Accumulated Clonal Genetic Alterations in Familial and Sporadic Colorectal Carcinomas with Widespread Instability in Microsatellite Sequences (original) (raw)
Abstract
A subset of hereditary and sporadic colorectal carcinomas is defined by microsatellite instability (MSI), but the spectra of gene mutations have not been characterized extensively. Thirty-nine hereditary nonpolyposis colorectal cancer syndrome carcinomas (HNPCCa) and 57 sporadic right-sided colonic carcinomas (SRSCCa) were evaluated. Of HNPCCa, 95% (37/39) were MSI-positive as contrasted with 31% (18/57) of SRSCCa (P < 0.000001), but instability tended to be more widespread in SRSCCa (P = 0.08). Absence of nuclear hMSH2 mismatch repair gene product by immunohistochemistry was associated with germline hMSH2 mutation (P = 0.0007). The prevalence of K-ras proto-oncogene mutations was similar in HNPCCa and SRSCCa (30% (11/37) and 30% (16/54)), but no HNPCCa from patients with germline hMSH2 mutation had codon 13 mutation (P = 0.02), and two other HNPCCa had multiple K-ras mutations attributable to subclones. 18q allelic deletion and p53 gene product overexpression were inversely related to MSI (P = 0.0004 and P = 0.0001, respectively). Frameshift mutation of the transforming growth factor β type II receptor gene was frequent in all MSI-positive cancers (85%, 46/54), but mutation of the E2F-4 transcription factor gene was more common in HNPCCa of patients with germline hMSH2 mutation than in those with germline hMLH1 mutation (100% (8/8) versus 40% (2/5), P = 0.04), and mutation of the Bax proapoptotic gene was more frequent in HNPCCa than in MSI-positive SRSCCa (55% (17/31) versus 13% (2/15), P = 0.01). The most common combination of mutations occurred in only 23% (8/35) of evaluable MSI-positive cancers. Our findings suggest that the accumulation of specific genetic alterations in MSI-positive colorectal cancers is markedly heterogeneous, because the occurrence of some mutations (eg, ras, E2F-4, and Bax genes), but not others (eg, transforming growth factor β type II receptor gene), depends on the underlying basis of the mismatch repair deficiency. This genetic heterogeneity may contribute to the heterogeneous clinical and pathological features of MSI-positive cancers.
The molecular genetics of colorectal carcinoma are among the best understood of the common human neoplasms (reviewed in Refs. 1 to 3 ). In most colorectal carcinomas, inactivation of the APC (adenomatous polyposis coli) gene initiates colorectal neoplasia leading to dysplasia, commonly in the form of an adenoma. In patients with familial adenomatous polyposis, germline inactivation of APC appears to be followed by its somatic inactivation in colorectal epithelium, typically leading to large numbers of adenomas. During progression through the adenoma-adenocarcinoma sequence, additional alterations accumulate in proto-oncogenes including ras and in tumor suppressor genes on chromosome 18q (DCC, Smad2, or Smad4, reviewed in Refs. 4 to 6 ) and 17p (p53). The alterations each appear to provide a selective growth advantage. These alterations are found in various combinations in usual colorectal carcinomas, and extensive allelic deletions and altered total DNA content by flow cytometry or related methods are frequent.
About 15% of colorectal cancers are characterized by microsatellite instability (MSI), also termed DNA replication errors or ubiquitous somatic mutations (reviewed in Refs. 7 to 16 ). Inactivation of one of a group of genes whose products participate in postreplicative repair of nucleotide mismatches leads to insertions and deletions of nucleotides in intrinsically unstable repeated sequences (ie, microsatellites) throughout the genome because of defective repair of the slippage mistakes made by DNA polymerases. MSI-positive tumors thus accumulate numerous frameshift mutations but also have a mutator phenotype that increases both base substitution mutations and frameshift mutations in expressed genes. In patients with hereditary nonpolyposis colorectal cancer syndrome (HNPCC, Warthin-Lynch syndrome; reviewed in Refs. 17 and 18 ), germline mutation of hMSH2 (human MutS homolog 2), hMLH1 (human MutL homolog 1), hPMS1 or hPMS2 (human postmeiotic segregation 1 and 2), or the GTBP (guanine/thymidine mismatch-binding protein)/hMSH6 gene predispose to tumorigenesis. In addition to germline and somatic alterations in these genes in HNPCC, somatic inactivation alone of mismatch repair genes have been identified as a cause of MSI in sporadic tumors. Loss of immunohistochemical expression of hMSH2 and hMLH1 gene products in MSI-positive tumors has been reported. 19-23 MSI-positive colorectal carcinomas in both the inherited and sporadic settings have unusual pathological manifestations, including right-sided predominance and high frequency of large size; poorly differentiated, medullary, or mucinous histopathological type; and prominent lymphoid inflammatory response. 24-31
Tumors with widespread MSI have extensive subtle alterations in repeated nucleotide sequences, including those within the coding regions of genes. Inactivation of the APC gene by subtle mutation is common in MSI-positive neoplasms, 32 although some studies have reported low rates of APC mutation. 33,34 In clear contrast to usual colorectal cancer, mutations are frequent in MSI-positive tumors among mononucleotide or other small repeats within the gene for the transforming growth factor β type II receptor (TGFβ RII), 33-43 the E2F-4 transcription factor gene (reviewed in Ref. 44 ), 45-47 the insulin-like growth factor II receptor gene, 48,49 the hMSH3 and hMSH6 mismatch repair genes, 47,50,51 and the Bax gene for a BCL-2-related protein 50,52-54 that promotes apoptosis (reviewed in Ref. 55 ). Because of the high frequency of mutation in microsatellite sequences throughout the genome, it is not clear that the intragenic mutations are causally related to tumor progression, and many genes with repeat sequences do not show instability. 54,56 In addition, allelic losses are infrequent, and total DNA content of tumor cells is typically in the normal range. Little is known, however, about the pattern in MSI-positive tumors of accumulated alterations in the oncogenes and suppressor genes that are important in the progression of usual colorectal neoplasia, because only small numbers of MSI-positive tumors have been studied, and conflicting results have been reported. For example, ras proto-oncogene mutations in MSI-positive colorectal carcinomas were reported to occur at frequencies similar to microsatellite-stable (MSS) cancers in some series 28,34,40,57,58 but at low frequency in others. 33,43,59-61 For p53 alterations, conflicting reports of similar 27,42,57,58 or lower 28,30,33,34,43,55,59-63 frequencies of abnormalities have also appeared. Furthermore, alterations such as loss of heterozygosity of chromosome 8p have been reported at different frequency in sporadic MSI-positive tumors than in HNPCC MSI-positive tumors, 33 and frequency of mutation of some genes (eg, E2F-4) differed with severity of MSI. 46
To address these uncertainties about genetic alterations in MSI-positive tumors, we studied the spectra of mutations in a large series of colorectal carcinomas from patients with HNPCC or sporadic colonic cancer. We evaluated MSI status; immunohistochemical expression of hMSH2 and hMLH1 gene products; mutation of the Kirsten ras proto-oncogene; allelic deletion of the long arm of chromosome 18q where the DCC, DPC4/Smad4, and JV-18/MADR2/Smad2 genes reside; overexpression of p53 gene product and mutation of the p53 gene; and mutations in nucleotide repeat sequences in the coding regions of the TGFβ RII, E2F-4, and Bax genes. The results have implications for the diagnosis and treatment of MSI-positive tumors as well as understanding of the biology of colorectal neoplasia.
Materials and Methods
Patients and Tumor Specimens
We studied 39 colorectal cancers from patients in 20 families, that met International Collaborative Group criteria for HNPCC 64 and/or had germline mutation of hMSH2 or hMLH1, 65,66 and 57 sporadic right-sided colonic cancers located proximal to the splenic flexure (Table 1) ▶ . The HNPCC cancers (HNPCCa) were obtained for collaborative studies of the genetics of HNPCC from registries at the Department of Preventive Medicine/Public Health, Creighton University School of Medicine (Omaha, NE) (n = 19); Department of Pathology, University of Auckland School of Medicine (Auckland, NZ) (n = 5); Memorial University of Newfoundland (St. John’s, Newfoundland, Canada) (n = 5); and The Johns Hopkins University School of Medicine (Baltimore, MD) (n = 10). Germline mutation of the known mismatch repair genes in an affected family member was evaluated previously by in vitro synthetic protein assays and by sequencing of hMSH2, hMLH1, hPMS1, hPMS2, and GTBP in 17 of the families: 7 had hMSH2 mutation, 5 had hMLH1 mutation, and 5 had no mutation found. 67
Table 1.
Summary of Patients
| HNPCCa (n = 39) | SRSCCa (n = 57) | |
|---|---|---|
| Mean age± SD | 42± 16 | 68± 14 |
| Age range | 19 to 76 | 25 to 90 |
| Gender ratio, M/F (% male) | 26/13 (67) | 25/32 (44) |
| Location of cancer: cecum through splenic flexure (%) | 65 | 100 |
| Histopathology of cancer: poorly differentiated or mucinous (%) | 57 | 21 |
| Stage II or III (% of patients with available data) | 89 | 100 |
Sporadic right-sided colonic cancers (SRSCCa) of stage II (direct extension through the muscularis propria without identified metastasis to regional lymph nodes or distant sites) and stage III (metastasis to regional lymph nodes but not distant sites) were chosen for comparison with HNPCCa. This control group was selected because of the strong right-sided predominance of colorectal cancer in HNPCC; 17,18 the known differences in phenotype and genotype between right- and left-sided sporadic colonic cancers, including higher frequency of MSI in right-sided tumors; 24-26,59,68,69 and the better prognosis of patients with HNPCCa. 17,70-73 SRSCCa were identified prospectively and consecutively for the Bowel Tumor Working Group Bank at The Johns Hopkins Hospital between 1986 and 1990 for a study of prognostic markers, as reported previously. 74
Three colon cancer cell lines with known MSI status and mismatch repair gene mutation status were used as controls in various analyses: LoVo is MSI positive with reported homozygous deletion of the hMSH2 gene, 75 HCT116 is MSI positive with mutation of hMLH1, and SW480 is MSS with no known mutation of either mismatch repair gene.
Microdissection and DNA Extraction
For all tumors, areas of carcinoma and nonneoplastic control tissue were microdissected from numbered, routine buffered, formalin-fixed, paraffin-embedded tissue sections. Areas of cellular tumor with minimal stroma and inflammatory cells were selected, and multiple areas with high cellularity from each slide were combined, favoring detection of clonal abnormalities. DNA was extracted as in our previous studies. 74,76 Frozen tissue was available for 11 MSI-positive SRSCCa; cryostat microdissection and DNA extraction for restriction fragment length polymorphism analysis were done as described previously. 77,78 DNA was prepared from fresh aliquots and from histopathological sections of formalin-fixed, paraffin-embedded cell blocks of LoVo, HCT116, and SW480.
MSI Analysis
Five noncoding polymorphic dinucleotide repeat sequences on the long arm of chromosome 18 (D18S69, D18S64, D18S55, D18S61, and D18S58 from centromere to telomere) and the noncoding nonpolymorphic polyadenine (poly(A)) tract in the fifth intron of the hMSH2 gene (BAT26) were chosen for determination of MSI status by polymerase chain reaction (PCR) amplification, as in our previous studies. 36,74 The normal alleles were usually represented by a major band accompanied by a few minor bands. Mobility shift of PCR products from tumor DNA as compared with corresponding nonneoplastic tissue was determined for each marker independently (Figures 1 and 2) ▶ ▶ . MSI-positive status of a tumor was defined by mobility shift in at least two of five or six markers. The MSI status of the SRSCCa based on the dinucleotide repeat sequences was included in our previous publications. 26,74
Figure 1.
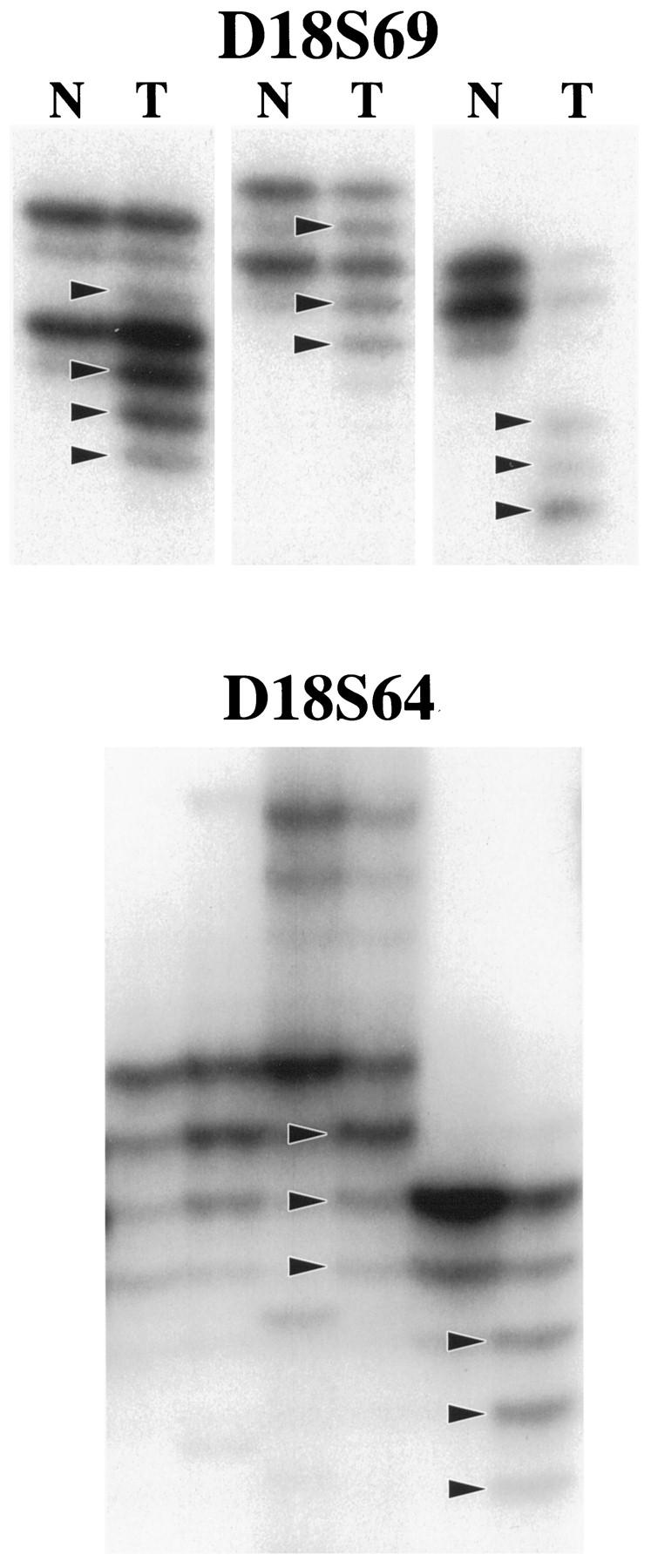
Illustration of MSI evaluation with two noncoding polymorphic CA dinucleotide repeat sequences on the long arm of chromosome 18 (D18S69 and D18S64). Alleles with shifts of size due to deletion of repeats in tumor DNA (T lanes) as compared with matched nonneoplastic mucosal DNA (N lanes) are indicated (arrowheads).
Figure 2.

Illustration of MSI evaluation with the noncoding nonpolymorphic BAT26 poly(A) tract in the fifth intron of the hMSH2 gene. Alleles with shifts of size due to deletion of nucleotides in tumor DNA are indicated (arrowheads). Note the consistent size of the wild-type allele (wt) in these tumor specimens.
Immunohistochemistry for hMSH2 and hMLH1 Gene Products
Sections (6 μm) of formalin-fixed, paraffin-embedded cancers were deparaffinized with xylenes for 30 minutes and rehydrated using graded ethanols. Antigen retrieval was performed using a heat-induced epitope retrieval method. 79 Immunoperoxidase staining using diaminobenzidine as chromogen was performed with the TechMate 1000 automatic staining system (Ventana/BioTek Solutions, Tucson, AZ). Immunoglobulin G mouse monoclonal antibody to hMSH2 gene product (Ab-2, Oncogene Science, Cambridge, MA) and two immunoglobulin G mouse monoclonal antibodies to hMLH1 gene product (Ab-1, Pharmingen, San Diego, CA, and NA28, Calbiochem, Cambridge, MA) were used. Counterstain was light hematoxylin.
To establish utility of antibodies and dilutions for use, histopathological sections of buffered formalin-fixed, paraffin-embedded cell blocks of the LoVo, HCT116, and SW480 colon cancer cell lines were prepared. The sections were stained along with eight MSI-positive HNPCCa from patients with known germline mutation status of the hMSH2 and hMLH1 mismatch repair genes (four with hMSH2 mutation and four with hMLH1 mutation). Nuclear staining for hMSH2 gene product was absent from the LoVo cell line (Figure 3) ▶ with reported homozygous deletion of the hMSH2 gene 75 and absence of hMSH2 gene product expression by Western blot, 80 and from the four HNPCCa of patients with known germline hMSH2 mutation. By contrast, nuclear staining for hMSH2 gene product was retained in the HCT116 cell line with hMLH1 mutation, the SW480 cell line without MSI, and the four HNPCCa from patients with germline hMLH1 mutation. Dilution of 1:30 was selected for use. By contrast, nuclear staining for hMLH1 gene product was retained unexpectedly over a range of dilutions in the HCT116 cell line and the four HNPCCa from patients with known germline hMLH1 mutation. Therefore, the hMLH1 antibodies were not used further in this study.
Figure 3.
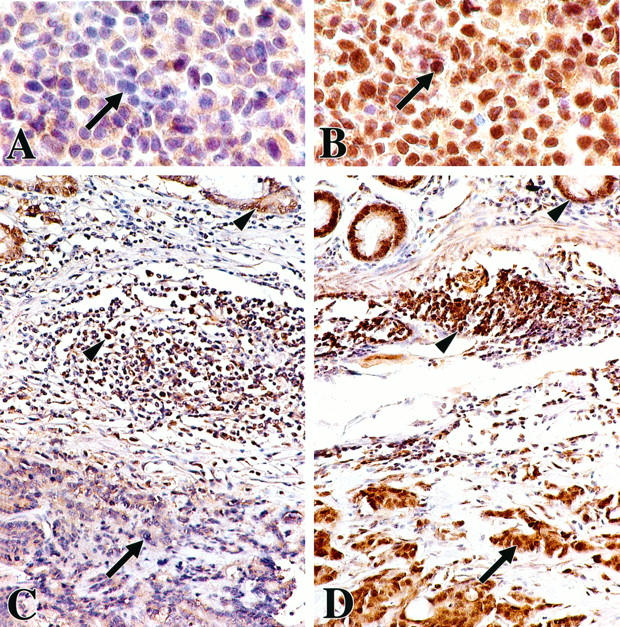
Immunohistochemistry for hMSH2 gene product expression. Nuclear staining is absent in the LoVo colon cancer cell line (A) with reported homozygous deletion of the hMSH2 gene 75 and absence of expression of hMSH2 gene product by Western blotting. 80 By contrast, nuclear staining is intense in the HCT116 colon cancer cell line with known hMHL1 mutation (B). Cytoplasmic staining is evident in both cell lines. Poorly differentiated colonic carcinomas and overlying mucosa from HNPCC patients with known germline hMSH2 mutation (C) and known germline hMLH1 mutation (D) show absence of nuclear staining of cancer cells (arrow) in C but intense staining (arrow) in D. Cytoplasmic staining is evident in both cancers. Nuclear staining of crypt epithelial cells in the proliferative compartment and of cells in germinal centers of lymphoid nodules (arrowheads) serve as internal positive controls.
Staining for hMSH2 gene product in tumor cell nuclei and in cytoplasm were each evaluated as present or absent in numbered slides by a gastrointestinal pathologist (SRH) who had no knowledge of the results of genetic analysis. Nuclear staining in crypt epithelial cells of the proliferative compartment and/or lymphoid cells in the germinal center of lymphoid nodules in each slide served as internal positive controls for comparison with cancer nuclei. Absence of staining in the smooth muscle of muscularis propria defined satisfactory negative background.
The immunohistochemistry slides were interpreted on two occasions 3 months apart. The results were concordant in 89% (67/75) of all cases (κ statistic 0.61, good reproducibility) and in 81% (39/48) of MSI-positive cancers (κ statistic 0.53, good reproducibility). For the discordant cases, all of which had intratumoral heterogeneity and were found to be MSI positive when the classification was uncoded, immunohistochemistry was repeated and the discrepancies were resolved by re-review of the slide sets for final classification and data entry before the MSI status and germline mutation status were known.
K-ras Proto-Oncogene Mutations
All possible sequence alterations of the K-ras proto-oncogene in codons 12 and 13 were determined. Exon 1 was amplified as in our previous studies, 81,82 and the resulting PCR product from each tumor was sequenced using the SequiTherm EXCEL DNA sequencing kit (Perkin-Elmer Corp., Norwalk, CT).
Chromosome 18q Allelic Loss
Loss of the long arm of chromosome 18 in MSS tumors was defined by the complete or partial loss of the polymorphic alleles in dinucleotide repeats on the long arm of chromosome 18, as described previously. 74 The results for the SRSCCa were included in our previous publication. 74 Because 18q allelic loss cannot be accurately assessed by use of dinucleotide repeats in MSI-positive cancers, chromosome 18q loss was examined by loss of heterozygosity in restriction fragment length polymorphisms by Southern blot analysis of DNA isolated from cryostat sections of frozen tissue available in 11 MSI-positive SRSCCa. DCC 1.9, SAM 1.1, Josh 4.4, and p15–65 were used after digestion with _Hin_dIII, _Eco_RI, _Pst_I, and _Eco_RI-_Sac_I restriction endonucleases, respectively, as described previously. 77,78
Immunostaining for p53 Gene Product
Detection of p53 gene product overexpression by immunohistochemistry serves as an indicator of p53 gene mutation with approximately 75% overall accuracy in colorectal neoplasms (reviewed in Ref. 83 ) because of the prolonged half-life of most mutated proteins in these tumors. Mouse monoclonal antibody DO7 against p53 (DAKO Corp., Carpinteria, CA) was used at 1:100 dilution in the TechMate 1000, and p53 labeling index was evaluated by computerized image analysis (CAS 200 system, Becton-Dickinson, Elmhurst, IL), as in our previous study. 84
Sequencing of p53 Gene
A subset of p53 mutations is characterized by intragenic insertions and deletions that truncate the protein product and prevent accumulation of the mutated protein. 85 Because insertions and deletions are common alterations in MSI-positive tumors, sequencing of exons 5 through 8 of the p53 gene was attempted in DNA extracted from 16 MSI-positive HNPCCa and 11 MSI-positive SRSCCa. A single 1.8-kb genomic fragment was generated by PCR using sense and antisense primers, as in our previous studies, 86,87 and the PCR products were sequenced using the SequiTherm EXCEL DNA sequencing kit (Perkin-Elmer). Nucleotide sequence alterations could not be characterized because of the presence of only subtle abnormal bands that were difficult to interpret as mutations. Interpretation was complicated by the expected retention of the wild-type allele in MSI-positive cancers, which have very infrequent allelic loss, and by the expected contribution of wild-type DNA from contaminating intraepithelial lymphocytes, which were evident by histopathology of many of the microdissected specimens.
Mutation in the Gene for TGFβ RII
Analysis of the poly(A) tract region in the fourth exon was done as reported previously. 36 A mutated allele was represented by a major band shifted by 1 or 2 bp from the major wild-type band with intensity equal to or greater than that of the wild-type band.
E2F-4 Gene Mutation
The CAG trinucleotide repeat sequence region was analyzed in available HNPCCa and MSI-positive SRSCCa (no mutations have been reported in MSS tumors) using a previously reported method. 46 A mutated allele was represented by a major band, which was shifted in position in comparison to the wild-type band. Amplification of the approximately 300-bp segment was obtained in only 66% (31/47) of DNA specimens from the formalin-fixed, paraffin-embedded cancers (P = 0.01 versus frequency of successful amplification of Bax and P < 0.000001 versus amplification of TGFβ RII). Sequencing of the region containing the trinucleotide repeat was done by the previously reported method in one SRSCCa with an allelic shift and available frozen tissue.
Bax Gene Mutation
The polydeoxyguanosine tract region in the third exon was analyzed in available HNPCCa and MSI-positive SRSCCa (no mutations have been reported in MSS tumors) by the method reported previously. 52 A mutated allele was represented by a major band that was shifted by 1 bp from the normal major band with intensity equal to or greater than that of the wild-type band.
Statistical Analysis
The sensitivity, specificity, predictive values of a positive and of a negative, and overall accuracy for MSI status of each of the noncoding dinucleotide and poly(A) tract markers were determined individually. Similar analysis was done for loss of nuclear staining for hMSH2 gene product in HNPCCa from kindreds previously tested for germline status of the hMSH2 and hMLH1 genes. Differences in frequencies were evaluated by Fisher’s exact probability test. Differences in means were evaluated by Mann-Whitney U test. True Epistat (Richardson, TX) statistical software was used. All reported P values are two-tailed.
Results
MSI Status
Of 39 HNPCCa, 95% (37/39) were MSI-positive as contrasted with 31% (18/57) of SRSCCa (P < 0.000001). The extent of instability, however, tended to be greater in MSI-positive SRSCCa than in HNPCCa: 94% (16/17) of MSI-positive SRSCCa had at least two-thirds of the dinucleotide markers shifted as contrasted with 69% (24/35) of MSI-positive HNPCCa (P = 0.08). HNPCCa from families with germline hMSH2 mutation, germline hMLH1 mutation, or no identified germline mutation had no statistically significant differences in the frequency of shifted markers. There was no evidence of subtle MSI in the two MSS HNPCCa: no shift was found in any of the markers tested.
The CA dinucleotide repeats on chromosome 18q (Figure 1) ▶ and the poly (A) tract in BAT26 (Figure 2) ▶ showed no statistically significant differences in their sensitivities (67 to 88%), specificities (95 to 100%), predictive values of a positive (96 to 100%), predictive values of a negative (77 to 87%), and overall accuracies (84 to 92%) for identifying MSI-positive carcinomas. D18S61 was least frequently shifted in MSI-positive tumors, and BAT26 was the only marker shifted in otherwise MSS tumors (n = 2, both SRSCCa). Four MSI-positive HNPCCa and one MSI-positive SRSCCa (9% of all MSI-positive cancers) had shifts only in dinucleotide repeats of the noncoding markers.
Expression of hMSH2 Gene Product
Twenty-nine MSI-positive HNPCCa from families previously evaluated for germline mutation status of hMSH2 and hMLH1 were studied by immunohistochemistry (Figure 3 ▶ , Table 2 ▶ ). Absence of nuclear expression of hMSH2 gene product, which was usually accompanied by retention of cytoplasmic staining (Figure 3) ▶ , was associated with germline hMSH2 mutation: 12 of 14 cancers in patients with germline hMSH2 mutation lacked nuclear expression as contrasted with 0 of 6 cancers in patients with germline hMLH1 mutation (P = 0.0007) and 1 of 9 cancers in patients with no identified germline mutation (P = 0.0007). The two cancers that retained nuclear expression of hMSH2 gene product in patients with germline hMSH2 mutation were from families with other members whose cancers had lost expression, indicating that the nature of the germline mutation was not responsible for the differences in expression. Absence of nuclear staining for hMSH2 gene product in MSI-positive cancers from patients evaluated for germline status had sensitivity for germline mutation of hMSH2 of 86%, specificity of 93%, predictive value of a positive of 92%, predictive value of a negative of 88%, and overall accuracy of 90%. All MSI-positive SRSCCa and all MSS cancers had intense nuclear expression of hMSH2 gene product accompanied by cytoplasmic staining.
Table 2.
Immunohistochemistry for hMSH2 Mismatch Repair Gene Product
| Percentage of carcinomas with absence of hMSH2 gene product expression in tumor nuclei (frequency) | |
|---|---|
| MSI+ HNPCCa | |
| Germline hMSH2 mutation | 86 (12/14) |
| Germline hMLH1 mutation | 0 (0/6) |
| No germline hMSH2 or hMLH1 mutation identified | 11 (1/9) |
| Germline not tested | 50 (1/2) |
| MSS HNPCCa | |
| Germline hMSH2 mutation | 0 (0/1) |
| MSI+ SRSCCa | 0 (0/17) |
| MSS SRSCCa | 0 (0/27) |
K-ras Proto-Oncogene Mutation
The prevalence of carcinomas with K-ras mutation in codons 12 or 13 was similar in HNPCC and sporadic patients: 30% (11/37) and 30% (16/54), respectively. MSI-positive SRSCCa had the lowest frequency of K-ras mutations (18% (3/17); Table 3 ▶ ), but the differences in frequencies between MSI-positive and MSS carcinomas were not statistically significant.
Table 3.
Summary of Genetic Analyses
| Percentage of carcinomas with genetic alteration (fraction) | ||||
|---|---|---|---|---|
| MSI+ HNPCCa (n = 37) | MSS HNPCCa (n = 2) | MSI+ SRSCCa (n = 18) | MSS SRSCCa (n = 39) | |
| K-ras mutation in codons 12 or 13 | 31 (11/35)*† | 0 (0/2)† | 18 (3/17)† | 35 (13/37)† |
| 18q allelic deletion | ND‡ | 0 (0/2)†§¶ | 0 (0/11)**†† | 62 (24/39)§¶†† |
| p53 gene product overexpression (>50% labeling index) | 5 (2/37)‡‡ | 50 (1/2)† | 0 (0/18)§§ | 33 (13/39)‡‡§§ |
| Mutation in repeat sequences | ||||
| In TGFβ RII | 83 (30/36)¶¶∥∥ | 0 (0/2)† | 89 (16/18)***††† | 0 (0/39)¶¶*** |
| In E2F-4 | 71 (12/17)† | 0 (0/1)† | 57 (8/14)† | ND‡ |
| In Bax | 55 (17/31)∥∥‡‡‡ | 0 (0/2)† | 13 (2/15)†††‡‡‡ | ND‡ |
The types of K-ras mutations differed with the clinical setting. Transition from G to A in the second nucleotide of codon 13 was the most frequent K-ras mutation in HNPCCa (55%, 6/11 cancers with K-ras mutation; Table 4 ▶ ), but no HNPCCa from patients with germline hMSH2 mutation had a codon 13 K-ras mutation (P = 0.02; Table 5 ▶ ). Transition from G to A in the second nucleotide of codon 12 was most common in SRSCCa (56%, 9/16 cancers with K-ras mutation; Table 4 ▶ ), but the differences in frequency between HNPCCa and SRSCCa were not statistically significant.
Table 4.
ras Gene Mutations
| Type of ras mutations in percentage (number of mutations)* | |||
|---|---|---|---|
| MSI+ HNPCCa (n = 11) | MSS HNPCCa (n = 0) | MSI+ SRSCCa (n = 3) | MSS SRSCCa (n = 13) |
| Codon 12 GGT, Gly | |||
| AGT, Ser | 27 (3†‡) | 0 | 8 (1) |
| GAT, Asp | 45 (5‡) | 67 (2) | 54 (7) |
| GCT, Val | 0 | 0 | 8 (1) |
| GTT, Ala | 0 | 0 | 8 (1) |
| Codon 13 GGC, Gly | |||
| GAT, Asp | 55 (6†‡) | 33 (1) | 23 (3) |
Table 5.
Genetic Analyses of MSI-positive HNPCCa Compared to Mismatch Repair Gene with Germline Mutation
| Percentage of carcinomas with genetic alteration (fraction) | |||
|---|---|---|---|
| Germline hMSH2 mutation (n = 17) | Germline hMLH1 mutation (n = 7) | No germline mutation found (n = 11) | |
| K-ras mutation | |||
| In codons 12 or 13* | 18 (3/17) | 57 (4/7)† | 44 (4/9) |
| In codon 12* | 18 (3/17) | 29 (2/7) | 22 (2/9) |
| In codon 13 | 0 (0/17)ठ| 43 (3/7) | 33 (3/9) |
| p53 gene product overexpression (>50% labeling index)* | 6 (1/17) | 0 (0/7) | 9 (1/11) |
| Mutation in repeat sequences | |||
| In TGFβ RII* | 88 (15/17) | 83 (5/6) | 82 (9/11) |
| In E2F-4 | 100 (8/8) | 40 (2/5)¶ | 67 (2/3) |
| In Bax* | 65 (11/17) | 60 (3/5) | 29 (2/7) |
Two MSI-positive HNPCCa had more than one ras mutation. One carcinoma had a mutation in codons 12 and 13, and the other had three mutations, ie, two in codon 12 and one in codon 13 (Table 4) ▶ . The case with three mutations had a histopathological section available for microdissection to analyze the topography of the ras mutations in samples of approximately 1 mm2. Regions with two, one, and no ras mutations were identified (Figure 4) ▶ , indicating the presence of multiple subclones with different ras mutation status within the cancer. Multiple ras mutations were not found in any SRSCCa with ras mutation (P = 0.16 versus HNPCCa).
Figure 4.

ras proto-oncogene sequence analysis in MSI-positive HNPCCa with three different ras mutations. Codons 12 and 13 are shown, and mutations are indicated by arrowheads. G-to-A transitions are evident in the first nucleotide of codon 12 and the second nucleotide of codon 13 in the initial tumor specimen. In microdissected specimens of approximately 1 mm 2 , G-to-A transitions are evident in the second nucleotide positions of codons 12 and 13 in region 4 and in the second nucleotide position of codon 13 alone in region 6. Other microdissected regions showed no ras mutation (not illustrated). The findings indicate the presence of heterogeneous subclones in the MSI-positive cancer.
18q Allelic Deletion
In HNPCCa, loss could be evaluated by microsatellite analysis in the two MSS cancers only; neither showed deletion (Table 3) ▶ . Allelic deletion was found in 48% (24/50) of SRSCCa, and loss was inversely related to MSI; none of 11 MSI-positive SRSCCa evaluated by restriction fragment length polymorphism analysis had deletion, as contrasted with 62% (24/39) of all MSS carcinomas evaluated with microsatellite markers (P = 0.0004; Table 3 ▶ ). Loss involved all evaluable markers in 22 of the 24 carcinomas with deletion, and the centromeric markers in an additional cancer. Thus, the entire DCC/DPC4-Smad4/JV-18-MADR2-Smad2 region was lost in 96% (23/24) of cancers with an 18q allelic deletion.
p53 Gene Product Overexpression
The labeling indices for p53 immunohistochemistry had a bimodal distribution with all values either above 70% or below 45% (Figure 5) ▶ . Extensive overexpression of the type associated with p53 gene mutation was found in 8% (3/39) of HNPCCa as contrasted with 23% (13/57) of SRSCCa (P = 0.06; Table 3 ▶ ). High p53 labeling index was inversely related to MSI positivity: only 4% (2/55) of MSI-positive HNPCCa and SRSCCa had extensive overexpression as contrasted with 34% (14/41) of MSS cancers (P = 0.0001).
Figure 5.

Labeling indices for p53 gene product overexpression by immunohistochemistry and computerized image analysis. A bimodal distribution is evident with all indices above 70% or below 45%. High p53 labeling index of the type associated with p53 gene mutation is inversely related to MSI positivity (P = 0.0001).
TGFβ RII Gene Mutation
Frameshift mutation resulting in loss or gain of 1 or 2 bp within the 10-bp adenine repeat occurred in 79% (30/38) of HNPCCa and 28% (16/57) of SRSCCa (P = 0.000001; Table 3 ▶ and Figure 6 ▶ ). All mutations were in MSI-positive carcinomas, with high frequencies in both HNPCCa and SRSCCa: 83% (30/36) of MSI-positive HNPCCa showed mutation, as did 89% (16/18) of MSI-positive SRSCCa.
Figure 6.

Analysis of tumor DNA for frameshift mutation in the nonpolymorphic poly(A) tract in the fourth exon of the TGFβ RII gene. Alleles with shifts of size due to deletion of nucleotides are indicated by arrowheads. Note the consistent size of the wild-type allele (wt) in these tumors. The microdissected tumor in the far right lane has two mutations with loss of 1 bp (upper arrowhead) and 2 bp (lower arrowhead), respectively, and presence of the wild-type allele.
E2F-4 Gene Mutation
Mutation resulting in loss or gain of 3 bp occurred at high frequency in both MSI-positive HNPCCa and MSI-positive SRSCCa (71% (12/17) and 57% (8/14), respectively; Table 3 ▶ ). Of note, E2F-4 mutation was more frequent in HNPCC patients with germline hMSH2 gene mutation than those with germline hMLH1 mutation (100% (8/8) versus 40% (2/5), P = 0.04; Table 5 ▶ ). Sequencing of an SRSCCa specimen with one altered allele of a size 3 bp smaller than wild type (fourth lane in Figure 7 ▶ ) and available frozen tissue confirmed loss of one of the CAG trinucleotide repeat sequences in codons 306 to 318 as compared with matched control DNA (Figure 8 ▶ ; lowercase letters indicate nucleotides with detectably altered mobility as compared with control): wild type, (CAG)12 CAG CAA CAG TAA CAG CAG CAG TTC GTC; tumor, (CAG)12 CAa CAg tAa cAg CAG CAG ttc gTC.
Figure 7.

Analysis for mutation in the CAG trinucleotide repeat sequence in the E2F-4 transcription factor gene. Alleles with shifts of size in tumor DNA (T lanes) as compared with matched nonneoplastic mucosal DNA (N lanes) are indicated by arrowheads. Note the consistent size of the wild-type allele (wt) in neoplastic as well as nonneoplastic specimens. The MSI-positive LoVo colon cancer cell line (LoVo lane) has mutations in both alleles, one with insertion of a trinucleotide (upper arrowhead) and the other with deletion (lower arrowhead).
Figure 8.

Sequence analysis of the E2F-4 transcription factor gene in an SRSCCa specimen with allelic shift to a size 3 bp smaller in one allele (fourth lane in Figure 7 ▶ ) and available frozen tissue. Shortening of the length of the CAG trinucleotide repeat from 13 to 12 was found: wild type, (CAG)12 CAG CAA CAG TAA CAG CAG CAG TTC GTC; tumor, (CAG)12 CAa CAg tAa cAg CAG CAG ttc gTC. Nucleotides with detectably shifted position (lowercase letters) in tumor DNA are indicated by arrowheads on the photograph.
Bax Gene Mutation
Frameshift mutation within the homopolymeric region occurred with higher frequency in MSI-positive HNPCCa as compared with MSI-positive SRSCCa (55% (17/31) versus 13% (2/15), respectively, P = 0.01; Table 3 ▶ and Figure 9 ▶ ). No tumor had mutation of both Bax alleles. The frequency of Bax mutation was significantly lower than that of TGFβ RII mutation in MSI-positive carcinomas (Table 3) ▶ .
Figure 9.

Analysis of tumor DNA for frameshift mutation in the nonpolymorphic polydeoxyguanosine tract in the third exon of the proapoptotic Bax gene. Shifts of allelic size due to deletions are indicated by arrowheads. Note the consistent size of the wild-type allele (wt) in these tumor specimens.
Mutational Spectra
Our evaluation of a panel of genetic alterations permitted assessment of MSI-positive carcinomas for mutational spectra relative to clinical setting (HNPCC versus sporadic) and germline mutation in families with HNPCC (Figure 10) ▶ . Heterogeneity of the mutational spectra was prominent. The most frequent pattern was characterized by TGFβ RII and E2F-4 mutation without Bax mutation, ras mutation, or p53 gene product overexpression, but this combination occurred in only 23% (8 of 35) of evaluable cancers. This pattern was found in two HNPCCa from patients with germline hMSH2 mutation representing two different families (patients 11 and 12 in Figure 10 ▶ ) and in six SRSCCa (patients 40 to 45 in Figure 10 ▶ ).
Figure 10.

Mutational spectra of MSI-positive HNPCCa and SRSCCa arranged according to clinical setting and frequency of genetic alterations. Solid and open squares indicate that the feature was present or not found, respectively, and slashed squares indicate that the feature was not evaluated. Striking heterogeneity of the accumulated alterations is evident, including in members of the same HNPCC family who shared identical germline mutations (indicated by letters). The most common pattern of mutations was found in only 23% of the MSI-positive cancers (8 of 35 evaluable tumors; patients 11, 12, and 40 to 45). An inverse association between E2F-4 mutation and mutation in codon 13 of K-ras in HNPCCa from patients with germline hMSH2 mutation is evident in patients 1 to 17. Mutational status of TGFβ RII and E2F-4 had the most frequent concordance (71%, 22 of 31 evaluable cancers).
Discussion
Our study has practical implications for the definition of MSI-positive colonic adenocarcinomas and for understanding the molecular events involved in colorectal carcinogenesis. Identification of MSI in colorectal cancers has been proposed as a screening test for HNPCC to serve as a prelude to expensive and complicated germline testing of the five mismatch repair genes known to be responsible for the syndrome. 88 In addition, MSI-positive colorectal cancers have been reported to have improved stage-specific prognosis and better response to chemotherapy than MSS cancers. 70-73,89,90 Until recently, there have been no consensus criteria for classification of a tumor as MSI-positive, and many definitions have been reported in the literature. 91,92 Variables for microsatellite assays include number tested, type (mainly dinucleotide versus mononucleotide, noncoding DNA versus exon), and number or fraction with shifts needed for the tumor to be considered MSI-positive. A recent consensus conference sponsored by the National Cancer Institute has formulated guidelines for evaluation of MSI. 93 The proposed reference panel for initial studies included three dinucleotide markers (D2S123, D5S346, and D17S250) and two mononucleotide markers (BAT25 and BAT26). Shift of allelic size in one marker was interpreted as low MSI, whereas shifts in two or more markers were classified as high MSI.
When all of the markers and gene mutations evaluated in our study were considered, there was no infallible single assay for MSI. As a consequence, our study supports the proposed use of a panel of assays but suggests use of markers in addition to those recommended by the consensus conference. The poly(A) tract in the coding region of the TGFβ RII gene had high sensitivity, specificity, predictive values, and overall accuracy for MSI status in our study. This coding sequence and similar mononucleotide tracts have the practical advantage of showing no evident polymorphism in nonneoplastic tissue in our and previous studies. 94,95 As a consequence, these assays can be used for microdissected tumors even if control DNA is not available, in contrast to assays that evaluate polymorphic markers and therefore require matched nonneoplastic control DNA for comparison. Mutations in the coding regions of E2F-4 and Bax occurred at lower frequency than the TGFβ RII coding region mutation in our study, and the E2F-4 assay was often unsatisfactory in DNA from formalin-fixed, paraffin-embedded tissue, probably because of the relatively large size of the PCR product. The poly(A) tract in BAT26 showed shifts in two SRSCCa with no other evidence of MSI in our study, possibly because of somatic mutation in the GTBP/hMSH6 gene, which results in susceptibility to shifts in mononucleotide repeats but not in dinucleotide microsatellites, 96-98 although contradictory results have been reported. 99 Therefore, in screening for HNPCC, the BAT26 assay may have occasional false positives, and the importance for prognosis and therapeutic response of minor MSI confined to mononucleotide microsatellites is as yet unknown. Of note, shift in the poly(A) tract in the TGFβ RII gene in our study was specific (100%) for MSI-positive cancers, although rare exceptions have been reported, 54 and highly accurate (85%), so that assay of this gene is an excellent initial test for MSI. The 9% of MSI-positive cancers that lacked shifts in either poly(A) tract in our study did have shifts in various dinucleotide repeats. Thus, our data support the proposal that assay of dinucleotide tracts is needed to evaluate the MSI status of colorectal carcinomas that do not have a shift in the TGFβ RII gene. Use of polymorphic dinucleotide markers for chromosomal regions of interest for allelic loss (eg 18q as in our study) provides useful information in MSS tumors as well as identifying MSI-positive tumors. Similar studies for methodological evaluation of MSI are needed in tumors of other organ systems.
Two HNPCCa in our study were MSS with no evidence of a shift in any marker. Both of the cancers were from families with known germline hMSH2 mutation. In the first patient, a 72-year-old woman, nuclear expression of hMSH2 gene product was maintained in the cancer available for immunohistochemistry, in contrast to absence of nuclear staining in most MSI-positive HNPCCa in such families (Table 2) ▶ . This patient had not undergone germline testing, but the cancer was probably a sporadic tumor occurring in an older patient in an HNPCC family, ie a phenocopy. The second patient, a 42-year-old woman, had an identified germline mutation of hMSH2, and her MSS cancer had histopathological characteristics associated with MSI, including poor differentiation and the presence of numerous signet ring cells and prominent lymphoid response. No tissue was available for immunohistochemistry for hMSH2 gene product, and the explanation of the absence of identifiable MSI in repeated analyses of this cancer is uncertain. Adenomas in HNPCC patients have a lower prevalence of MSI than do HNPCCa, 53,100-104 and this cancer may have been early in the process of tumorigenesis, analogous to adenomas.
We found that hMSH2 gene product was usually absent from the nucleus of MSI-positive cancers of patients in families with known germline mutation in this mismatch repair gene, as reported previously, 19,20 and in a cell line (LoVo) with reported homozygous deletion of the hMSH2 gene 75 and absence of hMSH2 gene product expression on Western blot. 80 Intratumoral heterogeneity in nuclear staining was often evident in these cancers (Figure 3) ▶ . This unexplained heterogeneity adversely impacts the diagnostic utility of immunohistochemistry by producing difficulty in its interpretation in MSI-positive cancers. In addition, there was retention of staining for hMSH2 gene product in the cytoplasm of many tumors from patients with germline hMSH2 gene mutation (Figure 3) ▶ , raising the possibility of abnormal translocation of the gene product to the nucleus rather than complete absence of the protein. In previous studies, allelic loss of the wild-type allele of hMSH2 was uncommon in HNPCCa, 33,105 so that gene product expression could be retained, but background staining could also explain the cytoplasmic findings. The nature of the underlying germline hMSH2 mutation did not explain the heterogeneity, because patients from the same kindred with the same germline mutation had different immunohistochemical results (Figure 10) ▶ . No MSI-positive cancers from patients with germline hMLH1 mutation, MSI-positive sporadic cancers, or MSS cancers showed loss of hMSH2 gene product expression in nuclei (Table 2) ▶ . Thus, immunohistochemistry appears to be useful in the identification of most patients who have germline hMSH2 mutation, thereby indicating the gene to be addressed by formal mutation analysis. Loss of hMSH2 gene product expression in MSI-positive HNPCCa from patients with unknown germline mutation of a mismatch repair gene and in sporadic MSI-positive colonic carcinomas was infrequent in our study, suggesting infrequent occurrence of germline hMSH2 mutation in these patient subsets. In agreement with this interpretation, somatic mutation of hMSH2 was rarely found in MSI-positive colonic cancers in previous studies of patients without HNPCC. 106-108 By contrast with the close relationship between loss of nuclear hMSH2 expression and germline mutation of the hMSH2 gene, loss of hMLH1 gene product expression can occur as a result of somatic hypermethylation of the gene promoter, leading to transcriptional silencing of expression in the absence of germline mutation of hMLH1. 109-111
The frequency of ras proto-oncogene mutation in MSI-positive colorectal cancers is usually reported in the literature to be lower than in MSS cancers. 33,43,59-61 By contrast, and in agreement with other studies, 28,34,40,57,58 we found a frequency in MSI-positive HNPCCa that was similar to MSS SRSCCa and a lower frequency in MSI-positive SRSCCa that was not statistically significantly different from MSS cancers (Table 3) ▶ . Two aspects of the ras mutations are of note. First, there was a suggestion that the types of ras mutations differed between MSI-positive cancers in the hereditary and sporadic settings: codon 12 mutations were predominant in MSI-positive SRSCCa whereas codon 13 mutations were somewhat more common in MSI-positive HNPCCa (Table 4) ▶ but were absent in the subset of HNPCC patients with germline hMSH2 gene mutation (Table 5) ▶ . As a consequence, the underlying mismatch repair gene defect appears to influence the position of the G-to-A transition in the ras proto-oncogene. This finding suggests complex relationships between defects in specific components of the mismatch repair system and the sequence context or DNA conformation of specific mispairs. Secondly, two MSI-positive HNPCCa had multiple ras mutations (Figure 4 ▶ and Table 4 ▶ ), which appeared to result from the presence of subclones with different mutation. Intratumoral heterogeneity has also been reported for loss of heterozygosity in the hMSH2 and hMLH1 loci of MSI-positive sporadic colon cancers. 42 Our finding of heterogeneity for ras mutations is similar to that reported in MSS colorectal adenomas, but the MSS cancers associated with the adenomas had only one of the mutations, indicating clonal dominance of the cancer by one of the subclones from the adenoma. 112 The presence of multiple ras mutations in some HNPCCa may reflect the effects of the mutator phenotype and also favors the concept that ras mutations provide weaker clonal advantages in HNPCCa with MSI as compared with MSS cancers.
Previous studies have shown low frequency of allelic losses in MSI-positive colorectal carcinomas. 33,59 Our study shows that chromosome 18q, the site of the DCC, DPC4/Smad4, and JV-18/MADR2/Smad2 genes that are commonly lost in MSS tumors, was retained in MSI-positive SRSCCa. Frozen specimens to permit restriction fragment length polymorphism analysis of HNPCCa were not available, but similar retention of 18q is expected. Previous cytogenetic studies also showed retention of 18q in the karyotypes of MSI-positive colon cancer cell lines. 113 These findings further emphasize the differing molecular pathogenesis between MSI-positive and MSS colonic cancers.
In agreement with previous studies of MSI-positive colonic carcinomas in both the hereditary and sporadic settings, 26,28,30,62 we found a low rate of p53 gene product overexpression of the type seen with p53 gene mutation (Figure 5) ▶ . p53 mutations were reported to be uncommon in MSI-positive colonic cancers, 33,34,53,59-61,63 although a recent study reported p53 gene mutations in four of nine MSI-positive colon cancer cell lines (two homozygous and two hemizygous). 112 The reason that p53 mutation status in MSI-positive tumors seems to vary so much from study to study is unclear. MSI-positive tumors are often heavily contaminated with nonneoplastic cells, especially tumor-infiltrating lymphocytes. 31 In addition, loss of heterozygosity is uncommon so that retention of both alleles of p53 would be expected. These factors make sequence analysis difficult, as occurred in our study, because of the dilution by wild-type DNA of any DNA with p53 nucleotide sequence alteration. Subtle or unusual mutations could also make it difficult to detect some mutations, such as those that reduce expression. In addition, it has been reported that there is a difference in p53 alteration status between right-sided and left-sided colorectal cancers, regardless of MSI status. 27,69 Therefore, right-sided MSI-positive cancers could have a low rate of p53 alteration because of their location alone. Our study comparing sporadic MSS right-sided colonic cancers with MSI-positive cancers favors genetic rather than anatomical explanations of the inverse relationship between MSI and p53 alteration.
Genes with nucleotide repeats in the coding region are expected to show high rates of mutation in those repeated sequences in MSI-positive cancers because of disordered mismatch repair and the presence of associated mutator phenotypes. 7-9 Our study, however, confirms the findings in previous studies that the frequency of mutations in repeat sequences varies greatly among potential target genes: the TGFβ RII gene was usually mutated, but the proapoptotic Bax gene had significantly lower rates of mutation and no tumor showed mutation of both Bax alleles, in contrast to TGFβ RII. The differences in frequencies of mutations suggest differences in the selective growth advantage for the tumor cells conferred by the various mutations. Of particular note, the frequency of mutation of the E2F-4 gene was higher in HNPCC patients with germline hMSH2 mutation than those with germline hMLH1 mutation, and the frequency of mutation in the Bax gene was higher in MSI-positive HNPCCa than in MSI-positive SRSCCa (Table 3) ▶ . Recent studies showed that E2F-4 mutations were common in a subset of colorectal carcinomas with extensive MSI 45-47 and were accompanied by frameshift mutations in a poly(A) repeat within the seventh exon of the hMSH3 gene. 47 In contrast to our study, MSI in Japanese HNPCCa was not associated with mutation of the Bax gene. 114 The importance of the molecular basis of mismatch repair deficiency in affecting subsequent gene mutations is also evident in cell lines that showed varying patterns of mutations associated with mutations in different mismatch repair genes. 115,116 Similarly, the basis of mismatch repair deficiency affects tumorigenesis in mouse models: germline mutation of the human homologs of MLH1, PMS1, or PMS2 results in very different types of murine tumors. 117
Our evaluation of a panel of genetic alterations permitted identification of the striking heterogeneity in the combinations of mutations in individual MSI-positive colonic cancers, analogous to the intertumoral heterogeneity of oncogene mutations and suppressor gene alterations in MSS cancers. 76,118 The heterogeneity of the gene mutational spectra in MSI-positive tumors may be explained in part by the underlying mechanisms of defective mismatch repair, because alterations of ras and Bax genes had evidence of differences between HNPCCa and SRSCCa, and E2F-4 mutation frequency differed in HNPCC patients with germline mutation of hMSH2 as compared with hMLH1. In turn, the heterogeneity in mutations may impact on the clinical behavior of MSI-positive cancers. For example, studies have shown improved prognosis in HNPCCa patients as compared to patients with sporadic colorectal cancer of equivalent stage. 70-73 Similarly, patients with sporadic MSI-positive cancers are reported to have better outcome than MSS cases. 28,89,90 Nonetheless, colorectal cancer is a major cause of death in HNPCC families, and many patients with sporadic MSI-positive cancers do die of their disease. The variability of the accumulated mutations and alterations in gene expression 119,120 may help to explain the variability of the tumor phenotype. Additional studies are needed to define the crucial alterations that may then serve as prognostic markers. These genetic alterations may eventually define targets for unique therapies based on understanding of the specific pathways that are altered by mutations in the molecular subsets of colorectal cancer.
Acknowledgments
The manuscript was typed by Mrs. Nancy Folker. Ellen Winslow assisted with graphics. The authors appreciate the assistance of Ms. C. Rahj Robinson.
Footnotes
Address reprint requests to Dr. Stanley R. Hamilton, Division of Pathology and Laboratory Medicine, University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030. E-mail: shamilto@notes.mdacc.tmc.edu.
Supported in part by the Clayton Fund and grants CA47527 and CA62924 from the National Cancer Institute, National Institutes of Health. HTL was supported by CA74684 and American Cancer Society grant EDT-84A.
References
- 1.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 2.Tomlinson I, Ilyas M, Novelli M: Molecular genetics of colon cancer. Cancer Metastasis Rev 1997, 16:67-79 [DOI] [PubMed] [Google Scholar]
- 3.Gryfe R, Swallow C, Bapat B, Redston M, Gallinger S, Couture J: Molecular biology of colorectal cancer. Curr Probl Cancer 1997, 21:233-300 [DOI] [PubMed] [Google Scholar]
- 4.Hartsough MT, Mulder KM: Transforming growth factor-β signaling in epithelial cells. Pharmacol Ther 1997, 75:21-41 [DOI] [PubMed] [Google Scholar]
- 5.Heldin CH, Miyazono K, ten Dijke P: TGFβ signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390:465-471 [DOI] [PubMed] [Google Scholar]
- 6.White RL: Tumor suppressing pathways. Cell 1998, 92:591-592 [DOI] [PubMed] [Google Scholar]
- 7.Perucho M: Cancer of the microsatellite mutator phenotype. Biol Chem 1996, 377:675-684 [PubMed] [Google Scholar]
- 8.Umar A, Kunkel TA: DNA replication fidelity, mismatch repair and genome instability in cancer cells. Eur J Biochem 1996, 238:297-307 [DOI] [PubMed] [Google Scholar]
- 9.Marra G, Boland CR: DNA repair and colorectal cancer. Gastroenterol Clin North Am 1996, 25:755-772 [DOI] [PubMed] [Google Scholar]
- 10.Jiricny J: Mismatch repair and cancer. Cancer Surv 1996, 28:47-68 [PubMed] [Google Scholar]
- 11.Modrich P, Lahue R: Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 1996, 65:101-133 [DOI] [PubMed] [Google Scholar]
- 12.Eshleman JR, Markowitz SD: Mismatch repair defects in human carcinogenesis. Hum Mol Genet 1996, 5:1489-1494 [DOI] [PubMed] [Google Scholar]
- 13.Arnheim N, Shibata D: DNA mismatch repair in mammals: role in disease and meiosis. Curr Opin Genet Dev 1997, 7:364-370 [DOI] [PubMed] [Google Scholar]
- 14.Lothe RA: Microsatellite instability in human solid tumors. Mol Med Today 1997, 3:61-68 [DOI] [PubMed] [Google Scholar]
- 15.Cho KR, Hedrick L: Genetic alterations in human tumors. Genetic Instability and Tumorigenesis. Edited by MB Kastan. Current Topics in Microbiology and Immunology, vol 221. Berlin, Springer-Verlag, 1997, pp 149–180 [DOI] [PubMed]
- 16.Hoffmann J-S, Cazaux C: DNA synthesis, mismatch repair, and cancer (review). Int J Oncol 1998, 12:377-382 [PubMed] [Google Scholar]
- 17.Lynch HT, Smyrk T, Lynch J: An update of HNPCC (Lynch syndrome). Cancer Genet Cytogenet 1997, 93:84-99 [DOI] [PubMed] [Google Scholar]
- 18.Marra G, Boland CR: Hereditary nonpolyposis colorectal cancer: the syndrome, the genes, and historical perspectives. J Natl Cancer Inst 1995, 87:1114-1125 [DOI] [PubMed] [Google Scholar]
- 19.Leach FS, Polyak K, Burrell M, Johnson KA, Hill D, Dunlop MG, Wyllie AH, Peltomaki P, de la Chapelle A, Hamilton SR, Kinzler KW, Vogelstein B: Expression of the human mismatch repair gene hMSH2 in normal and neoplastic tissues. Cancer Res 1996, 56:235-240 [PubMed] [Google Scholar]
- 20.Thibodeau SN, French AJ, Roche PC, Cunningham JM, Tester DJ, Lindor NM, Moslein G, Baker SM, Liskay RM, Burgart LJ, Honchel R, Halling KC: Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res 1996, 56:4836-4840 [PubMed] [Google Scholar]
- 21.Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J: Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 1997, 57:4749-4756 [PubMed] [Google Scholar]
- 22.Kim H, Piao Z, Kim JW, Choi JS, Kim NK, Lee JM, Park JH: Expression of hMSH2 and hMLH1 in colorectal carcinomas with microsatellite instability. Mod Pathol 1998, 194:3-10 [DOI] [PubMed] [Google Scholar]
- 23.Thibodeau SN, French AJ, Cunningham JM, Tester D, Burgart LJ, Roche PC, Mcdonnell SK, Schaid DJ, Vockley CW, Michels VV, Farr GH, Jr, O’Connell MJ: Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res 1998, 58:1713-1718 [PubMed] [Google Scholar]
- 24.Thibodeau SN, Bren G, Schaid D: Microsatellite instability in cancer of the proximal colon. Science 1993, 260:816-819 [DOI] [PubMed] [Google Scholar]
- 25.Lothe RA, Peltomaki P, Meling GI, Aaltonen LA, Nystrom-Lahti M, Pylkkanen L, Heimdal K, Andersen TI, Moller P, Rognum TO, Fossa SD, Haldorsen T, Langmark F, Brogger A, de la Chapelle A, Borresen A-L: Genomic instability in colorectal cancer: relationship of clinicopathological variables and family history. Cancer Res 1993, 53:5849-5852 [PubMed] [Google Scholar]
- 26.Kim H, Jen J, Vogelstein B, Hamilton SR: Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994, 145:148-156 [PMC free article] [PubMed] [Google Scholar]
- 27.Ilyas M, Tomlinson IPM, Novel MR, Hanby A, Bodmer WF, Talbot IC: Clinico-pathological features and p53 expression in left-sided sporadic colorectal cancers with and without microsatellite instability. J Pathol 1996, 179:370-375 [DOI] [PubMed] [Google Scholar]
- 28.Ruschoff J, Dietmaier W, Luttges J, Seitz G, Bocker T, Zimgibl H, Schlegel J, Schackert HK, Jauch KW, Hofstaedter F: Poorly differentiated colonic adenocarcinoma, medullary type: clinical, phenotypic, and molecular characteristics. Am J Pathol 1997, 150:1815-1825 [PMC free article] [PubMed] [Google Scholar]
- 29.Senba S, Konishi F, Okamoto T, Kashiwagi H, Kanazawa K, Miyaki M, Konishi M, Tsukamoto T: Clinicopathologic and genetic features of nonfamilial colorectal carcinomas with DNA replication errors. Cancer 1998, 82:279-285 [PubMed] [Google Scholar]
- 30.Forster S, Sattler HP, Hack M, Romanakis K, Rohde V, Seitz G, Wullich B: Microsatellite instability in sporadic carcinomas of the proximal colon: association with diploid DNA content, negative protein expression of p53, and distinct histomorphologic features. Surgery 1998, 123:13-18 [PubMed] [Google Scholar]
- 31.Jass JR, Do K-A, Simms LA, Iino H, Wynter C, Pillay SP, Searle J, Radford-Smith G, Young J, Leggett B: Morphology of sporadic colorectal cancer with DNA replication errors. Gut 1998, 42:673-679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JKV, Markowitz SD, Morin P, Kinzler KW, Vogelstein B, Dunlop MG: APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci USA 1996, 93:9049-9054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konishi M, Kikuchi-Yanoshita R, Tanaka K, Muraoka M, Onda A, Okumura Y, Kishi N, Iwama T, Mori T, Koike M, Ushio K, Chiba M, Nomizu S, Konishi F, Utsunomiya J, Miyaki M: Molecular nature of colon tumors in hereditary nonpolyposis colon cancer, familial polyposis, and sporadic colon cancer. Gastroenterology 1996, 111:307-317 [DOI] [PubMed] [Google Scholar]
- 34.Olschwang S, Hamelin R, Laurent-Puig P, Thuille B, DeRycke Y, Li Y-J, Muzeau F, Girodet J, Salmon R-J, Thomas G: Alternative genetic pathways in colorectal carcinogenesis. Proc Natl Acad Sci USA 1997, 94:12122-12127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L-Z, Lutterbaugh J, Fan RS, Aborowska E, Kinzler KW, Vogelstein B, Brattain M, Willson JKV: Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 1995, 268:1336-1338 [DOI] [PubMed] [Google Scholar]
- 36.Parsons R, Myeroff LL, Liu B, Willson JKW, Markowitz SD, Kinzler KW, Vogelstein B: Microsatellite instability and mutations of the transforming growth factor β type II receptor gene in colorectal cancer. Cancer Res 1995, 55:5548-5550 [PubMed] [Google Scholar]
- 37.Souza RF, Garrigue-Antar L, Lei J, Yin J, Appel R, Vellucci VF, Zou TT, Zhou X, Wang S, Rhyu MG, Cymes K, Chan O, Park WS, Krasna MJ, Greenwald BD, Cottrell J, Abraham JM, Simms L, Leggett B, Young J, Harpaz N, Reiss M, Meltzer SJ: Alterations of transforming growth factor-β 1 receptor type II occur in ulcerative colitis-associated carcinomas, sporadic colorectal neoplasms, and esophageal carcinomas, but not in gastric neoplasms. Hum Cell 1996, 9:229-236 [PubMed] [Google Scholar]
- 38.Togo G, Toda N, Kanai F, Kato N, Shiratori Y, Kishi K, Imazeki F, Makuuchi M, Omata M: A transforming growth factor β type II receptor gene mutation common in sporadic cecum cancer with microsatellite instability. Cancer Res 1996, 56:5620-5623 [PubMed] [Google Scholar]
- 39.Akiyama Y, Iwanaga R, Ishikawa T, Sakamoto K, Nishi N, Nihei Z, Iwama T, Saitoh K, Yuasa Y: Mutations of the transforming growth factor-β type II receptor gene are strongly related to sporadic proximal colon carcinomas with microsatellite instability. Cancer 1996, 78:2478-2484 [DOI] [PubMed] [Google Scholar]
- 40.Tannergard P, Liu T, Weger A, Nordenskjold M, Lindblom A: Tumorigenesis in colorectal tumors from patients with hereditary non-polyposis colorectal cancer. Hum Genet 1997, 101:51-55 [DOI] [PubMed] [Google Scholar]
- 41.Jiang W, Tillekeratne MPM, Brattain MG, Banerje SS: Decreased stability of transforming growth factor β type II receptor mRNA: RER+ human colon carcinoma cells. Biochemistry 1997, 36:14786-14793 [DOI] [PubMed] [Google Scholar]
- 42.Habano W, Sugai T, Nakamura S-I: Mismatch repair deficiency leads to a unique mode of colorectal tumorigenesis characterized by intratumoral heterogeneity. Oncogene 1998, 16:1259-1265 [DOI] [PubMed] [Google Scholar]
- 43.Iacopetta BJ, Welch J, Soong R, House AK, Zhou X-P, Hamelin R: Mutation of the transforming growth factor β type II receptor gene in right-sided colorectal cancer: relationship to clinicopathological features and genetic alterations. J Pathol 1998, 184:390-395 [DOI] [PubMed] [Google Scholar]
- 44.Sladek TL: E2F transcription factor action, regulation, and possible role in human cancer. Cell Proliferation 1997, 30:97-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshitaka T, Matsubara N, Ikeda M, Tanino M, Hanafusa H, Tanaka N, Shimizu K: Mutations of E2F-4 trinucleotide repeats in colorectal cancer with microsatellite instability. Biochem Biophys Res Commun 1996, 227:553-557 [DOI] [PubMed] [Google Scholar]
- 46.Souza RF, Yin J, Smolinski KN, Zou TT, Wang S, Shi YQ, Rhyu MG, Cottrell J, Abraham JM, Biden K, Simms L, Leggett B, Bova GS, Frank T, Powell SM, Sugimura H, Young J, Harpaz N, Shimizu K, Matsubara N, Meltzer SJ: Frequent mutation of the E2F-4 cell cycle gene in primary human gastrointestinal tumors. Cancer Res 1997, 57:2350-2353 [PubMed] [Google Scholar]
- 47.Ikeda M, Orimo H, Moriyama H, Nakajima E, Matsubara N, Mibu R, Tanaka N, Shimada T, Kimura A, Shimizu K: Close correlation between mutations of E2F4 and hMSH3 genes in colorectal cancers with microsatellite instability. Cancer Res 1998, 58:594-598 [PubMed] [Google Scholar]
- 48.Souza RF, Appel R, Yin J, Wang S, Smolinski KN, Abraham JM, Zou TT, Shi YQ, Lei J, Cottrell J, Cymes K, Biden K, Simms L, Leggett B, Lynch PM, Frazier M, Powell SM, Harpaz N, Sugimura H, Young J, Meltzer SJ: Microsatellite instability in the insulin-like growth factor II receptor gene in gastrointestinal tumours. Nat Genet 1996, 14:255-257 [DOI] [PubMed] [Google Scholar]
- 49.Ouyang H, Shiwaku HO, Hagiwara H, Miura K, Abe T, Kato Y, Ohtani H, Shiiba K, Souza RF, Meltzer SJ, Horii A: The insulin-like growth factor II receptor gene is mutated in genetically unstable cancers of the endometrium, stomach, and colorectum. Cancer Res 1997, 57:1851-1854 [PubMed] [Google Scholar]
- 50.Yamamoto H, Sawai H, Perucho M: Frameshift somatic mutations in gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res 1997, 57:4420-4426 [PubMed] [Google Scholar]
- 51.Yin J, Kong D, Wang S, Zou T-T, Souza RF, Smolinski KN, Lynch PM, Hamilton SR, Sugimura H, Powell SM, Young J, Abraham JM, Meltzer SJ: Mutation of hMSH3 and hMSH6 mismatch repair genes in genetically unstable human colorectal and gastric carcinomas. Hum Mutat 1997, 10:474-478 [DOI] [PubMed] [Google Scholar]
- 52.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M: Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275:967-969 [DOI] [PubMed] [Google Scholar]
- 53.Yagi OK, Akiyama Y, Nomizu T, Iwama T, Endo M, Yuasa Y: Proapoptotic gene BAX is frequently mutated in hereditary nonpolyposis colorectal cancers but not in adenomas. Gastroenterology 1998, 114:268-274 [DOI] [PubMed] [Google Scholar]
- 54.Ouyang H, Furukawa T, Abe T, Kato Y, Horii A: The BAX gene, the promoter of apoptosis, is mutated in genetically unstable cancers of the colorectum, stomach, and endometrium. Clin Cancer Res 1998, 4:1071-1074 [PubMed] [Google Scholar]
- 55.Potten CS, Wilson JW, Booth C: Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells 1997, 15:82-93 [DOI] [PubMed] [Google Scholar]
- 56.Simms LA, Zou TT, Young J, Shi YQ, Appel R, Rhyu MG, Sugimura H, Chenevix-Trench G, Souza RF, Meltzer SJ, Leggett BA: Apparent protection from instability of repeat sequences in cancer-related genes in replication error positive gastrointestinal cancers. Oncogene 1997, 14:2613-2618 [DOI] [PubMed] [Google Scholar]
- 57.Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, Petersen GM, Kinzler KW, Vogelstein B, de la Chapelle A: Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260:812-816 [DOI] [PubMed] [Google Scholar]
- 58.Craanen ME, Blok P, Offerhaus GJ, Tytgat GN: Recent developments in hereditary nonpolyposis colorectal cancer. Scand J Gastroenterol 1996, 218(suppl):92-97 [DOI] [PubMed] [Google Scholar]
- 59.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M: Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363:558-561 [DOI] [PubMed] [Google Scholar]
- 60.Breivik J, Lothe RA, Meling GI, Rognum TO, Borresen-Dale AL, Gaudernack G: Different genetic pathways to proximal and distal colorectal cancer influenced by sex-related factors. Int J Cancer 1997, 74:664-669 [DOI] [PubMed] [Google Scholar]
- 61.Losi L, Ponz de Leon M, Jiricny J, Di Gregorio C, Benatti P, Percesepe A, Fante R, Roncucci L, Pedroni M, Benhattar J: K-ras and p53 mutations in hereditary non-polyposis colorectal cancers. Int J Cancer 1997, 74:94-96 [DOI] [PubMed] [Google Scholar]
- 62.Muta H, Noguchi M, Perucho M, Ushio K, Sugihara K, Ochiai A, Nawata H, Hirohashi S: Clinical implications of microsatellite instability in colorectal cancers. Cancer 1996, 77:265-270 [DOI] [PubMed] [Google Scholar]
- 63.Cottu PH, Muzeau F, Estreicher A, Flejou JF, Iggo R, Thomas G, Hamelin R: Inverse correlation between RER+ status and p53 mutation in colorectal cancer cell lines. Oncogene 1996, 13:2727-2730 [PubMed] [Google Scholar]
- 64.Vasen HFA, Mecklin JP, Kahn PM, Lynch HT: Hereditary non-polyposis colorectal cancer. Lancet 1991, 338:877. [DOI] [PubMed] [Google Scholar]
- 65.Wijnen J, Khan PM, Vasen H, van der Klift H, Mulder A, van Leeuwen-Cornelisse I, Bakker B, Losekoot M, Moller P, Fodde R: Hereditary nonpolyposis colorectal cancer families not complying with the Amsterdam criteria show extremely low frequency of mismatch repair gene mutations. Am J Hum Genet 1997, 61:329-335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Genuardi M, Anti M, Capozzi E, Leonardi F, Fornasarig M, Novella E, Bellacosa A, Valenti A, Gasbarrini GB, Roncucci L, Benatti P, Percesepe A, Ponz de Leon M, Coco C, de Paoli A, Valentini M, Boiocchi M, Neri G, Viel A: MLH1 and MSH2 constitutional mutations in colorectal cancer families not meeting the standard criteria for hereditary nonpolyposis colorectal cancer. Int J Cancer 1998, 75:835-839 [DOI] [PubMed] [Google Scholar]
- 67.Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT, Watson P, Jass JR, Dunlop M, Wyllie A, Peltomaki P, de la Chapelle A, Hamilton SR, Vogelstein B, Kinzler KW: Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nat Med 1996, 2:169-174 [DOI] [PubMed] [Google Scholar]
- 68.Bufill JA: Colorectal cancer: evidence for distinct genetic categories based on proximal or distal tumor location. Ann Intern Med 1990, 113:779-788 [DOI] [PubMed] [Google Scholar]
- 69.Watatani M, Yoshida T, Kuroda K, Ieda S, Yasutomi M: Allelic loss of chromosome 17p, mutation of the p53 gene, and microsatellite instability in right- and left-sided colorectal cancer. Cancer 1995, 77(suppl 8):1688-1693 [DOI] [PubMed] [Google Scholar]
- 70.Fujita S, Moriya Y, Sugihara K, Akasu T, Ushio K: Prognosis of hereditary nonpolyposis colorectal cancer (HNPCC) and the role of Japanese criteria for HNPCC. Jpn J Clin Oncol 1996, 26:351-355 [DOI] [PubMed] [Google Scholar]
- 71.Sankila R, Aaltonen LA, Jarvinen HJ, Mecklin JP: Better survival rates in patients with MLH1-associated hereditary colorectal cancer. Gastroenterology 1996, 110:682-687 [DOI] [PubMed] [Google Scholar]
- 72.Percesepe A, Benatti P, Roncucci L, Sassatelli R, Fante R, Ganazzi D, Bellacosa A, Genuardi M, Neri G, Viel A, Ponz de Leon M: Survival analysis in families affected by hereditary non-polyposis colorectal cancer. Int J Cancer 1997, 71:373-376 [DOI] [PubMed] [Google Scholar]
- 73.Myrhoj T, Bisgaard ML, Bernstein I, Svendsen LB, Sondergaard JO, Bulow S: Hereditary non-polyposis colorectal cancer: clinical features and survival. Results from the Danish HNPCC Register. Scand J Gastroenterol 1997, 32:572-576 [DOI] [PubMed] [Google Scholar]
- 74.Jen J, Kim H, Piantadosi S, Liu Z-F, Levitt RC, Sistonen P, Kinzler KW, Vogelstein B, Hamilton SR: Allelic loss of chromosome 18q and prognosis of colorectal cancer. N Engl J Med 1994, 331:213-221 [DOI] [PubMed] [Google Scholar]
- 75.Umar A, Boyer JC, Thomas DC, Nguyen DC, Risinger JI, Boyd J, Ionov Y, Perucho M, Kunkel TA: Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J Biol Chem 1994, 269:14367-14370 [PubMed] [Google Scholar]
- 76.Moskaluk CA, Kern SE: Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 77.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AMM, Bos JL: Genetic alterations during colorectal-tumor development. N Engl J Med 1988, 319:525-532 [DOI] [PubMed] [Google Scholar]
- 78.Vogelstein B, Fearon ER, Kern SE, Hamilton SR, Preisinger AC, Nakamura Y, White R: Allelotype of colon carcinomas. Science 1989, 244:207-211 [DOI] [PubMed] [Google Scholar]
- 79.Bankfalvi A, Navabi H, Bier B, Brocker W, Jasani B, Schmid KW: Wet autoclave pretreatment for antigen retrieval in diagnostic immunohistochemistry. J Pathol 1994, 174:223-228 [DOI] [PubMed] [Google Scholar]
- 80.Drummond JT, Li GM, Longley MJ, Modrich P: Isolation of an hMSH2–p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 1995, 268:1909-1912 [DOI] [PubMed] [Google Scholar]
- 81.Jen J, Powell SM, Papadopoulos N, Smith KJ, Hamilton SR, Vogelstein B, Kinzler KW: Molecular determinants of dysplasia in colorectal lesions. Cancer Res 1994, 54:5523-5526 [PubMed] [Google Scholar]
- 82.Nucci MR, Robinson CR, Longo P, Campbell P, Hamilton SR: Phenotypic and genotypic characteristics of aberrant crypt foci in human colorectal mucosa. Hum Pathol 1997, 28:1396-1407 [DOI] [PubMed] [Google Scholar]
- 83.Mulder JW, Wielenga VJ, Pals ST, Offerhaus GJ: p53 and CD44 as clinical markers of tumour progression in colorectal carcinogenesis. Histochem J 1997, 29:439-452 [DOI] [PubMed] [Google Scholar]
- 84.Bass IO, Mulder J-W, Offerhaus GJA, Vogelstein B, Hamilton SR: An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol 1994, 172:5-12 [DOI] [PubMed] [Google Scholar]
- 85.Greenblatt MS, Grollman AP, Harris CC: Deletions and insertions in the p53 tumor suppressor gene in human cancers: confirmation of the DNA polymerase slippage/misalignment model. Cancer Res 1996, 56:2130-2136 [PubMed] [Google Scholar]
- 86.Boyle JO, Hakim J, Koch W, van der Riet P, Hruban RH, Roa A, Correo R, Eby YJ, Ruppert JM, Sidransky D: The incidence of p53 mutations increases with progression of head and neck cancer. Cancer Res 1993, 53:4477-4480 [PubMed] [Google Scholar]
- 87.Moskaluk CA, Heitmiller R, Zahurak M, Schwab D, Sidransky D, Hamilton SR: p53 and p21WAF1/CIP1/SDI1 gene products in Barrett esophagus and adenocarcinoma of the esophagus and esophagogastric junction. Hum Pathol 1996, 27:1211-1220 [DOI] [PubMed] [Google Scholar]
- 88.Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, Srivastava S: A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997, 89:1758-1762 [DOI] [PubMed] [Google Scholar]
- 89.Bubb VJ, Curtis LJ, Cunningham C, Dunlop MG, Carothers AD, Morris RG, White S, Bird CC, Wyllie AH: Microsatellite instability and the role of hMSH2 in sporadic colorectal cancer. Oncogene 1996, 12:2641-2649 [PubMed] [Google Scholar]
- 90.Lukish JR, Muro K, DeNobile J, Katz R, Williams J, Cruess DF, Drucker W, Kirsch I, Hamilton SR: Prognostic significance of DNA replication errors in young patients with colorectal cancer. Ann Surg 1998, 227:51-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bocker T, Schlegel J, Kullmann F, Stumm G, Zirngibl H, Epplens JT, Ruschoff J: Genomic instability in colorectal carcinomas: comparison of different evaluation methods and their biological significance. J Pathol 1996, 179:15-19 [DOI] [PubMed] [Google Scholar]
- 92.Bocker T, Diermann J, Friedl W, Gebert J, Holinski-Feder E, Karner-Hanusch J, von Knebel-Doeberitz M, Koelble K, Moeslein G, Schackert H-K, Wirtz H-C, Fishel R, Ruschoff J: Microsatellite instability analysis: a multicenter study for reliability and quality control. Cancer Res 1997, 57:4739-4743 [PubMed] [Google Scholar]
- 93.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Fodde R, Rodriguez-Bigas MA, Ranzani GN, Srivastava S: A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res (in press) [PubMed]
- 94.Hoang JM, Cottu PH, Thuille B, Salmon RJ, Thomas G, Hamelin R: BAT-26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res 1997, 57:300-303 [PubMed] [Google Scholar]
- 95.Zhou XP, Hoang JM, Cottu P, Thomas G, Hamelin R: Allelic profiles of mononucleotide repeat microsatellites in control individuals and in colorectal tumors with and without replication errors. Oncogene 1997, 15:1713-1718 [DOI] [PubMed] [Google Scholar]
- 96.Palombo F, Gallinari P, Iaccarino I, Lettieri T, Hughes M, D’Arrigo A, Truong O, Hsuan JJ, Jiricny J: GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science 1995, 268:1912-1914 [DOI] [PubMed] [Google Scholar]
- 97.Papadopoulos N, Nicolaides NC, Liu B, Parsons R, Lengauer C, Palombo F, D’Arrigo A, Markowitz S, Willson JKV, Kinzler KW, Jiricny J, Vogelstein B: Mutations of GTBP in genetically unstable cells. Science 1995, 268:1915-1917 [DOI] [PubMed] [Google Scholar]
- 98.Ohzeki S, Tachibana A, Tatsumi K, Kato T: Spectra of spontaneous mutations at the hprt locus in colorectal carcinoma cells lines defective in mismatch repair. Carcinogenesis 1997, 18:1127-1133 [DOI] [PubMed] [Google Scholar]
- 99.Ku JL, Park JG: Microsatellite instability (MSI) in mononucleotide repeat marker BAT-26 is not a good indicator of hMSH6 gene mutations in colorectal cancer cell lines (abstract). Proc Am Assoc Cancer Res 1998, 39:535 [Google Scholar]
- 100.Young J, Leggett B, Gustafson C, Ward M, Searle J, Thomas L, Buttenshaw R, Chenevix-Trench G: Genomic instability occurs in colorectal carcinomas but not in adenomas. Hum Mutat 1993, 2:351-354 [DOI] [PubMed] [Google Scholar]
- 101.Aaltonen LA, Peltomaki P, Mecklin J-P, Jarvinen H, Jass JR, Green JS, Lynch HT, Watson P, Tallqvist G, Juhola M, Sistonen P, Hamilton SR, Kinzler KW, Vogelstein B, de la Chapelle A: Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res 1994, 54:1645-1648 [PubMed] [Google Scholar]
- 102.Jacoby RF, Marshall DJ, Kailas S, Schlack S, Harms B, Love R: Genetic instability associated with adenoma to carcinoma progression in hereditary nonpolyposis colon cancer. Gastroenterology 1995, 109:73-82 [DOI] [PubMed] [Google Scholar]
- 103.Samowitz WS, Slattery ML: Microsatellite instability in colorectal adenomas. Gastroenterology 1997, 112:1515-1519 [DOI] [PubMed] [Google Scholar]
- 104.Samowitz WS, Slattery ML: Transforming growth factor-β receptor type 2 mutations and microsatellite instability in sporadic colorectal adenomas and carcinomas. Am J Pathol 1997, 151:33-35 [PMC free article] [PubMed] [Google Scholar]
- 105.Lu SL, Akiyama Y, Nagasaki H, Nomizu T, Ikeda E, Baba S, Ushio K, Iwama T, Maruyama K, Yuasa Y: Loss or somatic mutations of hMSH2 occur in hereditary nonpolyposis colorectal cancers with hMSH2 germline mutations. Jpn J Cancer Res 1996, 87:279-287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moslein G, Tester DJ, Lindor NM, Honchel R, Cunningham JM, French AJ, Halling KC, Schwab M, Goretzki P, Thibodeau SN: Microsatellite instability and mutation analysis of hMSH2 and hMLH1 in patients with sporadic, familial and hereditary colorectal cancer. Hum Mol Genet 1996, 5:1245-1252 [DOI] [PubMed] [Google Scholar]
- 107.Tomlinson IP, Ilyas M, Bodmer WF: Allele loss occurs frequently at hMLH1, but rarely at hMSH2, in sporadic colorectal cancers with microsatellite instability. Br J Cancer 1996, 74:1514-1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herfarth KK, Kodner IJ, Whelan AJ, Ivanovich JL, Bracamontes JR, Wells SA, Jr, Goodfellow PJ: Mutations in MLH1 are more frequent than in MSH2 in sporadic colorectal cancers with microsatellite instability. Genes Chromosomes Cancer 1997, 18:42-49 [DOI] [PubMed] [Google Scholar]
- 109.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R: Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997, 57:808-811 [PubMed] [Google Scholar]
- 110.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa J-P, Markowitz S, Willson JKV, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB: Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998, 95:6870-6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP: Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 1998, 72:141-196 [PubMed] [Google Scholar]
- 112.Shibata D, Schaeffer J, Liz ZH, Capella G, Perucho M: Genetic heterogeneity of the c-K-ras locus in colorectal adenomas but not in adenocarcinomas. J Natl Cancer Inst 1993, 85:1058-1063 [DOI] [PubMed] [Google Scholar]
- 113.Eshleman JR, Casey G, Kochera ME, Sedwick WD, Swinler SE, Veigl ML, Willson JKV, Schwartz S, Markowitz SD: Chromosome number and structure both are markedly stable in RER colorectal cancers and are not destabilized by mutation of p53. Oncogene 1998, 17:719-726 [DOI] [PubMed] [Google Scholar]
- 114.Sakakibara T, Nakamura T, Yamamoto M, Matsuo M: Microsatellite instability in Japanese hereditary non-polyposis colorectal cancer does not induce mutation of a simple repeat sequence of the bax gene. Cancer Lett 1998, 124:193-198 [DOI] [PubMed] [Google Scholar]
- 115.Eshleman JR, Markowitz SD, Donover PS, Lang EZ, Lutterbaugh JD, Li GM, Longley M, Modrich P, Veigl ML, Sedwick WD: Diverse hypermutability of multiple expressed sequence motifs present in a cancer with microsatellite instability. Oncogene 1996, 12:1425-1432 [PubMed] [Google Scholar]
- 116.Malkhosyan S, McCarty A, Sawai H, Perucho M: Differences in the spectrum of spontaneous mutations in the hprt gene between tumor cells of the microsatellite mutator phenotype. Mutat Res 1996, 316:249-259 [DOI] [PubMed] [Google Scholar]
- 117.Prolla TA, Baker SM, Harris AC, Tsao J-L, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM: Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet 1998, 18:276-279 [DOI] [PubMed] [Google Scholar]
- 118.Boland CR, Sato J, Appelman HD, Bresalier RS, Feinberg AP: Microallelotyping defines the sequence and tempo of allelic losses at tumour suppressor gene loci during colorectal cancer progression. Nat Med 1995, 1:902-909 [DOI] [PubMed] [Google Scholar]
- 119.Ahuja N, Mohan AL, Li Q, Stolker JM, Herman JG, Hamilton SR, Baylin SB, Issa J-P: Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res 1997, 57:3370-3374 [PubMed] [Google Scholar]
- 120.Ricote M, Geller P, Perucho M: Frequent alterations in gene expression in colon tumor cells of the microsatellite mutator phenotype. Mutat Res 1997, 374:153-167 [DOI] [PubMed] [Google Scholar]