Cyclooxygenase-2 Expression in Helicobacter pylori-Associated Premalignant and Malignant Gastric Lesions (original) (raw)
Abstract
Expression of cyclooxygenase-2 (COX-2) in various stages of the _Helicobacter pylori-_associated gastric carcinogenesis pathway has not been elucidated. We investigated the distribution and intensity of COX-2 expression in premalignant and malignant gastric lesions, and monitored the changes after H. pylori eradication. Gastric biopsies from _H. pylori_-infected patients with chronic active gastritis, gastric atrophy, intestinal metaplasia (IM), gastric adenocarcinoma, and noninfected controls were studied. Expression of COX-2 was evaluated by immunohistochemistry and in situ hybridization. Endoscopic biopsies were repeated 1 year after successful eradication of H. pylori in a group of IM patients for comparing COX-2 expression and progression of IM. In all _H. pylori_-infected patients, COX-2 expression was predominantly found in the foveolar and glandular epithelium and, to a lesser extent, in the lamina propria. In the noninfected group, only 35% of cases demonstrated weak COX-2 expression. Intensity of COX-2 was not significantly different between the chronic active gastritis, gastric atrophy, IM, and gastric adenocarcinoma groups. In 17 patients with IM, COX-2 expressions in the epithelial cells and stromal cells were reduced 1 year after H. pylori eradication. However, the changes in COX-2 expression did not correlate with progression/regression of IM. Both premalignant and malignant gastric lesions demonstrate strong COX-2 expression. Successful eradication of H. pylori leads to down-regulation of COX-2 expression but failed to reverse IM at 1 year.
There is a wealth of evidence to support that cyclooxygenase-2 (COX-2), an inducible isoform of cyclooxygenase enzyme, which converts arachidonic acid to prostanoids, is strongly expressed in gastrointestinal tumors. Expression of COX-2 had been reported in colorectal cancers, 1,2 pancreatic cancer, 3 hepatocellular carcinoma, 4,5 esophageal cancers, 6,7 and gastric cancers. 8,9 Up-regulation of this inducible COX isoform in malignancies has also been found to associate with increased invasiveness and metastatic potentials of the tumors. 10,11 Recently, there are also studies demonstrating COX-2 expression in premalignant lesions. These include colonic adenoma, 1 Barrett’s esophagus, 12 and atypical alveolar epithelium in asbestosis and idiopathic fibrosing alveolitis, 13 implying that this enzyme may be involved in the early process of carcinogenesis. Although the exact mechanism of how COX-2 contributes to cancer development is still unclear, existing data suggests that inhibition of COX-2 induces apoptosis, suppresses cellular proliferation, down-regulates bcl-2 expression, 14-18 enhances the growth of H-ras-transformed cells, 19 and induces expression of epidermal growth factor. 20
H. pylori has been classified as class 1 carcinogen for gastric cancer by the International Agency for Research on Cancer in 1994. 21 According to Correa’s 22 model, gastric cancer develops in a multistep process from chronic active gastritis (CAG), gastric atrophy (GA), intestinal metaplasia (IM), dysplasia, and finally, gastric cancer. Recent studies also showed that H. pylori induced COX-2 expression in human gastric mucosa. 23-26 Furthermore, McCarthy et al 26 showed that COX-2 expression in antral mucosa was reduced but not eliminated in the epithelium after successful eradication of H. pylori. The role of _H. pylori_-induced COX-2 expression in gastric carcinogenesis is not clear. This study attempted to address the issue from two perspectives. First, we studied COX-2 expression at different stages of gastric carcinogenesis from inflammation, premalignant lesions, to gastric cancer. Second, we followed the changes of COX-2 expression after eradication of H. pylori infection and attempted to correlate these changes with improvement or worsening of gastric premalignant lesions.
Patients and Methods
Gastric biopsy tissues from _H. pylori_-infected patients with diagnoses of GA (n = 20), IM (n = 51), and gastric adenocarcinoma (n = 25) were studied. None of these patients were taking aspirin or NSAID. Intensity of COX-2 expression was examined by immunohistochemistry and was graded as 0 (negative), 1 (<5% cells with positive staining), 2 (5 to 30%), 3 (30 to 60% and with strong staining), and 4 (>60% and with very strong staining). Scoring was done by two independent investigators and the mean score was taken in each case. The localization of COX-2 protein detected by immunohistochemistry was verified by in situ hybridization. The scorings of COX-2 expression in various histological types were compared to patients with CAG (n = 40) and noninfected controls (n = 40).
In a group of patients with IM, endoscopic biopsy and immunohistochemistry were repeated at 1 year after 1 week of anti-Helicobacter therapy (omeprazole, amoxicillin, and clarithromycin). Antral biopsies were taken from the greater and lesser curvature within 2 to 3 cm from the pylorus for assessment of COX-2 staining and grading of IM as stipulated by the Updated Sydney Grading System. 27 Eradication of H. pylori was confirmed by negative results on the rapid urease test and histology.
Immunohistochemistry
Tissues were fixed in 10% buffered formalin and embedded in paraffin. Five-micrometer-thick sections were deparaffinized, and endogenous peroxidase activity was blocked with 3% H2O2 in Tris-buffered saline (TBS). Nonspecific binding was blocked with 5% rabbit serum (DAKO, Glostrup, Denmark) in TBS, and the tissues were incubated with antibody to COX-2 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA) in TBS containing 2% rabbit serum and 1% bovine serum albumin for 2 hours. This was followed by incubation with biotinylated rabbit anti-goat immunoglobulins (1:400; DAKO) for 45 minutes and then with avidin-biotin-peroxidase complex (DAKO) for 45 minutes. The color was developed in diaminobenzidine solution (Sigma Chemical Co., St. Louis, MO). The sections were then counterstained with Mayer’s hematoxylin. Tissues were incubated with TBS containing 2% rabbit serum and 1% bovine serum albumin without the primary antibody as control. To confirm the cellular localization of COX-2, the immunohistochemical staining was repeated with another primary antibody against COX-2 (Cayman, Ann Arbor, MI) in a subset of samples (n = 25).
Generation of COX-2 RNA Probes
Human COX-2 cDNA fragment was prepared by reverse transcription-polymerase chain reaction of gastric carcinoma. Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA). Five micrograms of total RNA was reverse-transcribed with the SuperScript Preamplification System for First Strand cDNA Synthesis (Life Technologies, Inc., Gaithersburg, MD) using random oligonucleotide priming in a 20-μl reaction. COX-2-specific primers (forward: TTCAAATGAGATTGTGGGAAAATTGCT; reverse: AGATCATCTCTGCCTGAGTATCTT) were used to amplify a fragment of 305 bp. Polymerase chain reaction was performed for 40 cycles at 94°C for 35 seconds, 55°C for 35 seconds, and 72°C for 30 seconds in a 25-μl volume containing 0.2 mmol/L dNTPs, 1× polymerase chain reaction buffer (20 mmol/L Tris-HCl, pH 8.4, 50 mmol/L KCl), 1.5 mmol/L MgCl2, and 0.625 U of Taq DNA-polymerase (Life Technologies, Inc.). The polymerase chain reaction products were analyzed in 2% agarose gel. The desired fragments were cut and cloned in pMOSBlue vectors using the pMOSBlue Blunt-Ended Cloning Kit (Amersham, Buckinghamshire, UK). The DNA fragment was sequenced to confirm identity and orientation of the insert. The anti-sense RNA probe was then synthesized using the DIG (digoxigenin) RNA Labeling Kit (Boehringer Mannheim, Germany). The integrity of the probe was determined by electrophoresis through an ethidium bromide-stained 2% agarose gel. Labeling efficiency was checked by dot blots as described by the manufacturer. A positive control experiment was conducted using glyceraldehyde-3-phosphate dehydrogenase RNA probe. For the negative control experiment, COX-2 sense RNA probe was prepared.
In Situ Hybridization
The method was modified from Komhoff et al. 28 Cryostat sections (8-μm thick) were mounted onto 3-aminopropyltriethoxysilane-coated slides (Marienfeld, Badmergentheim, Germany) and fixed for 10 minutes in 4% formaldehyde in phosphate-buffered saline (PBS), pH 7.4. After treatment in 0.2 N HCl for 20 minutes, the slides were incubated with 5 μg/ml of proteinase K (Sigma) at 37°C for 15 minutes. Sections were then acetylated in 0.1 mol/L of triethanolamine with 0.25% acetic anhydride for 5 minutes and then 0.5% acetic anhydride for an additional 5 minutes. Prehybridization was performed with hybridization solution containing 50% formamide, 1× Denhardt’s solution, 10% dextran sulfate, 0.2 mg/ml of salmon sperm DNA, 1 mg/ml of tRNA, 1 mmol/L of ethylenediaminetetraacetic acid, and 2× standard sodium citrate (SSC) devoid of cDNA probes for 1 hour. Hybridization was performed with 1 ng/μl digoxygenin-labeled COX-2 anti-sense RNA probe, and incubated at 65°C for 18 hours in a moist chamber. The slides were washed in 2× SSC at room temperature for 1 hour, 1× SSC at room temperature for 45 minutes, 0.1× SSC at 60°C for 30 minutes, and 0.1× SSC at room temperature for 15 minutes. Immunological detection was conducted using the DIG Nuclear Acid Detection Kit (Boehringer Mannheim). After the color development, the slides were counterstained with nuclear fast red. For negative controls, sections were hybridized with COX-2 sense RNA probe or treated with RNase before hybridization.
Statistical Analysis
Scoring of COX-2 staining between the five study groups (CAG, GA, IM, gastric adenocarcinoma, and control) were compared by Kruskal-Wallis nonparametric analysis of variance test, using Dunn’s multiple comparison test for post hoc comparison. Expression of COX-2 and grading of IM before and at 1 year after eradication of H. pylori was compared by the Mann-Whitney U test. Statistical significance was taken at P < 0.05.
Results
COX-2 expression was found in all gastric biopsies of _H. pylori_-infected patients, including 40 cases of CAG, 20 cases of GA, 51 cases of IM, and 25 cases of CA, with intensity scoring ranged from 1 to 4 (Table 1) ▶ . _H. pylori_-associated gastritis exhibited strong expression of COX-2 in foveolar and glandular epithelium but with a lower intensity in mononuclear inflammatory cells, myofibroblasts, and endothelial cells (Figure 1, A and B) ▶ . Strong expression of COX-2 was also found on intestinal epithelium of GA (Figure 1C) ▶ and IM (Figure 1, D and E) ▶ . Both diffuse and intestinal types of gastric cancer cells expressed COX-2 protein on immunohistochemistry and there was no significant difference in intensity of expression (median score, 2 versus 2; Figure 1, F and G ▶ ). The localization of COX-2 was confirmed by the second polyclonal antibody (Cayman antibody; Table 2 ▶ ) as well as by in situ hybridization (Figure 2) ▶ . On the contrary, only 14 (35%) of noninfected patients expressed COX-2 protein in the gastric biopsy. The intensity of COX-2 expression was significantly higher in the _H. pylori-_infected group when compared to the noninfected control (P < 0.0001). However, there was no difference in the COX-2 expression between the four groups of patients with H. pylori infection (Table 1) ▶ .
Table 1.
Scoring of COX-2 Expression in Noninfected Patients (Control) and _H. pylori_-Infected Patients with Chronic Active Gastritis (CAG), Gastric Atrophy (GA), Intestinal Metaplasia (IM), and Gastric Cancer (CA)
| Score | Control (n = 40) | CAG (n = 40) | GA (n = 20) | IM (n = 51) | CA (n = 25) |
|---|---|---|---|---|---|
| 0 | 26 | 0 | 0 | 0 | 0 |
| 1 | 14 | 6 | 4 | 21 | 11 |
| 2 | 0 | 32 | 11 | 24 | 4 |
| 3 | 0 | 2 | 4 | 6 | 9 |
| 4 | 0 | 0 | 1 | 0 | 1 |
Figure 1.
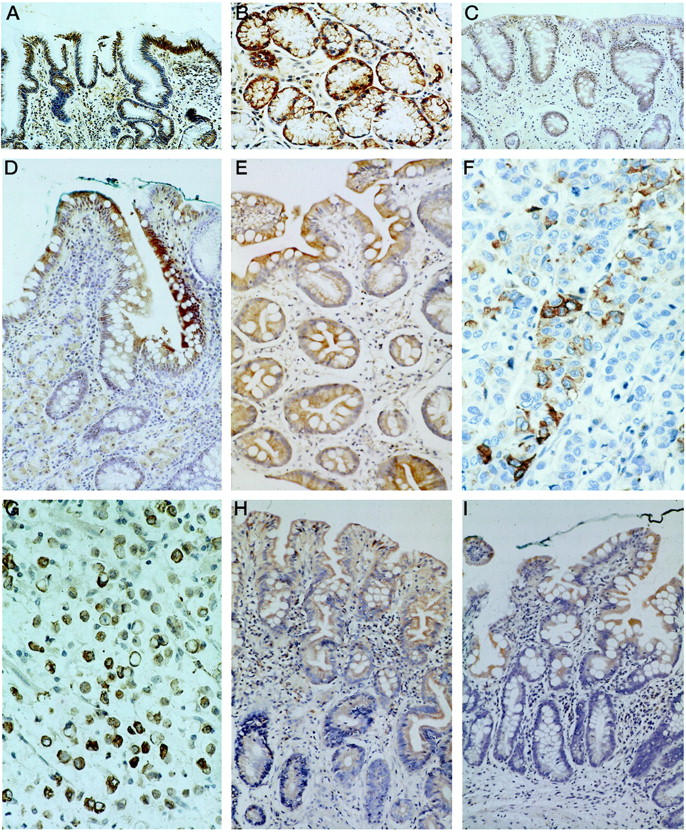
Immunohistochemical staining of COX-2 in: CAG showing strong expression in foveolar and glandular epithelium (A) and mild expression in mononuclear inflammatory cells, myofibroblasts, and endothelial cells in the lamina propria (B); GA showing strong expression in the atrophic glands of the gastric body (C); IM showing strong expression in intestinal epithelium and goblet cells (D and E); gastric cancer showing strong expression in cancer cells of both intestinal type (F) and diffuse type (G); IM before (H) and 1 year after eradication of H. pylori (I). There was an overall reduction in COX-2 expression with more dramatic changes in the glandular epithelium and stromal cells in the lamina propria. Residual expression of COX-2 was still present in the foveolar epithelium.
Table 2.
Cellular Localization of COX-2 by the Use of Two Different Primary Antibodies (Santa Cruz; Santa Cruz, CA, and Cayman; Ann Arbor, MI) in Gastric Epithelium
| Santa Cruz antibody | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Foveolar epithelium | Glandular epithelium | Stroma | Intestinal metaplasia | Tumor | |||||||
| + | − | + | − | + | − | + | − | + | − | ||
| Cayman antibody | + | 10 | 0 | 8 | 3 | 10 | 1 | 5 | 0 | 5 | 0 |
| − | 4 | 1 | 0 | 4 | 0 | 4 | 0 | 0 | 0 | 0 |
Figure 2.

A: In situ hybridization demonstrated that mRNA of COX-2 was predominantly located in the epithelial cells of the foveolar and glandular epithelium. B: Negative control by using sense COX-2 probe was shown.
Seventeen patients with IM and successful eradication of H. pylori were followed-up for 1 year with repeated endoscopy. Comparing the paired samples from the same patient before and after H. pylori eradication reviewed significant reduction of COX-2 expression in the epithelial cells (P = 0.049) and stromal cells (P = 0.001) (Table 3) ▶ . Changes in COX-2 expression in individual cases are shown in Figure 3 ▶ . Although there was an overall reduction in COX-2 expression at 1 year, changes in the foveolar epithelium failed to reach statistical significance. Furthermore, in all 17 cases, COX-2 expression was still detected in the epithelium albeit reduced expression. Reduction in COX-2 expression was more prominent in the stromal cells (Figure 1, H and I) ▶ . In seven cases, total disappearance of COX-2 in the laminar propria was documented at follow-up. Changes in IM before and after eradication of H. pylori were highly diversified in these 17 patients. Progression of IM at 1-year follow-up was found in six patients, regression of IM in five, and unchanged histology in six. Overall, there was no significant change in the severity of IM after H. pylori eradication. There was no correlation between changes in IM and COX-2 down-regulation in this group of patients.
Table 3.
COX-2 Expression in 17 Patients with Intestinal Metaplasia before (HP+) and at 1-Year after (HP−) Eradication of H. pylori
| Score | Epithelial cells | Stromal cells | ||
|---|---|---|---|---|
| HP+ | HP− | HP+ | HP− | |
| 0 | 0 | 0 | 0 | 7 |
| 1 | 1 | 7 | 7 | 9 |
| 2 | 5 | 6 | 10 | 1 |
| 3 | 11 | 3 | 0 | 0 |
| 4 | 0 | 1 | 0 | 0 |
Figure 3.

A: Changes in COX-2 expression in foveolar epithelium, glandular epithelium, and stromal cells of the stomach before (HP+) and at 1 year after (HP−) eradication of H. pylori in 17 patients with IM. Thickness of each line represents number of patients in that category (with numbers indicated). COX-2 expression was significantly reduced in glandular epithelium (P = 0.036) and stromal cells (P = 0.0012). No significant reduction in foveolar epithelium (P = 0.099) could be demonstrated. B: The percentage of samples showing increased, unchanged, and reduced in COX-2 expression at 1-year follow-up.
Discussion
Although previous studies confirmed that COX-2 expression in gastric mucosa is found in the presence of _H. pylori_-induced gastritis, 23-26 their localization in the gastric tissue varies between different studies. In our study, COX-2 staining was predominantly found in the foveolar and glandular epithelium which was in keeping with that reported by Sawaoka et al. 23 On the contrary, Fu et al 24 reported that in _H. pylori_-positive gastritis, COX-2 staining is primarily localized in the mononuclear cells and myofibroblast in the laminar propria with no detectable staining in the epithelium. The difference in localization of COX-2 staining could be the result of variations in antibodies used in different studies and patient heterogeneity.
However, we have confirmed the cellular localization of COX-2 with a second set of antibodies (Table 2) ▶ and also by in situ hybridization (Figure 2) ▶ which strongly supported the validity of our findings.
In this study, we have shown that COX-2 is expressed in the epithelial lining of the stomach in the whole spectrum of _H. pylori-_associated gastric carcinogenesis pathway from CAG, to GA and IM, and finally to gastric cancer. This finding implies that COX-2 might be involved in the early stages of gastric cancer development ie, GA and IM. Similar phenomena have been reported in colonic adenoma and colorectal cancer as well as Barrett’s esophagus and esophageal cancers. 1,12 In a study on colorectal cancers, Hau et al 29 reported enhanced COX-2 expression in colonic adenoma when compared with normal mucosa but there was an insignificant difference between adenoma and adenocarcinoma of the colon. This observation parallels the relationship between GA, IM, and gastric cancer. Although the mechanism of COX-2 expression in carcinogenesis is still unclear, studies suggest that overexpression of COX-2 is associated with cellular resistance to apoptosis whereas treatment with specific COX-2 inhibitor suppresses cellular proliferation and induces apoptosis. 6,14-18 This is postulated to be the possible mechanism of chemoprevention of colorectal cancer by nonsteroidal anti-inflammatory drugs. Moreover, prostaglandin, the product of cyclooxygenase, may also be related to the carcinogenesis process as PGE2 levels are increased in colorectal tumors 30 and eicosanoids stimulate the proliferation of colonocytes. 31 It would be helpful to study the concentration of prostaglandin 32 in gastric samples. Because of the unavailability of fresh samples, we were not able to examine the prostaglandin levels in this study. Interestingly, the addition of prostaglandins do not seem to affect the actions of NSAID in colon and gastric cancer cells, which argue against a role for the eicosanoids in tumorigenesis. 33,34
On the other hand, COX-2- or prostaglandin-independent pathways are also likely to have a role to play in chemoprevention. It has been shown that salicylic acid and sulindac sulfone, which do not appreciably affect the catalytic activity of COX, also induce apoptosis in colonic cell lines. 33 Recently, other potential mechanisms for NSAID-related cancer prevention have also been suggested, which include alterations in cell cycle-machinery such as p21. 35
Because COX-2 expression is induced by H. pylori infection, we intended to investigate whether eradication of the bacterium, by resolution of inflammation, would lead to down-regulation of COX-2 expression and plausibly reverse the carcinogenesis process. Previous studies have shown that COX-2 expression in the antral epithelium is reduced but not eliminated after successful eradication of H. pylori. 26 In that study, biopsies were taken from patients with chronic gastritis and comparison was made at 4 weeks after antibiotic therapy. The present study examines a wider spectrum of gastric histology and a much longer follow-up period of 1 year after successful eradication of H. pylori. We found that in patients with IM, despite reduction of COX-2 expression at 1 year after eradication of H. pylori, there was no evidence of regression of IM or hence, the carcinogenic process. Specifically, there was no correlation between down-regulation of COX-2 and regression of IM. The most convenient explanation would be that 1-year follow-up is not sufficiently long enough for the regression of IM in the gastric tissue to be detected. In fact, most studies following patients after H. pylori eradication do not show significant improvement in premalignant gastric lesions, such as atrophy and IM. 36-38 However, the follow-up period of these studies may not be long enough to show any significant change in histology. Down-regulation of COX-2 and regression of IM, if it ever occurs, is likely to be a slow process.
Results from this study also showed that despite eradication of H. pylori in the stomach, there was only a modest reduction of COX-2 in the gastric epithelium. On the contrary, COX-2 expression in the lamina propria was markedly reduced. With the resolution of mucosal inflammation, COX-2 expression in mononuclear cells, myofibroblasts, and endothelial cells were greatly diminished. This dissociation in COX-2 expression between the epithelium and the lamina propria implies that expression of COX-2 in the former is not solely caused by inflammation. Other factors in the gastric milieu might contribute to perpetuation of COX-2 overexpression after successful H. pylori eradication. Thus, treatment of H. pylori infection alone, with the associated improvement in gastric inflammation, may not be sufficient to reverse the gastric carcinogenesis process. It would be interesting to examine the role of NSAID, or selective COX-2 inhibitors, on changes of COX-2 expression in premalignant gastric lesions after H. pylori eradication. However, human studies are lacking in this aspect. In addition to its role in carcinogenesis, animal studies suggested inhibition of COX-2 may impair ulcer healing 39 and resolution of allergic edema. 40 We had recently demonstrated the expression of both COX-1 and COX-2 in _H. pylori_-associated gastric ulcer. 41 Whether the use of a COX-2 inhibitor, which is generally thought to be safe for the stomach, will actually impair the healing of ulcer in human still await further evaluation.
In conclusion, COX-2 protein is expressed in the gastric epithelium throughout the multistep gastric carcinogenesis cascade. Curing of H. pylori infection leads to down-regulation of COX-2 in the lamina propria but less so in the gastric epithelium. Hence, eradication of H. pylori may not be sufficient for the prevention of gastric cancer. Specific COX-2 inhibition should be tested for its efficacy in halting the process of carcinogenesis.
Footnotes
Address reprint requests to Joseph J. Y. Sung, M.D., Department of Medicine & Therapeutics, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong. E-mail: joesung@cuhk.edu.hk.
Supported by Research Grants Council of Hong Kong (No. RGC 2140129).
References
- 1.Ebherhat CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN: Up-regulation of cyclooxygenase-2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107:1183-1188 [DOI] [PubMed] [Google Scholar]
- 2.Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asia K, Kimura S, Kato H, Kondo M, Hla T: Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res 1995, 55:3785-3789 [PubMed] [Google Scholar]
- 3.Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT, Fahey TJ: Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res 1999, 59:589-590 [PubMed] [Google Scholar]
- 4.Koga H, Sakisaka S, Ohishi M, Kawaguchi T, Taniguchi E, Sasatonmi K, Harada M, Kusaba T, Tanaka M, Kimura R, Nakashima Y, Nakashima O, Kojiro M, Kurohiji T, Sata M: Expression of cyclooxygenase-2 in human hepatocellular carcinoma: relevance to tumor dedifferentiation. Hepatology 1999, 29:688-696 [DOI] [PubMed] [Google Scholar]
- 5.Shiota G, Okubo M, Noumi T, Noguchi N, Oyama K, Takano Y, Yashima K, Kishimoto Y, Kawasaki H: Cyclooxygenase-2 expression in hepatocellular carcinoma. Hepatogastroenterology 1999, 46:407-412 [PubMed] [Google Scholar]
- 6.Zimmermann KC, Sarbia M, Weber AA, Borchard F, Gabbert HE, Schror K: Cyclooxygenase-2 expression in human esophageal carcinoma. Cancer Res 1999, 59:198-204 [PubMed] [Google Scholar]
- 7.Ratnasinghe D, Tangrea J, Roth MJ, Dawsey S, Hu M, Anver M, Wang QH, Taylor PR: Expression of cyclooxygenase-2 in human squamous cell carcinoma of the esophagus: an immunohistochemical survey. Anticancer Res 1999, 19:171-174 [PubMed] [Google Scholar]
- 8.Ristimaki A, Honkanen N, Jankala H, Sipponen P, Harkonen M: Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res 1997, 57:1276-1280 [PubMed] [Google Scholar]
- 9.Uefuji K, Ichikura T, Mochizuki H, Shinomiya N: Expression of cyclooxygenase-2 protein in gastric carcinoma. J Surg Oncol 1998, 69:168-172 [DOI] [PubMed] [Google Scholar]
- 10.Murata H, Kawano S, Tsuji S, Tsuji M, Sawaoka H, Kimura Y, Shiozaki H, Hori M: Cyclooxygenase-2 over-expression enhances lymphatic invasion and metastasis in human gastric carcinoma. Am J Gastroenterol 1999, 94:451-455 [DOI] [PubMed] [Google Scholar]
- 11.Tsuji M, Kawano S, DuBois RN: Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA 1997, 94:3336-3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson KT, Fu S, Ramanujam KS, Meltzer SJ: Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in Barrett’s esophagus and associated adenocarcinoma. Cancer Res 1998, 58:2929-2934 [PubMed] [Google Scholar]
- 13.Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A: Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res 1998, 58:4997-5001 [PubMed] [Google Scholar]
- 14.Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN: Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res 1998, 58:362-366 [PubMed] [Google Scholar]
- 15.Sawaoka H, Kawano S, Tsuji S, Tsujii M, Gunawan ES, Takei Y, Nagano K, Hori M: Cyclooxygenase-2 inhibitors suppress the growth of gastric cancer xenografts via induction of apoptosis in nude mice. Am J Physiol 1998, 274:G1061-G1067 [DOI] [PubMed] [Google Scholar]
- 16.Battu S, Rigaud M, Beneytout JL: Resistance to apoptosis and cyclooxygenase-2 expression in a human adenocarcinoma cell line HT29 CL.19A. Anticancer Res 1998, 18:3579-3583 [PubMed] [Google Scholar]
- 17.Erickson BA, Longo WE, Panesar N, Mazuski JE, Kaminski DL: The effect of selective cyclooxygenase inhibitors on intestinal epithelial cell mitogenesis. J Surg Res 1999, 81:101-107 [DOI] [PubMed] [Google Scholar]
- 18.Liu XH, Yao S, Kirschenbaum A, Levine AC: NS398, a selective cyclooxygenase-2 inhibitor, induces apoptosis and down-regulates bcl-2 expression in LNCaP cells. Cancer Res 1998, 58:4245-4249 [PubMed] [Google Scholar]
- 19.Sheng GG, Shao J, Sheng H, Hooton EB, Isakson PC, Morrow JD, Coffey Jr RJ, DuBois RN: A selective cyclooxygenase-2 inhibitor suppresses the growth of H-ras-transformed rat intestinal epithelial cells. Gastroenterology 1997, 113:1883–1891 [DOI] [PubMed]
- 20.Kinoshita T, Takahashi Y, Sakashita T, Inoue H, Tanabe T, Yoshimoto T: Growth stimulation and induction of epidermal growth factor receptor by overexpression of cyclooxygenase-1 and -2 in human colon carcinoma cells. Biochim Biophys Acta 1999, 1438:120-130 [DOI] [PubMed] [Google Scholar]
- 21.International Agency for Research on Cancer Working Group on the Evaluation of Carcinogenic Risks to Humans: Helicobacter pylori. Schistosomes, Liver Flukes and Helicobacter pylori: Views and Expert Opinions of an IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, IARC, 1994, pp 177–240
- 22.Correa P: Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 1995, 19(suppl 1):S37-S43 [PubMed] [Google Scholar]
- 23.Sawaoka H, Kawano S, Tsuji S, Tsuji M, Sun W, Gunawan ES, Hori M: Helicobacter pylori infection induces cyclooxygenase-2 expression in human gastric mucosa. Prostaglandins Leukot Essent Fatty Acids 1998, 59:313-316 [DOI] [PubMed] [Google Scholar]
- 24.Fu S, Ramanujam KS, Wong A, Fantry GT, Drachenberg CB, James SP, Meltzer SK, Wilson KT: Increased expression and cellular localization of inducible nitric oxide synthase and cyclo-oxygenase 2 in Helicobacter pylori gastritis. Gastroenterology 1999, 116:1319-1329 [DOI] [PubMed] [Google Scholar]
- 25.To KF, Chan FKL, Leung WK, Sung JJY: Expression of cyclo-oxygenase 1 (Cox-1) and 2 (Cox-2) in H. pylori associated gastritis. Gastroenterology 1998, 114:A310
- 26.McCarthy CJ, Crofford LJ, Greenson J, Scheiman JM: Cyclooxygenase 2 expression in gastric antral mucosa before and after eradication of Helicobacter pylori. Am J Gastroenterol 1999, 94:1218-1223 [DOI] [PubMed] [Google Scholar]
- 27.Dixon MF, Genta RM, Yardley JH, Correa P: Classification and grading of gastritis: the updated Sydney system. International Workshop on the Histopathology of Gastritis, Houston, TX, 1994. Am J Surg Pathol 1996, 20:1161-1181 [DOI] [PubMed] [Google Scholar]
- 28.Komhoff M, Hermann-Josef G, Klein T, Swyberth HW, Nusing RM: Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: implication for renal function. Am J Physiol 1997, 272:F460-F468 [DOI] [PubMed] [Google Scholar]
- 29.Hau X, Bishop AE, Wallace M, Wang H, Willcocks TC, Maclouf J, Polak JM, Knight S, Talbot IC: Early expression of cyclo-oxygenase 2 during sporadic colorectal carcinogenesis. J Pathol 1999, 187:295-301 [DOI] [PubMed] [Google Scholar]
- 30.Rigas B, Goldman IS, Levine L: Altered eicosanoid levels in human colon cancer. J Lab Clin Med 1993, 122:518-523 [PubMed] [Google Scholar]
- 31.Qiao L, Kozoni V, Tsioulias GJ, Koutsos MI, Hanif R, Shiff SJ, Rigas B: Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochem Biophys Acta 1995, 1258:215-223 [DOI] [PubMed] [Google Scholar]
- 32.Gronert K, Gewirtz A, Madara JL, Sherdan CN: Identification of a human enterocyte lipoxin A4 receptor that is regulated by interleukin (IL)-13 and interferon γ and inhibits tumor necrosis factor α-induced IL-8 release. J Exp Med 1998, 187:1285-1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanif R, Pittas A, Feng Y, Koutsos MI, Staiano-Coico L, Shiff SJ, Rigas B: Effects of nonsteroidal antiinflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol 1996, 52:237-245 [DOI] [PubMed] [Google Scholar]
- 34.Fujiwara Y, Tarnawski A, Fujiwara K, Arakawa T, Kobayashi K: Inhibitory effects of indomethacin on growth and proliferation of gastric carcinoma cells KATO III. J Physiol Pharmacol 1993, 44:147-154 [PubMed] [Google Scholar]
- 35.Goldberg Y, Nassif II, Pittas A, Tsai LL, Dynlacht BD, Rigas B, Shiff SJ: The anti-proliferative effect of sulindac and sulindac sulfide on HT-29 colon cancer cells: alterations in tumor suppressor and cell cycle-regulatory proteins. Oncogene 1996, 12:893-901 [PubMed] [Google Scholar]
- 36.Ierardi E, Balzano I, Traversa A, Principi M, Sgarro C, Passaro S, Francavilla A: Lack of reversibility of epithelial proliferative changes after H. pylori eradication in areas of intestinal metaplasia. Gut 1996, 39(Suppl 2):A18
- 37.Forbes GM, Warren JR, Glaser ME, Cullen DJE, Marshall BJ, Collins BJ: Long term follow up of gastric histology after H. pylori eradication. J Gastroenterol Hepatol 1996, 11:670-673 [DOI] [PubMed] [Google Scholar]
- 38.Van Der Hulst RWM, Van Der Ende A, Dekker FW, Ten Kate FJW, Weel JFL, Keller JJ, Kruizinga SP, Dankert J, Tytgat GNJ: Effect of Helicobacter pylori eradication on gastritis in relation to cagA: a prospective 1-year follow-up study. Gastroenterology 1997, 113:25-30 [DOI] [PubMed] [Google Scholar]
- 39.Jones MK, Wang H, Peskar BM, Levin E, Itani RM, Sarfeh IJ, Tarnawski AS: Inhibition of angiogenesis by non-steroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat Med 1999, 5:1418-1423 [DOI] [PubMed] [Google Scholar]
- 40.Bandeira-Melo C, Serra MF, Diaz BL, Cordeiro RS, Silva PM, Lenzi HL, Bakhle YS, Serhan CN, Martins MA: Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in _Angiostrongylus costaricensis_-infected rats: relationship with current eosinophilia. J Immunol 2000, 164:1029-1036 [DOI] [PubMed] [Google Scholar]
- 41.Chan FKL, To KF, Cheng ASL, Ng YB, Lee TL, Chung SCS, Sung JY: Expression of cyclooxygenase-1 and -2 in gastric ulcers. Gut 1999, 45(Suppl 3):A49 [Google Scholar]