Immunization with a Nontoxic/Nonfibrillar Amyloid-β Homologous Peptide Reduces Alzheimer’s Disease-Associated Pathology in Transgenic Mice (original) (raw)
Abstract
Transgenic mice with brain amyloid-β (Aβ) plaques immunized with aggregated Aβ1-42 have reduced cerebral amyloid burden. However, the use of Aβ1-42 in humans may not be appropriate because it crosses the blood brain barrier, forms toxic fibrils, and can seed fibril formation. We report that immunization in transgenic APP mice (Tg2576) for 7 months with a soluble nonamyloidogenic, nontoxic Aβ homologous peptide reduced cortical and hippocampal brain amyloid burden by 89% (P = 0.0002) and 81% (P = 0.0001), respectively. Concurrently, brain levels of soluble Aβ1-42 were reduced by 57% (P = 0.0019). Ramified microglia expressing interleukin-1β associated with the Aβ plaques were absent in the immunized mice indicating reduced inflammation in these animals. These promising findings suggest that immunization with nonamyloidogenic Aβ derivatives represents a potentially safer therapeutic approach to reduce amyloid burden in Alzheimer’s disease, instead of using toxic Aβ fibrils.
Evidence that amyloid plays an important role in the early pathogenesis of Alzheimer’s disease (AD) comes from various studies. 1-3 Genetic analyses of families with early-onset AD have revealed mutations in chromosome 21, near or within the Aβ sequence, in addition to mutations within the presenilin 1 and 2 genes. Most of these mutations lead to an increased production of Aβ1-42 and/or total Aβ. Also, Down’s syndrome patients have three copies of the amyloid precursor protein (APP) gene and develop AD neuropathology at an early age. In addition, Aβ fibrils are toxic in neuronal culture 4 and to some extent when injected into animal brains. 5,6
To date there is no cure or effective therapy for reducing a patient’s amyloid burden or preventing amyloid deposition in AD. Amyloid-related therapeutic strategies include the use of compounds that affect processing of the amyloid-β precursor protein (APP), 7 interfere with fibril formation, or that promote fibril disassembly. 8-10 In vitro studies have shown that monoclonal antibodies raised against the N-terminal region of Aβ can disaggregate Aβ fibrils, maintain Aβ solubility, and prevent Aβ toxicity in cell culture. 11,12 Schenk and colleagues 13 extended these studies to an in vivo situation by vaccinating transgenic (Tg) APP mice (PDAPP) with aged, fibrillar Aβ1-42 in which the treatment resulted in a reduction of amyloid burden and associated pathology. This effect is thought to be mediated by antibodies, because peripherally administered antibodies against Aβ have been shown to reduce brain parenchymal amyloid burden. 14 In addition, intranasal immunization with freshly solubilized Aβ1-40 reduces cerebral amyloid burden. 15 Recently, two studies demonstrated that a vaccination-induced reduction in brain amyloid deposits resulted in cognitive improvements. 16,17 Both groups used the same protocol as Schenk and colleagues, 13 which is injection of fibrillar Aβ1-42 peptide mixed with Freund’s adjuvant. Although these results provide promise for using immunomodulation as a general approach to treat AD, immunization with intact Aβ may not be feasible for humans because of potential toxicity. First, Aβ crosses the blood brain barrier in experimental animals, 18 forms toxic fibrils, 4 and further it can seed fibril formation. 19 Therefore, in humans, it is possible that Aβ1-42 can co-deposit on existing amyloid plaques leading to increased toxicity, and may actually promote plaque formation. Secondly, although autoimmunity has not been observed in the mice vaccinated with human Aβ, it is more likely to occur in humans vaccinated with the human form of Aβ. Overall, the use of peptides that are not fibrillogenic/toxic and not identical to endogenous Aβ should reduce the likelihood of these serious side effects.
We have in earlier reports demonstrated that LPFFD, a nonamyloidogenic peptide with sequence homology to Aβ, blocks fibril formation, 8 and induces in vivo disassembly of fibrillar Aβ deposits. 9 Extension of peptides with these properties to include the major immunogenic sites of Aβ takes advantage of their lack of amyloidogenicity/toxicity while resulting in antibody generation against Aβ, that leads to clearance of Aβ plaques.
We have now designed a highly soluble nonamyloidogenic peptide homologous to Aβ that has a reduced ability to adopt a β-sheet conformation and, therefore, a much lower risk of toxicity in humans. This peptide was designed to have reduced fibrillogenic potential and enhanced immunogenicity while maintaining the major immunogenic sites of Aβ peptides. Accordingly, the peptide contains the first 30 amino acid residues of Aβ with six lysine residues at the N-terminus. Poly-l-lysine enhances immunogenicity, 20 and the coupling of lysine residues to the C-terminus of short Aβ sequences within the 15 to 25 domain of Aβ has recently been proposed by Pallitto and colleagues 21 in the design of anti-β-sheet peptides or Aβ fibrillogenesis inhibitors. The use of potential peptide inhibitors of fibril formation as immunogens has never been previously proposed.
Materials and Methods
Peptides and Other Chemicals
The peptides used (Aβ1-40, Aβ1-42, Aβ1-30-NH2, and K6Aβ1-30-NH2) were synthesized at the Keck Foundation (Yale University, New Haven, CT), as described previously. 9 The peptide used for the immunizations, K6Aβ1-30-NH2, maintains the two major immunogenic sites of Aβ peptides, which are residues 1 to 11 and 22 to 28 of Aβ1-42 based on the antigenic index of Jameson and Wolf, 22 and on preliminary results obtained in our laboratory. All other chemicals were from Sigma, St. Louis, MO, unless otherwise stated.
Secondary Structure Studies
Secondary structure (α-helix, β-sheet, and random coil) of the peptides was evaluated by circular dichroism as described previously. 8,23 Results are expressed as molar ellipticity in units of degree cm 2 dmol−1, and the data were analyzed by the Lincomb and convex constraint algorithms 24 to obtain the percentages of different types of secondary structure.
Study of Amyloid Fibril Formation in Vitro
Aliquots of the peptides prepared in 0.1 mol/L Tris, pH 7.4, were incubated for different times, and their fibril formation compared to that of Aβ1-30-NH2 and Aβ1-42. In vitro fibrillogenesis was evaluated by a fluorometric assay based on the fluorescence emission by thioflavin T, as we have previously described. 8,25 Thioflavin T binds specifically to amyloid and this binding produces a shift in its emission spectrum and a fluorescent enhancement proportional to the amount of amyloid formed. 26
Neurotoxicity
The potential neurotoxicity of K6Aβ1-30-NH2 (1 to 100 μmol/L) was evaluated at 2 and 6 days in a human neuroblastoma cell line (SK-N-SH) using the standard MTT assay as described by the manufacturer (Roche Molecular Biochemicals, Indianapolis, IN). Aβ1-30-NH2, Aβ1-40, and Aβ1-42 were used as control peptides. Briefly, cells were plated at 10,000 cells/100 μl culture medium per well in flat-bottom, 96-well microtiter plates. The cells were allowed to attach to the plate overnight in an incubator (37°C, 5.0% CO2), and then 10 μl of freshly prepared peptide solution (in nanopure H2O) was added. Aβ1-42 was only partially soluble at 100 μmol/L and was, therefore, added as a suspension at that concentration. Subsequent steps were as described in the assay protocol.
Animals
The vaccination was performed in the Tg2576 APP mouse model developed by Karen Hsiao and colleagues. 27 These mice develop Aβ plaques as early as at 11 to 13 months of age. We chose this model over the double Tg APP/PS1 model, 28 because the age of onset and progression of Aβ deposition in the single Tg APP mice more closely resembles that of AD. Age-matched vehicle-treated Tg mice and non-Tg littermates receiving K6Aβ1-30-NH2 were used as controls, and the animals received their first injection at 11 to 13 months, at which time few plaques should already be present. Four mice were in each group. The animals were maintained on a 12-hour light-dark cycle, and had access to food and water ad libitum. The animal care was in accordance with institutional guidelines.
Vaccine Administration
K6Aβ1-30-NH2 was supplied as trifluoroacetic acid salt. The immunization procedure was performed as previously described by Schenk and colleagues 13 except that the peptide was not incubated overnight at 37°C before injection. Briefly, the peptide was dissolved in phosphate-buffered saline (PBS) at a concentration of 2 mg/ml and then mixed 1:1 (v/v) with the adjuvant or PBS. Complete Freund’s adjuvant was used for the first injection, incomplete Freund’s adjuvant for the next three injections, and PBS from the fifth injection forward. The mice received a subcutaneous injection of 100 μl of the mixture (ie, 100 μg/100 μl) followed by a second injection 2 weeks later, and then monthly thereafter.
Antibody Titers
Antibody titers were determined by serial dilutions of sera using an enzyme-linked immunosorbent assay (ELISA) as described previously, 29 in which Aβ or its derivative is coated onto microtiter wells. The titer, defined as the dilution yielding 50% of the maximum signal, was detected by a goat anti-mouse IgG linked to a horseradish peroxidase (Amersham Pharmacia Biotech, Piscataway, NJ), and tetramethyl benzidine (Pierce, Rockford, IL) was the substrate.
Histology
Mice were anesthetized with sodium pentobarbital (150 mg/kg, intraperitoneally), perfused transaortically with phosphate buffer and the brains processed as previously described. 5 The right hemisphere was immersion-fixed in periodate-lysine-paraformaldehyde, whereas the left hemisphere was snap-frozen for measurements of Aβ levels using established ELISA methods. 30,31 Serial coronal sections (40 μm) were cut and five series of sections at 0.2-mm intervals were saved for histological analysis of 1) 6E10-, 2) Congo Red-, 3) interleukin-1β (IL-1β-OX42/tomato lectin-, 4) GFAP-, and 5) cresyl violet-stained sections. 6E10 recognizes Aβ and stains both pre-amyloid and Aβ plaques. 32 Congo Red staining was performed to identify amyloid lesions in these animals. GFAP is a component of the glial intermediate filaments that form part of the cytoskeleton and is found predominantly in astrocytes. Microglia seem to be the major source of IL-1 within the central nervous system, 33 and OX-42 recognizes CD11b on microglia, a rat equivalent of the human C3bi receptor. 34 Tomato lectin binds to poly-_N_-acetyl lactosamine residues and has in neural tissue specific affinity for microglial cells. 35 Both astrocytes and microglia are associated with Aβ deposits. Staining with cresyl violet was performed to determine whether the immunization was causing neuronal shrinkage and/or cell loss in these animals. After sectioning, the series were placed in ethylene glycol cryoprotectant and stored at −20°C until used.
Cresyl Violet and Congo Red: Mounted sections were defatted in xylene and hydrated in a gradient of ethyl alcohol and water series. Staining was performed as previously described. 5,6,8
6E10, GFAP, IL-1β , and OX-42: Staining was performed as previously described. 5,8 Briefly, sections were incubated in 6E10 (kindly provided by Richard Kascsak, Institute for Basic Research) primary antibody that selectively binds to human Aβ at a 1:1000 dilution. A mouse-on-mouse immunodetection kit (Vector Laboratories, Burlingame, CA) was used in which the anti-mouse IgG secondary antibody was used at a 1:2000 dilution. GFAP (1:500; DAKO, Glostrup, Denmark), IL-1β (1:250; Endogen, Rockford, IL), and OX-42 (1:250; Biosource Int., Camarillo, CA) staining was performed the same way as the 6E10 staining, except the secondary antibody was diluted 1:1300. The sections were reacted in 3,3′-diaminobenzidine tetrahydrochloride (DAB) with or without nickel ammonium sulfate (Ni; Mallinckrodt, Paris, KY) intensification. For double-labeling of IL-1β and Aβ plaques, sections were first stained for IL-1β (DAB/Ni; black) in which peroxidase was the enzyme. The plaques (6E10) were then stained using the Vector Red alkaline-phosphatase substrate kit I (Vector).
Tomato Lectin: Sections removed from the cryoprotectant were washed in PBS, 0.3% Triton-X-100 in PBS (PBS-Tx), and then incubated for 30 minutes in 0.3% hydrogen peroxide in PBS to quench endogenous peroxidase activity. After 2 hours of incubation with tomato lectin (10 μg/ml PBS; Vector), sections were washed in PBS-Tx and then reacted with avidin-horseradish peroxidase (Vector) for 1 hour. Subsequent steps were as those used for the antibody staining.
Image Analysis
Immunohistochemistry of tissue sections was quantified with a Bioquant image analysis system, and unbiased sampling was used. 36 All procedures were performed by an individual blind to the experimental condition of the study. Cortical area analyzed was dorsomedially from the cingulate cortex and extended ventrolaterally to the rhinal fissure within the right hemisphere. The area of the grid was 800 × 800 μm 2 and amyloid load was measured in 10 frames per mouse (each: 640 × 480 μm2), chosen randomly. Hippocampal measurements were performed on the entire hippocampus in a similar manner as the cortical analysis. The Aβ burden is defined as the percentage of area in the measurement field occupied by reaction product.
Sandwich ELISA Assay for Soluble Aβ Levels
Before extraction of Aβ from brain tissue, 10% (w/v) homogenates were prepared in tissue homogenization buffer (20 mmol/L Tris, pH 7.4, 250 mmol/L sucrose, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L EGTA). Immediately before use, 1/100 volume of 100 mmol/L phenylmethylsulfonyl fluoride stock solution (in ethanol) and 1/1000 volume of LAP (5 mg each of leupeptin, antipain, and pepstatin A per ml of _N-N_-dimethylformamide) were added to the homogenization buffer. The homogenate was then thoroughly mixed with an equal volume of 0.4% diethylamine/100 mmol/L NaCl, then spun at 135,000 × g for 1 hour at 4°C, and subsequently neutralized with 1/10 volume 0.5 mol/L Tris, pH 6.8. The samples were then aliquoted, flash-frozen on dry ice, and stored at −80°C until loaded onto plates. Soluble Aβ levels were measured in the left hemisphere using monoclonal antibody 6E10 (specific to an epitope present on 1 to 16 amino acid residues of Aβ), rabbit antiserum R162 (specific for Aβ40), and rabbit antiserum 165 (specific for Aβ42) in a double-antibody sandwich ELISA as described previously. 30,31 The optical density (OD) was measured at 450 nm in a micro-ELISA reader. The relationship between OD and Aβ40 or Aβ42 concentrations was determined by a four-parameter logistic log function. Nonlinear curve fitting was performed with KlinetiCalc program (Biotek Instruments, Inc., Winooski, VT) to convert OD of plasma to estimated concentrations. All samples were coded, and the investigators were blinded to group assignment until levels were measured and recorded. The detection limit of the assay is 10 pg/ml for Aβ40 and Aβ42. The percent coefficient of variation normally ranges from 8 to 14% (interassay) and 10 to 18% (intraassay).
Data Analysis
The cell culture data were analyzed by one-way analysis of variance, followed by a Dunnett’s test for post hoc analysis (GraphPad Prism 3.0). An unbiased stereological image analysis system (Bioquant; R&M Biometrics Inc., Nashville, TN) was used to determine the amyloid burden in 6E10-stained brain sections. The data for the amyloid burden and the levels of soluble Aβ within the brain were analyzed by a Student’s _t_-test, two-tailed.
Results
Before conducting the vaccination study it was necessary to confirm that the prototype peptide, KKKKKK-Aβ1-30-NH2, had indeed less β-sheet structure, reduced fibrillogenicity compared to Aβ1-42, and that it was nontoxic in neuronal culture. The secondary structure of these peptides was determined by circular dichroism, and their ability to form amyloid fibrils by a thioflavin-T fluorometric assay. An additional control peptide was Aβ1-30-NH2.
Circular Dichroism Assay
Compounds with high β-sheet content are more toxic and more likely to form fibrils than compounds with low β-sheet content. 37 The peptide with the polylysine at the N-terminus had much less β-sheet content that the amidated Aβ1-30 or Aβ1-42 (Table 1) ▶ .
Table 1.
Circular Dichroism Assay at Different Time Points Shows the Percentages of α-Helix, β-Sheet, and Random Coil as Calculated by the Lincomb and Convex Constraint Algorithms
| Time (h) | Aβ1-42 | Aβ1-30-NH2 | K6-Aβ1-30-NH2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| α-Helix | β-Sheet | Random coil | α-Helix | β-Sheet | Random coil | α-Helix | β-Sheet | Random coil | |
| 0 | 9 | 36 | 55 | 5 | 37 | 58 | 2 | 18 | 79 |
| 24 | 9 | 40 | 51 | 8 | 36 | 56 | 5 | 16 | 78 |
| 96 | 5 | 55 | 40 | 7 | 49 | 44 | 34 | 16 | 50 |
Thioflavin T Assay
Aβ1-42 was already fibrillar at t = 0, whereas Aβ1-30-NH2 gradually formed fibrils throughout time (Figure 1A) ▶ . The relatively high degree of thioflavin T staining of the Aβ1-30-NH2 versus Aβ1-42 after 6 days reflects the known batch-to-batch variability of Aβ peptide fibril formation, 38 as well as some degree of pellet formation by the Aβ1-42 with prolonged incubation. K6Aβ1-30-NH2 did not form fibrils after incubation at 37°C for at least 15 days.
Figure 1.

A: Thioflavin T fluorometric assay. Fibril formation of Aβ1-42, Aβ1-30-NH2, and K6Aβ1-30-NH2 (KKKKKKDAEF… etc) was measured in vitro after incubation at 37°C. K6Aβ1-30-NH2 was the only peptide that did not form fibrils at any of the time points. B and C: Aβ40 and 42 were toxic to human neuroblastoma cells (SK-N-SH) in culture as determined by the MTT assay, whereas K6Aβ30-NH2 had no effect at 2 days (B) and was slightly trophic at 6 days (C). Aβ1-30-NH2 had no effect. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to VEH group (one-way analysis of variance).
Neurotoxicity
To further assess the safety of this vaccination approach the neurotoxicity of K6Aβ1-30-NH2 was determined. K6Aβ1-30-NH2 had no effect on cell viability at 2 days and was slightly trophic at 6 days (P < 0.05), whereas Aβ1-40 and Aβ1-42 were toxic (P < 0.05 to 0.001) to the human neuroblastoma cells (SK-N-SH), compared to vehicle group, as determined by the MTT assay (Figure 1, B and C) ▶ . Aβ1-30-NH2 had no effect. During the incubation period, aggregates were visible under the microscope only in culture wells containing Aβ1-42 (10 to 100 μmol/L).
Antibody Titer
Tg2576 and their non-Tg littermates were vaccinated with K6Aβ1-30-NH2 or vehicle. Almost all of the mice developed antibodies against the immunogen (K6Aβ1-30-NH2), that cross-reacted with Aβ1-40 and Aβ1-42. The titer, defined as the dilution yielding 50% of the maximum signal, ranged from a few hundreds to several thousands (data not shown). Vehicle-treated animals injected with the adjuvant and PBS did not develop antibodies against these three peptides (data not shown). Nontransgenic mice had generally higher titer against all three peptides, and the polyclonal antibodies had higher avidity for the immunogen compared to Aβ1-40 and Aβ1-42. These findings are as expected because the immunogen is based on the human sequence of Aβ that differs in three amino acids from the mouse Aβ, 39 and K6Aβ1-30-NH2 that elicited the immune response should have more binding motifs for antibodies than the intact Aβ peptides.
Amyloid Burden and Associated Histopathology
The mice were killed at 18 to 20 months of age after 7 months of treatment, and their right hemisphere was processed for histology as described. 5 The brain sections were stained with cresyl violet, Congo Red, tomato lectin, and with antibodies against: 1) human Aβ (6E10), microglia (OX-42, IL-1β), and GFAP (anti-GFAP). After K6Aβ1-30-NH2 vaccination, cortical and hippocampal amyloid burden in the Tg mice was reduced by 89 and 81%, respectively (Figure 2, A and B ▶ , and Figure 3, A and B ▶ ), as determined by stereological techniques. The total number of Congo Red-positive amyloid deposits was reduced in the immunized animals; however, the percentage of Aβ-immunoreactive lesions that were Congo Red-positive appeared to remain the same as in the nonimmunized Tg mice. The clearance of the amyloid deposits seemed to be similar in other brain regions. Selected brain sections from a control mouse with high amyloid burden and an immunized mouse with reduced amyloid burden were stained with sera from several immunized and control mice, whose antibody titer ranged from 0 to 3000. As expected, with increasing titer more plaques were stained and the pattern was similar in both mice (data not shown). There was no obvious difference between the Tg treatment groups in cresyl violet staining. Reactive astrocytes were observed associated with all amyloid plaques. Because the vehicle-treated Tg mice had a higher plaque burden, they had more clusters of astrocytes than immunized Tg mice. OX-42 staining of ramified rather than phagocytic (ameboid) microglia was predominantly observed associated with plaques. To verify that this lack of microglial phagocytes was not because of down-regulation of the CD11b receptor, the binding motif of OX-42, 34 sections from all treatment groups were stained with tomato lectin. This particular lectin binds to poly-_N_-acetyl lactosamine residues found predominantly in ramified and phagocytic microglial cells, in addition to endothelial and ependymal cells. 35 These two latter cell types were stained in all of the mice. The microglial lectin staining resembled the OX-42 staining. In other words, in both immunized and control Tg groups, the microglia did not have phagocytic morphology and number of ramified microglial processes per plaque seemed to be similar between immunized and nonimmunized mice (data not shown). On the other hand, IL-1β staining of ramified microglial cells was prominent surrounding the Aβ plaques in the control Tg mice (Figure 2C) ▶ , whereas virtually no IL-1β staining was observed in the immunized mice (Figure 2D) ▶ . Significantly, there was no indication of glomerulonephritis in hematoxylin and eosin-stained kidney sections from the K6Aβ1-30-NH2-treated mice, suggesting that the mice had not developed an autoimmune disorder at the time of sacrifice.
Figure 2.

Coronal sections (original magnification, ×50) stained with 6E10 against Aβ (brown), through the hippocampus and cortex in a Tg control (A) and K6Aβ1-30-treated (B) Tg mouse. C and D are adjacent sections (original magnifications, ×100) double-stained for interleukin-1β (black) that recognizes microglia, and Aβ (red). Note the reduction of amyloid burden in the immunized mouse (B), and the lack of ramified microglia (D) surrounding Aβ plaque in the same mouse, compared to a control mouse (A and C). Scale bars, 100 μm (A and C). Abbreviations: hip, hippocampus; cx, cortex; ml, corpus callosum.
Figure 3.
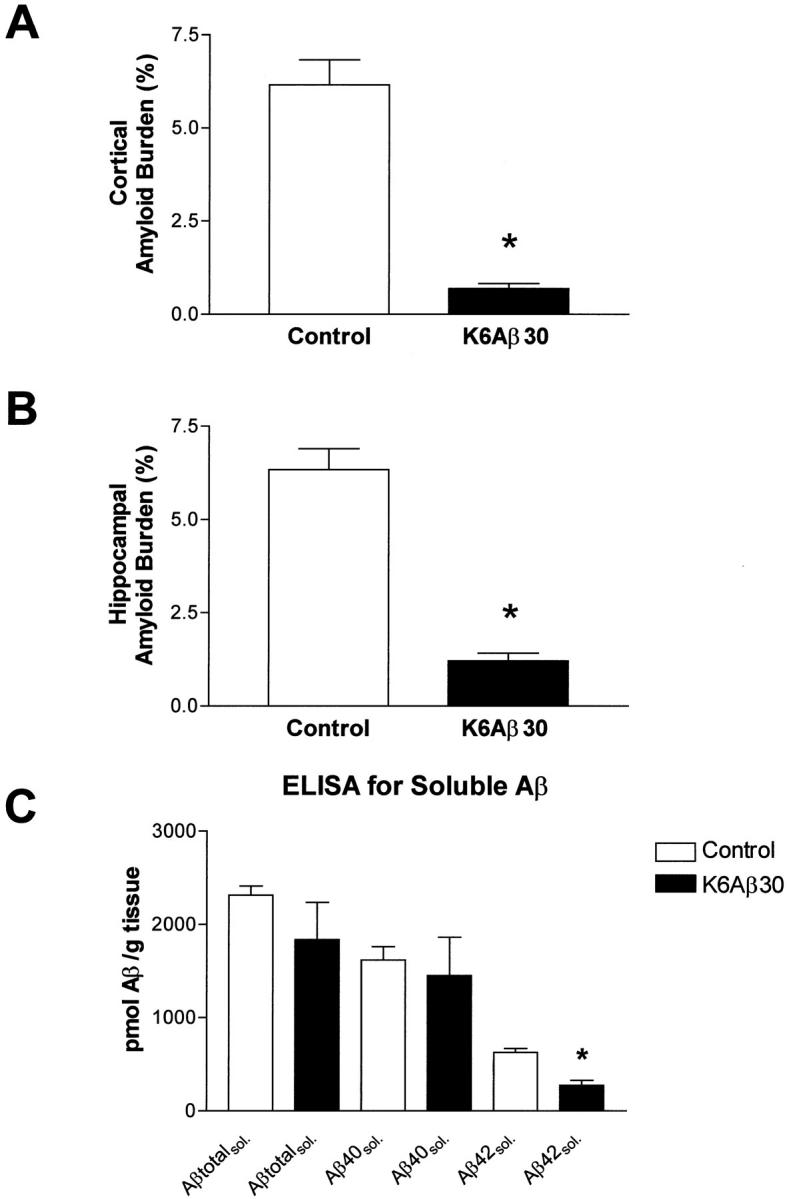
Reduction in cortical (A) and hippocampal (B) amyloid burden (6E10) after 7 months of treatment with K6Aβ1-30-NH2. There was an 89% reduction in cortical amyloid burden (*, P = 0.0002; _t_-test; n = 4 per group) and an 81% reduction in hippocampal amyloid burden (*, P = 0.0001). Soluble Aβ1-42 levels (C) were reduced by 57% within the brains of the vaccinated mice (*, P = 0.0019).
Soluble Aβ by ELISA
Measurements of soluble Aβ levels were performed on the left hemisphere of the mice whose right hemisphere was used for histology. Soluble Aβ1-42 was reduced by 57% after vaccination with K6Aβ1-30-NH2 for 7 months (P = 0.0019), compared to control group (Figure 3C) ▶ . Although there was a trend for reduced levels of soluble total Aβ and Aβ1-40 in the K6Aβ1-30-treated group, the values were not significantly different from the vehicle group.
Overall, immunization in Tg APP mice with nonamyloidogenic/non toxic (low β-sheet content) Aβ homologous peptide results in a similar reduction of amyloid burden as observed by Schenk and colleagues 13 in which they used a fibrillar/toxic (high β-sheet content) Aβ1-42.
Discussion
These findings demonstrate that Aβ aggregates/fibrils are not necessary to elicit a sufficient immune response that results in clearance of Aβ plaques. The use of soluble nonfibrillar/nontoxic Aβ homologous peptides, such as K6Aβ1-30-NH2, is likely to be a safer vaccination approach for humans.
The mechanism of the vaccination-induced reduction in cerebral amyloid burden is not fully understood. However, based on the passive vaccination study by Bard and colleagues 14 it is likely that antibodies have a pivotal role. Interestingly, they demonstrated that there was no correlation between antibody efficacy and affinity for soluble Aβ or binding to aggregated synthetic Aβ peptide. Effective antibodies were, however, able to bind to plaques in unfixed brain sections. Janus and colleagues, 17 using the same protocol as Schenk and colleagues 13 observed that the sera from Aβ-immunized mice preferentially stained dense core plaques rather than diffuse Aβ deposits suggesting that the antibodies may have a higher affinity for β-sheet Aβ. Based on these somewhat contradictory findings more studies are needed on Aβ-antibody interactions that may give insight into the mechanism of antibody-mediated Aβ clearance. It is unlikely that these antibodies are affecting the production of Aβ because they do not recognize APP. 15 It is more probable that the antibodies enhance clearance of Aβ through microglial activation after antibody binding to Aβ plaques. 13,14 Their effect may also in part be because of binding to soluble Aβ within the brain, that alters the equilibrium between deposited Aβ versus soluble Aβ. Given the numerous reports that show that Aβ can bi-directionally cross the blood brain barrier, 40-47 the vaccination effect may be in part mediated through binding of the antibodies to soluble Aβ in peripheral fluids. Subsequent reduction in peripheral Aβ levels may alter the equilibrium between Aβ found within and outside the central nervous system that may result in efflux of Aβ out of this compartment. This is particularly likely since most of the antibodies are circulating in peripheral tissues and the tg mice have high levels of Aβ not only within the central nervous system but also in the periphery. A recent report shows that in the Tg2576 mice, plasma levels of Aβ decrease as cerebral plaque burden increases. 48 This suggests an interaction between these two compartments that can be manipulated.
Interestingly, in the behavioral vaccination study by Morgan and colleagues, 16 they observed a partial reversal in cognitive deficits in APP/PS1 mice although cerebral amyloid burden as measured by immunohistochemistry was not significantly reduced. As pointed out by Morgan and colleagues, 16 soluble Aβ has been proposed to cause synapse loss in APP Tg mice, as some Tg lines have reduced synaptophysin staining in the dentate gyrus without Aβ deposits. 49 Therefore, a possible explanation for the cognitive improvement in the immunized mice in the absence of reduced plaque burden, was a decrease in soluble Aβ, although this potential connection was not measured in their study. 16 Our results show that after 7 months of treatment, the 81 to 89% reduction in amyloid plaque burden is associated with a 57% reduction in soluble Aβ1-42 within the brain, whereas the reduction in soluble total Aβ and Aβ1-40 was not significantly different from the control group. In other words, soluble Aβ is reduced less than plaque Aβ. However, detailed time course studies must be performed to determine further any changes in the equilibrium between plaque Aβ and soluble Aβ both within and outside the brain. Our findings indirectly demonstrate the importance of Aβ1-42 for plaque maintenance. Overall, it is likely that several different mechanisms may result in reduction of cerebral amyloid burden, depending on the animal model and the properties of the peptide used for immunization.
Numerous studies have suggested that amyloid deposition can activate inflammatory cascades in the brain, such as increased IL-1β production associated with neuronal injury and death. 6,50 It is possible that our immunization with Aβ homologous peptides could also stimulate such negative inflammatory pathways, along with amyloid reduction. However, we observed few phagocytic microglia in our immunized animals, as identified by OX-42 immunoreactivity or tomato lectin binding. This is not surprising because after 7 months of treatment most of the plaques have been cleared. Furthermore, in the immunized group of mice microglial IL-1β staining was virtually absent, whereas numerous ramified IL-1β-positive microglia were associated with the plaques in the control Tg group. We have previously reported a similar lack of IL-1β staining in a rat model of cerebral amyloidosis after treatment with a β-sheet breaker peptide. 9 However, in that acute study (16 days) this effect was associated with an extensive increase in phagocytic OX-42 staining, indicating that phagocytes do not express IL-1β. Our current observations may suggest that an important effect of the immunization is reduced inflammation within the brain.
We are currently further exploring the therapeutic potential of K6Aβ1-30-NH2 and related peptides by using aluminum-based adjuvants that are approved for use in humans, and by assessing the behavioral status of the mice before and during the treatment period. Overall, our approach has a much lower risk of leading to toxic effects in humans, than the use of Aβ1-40/42, while maintaining the therapeutic potential of immunization for AD.
Footnotes
Address reprint requests to Einar M. Sigurdsson, Ph.D., New York University School of Medicine, Room TH427, 550 First Ave., New York, NY 10016. E-mail: einar.sigurdsson@med.nyu.edu.
Supported by Metlife Award (to B. F.) and National Institutes of Health grants AG15408, AR02594, AG17617, and AG05891.
References
- 1.Golde TE, Eckman CB, Younkin SG: Biochemical detection of Aβ isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochim Biophys Acta 2000, 1502:172-187 [DOI] [PubMed] [Google Scholar]
- 2.Yankner BA: The pathogenesis of Alzheimer’s disease. Is amyloid β-protein the beginning or the end? Ann N Y Acad Sci 2000, 924:26-28 [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ: Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid β-protein. Ann N Y Acad Sci 2000, 924:17-25 [DOI] [PubMed] [Google Scholar]
- 4.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL: Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science 1989, 245:417-420 [DOI] [PubMed] [Google Scholar]
- 5.Sigurdsson EM, Lorens SA, Hejna MJ, Dong XW, Lee JM: Local and distant histopathological effects of unilateral amyloid-β 25-35 injections into the amygdala of young F344 rats. Neurobiol Aging 1996, 17:893-901 [DOI] [PubMed] [Google Scholar]
- 6.Sigurdsson EM, Lee JM, Dong XW, Hejna MJ, Lorens SA: Bilateral injections of amyloid-β 25-35 into the amygdala of young Fischer rats: behavioral, neurochemical, and time dependent histopathological effects. Neurobiol Aging 1997, 18:591-608 [DOI] [PubMed] [Google Scholar]
- 7.Dovey HF, John V, Anderson JP, Chen LZ, de Saint AP, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, Hu KL, Johnson-Wood KL, Kennedy SL, Kholodenko D, Knops JE, Latimer LH, Lee M, Liao Z, Lieberburg IM, Motter RN, Mutter LC, Nietz J, Quinn KP, Sacchi KL, Seubert PA, Shopp GM, Thorsett ED, Tung JS, Wu J, Yang S, Yin CT, Schenk DB, May PC, Altstiel LD, Bender MH, Boggs LN, Britton TC, Clemens JC, Czilli DL, Dieckman-McGinty DK, Droste JJ, Fuson KS, Gitter BD, Hyslop PA, Johnstone EM, Li WY, Little SP, Mabry TE, Miller FD, Ni B, Nissen JS, Porter WJ, Potts BD, Reel JK, Stephenson D, Su Y, Shipley LA, Whitesitt CA, Yin T, Audia JE: Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J Neurochem 2001, 76:173-181 [DOI] [PubMed] [Google Scholar]
- 8.Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B: β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer’s therapy. Nat Med 1998, 4:822-826 [DOI] [PubMed] [Google Scholar]
- 9.Sigurdsson EM, Permanne B, Soto C, Wisniewski T, Frangione B: In vivo reversal of amyloid-β lesions in rat brain. J Neuropath Exp Neurol 2000, 59:11-17 [DOI] [PubMed] [Google Scholar]
- 10.Findeis MA: Approaches to discovery and characterization of inhibitors of amyloid β-peptide polymerization. Biochim Biophys Acta 2000, 1502:76-84 [DOI] [PubMed] [Google Scholar]
- 11.Solomon B, Koppel R, Frankel D, Hanan-Aharon E: Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc Natl Acad Sci USA 1997, 94:4109-4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solomon B, Koppel R, Hanan E, Katzav T: Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer β-amyloid peptide. Proc Natl Acad Sci USA 1996, 93:452-455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P: Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400:173-177 [DOI] [PubMed] [Google Scholar]
- 14.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T: Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 2000, 6:916-919 [DOI] [PubMed] [Google Scholar]
- 15.Weiner HL, Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, Issazadeh S, Hancock WW, Selkoe DJ: Nasal administration of amyloid-β peptide decreases cerebral amyloid burden in a mouse model of Alzheimer’s disease. Ann Neurol 2000, 48:567-579 [PubMed] [Google Scholar]
- 16.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW: Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 2000, 408:982-985 [DOI] [PubMed] [Google Scholar]
- 17.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D: Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 2000, 408:979-982 [DOI] [PubMed] [Google Scholar]
- 18.Zlokovic B: Can blood-brain barrier play a role in the development of cerebral amyloidosis and Alzheimer’s disease pathology. Neurobiol Dis 1997, 4:23-26 [DOI] [PubMed] [Google Scholar]
- 19.Jarrett JT, Berger EP, Lansbury PTJ: The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32:4693-4697 [DOI] [PubMed] [Google Scholar]
- 20.Werdelin O: Fact and speculation on the function of immune response genes in antigen presentation. Scand J Immunol 1981, 14:623-629 [DOI] [PubMed] [Google Scholar]
- 21.Pallitto MM, Ghanta J, Heinzelman P, Kiessling LL, Murphy RM: Recognition sequence design for peptidyl modulators of β-amyloid aggregation and toxicity. Biochemistry 1999, 38:3570-3578 [DOI] [PubMed] [Google Scholar]
- 22.Jameson BA, Wolf H: The antigenic index: a novel algorithm for predicting antigenic determinants. Comput Appl Biosci 1988, 4:181-186 [DOI] [PubMed] [Google Scholar]
- 23.Soto C, Golabek A, Wisniewski T, Castano EM: Alzheimer’s β-amyloid peptide is conformationally modified by apolipoprotein E in vitro. Neuroreport 1996, 7:721-725 [DOI] [PubMed] [Google Scholar]
- 24.Perczel A, Park K, Fasman GD: Analysis of the circular dichroism spectrum of proteins using the convex constraint algorithm: a practical guide. Anal Biochem 1992, 203:83-93 [DOI] [PubMed] [Google Scholar]
- 25.Wisniewski T, Castano E, Ghiso J, Frangione B: Cerebrospinal fluid inhibits Alzheimer β-amyloid fibril formation in vitro. Ann Neurol 1993, 34:631-633 [DOI] [PubMed] [Google Scholar]
- 26.LeVine H: Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci 1993, 2:404-410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G: Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274:99-102 [DOI] [PubMed] [Google Scholar]
- 28.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K: Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 1998, 4:97-100 [DOI] [PubMed] [Google Scholar]
- 29.Jimenez-Huete A, Alfonso P, Soto C, Albar JP, Rabano A, Ghiso J, Frangione B, Mendez E: Antibodies directed to the carboxyl terminus of amyloid β-peptide recognize sequence epitopes and distinct immunoreactive deposits in Alzheimer’s disease brain. Alzheimers Rep 1998, 1:41-47 [Google Scholar]
- 30.Mehta PD, Dalton AJ, Mehta SP, Kim KS, Sersen EA, Wisniewski HM: Increased plasma amyloid β protein 1-42 levels in Down syndrome. Neurosci Lett 1998, 241:13-16 [DOI] [PubMed] [Google Scholar]
- 31.Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM: Plasma and cerebrospinal fluid levels of amyloid β proteins 1-40 and 1-42 in Alzheimer disease. Arch Neurol 2000, 57:100-105 [DOI] [PubMed] [Google Scholar]
- 32.Kim KS, Wen GY, Bancher C, Chen CMJ, Sapienza V, Hong H, Wisniewski HM: Detection and quantification of amyloid β-peptide with 2 monoclonal antibodies. Neurosci Res Comm 1990, 7:113-122 [Google Scholar]
- 33.Schobitz B, De Kloet ER, Holsboer F: Gene expression and function of interleukin 1, interleukin 6 and tumor necrosis factor in the brain. Prog Neurobiol 1994, 44:397-432 [DOI] [PubMed] [Google Scholar]
- 34.Robinson AP, White TM, Mason DW: Macrophage heterogeneity in the rat as delineated by two monoclonal antibodies MRC OX-41 and MRC OX-42, the latter recognizing complement receptor type 3. Immunology 1986, 57:239-247 [PMC free article] [PubMed] [Google Scholar]
- 35.Acarin L, Vela JM, Gonzalez B, Castellano B: Demonstration of poly-N-acetyl lactosamine residues in ameboid and ramified microglial cells in rat brain by tomato lectin binding. J Histochem Cytochem 1994, 42:1033-1041 [DOI] [PubMed] [Google Scholar]
- 36.West MJ: Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 1999, 22:51-61 [DOI] [PubMed] [Google Scholar]
- 37.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW: In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res 1991, 563:311-314 [DOI] [PubMed] [Google Scholar]
- 38.Soto C, Castano EM, Kumar RA, Beavis RC, Frangione B: Fibrillogenesis of synthetic amyloid-beta peptides is dependent on their initial secondary structure. Neurosci Lett 1995, 200:105-108 [DOI] [PubMed] [Google Scholar]
- 39.Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP: Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res Mol Brain Res 1991, 10:299-305 [DOI] [PubMed] [Google Scholar]
- 40.Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B: Blood-brain barrier transport of circulating Alzheimer’s amyloid β. Biochem Biophys Res Commun 1993, 197:1034-1040 [DOI] [PubMed] [Google Scholar]
- 41.Maness LM, Banks WA, Podlisny MB, Selkoe DJ, Kastin AJ: Passage of human amyloid β-protein 1-40 across the murine blood-brain barrier. Life Sci 1994, 55:1643-1650 [DOI] [PubMed] [Google Scholar]
- 42.Martel CL, Mackic JB, McComb JG, Ghiso J, Zlokovic BV: Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer’s amyloid β in guinea pigs. Neurosci Lett 1996, 206:157-160 [DOI] [PubMed] [Google Scholar]
- 43.Poduslo JF, Curran GL, Haggard JJ, Biere AL, Selkoe DJ: Permeability and residual plasma volume of human, Dutch variant, and rat amyloid β-protein 1-40 at the blood-brain barrier. Neurobiol Dis 1997, 4:27-34 [DOI] [PubMed] [Google Scholar]
- 44.Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV: Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid β peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem 1998, 70:210-215 [DOI] [PubMed] [Google Scholar]
- 45.Poduslo JF, Curran GL, Sanyal B, Selkoe DJ: Receptor-mediated transport of human amyloid β-protein 1-40 and 1-42 at the blood-brain barrier. Neurobiol Dis 1999, 6:190-199 [DOI] [PubMed] [Google Scholar]
- 46.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV: Clearance of Alzheimer’s amyloid-β(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000, 106:1489-1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ji Y, Permanne B, Sigurdsson EM, Holtzman DM, Wisniewski T: Amyloid β40/42 clearance across the blood-brain barrier following intra-ventricular injections in wild-type, apoE knock-out and human apoE3 or E4 expressing transgenic mice. J Alzheimer’s Dis 2001, 3:23-30 [DOI] [PubMed] [Google Scholar]
- 48.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG: Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 2001, 21:372-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L: High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000, 20:4050-4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T: Inflammation and Alzheimer’s disease. Neurobiol Aging 2000, 21:383-421 [DOI] [PMC free article] [PubMed] [Google Scholar]