. Forty-eight hours posttransfection, cell extracts were prepared as described previously (12). CAT assays and separation of reaction products were performed as previously described (29). Fold of trans-activation was quantitated by scintillation counting of the products separated from the reaction.
Figure 4.

Effect of human α-internexin expression on HTLV LTR in cotransfection assays. Approximately 1 × 105 HeLa (A) and SW13 (vim-) (B) cells were independently transfected with 0.5 μg of HTLV LTR CAT, 0.25 μg pTax, and increasing amounts of human α-internexin as shown by using the CaPO4 method. pcDNA3 was used as filler DNA to equalize the amount of DNA for each transfection. Forty-eight hours posttransfection, cell extracts were prepared and CAT assays were performed as described in Methods.
Indirect Immunofluorescence.
For the distribution and colocalization of TxBP-1 and Tax, HeLa and SW13 (vim-) cells were transfected with 10 μg each of expression plasmids and cells were cultivated on the glass slides which were precoated with poly l-lysine. To examine the endogenous α-internexin association with Tax, NT2 cells were transfected with 10 μg of Tax expression plasmid alone. Cells were prepared for immunofluorescence as described previously (12). Confocal microscopy was performed on a Bio-Rad MRC 600 laser-scanning confocal microscope attached to a Zeiss Aniovert 35M microscope and viewed by using a 40× 1.3-na oil objective.
RESULTS
Identification of a Brain-Specific Protein that Interacts with Tax.
The yeast two-hybrid system was used to identify cDNA clones from a human brain library that encode Tax-binding proteins. The hybrid plasmids, pGBT9 Tax, and the pGAD10 fusion library were cotransformed into the Saccharomyces cerevisiae host strain HF7c containing GAL4-responsive HIS3 and lacZ reporter genes. Of 5 × 105 colonies screened, only two transformants readily turned blue in the lacZ assays (<15-min incubation). One cDNA clone (TxBP-1) was further characterized. To examine whether TxBP-1 interacts specifically with Tax, we cotransformed TxBP-1 DNA with the corresponding Gal4-DNA-binding domain plasmid (pGBT9) lacking Tax cDNA, or with irrelevant fusion plasmids (12). TxBP-1 either alone or in combination with the DNA-binding domain vector failed to activate lacZ expression, indicating that TxBP-1 interacts specifically with Tax. For additional specificity studies, we have tested HIV-1 Tat, Vpr, and HIV-2 Nef (12) for binding to TxBP-1. TxBP-1 interacted only with Tax, used as the positive control, but not with other retroviral proteins tested (data not shown).
TxBP-1 Shows Homology with a Type IV Neuronal Intermediate Filament Protein and Is Expressed in Neuronal Cells.
Using 5′ and 3′ flanking primers of yeast activation domain vector (pGAD10) and internal primers of the cDNA clone, we have sequenced the TxBP-1 cDNA as described in Materials and Methods. The TxBP-1 cDNA contains a 1.2-kb insert with a 0.9 kb ORF at the N terminus. The ORF encodes a protein of 44 kDa, followed by untranslated sequences. TxBP-1 was expressed from the yeast activation domain AUG codon, indicating that the gene is truncated at the 5′ end. To determine the expression pattern of TxBP-1, we performed RT-PCR on RNA from five cell lines: MEL, a mouse erythroleukemic cells; H9, a human T cell line; PC12, a rat pheochromocytoma cell line; NIH 3T3, a mouse fibroblast cell line and U937, a human monocytic cell line (Fig. 1A). Only the neuron-like cell line PC12 expressed TxBP-1, suggesting that TxBP-1 may be a neuron-specific protein. A GenBank search revealed the greatest homology (96%) with a published sequence of human type IV neuronal intermediate filament (IF) protein α-internexin (Fig. 1B), with the alignment beginning at amino acid 200 of the 66-kDa human α-internexin (30). The two probably represent the same gene. TxBP-1 also shares extensive homology with vimentin, another intermediate filament protein. However, neither rat vimentin nor the intermediate filament NF-L binds to Tax (data not shown).
Figure 1.

(A) Distribution of TxBP-1 RNA in different cell lines. Total RNA isolated from MEL, mouse erythroleukemic cells; H9, human T cell line; PC12, undifferentiated rat pheochromocytoma cells; NIH 3T3, mouse fibroblastic cells; and U937, monocytic cell lines was subjected to reverse transcription. The reverse-transcribed cDNA was subjected to PCR by using β-actin and TxBP-1 primers as described in Methods. The amplified products were subjected to Southern blot analysis, and the membrane was hybridized with end-labeled 32p β-actin and TxBP-1 oligonucleotides. The membrane was exposed to x-ray film. Upper arrow denotes the TxBP-1; lower arrow denotes the β-actin and PC stands for positive control (β-actin). (B) Homology between the amino acid sequences of TxBP-1 and human α-internexin-NF66 (hNF-66).
TxBP-1 Binds to Tax in Vitro.
To test the in vitro binding of Tax to TxBP-1, we constructed GST fusion plasmids containing the sequences of both proteins. PCR-amplified Tax insert was cloned in-frame into the GST plasmid, and the resultant plasmid permitted the expression of the GST-Tax fusion protein. The _Bam_HI-_Bgl_II fragment of pGAD10-TxBP-1 was also cloned in-frame into the GST plasmid; transformation of this plasmid led to expression of the GST-TxBP-1 fusion protein. Treatment with thrombin failed to cleave GST-Tax, whereas GST-TxBP-1 was readily cleaved into GST and TxBP-1 (Fig. 2A). Cleaved TxBP-1 was mixed with GST-Tax beads, washed three times with column buffer, and boiled, eluant was analyzed by SDS/PAGE, and Western blot was performed by using rabbit polyclonal antibodies against rat α-internexin. As shown in Fig. 2B, TxBP-1 bound to GST-Tax but not to GST alone. These results suggest that TxBP-1 binds to Tax in vitro.
Figure 2.
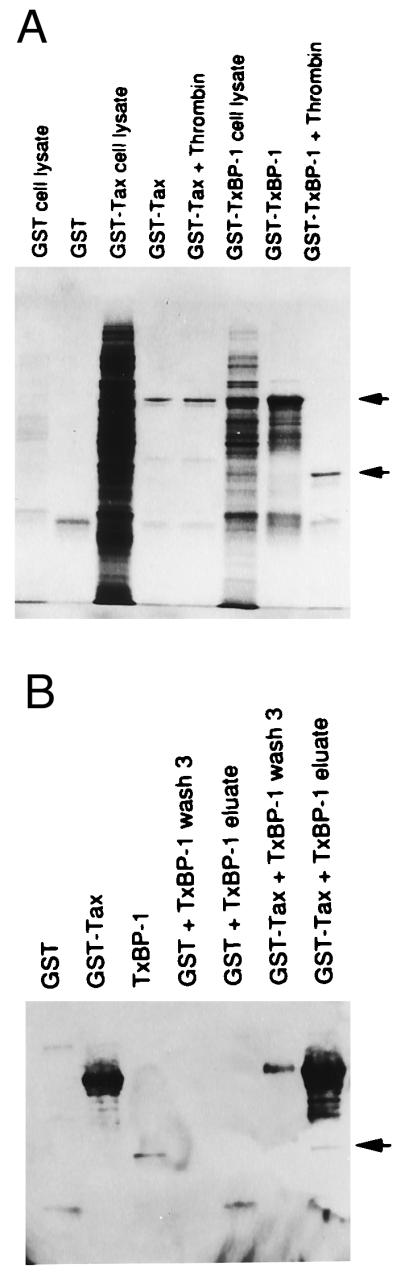
(A) Expression and cleavage of GST-TxBP-1 and GST-Tax fusion proteins. GST-TxBP-1 and GST-Tax fusion proteins expressed in E. coli was purified and cleaved with thrombin as described in Methods. Crude bacterial lysates and proteins purified on GST beads were electrophoresed on 12% SDS-polyacrylamide gels and stained with Coomassie blue. Upper arrow denotes the uncleaved GST-Tax in lane 4, and lower arrow denotes the cleaved TxBP-1. (B) In vitro binding of TxBP-1 to Tax. Purified GST-Tax fusion protein bound to GST beads was used as an affinity matrix to test the in vitro binding of TxBP-1. As a control, GST protein bound to G-Sepharose beads was used. The binding of TxBP-1 to Tax was analyzed by Western blotting by using the polyclonal antibodies raised in rabbit against rat α-internexin. Arrow denotes the bound TxBP-1.
Tax Interacts with the N-Terminal Region of TxBP-1.
To better define the region of TxBP-1 involved in complex formation with Tax, we constructed several progressive N- and C-terminal deletion mutants as shown in the Table 1. N-terminal deletion mutants have 150, 300, 480, and 660 bp deleted from their 5′ ends; the resultant constructs were allowed the expression of polypeptides containing 250, 200, 150, and 100 aa of the C-terminal end, respectively. N-terminal progressive deletion mutants were constructed by PCR amplification of the deleted TxBP-1 gene with suitable synthetic oligonucleotides primers and insertion of the amplified sequence in the frame of the yeast activation domain. The C-terminal deletion mutants were constructed by partial digestion of TxBP-1 cDNA with _Pst_I enzyme, followed by religation. Three C-terminal deletion mutants, Δ120–900, Δ234–900, and Δ348–900 allow the expression of polypeptides from the N-terminal 40, 78, and 116 aa, respectively (Table 1). TxBP-1 mutant plasmids and pGBT9 Tax were cotransformed into yeast. All N-terminal deletion mutants were incapable of activating the lacZ activity. In contrast, all C-terminal deletion mutants interacted with Tax, suggesting that the region that interacts with Tax is localized to the N-terminal 40 aa of TxBP-1 or the central rod region of α-internexin.
Table 1.
Binding characteristics of TxBP-1 and Tax mutants
| TxBP-1 | β-Gal assay | Tax* | β-Gal assay | % transactivation | |
|---|---|---|---|---|---|
| HTLV-1 LTR | HIV-1 LTR | ||||
| Δ 1–150 | − | WT | +++ | 100 | 100 |
| Δ 1–300 | − | S10-A | − | <5 | <10 |
| Δ 1–480 | − | C29-S | − | <5 | 70 |
| Δ 1–600 | − | H43-Q | +++ | 100 | <10 |
| Δ 120–900 | +++ | ΔH41-43 | − | <5 | <10 |
| Δ 234–900 | +++ | ΔY18-R62 | − | <5 | <10 |
| Δ 348–900 | +++ | S160-A | + | 35 | <10 |
| S274-A | − | <5 | 50 | ||
| P316-A | +++ | <5 | 70 | ||
| L320-G | − | <5 | 75 |
Tax Mutations that Abrogate the Complex Formation of Tax with TxBP-1.
The activation domains of Tax are poorly defined, because single mutations at discontiguous sites have been found to completely abolish Tax activity. We have selected three groups of mutants: those defective for transactivation of both HTLV-1 LTR and HIV-1 LTR (S10-A, S274-A, ΔH41–43, ΔY18-R62), HTLV-1 LTR alone (C29-S, P316-A, L320-G), or of HIV-1 LTR alone (ΔH41–43, H43-Q, S160-A) (31). The interaction of TxBP-1 with different Tax mutants was examined by the yeast two-hybrid assay. Although many of the mutant Tax proteins were simultaneously defective for binding TxBP-1 and transactivation of HTLV-1 LTR, mutant P316-A bound TxBP-1 as efficiently as wild-type Tax but was not able to transactivate the HTLV-1 LTR (Table 1). Binding of Tax mutants to TxBP-1 was also discordant for transactivation of the HIV-1 LTR. These results suggest that the binding domain(s) on Tax for TxBP-1 is distinct from those involved in interaction with CREB/ATF factors (8, 32).
Impairment of Self-Assembling Capacity of Human α-Internexin by Tax in Cells Lacking Vimentin.
The human adrenal tumor cell line, SW13 (vim-), was used to study the effect of Tax on the self-assembly of exogenously introduced human α-internexin. First, the cells were transfected separately with pTax and a vector expressing the complete human α-internexin gene. Forty-eight hours later, the cells were stained with the appropriate antibodies and examined under the confocal microscope. Tax was found to be localized in the nucleus as well as the cytoplasm (Fig. 3A), and human α-internexin was localized in the cytoplasm, which is consistent with the previous observation that α-internexin is a cytoplasmic protein (Fig. 3B; ref. 33). Furthermore, α-internexin forms a filamentous network in the transfected cells. When the SW13 (vim-) cells were cotransfected with pTax and α-internexin plasmids, a complete colocalization of the two proteins was again observed as a ring-like structure around the nucleus, with a concomitant disruption of the α-internexin filamentous network (Fig. 3 C_–_E).
Figure 3.

Disruption of human α-internexin self-assembly by Tax in cells lacking vimentin. Tax and human α-internexin expression vectors were transfected separately or together into SW13 (Vim-) cells, and 48 hr posttransfection cells were fixed, stained with anti-Tax and anti-α-internexin antibodies, and visualized by confocal microscopy. (A) Tax alone stained with anti-Tax antibody followed by secondary antibodies conjugated with rhodamine. (B) Human α-internexin stained with anti-α-internexin antibody followed by secondary antibodies conjugated with FITC (filamentous form with green fluorescence). (C_–_E) Cells expressing both exogenous α-internexin and Tax. (C) Rhodamine staining because of Tax. (D) Green fluorescence because of α-internexin. (E) Colocalization of α-internexin and Tax in the same cell as shown in C and D as visualized by both colors.
Inhibition of Transactivating Activity of Tax by Human α-Internexin.
To investigate whether this perturbed cellular localization would have any impact on Tax function, we expressed human α-internexin under the control of the cytomegalovirus promoter and studied its ability to affect Tax-mediated transactivation in HeLa and SW13 (vim-) cell lines in cotransfection assays. As shown in Fig. 4, the inhibitory effect of α-internexin on Tax-mediated transactivation was dose-dependent. Cotransfection of increasing concentrations of α-internexin in conjunction with pTax resulted in a significant decrease (80%) in transactivation of the HTLV LTR-driven CAT reporter gene. Inhibition of transactivating activity of Tax by α-internexin was higher in SW13 (vim-) cell lines (Fig. 4B) than in HeLa cells (Fig. 4A). α-Internexin alone, even at high concentrations, did not have any effect on the reporter plasmid.
Reorganization of Endogenous Human α-Internexin in Neuronal Precursor Cells Expressing Tax.
To examine the effect of Tax on the organization of endogenous α-internexin in its natural cell context, neuronal precursor cells were transfected with pTax expression vector, and 48 hr posttransfection, cells were immunostained and examined under the confocal microscope. Unlike the findings in HeLa cells, Tax was distributed in the nucleus as well as the cytoplasm of these neuronal precursor cells (Fig. 5A). The endogenous α-internexin (green fluorescence) in untransfected cells was present in a filamentous network in the cytoplasm (Fig. 5B). The expression of Tax in these cells resulted in the complete colocalization of Tax with endogenous α-internexin at the perinuclear region (Fig. 5 C_–_E, F_–_H, and I_–_K), and network formation was no longer observed in these Tax-expressing cells. These results suggest that the in vivo binding of Tax to endogenous α-internexin may alter and/or disrupt the assembly of α-internexin in human neuronal cells.
Figure 5.

Reorganization of endogenous α-internexin in neuronal precursor cells expressing Tax. Tax expression vector was transfected into neuronal precursor cells, and 48 hr posttransfection cells were fixed, stained with anti-Tax antibodies, and visualized by confocal microscopy. The endogenous a-internexin was identified by the staining of the untransfected cells with the anti-α-internexin antibody. For the colocalization of Tax and endogenous α-internexin, the cells transfected with Tax were stained with anti-Tax and anti-α-internexin antibodies. (A) Tax alone stained with anti-Tax antibody followed by secondary antibodies conjugated with rhodamine. (B) Endogenous α-internexin stained with anti-α-internexin antibody followed by secondary antibodies conjugated with FITC (filamentous form with green fluorescence). (C_–_K) Various cells expressing both endogenous α-internexin and Tax. (C, F, and I) Rhodamine staining because of Tax. (D, G_,_ and J) Green fluorescence because of endogenous α-internexin. (E, H, and K) Colocalization of endogenous α-internexin and Tax in the same cell as shown in C, F, and I and D, G, and J as visualized by both colors.
DISCUSSION
Tax has been shown to activate several cellular genes that ultimately may lead to ATL. So far, Tax seems to exert its function by protein–protein interaction with several cellular transcription factors, e.g., CREB/ATF and NF-κB proteins (5, 7, 12). However, the role of Tax in the induction and/or progression of the neurological disease TSP/HAM is not known. Our results presented here suggest that in addition to interacting with transcription factors that may lead to uncontrolled proliferation of T cells, Tax may also play a role in the pathogenesis of TSP/HAM through direct binding of a neuronal-specific intermediate filament protein. Mutagenesis studies suggest that the sites important for the binding of transcription factors are also important for transactivation by Tax (12). In contrast, the results of this study showed that the Tax domain(s) that bind to TxBP-1 can be separable from those involved in transactivation. TxBP-1/α-internexin belongs to the class IV intermediate filament protein family. It has been shown that α-internexin self-assembles as well as coassembles with the neurofilament triplet proteins into filament networks in transfected cells, and α-internexin is thought to play an important role in organizing the formation of neuronal filament networks in the developing nervous system in vivo (33). All IF proteins comprise three distinct regions, namely head, rod, and tail. The central rod region is highly conserved among all IF proteins and is required for the formation of a coiled-coil dimer, the first step in IF assembly (34). Several studies also demonstrated that the rod region of IF proteins is essential for their polymerization into the filamentous form (35–37). The TxBP-1 amino acid sequence alignment begins at the rod region of α-internexin, and our deletion analyses indicate that Tax binds to the N-terminal part of the central rod region (coil 1). The binding and displacement of the rod region of α-internexin by Tax could impede neurofilament assembly.
We further demonstrated an interaction of TxBP-1/α-internexin and Tax in different human cells. Human α-internexin expressed from a transgene in different cells is localized predominantly in the cytoplasm, in agreement with the previous observations made on transfected rat α-internexin, which self-assembles into cytoplasmic filament networks (33). Similarly, our detection of Tax in the nucleus is consistent with previous results (31). However, coexpression of Tax and TxBP-1 in HeLa cells resulted in a perturbed distribution of both proteins, which exhibited a perinuclear, often punctate distribution (data not shown). This may result from a push-and-pull mechanism attributed to the nuclear targeting and nuclear export signals of the two respective proteins. Coexpression of human α-internexin in conjunction with Tax resulted in a significant decrease in the transactivation of the HTLV LTR CAT reporter gene (Fig. 4) and the disruption of the cytoplasmic IF network. These results indicate that the interaction between these proteins have biological consequences. Because α-internexin has been shown to be important for the stabilization of axons and for the recruitment of other NFTPs (neurofilament triplet proteins) into the filamentous networks, the interaction between Tax and endogenous α-internexin may cause the slow axonal transport that resulted from the accumulation of neurofilaments and ultimately may lead to the destruction of axons.
The involvement of neuronal IFs in neurodegeneration has been suggested. For example, several motor neuron diseases caused by the excessive production of IF have been reported in the transgenic mouse model (26–28). In addition, it has been shown that neuronal filaments are intrinsic determinants of axonal diameter (38). Thus, we speculate that binding of Tax to neuronal filaments may alter its normal assembly process, subsequently leading to a motor neuron disease, namely TSP/HAM. Although no malformation of the myelin sheath was detected in transgenic mice deficient in neurofilament protein (39), the axons of mice overexpressing neurofilament proteins showed deformity in the myelination process (26, 28), which is the sentinel feature of TSP/HAM.
Although HTLV does not infect neurons directly, Tax is known to be able to function as an extracellular protein, e.g., to transactivate transcription from the HTLV LTR transcellularly (14, 15). Extracellular Tax has also been shown to up-regulate cytokine expression in adult human microglial cells (40). Using the PCR and in situ hybridization assays, the presence of Tax RNA has been demonstrated in lymphocytes and astrocytes in the central nervous system of TSP/HAM patients (24, 41, 42). In addition, Moritoyo, et al. (23) have detected Tax gene expression in infiltrating CD4+ T lymphocytes in areas of inflammation and white matter destruction in the spinal cord of TSP/HAM patients. The extracellular Tax secreted by these cells in the CNS may elicit its effect on the neuronal cells and reorganize the neurofilament protein networks. The expression of Tax in the central nervous system and its association with neuronal filament protein suggest a mechanism of HTLV-1-associated demyelination that is not necessarily mutually exclusive with the more commonly postulated immune-based mechanism. It may be of interest to investigate the potential role of Tax in the pathogenesis of neurological disorders resembling TSP/HAM in transgenic mice expressing the Tax gene product under the regulation of a neuron-specific promoter.
Acknowledgments
We thank Dr. Jun-zhou Li for help with the GenBank search, Dr. K.T. Jeang (National Institutes of Health) for the generous gift of wild-type and mutant Tax expression plasmids, and C. Z. Giam (Case Western University, Cleveland, OH) for antibodies against Tax. This work was supported in part by National Institutes of Health Grants RO1 AI31378 and U19 AI36612-02 awarded to F.W.-S. We also acknowledge support from the University of California at San Diego Center for AIDS Research and the National Center for Microscopy and Imaging Research (National Institutes of Health Grant RR04050; P.I., Mark. H. Ellisman).
ABBREVIATIONS
HTLV
human T-cell leukemia virus
TSP/HAM
tropical spastic paraparesis/HTLV-1 associated myelopathy
TxBP-1
Tax-binding protein-1
LTR
long terminal repeat
IF
intermediate filaments
ATL
adult T-cell leukemia
References
- 1.Poiesz B J, Ruscetti F, Gazdar A, Bunn P, Minna J, Gallo R C. Proc Natl Acad Sci USA. 1980;77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gessain A, Barin F, Vernant J C, Gout O, Maurs L, Calender A, de The G. Lancet. 1985;ii:407–409. doi: 10.1016/s0140-6736(85)92734-5. [DOI] [PubMed] [Google Scholar]
- 3.Tsujimoto A, Teruuchi T, Imamura J, Shimptohno K, Miyoshi I, Miwa M. Mol Biol Med. 1988;5:29–42. [PubMed] [Google Scholar]
- 4.Cullen B R. Microbiol Rev. 1992;56:375–394. doi: 10.1128/mr.56.3.375-394.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sodroski J G, Rosen C A, Haseltine W A. Science. 1984;226:177–179. [Google Scholar]
- 6.Yoshida M. AIDS Res Hum Retroviruses. 1994;10:1193–1197. doi: 10.1089/aid.1994.10.1193. [DOI] [PubMed] [Google Scholar]
- 7.Zhao L J, Giam C Z. Proc Natl Acad Sci USA. 1992;89:7070–7074. doi: 10.1073/pnas.89.15.7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner S, Green M R. Science. 1993;262:395–399. doi: 10.1126/science.8211160. [DOI] [PubMed] [Google Scholar]
- 9.Montagne J, Beraud C, Crenon I, Lombard-Platet G, Gazzolo L, Sergeant A, Jalinot P. EMBO J. 1990;9:957–964. doi: 10.1002/j.1460-2075.1990.tb08194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beimling P, Moelling K. Oncogene. 1992;7:257–262. [PubMed] [Google Scholar]
- 11.Bantignies F, Rousset R, Desbois C, Jalinet P. Mol Cell Biol. 1996;16:2174–2182. doi: 10.1128/mcb.16.5.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy T R, Tang H, Li X, Wong-Staal F. Oncogene. 1997;14:2785–2792. doi: 10.1038/sj.onc.1201119. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki T, Hirai H, Fujisawa J, Fujita T, Yoshida M A. Oncogene. 1993;8:2391–2397. [PubMed] [Google Scholar]
- 14.Lindholm P F, Marriott S J, Gitlin S D, Bohan C A, Brady J N. New Biol. 1990;2:1034–1043. [PubMed] [Google Scholar]
- 15.Marriott S J, Trinh D, Brady J N. Oncogene. 1992;7:1749–1755. [PubMed] [Google Scholar]
- 16.Nerenberg M, Hinrichs S H, Reynolds R K, Khoury G, Jay G. Science. 1987;237:1324–1329. doi: 10.1126/science.2888190. [DOI] [PubMed] [Google Scholar]
- 17.Hinrichs S H, Nerenberg M, Reynolds R K, Khoury G, Jay G. Science. 1987;237:1340–1343. doi: 10.1126/science.2888191. [DOI] [PubMed] [Google Scholar]
- 18.Osame M, Usuku K, Izumo S, Ijichi N, Amitani H, Igata A, Matsumoto M, Tara M. Lancet. 1986;i:1031–1032. doi: 10.1016/s0140-6736(86)91298-5. [DOI] [PubMed] [Google Scholar]
- 19.Shirabe S, Nakamura T, Tsujihata M, Nagataki S, Seiki M, Yoshida M. Arch Neurol. 1990;47:1258–1260. doi: 10.1001/archneur.1990.00530110120029. [DOI] [PubMed] [Google Scholar]
- 20.Renjifi B, Borrero I, Essex M. J Virol. 1995;69:2611–2616. doi: 10.1128/jvi.69.4.2611-2616.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubota R, Umehara F, Izumo S, Ijichi S, Matsumuro K, Yashiki S, Fujiyoshi T, Sonoda S, Osame M. J Neuroimmunol. 1994;53:23–29. doi: 10.1016/0165-5728(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson S, Shida H, McFarlin D, Fauchi A, Koenig S. Nature (London) 1990;348:245–248. doi: 10.1038/348245a0. [DOI] [PubMed] [Google Scholar]
- 23.Moritoyo T, Reinhart T A, Moritoyo H, Sato E, Izumo S, Osame M, Haase A T. Ann Neurol. 1996;40:84–90. doi: 10.1002/ana.410400114. [DOI] [PubMed] [Google Scholar]
- 24.Lehky T J, Fox C H, Koenig S, Levin M C, Flerlage N, Izumo S, Sato E, Raine C S, Osame M, Jacobson S. Ann Neurol. 1995;37:167–175. doi: 10.1002/ana.410370206. [DOI] [PubMed] [Google Scholar]
- 25.Marriott S J, Lindholm P F, Redi R L, Brady J N. New Biol. 1991;3:678–686. [PubMed] [Google Scholar]
- 26.Xu Z, Cork L C, Griffin J W, Cleveland D W. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]
- 27.Cote F, Collard J-F, Julien J-P. Cell. 1993;73:35–46. doi: 10.1016/0092-8674(93)90158-m. [DOI] [PubMed] [Google Scholar]
- 28.Collard J-F, Cote F, Julien J-P. Nature (London) 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 29.Seed B, Sheen J-Y. Gene. 1988;67:271–277. doi: 10.1016/0378-1119(88)90403-9. [DOI] [PubMed] [Google Scholar]
- 30.Chan S-O, Chiu F-C. Mol Brain Res. 1995;29:177–184. doi: 10.1016/0169-328x(94)00268-j. [DOI] [PubMed] [Google Scholar]
- 31.Semmes O J, Jeang K T. J Virol. 1992;66:7183–7192. doi: 10.1128/jvi.66.12.7183-7192.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perini G, Wagner S, Green M. Nature (London) 1995;376:602–605. doi: 10.1038/376602a0. [DOI] [PubMed] [Google Scholar]
- 33.Ching G Y, Liem R K H. J Cell Biol. 1993;122:1323–1335. doi: 10.1083/jcb.122.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinert P M, Roop D R. Annu Rev Biochem. 1988;57:593–625. doi: 10.1146/annurev.bi.57.070188.003113. [DOI] [PubMed] [Google Scholar]
- 35.Gill S R, Wong P C, Monteiro M J, Cleveland D W. J Cell Biol. 1990;111:2005–2019. doi: 10.1083/jcb.111.5.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong P C, Cleveland D W. J Cell Biol. 1990;111:1987–2003. doi: 10.1083/jcb.111.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chin S S M, Macioce P, Liem R K H. J Cell Sci. 1991;99:335–350. doi: 10.1242/jcs.99.2.335. [DOI] [PubMed] [Google Scholar]
- 38.Hoffman P N, Cleveland D W, Griffin J W, Landes P W, Cowan N J, Price D L. Proc Natl Acad Sci USA. 1987;84:3472–3476. doi: 10.1073/pnas.84.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eyer J, Peterson A. Neuron. 1994;12:389–405. doi: 10.1016/0896-6273(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 40.Dhib-Jalbut S, Hoffman P M, Yamabe T, Sun D, Xia J, Eisenberg H, Bergey G, Ruscetti F W. Ann Neurol. 1994;36:787–790. doi: 10.1002/ana.410360516. [DOI] [PubMed] [Google Scholar]
- 41.Kira J-I, Koyanagi Y, Yamada T, Itoyama Y, Goto I, Yamamoto N, Sasaki H, Sakaki Y. Ann Neurol. 1991;29:194–201. doi: 10.1002/ana.410290214. [DOI] [PubMed] [Google Scholar]
- 42.Levin M C, Lehky T J, Flerlage N, Katz D, Kinzma D W, Jaffe E S, Heiss J D, Patronas N, McFarland H F, Jacobson S. N Engl J Med. 1997;336:839–845. doi: 10.1056/NEJM199703203361205. [DOI] [PubMed] [Google Scholar]