Pharmacokinetics and pharmacodynamics of the leukotriene B4 receptor antagonist CP-105,696 in man following single oral administration (original) (raw)
Abstract
Aims
CP-105,696, (+)-1-(3S,4R)-[3-(4-phenylbenzyl)-4-hydroxy-chroman-7-yl] cyclopropane carboxylic acid is a potent, novel LTB4 receptor antagonist advanced to clinical trials to determine its efficacy in inflammatory diseases. The pharmacokinetics and pharmacodynamics of CP-105,696 were investigated in healthy male volunteers following oral administration of single doses of 5 to 640 mg.
Methods
Forty-eight subjects participated in a randomized, double-blind, parallel group study. Plasma and urine concentrations of CP-105,696 were determined at intervals after drug administration. As an indication of LTB4 receptor antagonism following oral administration of CP-105,696, the inhibiton of LTB4-induced upregulation of the neutrophil cell surface complement receptor (CR3), CD11b/CD18, was monitored at 4 h following drug administration using an ex vivo whole blood flow cytometry assay.
Results
_C_max and AUC(0,∞) increased in a dose-related manner. Respective mean _C_max values were 0.54 to 30.41 μg ml−1 following doses of 5 to 640 mg. Respective mean AUC(0,∞) values were 1337 to 16819 μg ml−1 h for the 40 to 640 mg dose groups. Plasma concentrations declined in a monoexponential manner, with terminal elimination half-lives ranging from 289 to 495 h. Group mean terminal elimination half-lives were dose-independent. Urinary excretion of unchanged drug accounted for <1% of the administered dose. A linear relationship was observed between CP-105,696 plasma concentrations and inhibition of LTB4-mediated CD11b upregulation on human neutrophils in whole blood. CP-105,696 plasma concentrations of 5–6 μg ml−1 were necessary to elicit a two-fold shift to the right of the LTB4 concentration response curve for CD11b upregulation.
Conclusions
These studies demonstrate pharmacologically significant LTB4-receptor antagonism following a single dose of CP-105,696 and pharmacokinetics consistent with once-daily dosing.
Keywords: leukotriene B4, receptor antagonist, CD11b, neutrophil, pharmacokinetics, pharmacodynamics
Introduction
Arachidonic acid is metabolized by 5-, 12- and 15-lipoxygenases to biologically active compounds which may play central roles in the pathophysiology of a number of inflammatory diseases including asthma, inflammatory bowel disease, rheumatoid arthritis, psoriasis and atopic dermatitis [1]. One 5-lipoxygenase product, leukotriene B4 (LTB4), is a potent in vitro and in vivo chemotactic agent for polymorphonuclear leukocytes, the primary inflammatory cells infiltrating plaques of patients with psoriasis and joints of patients with rheumatoid arthritis [2–5]. LTB4 is present at elevated concentrations in the psoriatic plaques and clinical efficacy of the 5-lipoxygenase inhibitor lonapalene correlates with inhibition of LTB4 biosynthesis [3, 6, 7]. LTB4 concentrations are also elevated in the synovial fluid of patients with rheumatoid arthritis and in the colonic mucosa of patients with inflammatory bowel disease, consistent with the infiltration of neutrophils into these tissues [5, 8, 9]. Furthermore, significant elevations in LTB4 concentrations are observed in bronchoalveolar lavage and/or arterial blood of subjects with symptomatic asthma and idiopathic pulmonary fibrosis [10, 11]. Together, these observations suggest that LTB4 receptor antagonism may alleviate the pathological sequelae of a number of diseases by inhibition of neutrophil recruitment and activation in sites of inflammation.
CP-105,696, (+)-1-(3S,4R)-[3-(4-phenylbenzyl)-4-hydroxy-chroman-7-yl] cyclopropane carboxylic acid, is a structurally novel and potent LTB4 receptor antagonist (Figure 1) [12]. Previous studies demonstrated that CP-105,696 is a potent antagonist of LTB4 binding to human neutrophil membranes (_IC_50 = 3.7 nm) and LTB4-induced human neutrophil chemotaxis (_IC_50 = 5.2 nm) in vitro [13]. It also inhibited the development and progression of murine collagen-induced arthritis in vivo at doses of 1–10 mg kg−1 day−1. These data indicate that CP-105,696 possesses potent in vitro and in vivo LTB4 receptor antagonistic properties suggestive of efficacy in inflammatory diseases, warranting the compound's further evaluation in humans.
Figure 1.
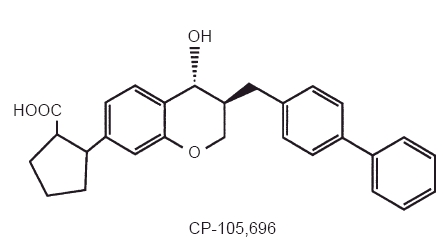
Structure of CP-105,696, (+)-1-(3S,4R)-[3-(4-phenylbenzyl)-4-hydroxy-chroman-7-yl] cyclopropane carboxylic acid.
The aim of this study was to investigate the pharmacokinetics of CP-105,696 in normal healthy male volunteers following oral administration at single doses of 5 to 640 mg. In addition, the pharmacodynamics of CP-105,696 were investigated by monitoring the inhibition of LTB4-induced upregulation of a neutrophil cell surface complement receptor, CD11b/CD18 (MAC-1), a process associated with activation of neutrophil adhesion and chemotaxis [14, 15]. CD11b expression was assayed using a quantitative ex vivo flow cytometric assay employing whole blood obtained from subjects following oral drug administration. Although previous studies demonstrated the influence of LTB4-receptor antagonism on CD11b upregulation in isolated human neutrophils using in vitro flow cytometric assays [16, 17], this study demonstrates that inhibiton of CD11b upregulation can be achieved and monitored in whole blood obtained from individuals following oral administration of a LTB4 receptor antagonist. Thus, CD11b upregulation is a pharmacological endpoint that can be monitored to assess the pharmacodynamics of this class of compounds in humans.
Methods
Drug administration
The study was conducted under medical supervision at Ohio State University College of Medicine (Columbus, OH., USA) following approval by the Institutional Review Board at that site. Forty-eight subjects gave written informed consent to participate in the study.
CP-105,696 was orally administered to healthy male volunteers at escalating single doses of 5, 10, 20, 40, 80, 160, 320 and 640 mg using a parallel-group design. CP-105,696 was prepared as a suspension and administered following an overnight fast. Each dose group consisted of six subjects, with four randomized to CP-105,696 and two to placebo in a double-blind manner.
Blood samples were obtained at 0 (pre-dose), 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 48 and 72 h for all dose groups, as well as at 96, 120, 144, 168 and 192 h post-dose for the 5, 10 and 20 mg dose groups, 120, 168, 216, 264, 312, 408, 504 and 600 h post-dose for the 40 mg dose group and 120, 168, 240, 312, 456, 600, 792 and 1008 h post-dose for the 80, 160, 320 and 640 mg dose groups. Additional blood samples were obtained from selected subjects at the discretion of the investigator. Plasma was prepared and stored at −20° C until analysis. Urine samples were obtained at 0–24 h post-dose. A 20 ml aliquot of the urine sample was stored at −20° C until analysis.
Assay for CP-105,696 plasma and urine concentrations
Plasma and urine concentrations of CP-105,696 were determined by reverse-phase high pressure liquid chromatography with ultraviolet detection. Following acidification of plasma or urine and extraction with methyl-t-butyl ether, chromatography was performed using a Waters Nova-Pak C18 column and a mobile phase consisting of acetonitrile: methanol:water:triethylamine (36:39:29:1, v/v/v/v) adjusted to pH 4.35 with phosphoric acid. CP-105,696 and the internal standard (an isopropyl analog of CP-105,696) were detected by ultraviolet absorbance at 280 nm. The plasma assay was validated over two concentration ranges resulting in dynamic ranges of 0.25 to 25 μg ml−1 and 10 to 500 μg ml−1. The dynamic range of the urine assay was 0.25 to 5 μg ml−1. Standard curves used weighted (1/x) least squares regression and were linear over their respective concentration ranges, with correlation coefficients of 0.996–1.000. The intra- and inter-day coefficients of variation for all assays were generally less than 10%.
Pharmacokinetic analyses
The pharmacokinetics of CP-105,696 were determined using noncompartmental analyses. The terminal phase rate constant (λz) was estimated using least squares regression analysis of the CP-105,696 plasma concentration-time data obtained over the terminal log-linear phase. Group mean half-lives were calculated as 0.693/mean λz. The area under the plasma concentration-time curve from 0 h post-dose to the last sampling time ((AUC(0,_t_last)) with a quantifiable concentration was calculated by the linear trapezoidal rule. The area from tlast to infinity was estimated by _C_est(_t_last)/λz, where _C_est(_t_last) represented the estimated concentration at _t_last based upon the aforementioned regression analysis. The area under the concentration-time curve from 0 to infinity (AUC(0,∞)) was estimated by the sum of AUC(0,_t_last) and AUC(_t_last-∞). Plasma clearance (CL/F) and volume of distribution (_V_z/F) were calculated assuming complete absorption of the drug (F = 1.00), using the equations Dose/AUC(0,∞) and CL/λz, respectively; the assumption of complete absorption is consistent with absolute bioavailability determinations of 80–100% in animals. The maximum CP-105,696 plasma concentration (_C_max) for each dose was obtained directly from the experimental data, with _t_max defined as the time of the first occurrence of _C_max.
Whole blood neutrophil CD11b/CD18 determinations
Whole blood was collected in EDTA vacutainer tubes prior to and 4 h after oral dosing with CP-105,696. After a 5 min incubation at 37° C, 100 μl of whole blood were added to tubes containing 10 μl of either buffer (phosphate buffered saline (PBS) supplemented with 0.2% bovine serum albumin and 10 mm EDTA, pH 7.25) or 10 times concentration of LTB4 (final concentration of LTB4 = 10−11 to 10−6 m). After a 10 min incubation, samples were placed in an ice-water bath and immediately diluted with 1 ml of PBS-EDTA containing 0.2% sodium azide and 2% heat inactivated fetal bovine serum. After centrifugation at 200 g for 10 min at 4° C, the supernatant was aspirated and the cells resuspended in the remaining volume by gentle shaking. The samples were then labeled with anti-human CD11b FITC (Bear 1 clone, Caltag) for 30 min at 4° C and processed as previously described [13]. The fluorescent intensity of the samples was measured on a Coulter Profile II flow cytometer. Forward and right angle light scatter were used to gate on the neutrophil population and to exclude monocytes, lymphocytes and dead cells. For each sample, 5000 events were collected. Data were collected as log fluorescence and expressed as mean channel fluorescence (MCF).
Analysis of flow cytometry data
For each patient, CD11b upregulation was expressed as a percent of the maximal MCF obtained for that curve and was calculated as follows:
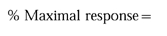

The data was then plotted with the LTB4 concentration on the X-axis and the percent maximal response on the Y-axis. The E_C_50 was determined using a graphics curve fitting program (Kaleidagraph), using a Hill logistic regression model:
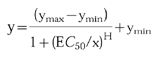
Where y is the value of the CD11b upregulation, ymax and ymin are the maximum and minimum allowable values, respectively, x is the concentration of LTB4, E_C_50 is the concentration of LTB4 at the half point between ymax and ymin and H is the maximum slope of the sigmoid curve.
The E_C_50 dose ratio was calculated as follows:
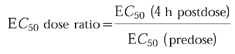
Statistical analyses
Differences in CP-105,696 pharmacokinetic parameters between dose groups were determined by single factor analysis of variance.
Determination of the CP-105,696 doses which resulted in a significant increase in the CD11b E_C_50 dose ratio were determined by comparing placebo- and drug-treated groups using analysis of variance with correction for multiple comparisons [18]. The test for significance used the following one-sided hypothesis:
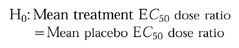
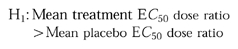
Contrast was used to measure simultaneously for a significant difference for each treatment group from placebo group (P < 0.05). An observation of significance in overall treatment effect was followed by individual comparisons of each treatment response to placebo response using a Bonferroni correction to preserve experiment-wise error. The Bonferroni adjusted significant P values were P < 0.01 (0.05/treatment groups). The statistical analysis was performed using SAS.
Results
Pharmacokinetics of CP-105,696
Oral administration of CP-105,696 to healthy male volunteers at single doses of 5 to 640 mg resulted in quantifiable plasma concentrations of the compound (Table 1, Figure 2). No quantifiable concentrations of CP-105,696 were detected in placebo-administered subjects or in the pre-dose plasma samples of drug-treated volunteers. Median _t_max estimates were 14, 6, 5, 5, 7, 6, 36 and 8 h following doses of 5, 10, 20, 40, 80, 160, 320 and 640 mg, respectively. Both mean _C_max and AUC(0,∞) increased with increasing dose (Figure 3). Mean terminal elimination half-life values were 346, 346, 433, 365 and 385 h for the 40, 80, 160, 320 and 640 mg dose groups, respectively. AUC(0,∞), λz and half-life were not calculated at doses of 5, 10 and 20 mg since the sampling length with quantifiable CP-105,696 plasma concentrations was too short relative to the projected half-lives. Oral clearance and volume of distribution were estimated assuming complete absorption of drug as has been observed in preclinical species (data not shown). Mean oral clearance and volume of distribution at the 40 to 640 mg dose levels ranged from 0.03 to 0.05 l h−1 and 15.9 to 28.1 l, respectively. There were no statistically significant (P < 0.05) differences in _k_el, clearance or volume of distribution between dose levels.
Table 1.
Pharmacokinetics of CP-105,696 in healthy male volunteers following oral administration at single doses of 5 to 640 mg.
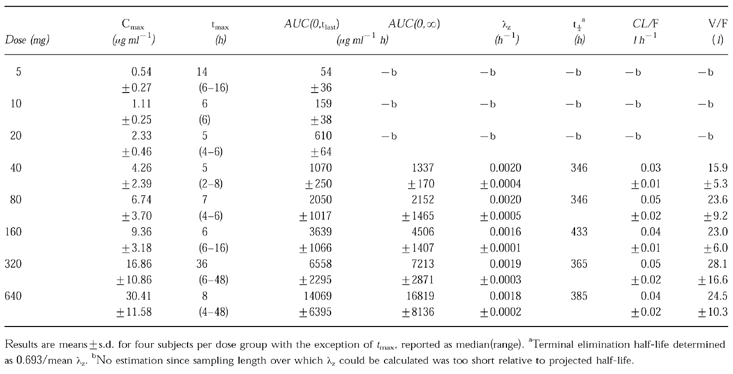
Figure 2.
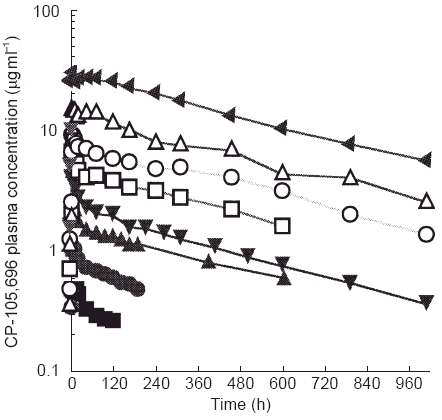
Mean plasma concentrations of CP-105,696 following oral administration to healthy male volunteers at single doses of 5 to 640 mg. (▪ 5 mg, • 10 mg, ▴ 20 mg, ▾ 40 mg, □ 80 mg, ○ 160 mg, ▵ 320 mg, ▸ 640 mg.
Figure 3.
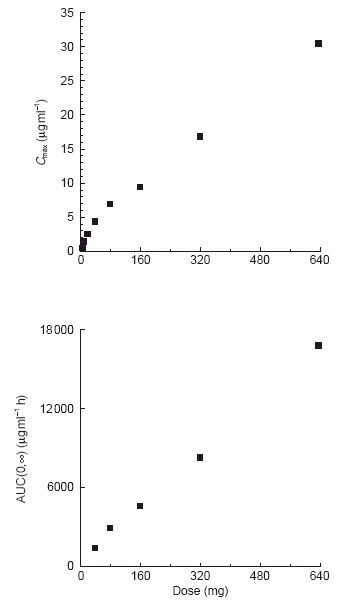
Relationship between mean _C_max or AUC and dose of CP-105,696 following oral administration to healthy human male volunteers at single doses of 5 to 640 mg.
Urinary concentrations of CP-105,696 were less than the lower limit of quantitation of the assay (0.25 μg ml−1) at the 640 mg dose level (data not shown). CP-105,696 excretion into urine was calculated as <1% of the administered dose based on the urine assay lower limit of quantitation. Therefore, the urine samples at lower dose groups were not assayed.
Pharmacodynamics
Whole blood was obtained at 4 h post-dose from each volunteer. The 4 h timepoint was used to assess the pharmacodynamics because it would allow for processing of pre-dose and post-dose samples on the same day and it was predicted to provide CP-105,696 plasma concentrations near the _C_max for the various dose levels. Expression of neutrophil CD11b/CD18 was determined ex vivo and correlated with CP-105,696 plasma concentrations (Table 2). The mean E_C_50 dose ratio for CD11b expression was 1.00 in control subjects following administration of placebo, indicating no placebo effect (Figure 4a). However, in subjects who received CP-105,696 the E_C_50 dose ratio increased in a linear fashion, indicating concentration-dependent inhibition of LTB4-mediated neutrophil activation. (Figure 4b, Figure 5). Statistically significant (P < 0.01) increases in the mean E_C_50 concentration ratio were achieved following doses of 320 and 640 mg.
Table 2.
Inhibition of LTB4-induced CD11b upregulation on human neutrophils following oral administration of CP-105,696.
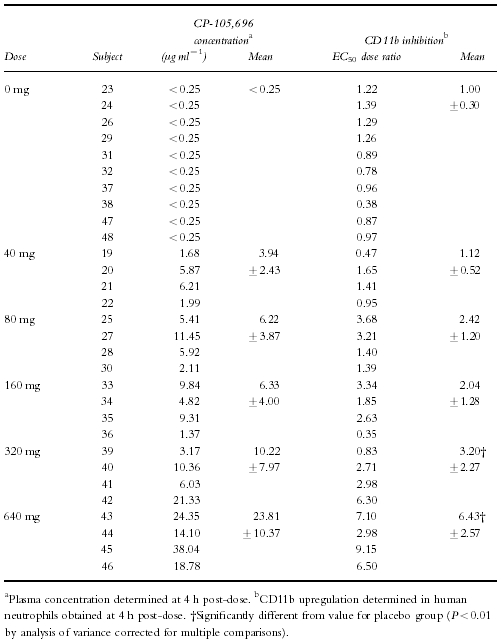
Figure 4.
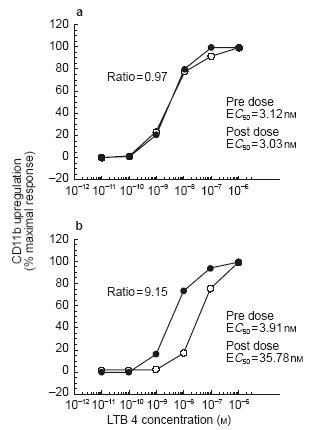
Examples of concentration-response curves demonstrating influence of circulating plasma concentrations of CP-105,696 on ex vivo LTB4-dependent CD11b upregulating in human whole blood. a) LTB4 concentration-response curves generated in human whole blood before (•) and after (○) administration of oral placebo to subject 48. b) LTB4 concentration-response curves generated in human whole blood before (•) and after (○) administration of a 640 mg dose of CP-105,696 to subject 45.
Figure 5.
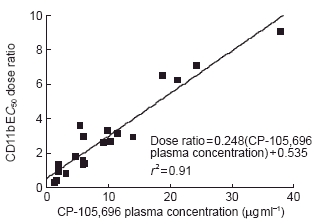
Relationship between E_C_50 dose ratio of LTB4-induced neutrophil CD11b upregulation and plasma concentration of CP-105,696 in normal human volunteers. CD11b expression in neutrophils was determined in whole blood using an ex vivo flow cytometric assay and CP-105,696 plasma concentrations were determined using h.p.l.c. (See Methods). E_C_50 dose ratio for LTB4-induced CD11b upregulation was determined by comparing the E_C_50 value in whole blood samples obtained at 4 h following oral administration of CP-105,696 to that in predose blood of the same subject.
Discussion
The terminal elimination half-life of CP-105,696 was prolonged, with half-lives of individual subjects ranging from 289–495 h and a mean half-life of 375 h for all subjects in the study. Both _C_max and AUC(0,∞) increased with increasing dose and terminal elimination half-life, oral clearance and volume of distribution were dose-independent. Although there was an apparent nonlinear relationship between mean _C_max and dose, the relationship between AUC(0,∞) and dose indicated that the overall disposition of CP-105,696 is linear. The nonlinearity in mean _C_max may be an artifact of the non-crossover design of this study and the variability in _t_max, which was most likely due to prolonged or delayed absorption resulting from administration of the compound in a suspension formulation. Less than 1% of the administered dose of CP-105,696 was detected in the urine of subjects in the 640 mg dose group, suggesting that biliary rather than renal excretion is the major route of elimination for unchanged drug. If that is the case, the prolonged half-life of this compound may be attributable in part to extensive enterohepatic recirculation. Supporting this hypothesis are preliminary studies in rats demonstrating that >90% of intravenously administered CP-105,696 is excreted via the bile and that non-recirculating cannulation of the bile duct results in marked reductions in the half-life of CP-105,696 in that species (data not shown). Further studies have been designed to evaluate the contribution of enterohepatic recirculation to the prolonged half-life of this compound in humans.
Other compounds with long half-lives include chloroquine, amiodarone, auronefin and gold sodium thiomalate, with half-lives ranging from 17–41 days [18–20]. The factor shared by these compounds that contributes to their prolonged half-lives is extensive deposition in tissues, rather than extremely low plasma clearance. For example, despite moderate plasma clearance values of 8 to 25 l h−1, the volumes of distribution of amiodarone and chloroquine are 4600 and ≥20 000 l, respectively, with tissue:plasma concentration ratios as high as 500. In contrast, the pharmacokinetics of CP-105,696 in humans are characterized by an extremely low plasma clearance in addition to a volume of distribution similar to plasma volume, both consistent with the compound's high protein binding. These differences would suggest that, unlike some other drugs with long half-lives, the pharmacokinetics of CP-105,696 may result principally from a combination of high protein binding and enterohepatic recirculation rather than extensive deposition into tissue.
Despite a high binding of CP-105,696 to human plasma proteins and a low volume of distribution, the compound retained significant activity in a pharmacological assay for LTB4-receptor antagonism, the inhibition of LTB4-mediated CD11b expression on blood neutrophils. A three-fold rightward shift in the concentration response curve of LTB4-induced upregulation of neutrophil CD11b was observed with mean plasma concentrations of CP-105,696 of ≥10.22 μg ml−1. Likewise, previous studies in mouse models of collagen-induced arthritis demonstrated that the compound retained significant in vivo activity despite high plasma protein binding and low volume of distribution in that species [13].
Statistically significant inhibition of CD11b upregulation was achieved following a single 320 mg dose of CP-105,696. Furthermore, the plasma concentrations necessary to significantly inhibit neutrophil CD11b upregulation were achieved throughout a 24 hour period following a 320 mg dose, suggesting that pharmacologically significant LTB4 receptor antagonism can be achieved with once-daily dosing of 320 mg CP-105,696. Considering the potential for significant drug accumulation following once-daily dosing of a compound with a 395 h half-life, we recognize that pharmacologically significant steady-state plasma concentrations of CP-105,696 may be achieved at relatively low daily doses following an oral loading dose. Further studies will be performed with this compound to assess its multiple dose pharmacokinetics and pharmacodynamics and its efficacy in inflammatory diseases in which LTB4 is believed to play a central role.
Acknowledgments
The assistance of Dan Septie, Mikhail Petrov and Ingrid Gienapp in the conduct of the clinical study and flow cytometric analyses is greatly appreciated. The assistance of David Raunig in statistical analyses is greatly appreciated.
References
- 1.Ford-Hutchinson AW. Leukotriene B4 in Inflammation. Crit Rev Immunol. 1990;10:1–12. [PubMed] [Google Scholar]
- 2.Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJH. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286:265–265. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- 3.Brain S, Camp R, Dowd P, Kobza Black A, Greaves M. The release of leukotriene B4-like material in biologically active amounts from the lesional skin of patients with psoriasis. J Invest Dermatol. 1984;83:70–73. doi: 10.1111/1523-1747.ep12261712. [DOI] [PubMed] [Google Scholar]
- 4.Camp R, Jones RR, Brain S, Woolard P, Greaves M. Production of intraepidermal microabscesses by topical application of leukotriene B4. J Invest Dermatol. 1984;82:202–204. doi: 10.1111/1523-1747.ep12259945. [DOI] [PubMed] [Google Scholar]
- 5.Jones A, Al-Janabi M, Solanki K, et al. In vivo leukocyte migration in arthritis. Arthritis Rheum. 1991;34:270–275. doi: 10.1002/art.1780340304. [DOI] [PubMed] [Google Scholar]
- 6.Duell EA, Ellis CN, Voorhees JJ. Determination of 5, 12, and 15-lipoxygenase products in keratomed biopsies of normal and psoriatic skin. J Invest Dermatol. 1988;91:446–450. doi: 10.1111/1523-1747.ep12476562. [DOI] [PubMed] [Google Scholar]
- 7.Black AK, Camp RDR, Mallet AI, Cunningham FM, Hofbauer M, Greaves MW. Pharmacologic and clinical effects of lonapalene (RS 43179), a 5-lipoxygenase inhibitor, in psoriasis. J Invest Dermatol. 1990;95:50–54. doi: 10.1111/1523-1747.ep12873300. [DOI] [PubMed] [Google Scholar]
- 8.Klickstein LB, Shapleigh C, Goetzl EJ. Lipoxygenation of arachidonic acid as a source of polymorphonuclear leukocyte chemotactic factors in synovial fluid and tissue in rheumatoid arthritis and spondyloarthritis. J Clin Invest. 1980;66:1166–1170. doi: 10.1172/JCI109947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharon P, Stenson WF. Enhanced synthesis of leukotriene B4 by colonic mucosa in inflammatory bowel disease. Gastroenterology. 1984;86:453–460. [PubMed] [Google Scholar]
- 10.Shindo K, Matsumoto Y, Hirai Y, et al. Measurement of leukotriene B4 in arterial blood of asthmatic patients during wheezing attacks. J Int Med. 1990;228:91–96. doi: 10.1111/j.1365-2796.1990.tb00200.x. [DOI] [PubMed] [Google Scholar]
- 11.Wardlaw AJ, Hay H, Cromwell O, Collins JV, Kay AB. Leukotrienes, LTC4 and LTB4, in bronchalveolar lavage in bronchial asthma and other respiratory diseases. J Allergy Clin Immunol. 1989;84:19–26. doi: 10.1016/0091-6749(89)90173-5. [DOI] [PubMed] [Google Scholar]
- 12.Koch K, Melvin LS, Reiter LA, et al. (+)-1-(3S,4R)-[3-(4-Phenylbenzyl)-4-hydroxychroman-7-yl]cyclopentane carboxylic acid, a hightly potent, selective leukotriene B4 antagonist with oral activity in the murine collagen-induced arthritis model. J Med Chem. 1994;37:3197–3199. doi: 10.1021/jm00046a001. [DOI] [PubMed] [Google Scholar]
- 13.Griffiths RJ, Pettipher ER, Koch K, et al. Leukotriene B4 plays a critical role in the progression of collagen-induced arthritis. Proc Natl Acad Sci. 1995;92:517–521. doi: 10.1073/pnas.92.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kishimoto TK, Jutila MA, Berg EL, Butcher EC. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science. 1989;245:1238–1241. doi: 10.1126/science.2551036. [DOI] [PubMed] [Google Scholar]
- 15.Jutila MA, Rott L, Berg EL, Butcher EC. Function and regulation of the neutrophil MEL-14 antigen in vivo: Comparison with LFA-1 and MAC-1. J Immunol. 1989;143:3318–3324. [PubMed] [Google Scholar]
- 16.Marder P, Schultz RM, Spaethe SM, Sofia MJ, Herron DK. Flow cytometric evaluation of the effects of leukotriene B4 receptor antagonists ( LY255283 and SC-41930) on calcium mobilization and integrin expression of activated human neutrophils. Prostaglandins Leukotrienes Essent Fatty Acids. 1992;46:265–270. doi: 10.1016/0952-3278(92)90033-f. [DOI] [PubMed] [Google Scholar]
- 17.Schultz RM, Marder P, Spaethe SM, Herron DK, Sofia MJ. Effects of two leukotrine B4 (LTB4) receptor antagonists ( LY255283 and SC-41930) on LTB4-induced human neutrophil adhesion and superoxide production. Prostaglandins, Leukotrienes Essent Fatty Acids. 1991;43:267–271. doi: 10.1016/0952-3278(91)90041-3. [DOI] [PubMed] [Google Scholar]
- 18.Neter J, Watterman W. Applied Linear Statistical Models. Homewood, IL: Irwin; 1974. [Google Scholar]
- 19.Latini R, Tognoni G, Kates RE. Clinical pharmacokinetics of amiodarone. Clin Pharmacokin. 1984;9:136–156. doi: 10.2165/00003088-198409020-00002. [DOI] [PubMed] [Google Scholar]
- 20.Frisk-Holmberg M, Bergqvist Y, Termond E, Domeij-Nyberg B. The single dose kinetics of chloroquine and its major metabolite desethylchloroquine in healthy subjects. Eur J Clin Pharmacol. 1984;26:521–530. doi: 10.1007/BF00542151. [DOI] [PubMed] [Google Scholar]
- 21.Blocka KLN, Paulus HE, Furst DE. Clinical pharmacokinetics of oral and injectable gold compounds. Clin Pharmacokin. 1986;11:133–143. doi: 10.2165/00003088-198611020-00003. [DOI] [PubMed] [Google Scholar]