Role of Endothelin-1 in Neovascularization of Ovarian Carcinoma (original) (raw)
Abstract
Endothelin-1 (ET-1) is overexpressed in ovarian carcinomas and acts, via ETA receptors (ETAR), as an autocrine growth factor. In this study we investigate the role of ET-1 in the neovascularization of ovarian carcinoma. Archival specimens of primary (n = 40) and metastatic (n = 8) ovarian tumors were examined by immunohistochemistry for angiogenic factor and receptor expression and for microvessel density using antibodies against CD31, ET-1, vascular endothelial growth factor (VEGF), and their receptors. ET-1 expression correlated with neovascularization and with VEGF expression. The localization of functional ETAR and ETAR mRNA expression, as detected by autoradiography and in situ hybridization, was evident in tumors and in intratumoral vessels, whereas ETBR were expressed mainly in endothelial cells. High levels of ET-1 were detected in the majority of ascitic fluids of patients with ovarian carcinoma and significantly correlated with VEGF ascitic concentration. Furthermore ET-1, through ETAR, stimulated VEGF production in an ovarian carcinoma cell line, OVCA 433, by an extent comparable to hypoxia. Finally, conditioned media from OVCA 433 as well as ascitic fluids caused an increase in endothelial cell migration and the ET-1 receptor blockade significantly inhibited this angiogenic response. These findings indicate that ET-1 could modulate tumor angiogenesis, acting directly and in part through VEGF.
Angiogenesis is essential for tumor growth and metastasis and is driven by the production of tumor and/or host-derived angiogenesis factors. 1 As for other solid cancers, the angiogenic potential of ovarian tumors, assessed by tumor microvessel density, directly correlates with a poor clinical outcome, suggesting that angiogenesis may contribute to disease progression. 2 Endothelin-1 (ET-1) is produced primarily in endothelial cells, in vascular smooth muscle cells, and in elevated amounts by many tumors. 3 ET-1 acts through two distinct subtypes of G protein-coupled receptors, ETA and ETB, expressed in a wide variety of tissues. 4 Because ET-1 stimulates proliferation and migration of endothelial cells through the ETB receptor (ETBR), 5-8 and is a potent mitogen for vascular smooth muscle and tumor cells through the ETAR, 9,10 it has been suggested that this peptide could stimulate angiogenesis.
We have previously demonstrated that expression of ET-1 is significantly increased in the majority of ovarian carcinomas compared with normal ovarian tissues. In these tumor cells ET-1 acts as an autocrine growth factor selectively through ETAR, as demonstrated by the inhibitory proliferative effects induced by a specific ETAR antagonist. 11-13 Moreover, the presence of ET-1 correlates with tumor vascularity and malignancy in well-vascularized brain tumors, 14 in colorectal cancer, 15 and ET-1 binding sites have also been characterized in the vessels of pulmonary tumors. 16 In addition, because ET-1, predominantly through ETAR, stimulates the synthesis of vascular endothelial growth factor (VEGF) in vascular smooth muscle cells and the VEGF-mediated angiogenic effects, 17 we hypothesized that the effect of ET-1 could be mediated by direct actions on tumor vessels and in part by VEGF stimulation.
VEGF a potent and specific mitogen for endothelial cells is also expressed in many tumors, including ovarian carcinoma, where it stimulates the cascade of events required for angiogenesis. 18-22 VEGF activity is mediated by two tyrosine kinase receptors, flt-1 expressed predominantly by endothelial cells 23,24 and KDR expressed in endothelial cells as well as by ovarian tumor cells. 25
To investigate the potential role of ET-1 in ovarian tumor angiogenesis, we performed immunohistochemical analysis of ET-1, VEGF, and their receptors in archival specimens of primary and metastatic human ovarian carcinomas (n = 48). By in situ hybridization and autoradiographic binding studies, we examined the localization of ET-1 receptor expression in ovarian tumor vessels. Furthermore we determined whether expression of ET-1 and its receptors is associated with vessel counts and with VEGF expression. Because ovarian cancer characteristically remains mainly confined to the peritoneal cavity, concentrations of ET-1 were measured in ascitic fluids. We have therefore investigated whether ET-1 released from ovarian carcinoma cells might modulate the production of VEGF and whether it could induce endothelial cell migration, a prerequisite for tumor neovascularization. All these findings, together with the high levels of ET-1 in neoplastic ascitic fluids, are consistent with the hypothesis that ET-1 plays an important role in ovarian cancer related-angiogenesis and represents a potential important target of anti-angiogenic therapy.
Materials and Methods
Cells, Tissues, and Ascitic Samples
Human ovarian carcinoma cell line, OVCA 433, a gift from Dr. G. Scambia (Catholic University School of Medicine, Rome, Italy), was cultured in Dulbecco’s modified Eagle’s medium and 10% fetal calf serum. Human endothelial cells were isolated from human umbilical vein (HUVECs) (Promocell, Heidelberg, Germany) and maintained in an endothelial cell growth medium kit containing with 2% fetal calf serum (Promocell). Tumor specimens were obtained with informed consent from 48 patients (age range, 27 to 65 years) undergoing surgery for ovarian carcinomas at the Regina Elena Cancer Institute. Primary tumors included 10 adenocarcinomas and 16 serous, eight mucinous, six endometrioid, and eight omental metastasis derived from five adenocarcinomas and three serous adenocarcinomas. Tissue samples were immediately snap-frozen in liquid nitrogen. From each specimen, 4-μm cryostat sections were obtained and fixed in absolute acetone for 10 minutes. Ascitic fluids were collected from an additional 20 patients with ovarian carcinoma, centrifuged at 4000 × g for 10 minutes, and then stored at −70°C in 1 ml aliquots. Control ascitic samples were obtained from five patients with nonneoplastic diseases.
Immunohistochemistry
Consecutive 4-μm sections were immunostained for VEGF, ET-1, CD31 (specific for endothelial cells), receptors for VEGF (KDR and flt-1), and receptors for ET-1 (ETAR and ETBR). Immunohistochemical staining was performed by the immunoperoxidase technique (Vector Laboratories, Burlingame, CA). Antibodies used were a rabbit polyclonal antibody (Ab) (Santa Cruz Biotechnology, Santa Cruz, CA) at a 1:200 dilution for VEGF; a mouse monoclonal Ab (clone TR.E.T.48.5; Affinity Bioreagents, Golden, CO) at 1:200 dilution for ET-1; two rabbit polyclonal antipeptide antibodies (a generous gift from Dr. R. Wu-Wong, Abbott, IL) at 1:20 dilution for ETAR and ETBR; a mouse monoclonal Ab (clone JC/70A; DAKO, Glostrup, Denmark) at a 1:200 dilution for CD31; two rabbit polyclonal antibodies (Santa Cruz Biotechnology) at a 1:100 dilution for KDR and flt-1. The VEGF (147) antibody is a rabbit polyclonal IgG raised against an amino-terminal epitope (1 to 140) common to all splice variants of VEGF. For ETAR, an Ab was raised against a decapeptide (DNPERYSTNL) of the extracellular NH2-terminal domain of ETAR, for ETBR an Ab was raised against a peptide (CGLSRIWGEERGFPPDRTP) of the NH2-terminal domain of ETBR. To ensure specificity, the primary Ab was preabsorbed for 12 hours at 4°C with a 50-fold excess of synthetic ET-1 (Peninsula Laboratories, Belmont, CA) or VEGF (Santa Cruz Biotechnology), omitted, or substituted with preimmune rabbit serum (negative control for ETAR and ETBR) or nonspecific IgG (negative control in all cases). Staining for positive controls was done on tissue from a colon cancer for VEGF; tissue from a breast cancer for ET-1; umbilical vein tissue from a bladder tumor for KDR, flt-1 and ETBR; vascular smooth muscle cells for ETAR. Nuclear counterstaining was performed with hematoxylin. The homogeneous staining for VEGF and ET-1 and their receptors, which was consistently present in all tumor cells, was scored into different grades as intensity of staining on an arbitrary scale of 0 to +3. Grade 0 represented cases in which tumor cells showed no detectable stain or traces (<20% of the tumor cells) of positive staining. Grade 1 represented cases that showed homogeneous reactivity in the majority of the tumor cells but with a weak staining intensity. Grade 2 represented cases that showed diffuse immunoreactivity in the majority of the tumor cells but with an intermediate intensity of staining. Grade 3 was assigned to cases in which the tumor cells homogeneously showed strong positive staining. Two independent observers performed tissue reading. The presence or absence of receptors was evaluated on both tumor endothelial cells and tumor epithelial cells. Vessel count was assessed by light microscopy in areas of the tumor containing the highest numbers of capillaries and small venules at the invasive edge. The highly vascular areas (hot spots) were identified by scanning tumor sections at low power and individual vessel count was performed by two independent observers on a ×200 field, according to the criteria of Weidner et al, 26 in which vessel lumen was not necessary for a structure to be defined as a vessel. Any immunostained endothelial cell clearly separated from adjacent microvessels, tumor cells, and other stromal elements, was considered a single microvessel.
In Situ Hybridization
Frozen tissue sections (6 μm) were collected onto slides and fixed in 4% paraformaldehyde for 20 minutes at 22°C, dehydrated through alcohols, and stored at −70°C. After rehydration for 15 minutes with phosphate-buffered saline 1×, 50 mmol/L MgCl2 and 15 minutes with 200 mmol/L Tris-HCl, pH 7.5, 100 mmol/L glycine, acetylation with 2× standard saline citrate (SSC), 100 mmol/L triethanolamine, 0.25% acetic anhydride, dehydration with alcohols and 0.6 mol/L ammonioacetate, and air-drying, slides were hybridized with buffer containing 50% formamide, 1 mg/ml salmon sperm DNA, 2× SSC, 10% dextran sulfate, 70 nmol/L dithiothreitol, and 10 to 13 × 10 3 cpm/μl of probe 35S-labeled CTP (40 mCi/ml; Amersham). Sense and anti-sense riboprobes for ETAR were synthesized from pBluescript SK− vectors with a full-length human ETAR cDNA insert. After hybridization for 16 to 20 hours at 50°C in a humid chamber, slides were washed 20 minutes in 2× SSC at room temperature, twice in 2× SSC-50% formamide at 45°C, once in 1× SSC-50% formamide at the same temperature, and in 0.1× SSC at room temperature. For autoradiography, slides were coated with NTB2 emulsion (Kodak, Rochester, NY) and exposed at 4°C for 2 weeks. After developing in Kodak D19, the sections were fixed and counterstained with hematoxylin.
Autoradiographic Binding Studies
Autoradiography was performed on five human ovarian carcinoma tissues as previously described. 27 After washing with 50 nmol/L Tris-HCl buffer, pH 7.4, containing 0.2% bovine serum albumin and 0.005% polyethylenimine (Sigma, St. Louis, MO), consecutive sections (10 μm) were incubated at 25°C for 60 minutes in a buffer supplemented with 40 μg/ml bacitracin (Sigma) in the presence of tracer, 0.1 nmol/L 125I-ET-1 (2,200 Ci/mmol; Dupont New England Nuclear Research Products, Wilmington, DE) in the absence (total binding) and in the presence of 1 μmol/L ET-1 (Peninsula Laboratories) (nonspecific binding). For total ETB binding sections were incubated in the presence of 1 μmol/L BQ 123 (Peninsula) and for total ETA binding with 1 μmol/L sarafotoxin S6c (Peninsula) or 1 μmol/L BQ 788 (Peninsula). After consecutive washes, slides were dipped in NTB2 emulsion and after 15 days developed in Kodak D19, fixed, and counterstained.
Assays of Angiogenic Factors
OVCA 433 cells were seeded at 1 × 10 6 cells/dish in complete medium and serum-starved for 24 hours. Cells were cultured at 37°C with 95% air/5% CO2 or incubated in aluminum chambers flushed with a gas mixture containing 5% CO2 and 95% N2 (hypoxia). After varying times, cell supernatants were collected, centrifuged, and frozen for subsequent use. Each conditioned media and ascitic sample was subjected to one freeze-thaw cycle only. All assays (ET-1 and VEGF) were performed in duplicate on microtiter plates by an enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. The working range in the enzyme-linked immunosorbent assay for VEGF assay was 31.2 to 2,000 pg/ml and for ET-1 was 0 to 120 pg/ml. When necessary, dilutions of the ascitic samples were made. Control ascitic samples from five patients with no malignant disease were also assayed.
Chemotaxis Assay
To examine the paracrine chemoattractant effects of the OVCA 433-conditioned medium and of the ascitic fluids on HUVEC migration, a 48-well modified Boyden chamber (Neuroprobe, Pleasanton, CA) and 0.01% gelatin-coated polyvinylpyrrolidone-free polycarbonate filters (8-μm pore, Nucleopore: Costar, New York, NY) were used. The lower compartment of the chamber was filled with chemoattractants or inhibitor (27 μl/well). Cells (5 × 10 5 cells/ml) were placed in the upper compartment (55 μl/well). Some of the ascitic fluid of OVCA 433-conditioned medium was preabsorbed with antibody (at a dilution of 1:50) to VEGF (Santa Cruz Biotechnology) for 2 hours before use. ET-1 receptor antagonists (BQ 123 and BQ 788; Peninsula Laboratories) were previously added to the HUVECs and preincubated for 15 minutes at 37°C. After 4 hours of incubation at 37°C, filter was removed, and cells on the upper side were scraped off. Migrated cells were fixed, stained with Diff-Quick (Baxter Diagnostics, Miami, FL), and counted in 10 high-power fields. Each experimental point was analyzed in triplicate.
Statistical Analysis
All correlations were examined by the Spearman count correlation coefficient. All statistical analyses were performed by the Inplot software system (GraphPad Software Inc., San Diego, CA).
Results
Expression of ET-1, ETAR, and ETBR
In a panel (n = 48) of primary and metastatic ovarian carcinomas we evaluated the expression of ET-1, ETAR, and ETBR. The presence of mature ET-1 was found in 84% of the ovarian carcinomas examined, including primary and metastatic lesions of different ovarian cancer histotype. ET-1 was localized in the cytoplasm of tumor and metastatic cells and in endothelial cells lining the tumor-feeding vessels (Figure 1A) ▶ . No staining was observed with anti-ET-1 Ab preabsorbed with an excess of blocking ET-1 (data not shown). Specific immunostaining identifying ETAR was seen primarily in the majority (89%) of cell cytoplasms in all areas of the tumors and metastases and in blood vessels adjacent to neoplastic cells (Figure 1B) ▶ . Immunostaining with an antipeptide antibody directed against ETBR was not detected on tumor cells, but positivity for ETBR occurred significantly on tumor endothelium (Figure 1C) ▶ . No staining was noted when preimmune rabbit IgG substituted the anti-ETA or anti-ETB for the primary Ab, as negative control in a human serous ovarian carcinoma (data not shown).
Figure 1.

Immunohistochemical staining of ET-1 (A), ETAR (B), ETBR (C), and CD31 (D) in an human serous ovarian carcinoma. Intense ET-1 and ETAR immunoreactivity (brown staining) was observed in all tumor cells (A and B). ETBR immunoreactivity was detected only in vascular elements (arrowheads, C). CD31 staining of endothelial cells (arrowheads) in the same ET-1-positive carcinoma tissues is shown (D). Original magnification, ×200; ABC-peroxidase, counterstained with hematoxylin.
In addition, the intracellular localization of ETAR mRNA was examined by in situ hybridization analysis. In situ hybridization using riboprobe with specificity for ETAR was performed on five specimens. ETAR mRNA strong expression was found to be a consistent feature of all tumors studied. A dense homogeneous accumulation of silver grains, identifying ETAR mRNA, was seen throughout the majority of tumor cells and vascular elements (Figure 2, A and C) ▶ . No specific labeling was seen with control sense probe (Figure 2, B and D) ▶ . The silver grain distribution was similar to the distribution of the ETAR immunostaining.
Figure 2.

In situ hybridization of ovarian carcinoma with antisense (A and C) and the sense (B and D) (control) riboprobe of ETAR. Positive signals for ETAR mRNA appear as black grain in the bright-field photographs. Intense labeling for ETAR mRNA in an ovarian serous carcinoma, is particularly localized in a nest of tumor cells (t) as well as in vascular elements of stromal microvessels (arrowheads). No specific labeling is noted in adjacent section with the sense riboprobe. Original magnification, ×200; counterstained with hematoxylin.
The predominance of ETA binding sites on the tumor cells was further confirmed by autoradiographic analysis of 125I-ET-1 binding assay (Figure 3) ▶ in eight ovarian carcinoma specimens. Autoradiographic localization of ETA binding was confirmed to be homogeneous within tumors without detectable hot spots. 125I-ET-1 binding was intense in tumor vessels on both their muscular and endothelial components (Figure 3A) ▶ . The addition of an excess of cold ET-1 displaced all 125I-ET-1 binding demonstrating the specificity of the binding (Figure 3B) ▶ . The addition of a potent ETBR antagonist, BQ 788, did not affect 125I-ET-1 binding to tumor cells, but displaced it from the vascular endothelium (Figure 3C) ▶ . The BQ 123, a potent ETAR antagonist, abolished labeling in the tumor cells, attenuated it in the vascular muscular component and showed only a decreased labeling related on the presence of ETB receptors on vascular cells (Figure 3D) ▶ . Sarafotoxin 6C (S6C), an ETBR agonist, determined results similar to those obtained with BQ 788 (data not shown). These results demonstrated the distribution of the ETAR and ETBR subtypes on tumor cells and vascular smooth muscle cells and on endothelial cells, respectively. No specific ETB binding sites were present in ovarian tumor cells.
Figure 3.
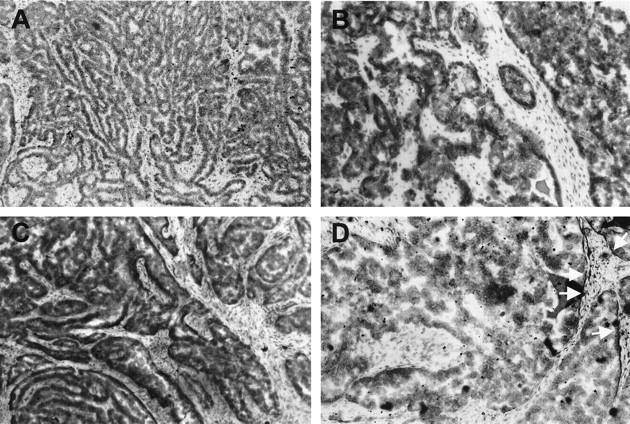
Autoradiographs of frozen sections of human serous ovarian carcinoma incubated with 125I-ET-1 (100 pmol/L). A: Binding is well distributed and intense in tumor cells. B: 125I-ET-1 is completely displaced by the addition of a 1 μmol/L cold ET-1. C: Total ETA binding: autoradiogram of a flanking tissue section incubated with 0.1 nmol/L 125I-ET-1 and 1 μmol/L BQ 788 showing radioactive ligand binding primarily to the tumor cells. D: Total ETB binding: autoradiogram of another flanking section incubated with 1 μmol/L BQ 123 completely displaces 125I-ET-1 binding to tumor cells, while it attenuates labeling to blood vessels. White arrows show only a decreased labeling in the blood vessels related to the restricted presence of ETBR on vascular cells. Original magnification, ×200
Expression of VEGF, KDR, and flt-1
A total of 38 out of 48 (79%) ovarian carcinomas, including primary and metastatic, showed VEGF immunoreactivity. As previously reported, 25,28,29 the intensity of VEGF staining was homogeneous within tumors, with no detectable hot-spot, and was mainly localized in the cytoplasm of all neoplastic cells (Figure 4A) ▶ . Specificity of VEGF immunostaining was confirmed by a complete loss of staining when the antibody was preneutralized with the corresponding control antigen (data not shown). We further characterized the expression of VEGF receptors in ovarian carcinoma by immunohistochemistry. The vascular cells in the ovarian carcinomas expressed high levels of KDR and a significant amount of flt-1 (Figure 4, B and C) ▶ .
Figure 4.

Immunohistochemical staining of VEGF (A), KDR (B), flt-1 (C), and CD31 (D) in a human serous ovarian carcinoma. Intense and homogeneous VEGF immunoreactivity (3+) was observed in two nests of tumor cells (t) whereas the portion of interstitium (*) included in the section was unstained (A). Intense KDR staining was observed in vascular cells (arrowheads, B). Flt-1 immunoreactivity was detected in the vascular elements of the tumor (arrowheads, C). CD31 staining was restricted to endothelial cells (arrowheads) in the same VEGF-positive carcinoma tissues (D). Original magnification, ×200; ABC-peroxidase, counterstained with hematoxylin.
Correlation between Vessel Count and ET-1, VEGF, and Receptor Expression
In the same panel of primary (n = 40) and metastatic (n = 8) ovarian carcinomas, we performed immunohistochemistry against CD31, a specific marker of endothelial cells, and the microvessel number was counted in the regions which included more microvessels than the rest of the tissue (hot-spot) (Figures 1D and 4D) ▶ ▶ . In the total patient population, the mean microvessel count in the hot-spot was 38.4 ± 23.7. The mean microvessel numbers of the ET-1-positive group and the ET-1-negative group were 57.6 ± 31.4 and 25.9 ± 24.0, respectively, indicating that there was greater neovascularization in the ET-1-positive tumors (P < 0.01; Table 1 ▶ ). The expression of ETAR in tumor blood vessels was higher in ET-1-positive tumors than in those negative for ET-1, whereas ETBR expression was similar between the two groups (Table 1) ▶ .
Table 1.
Correlation between Expression of ET-1, Vessel Count, and ET-1 Receptors in Human Ovarian Carcinomas
| ET-1 expression | Vessel count | Receptors for ET-1 | |
|---|---|---|---|
| ETAR expression* | ETBR expression* | ||
| Positive (n = 39) | 57.6 ± 31.4 P < 0.01 | 39 (100%) | 36 (92%) |
| Negative (n = 9) | 25.9 ± 24.0 | 5 (55%) | 8 (89%) |
As shown in Figure 5 ▶ , the analysis of the relation between ET-1 expression and the number of microvessels in the tumor areas of highest vascularization showed a statistical association between tumor microvessel count and ET-1 expression (r = 0.60; P < 0.0002). The blood vessel numbers (mean ± SD) in the VEGF-negative and VEGF-positive tumors were 23.8 ± 15.1 and 53 ± 33.2, respectively. This difference was statistically significant (P < 0.0001), indicating that there was greater neovascularization in the VEGF-expressing cancers (Table 2) ▶ . Therefore, a significant correlation among vessel count and VEGF expression was observed (r = 0.71; P < 0.0001). KDR was the predominantly expressed VEGF receptor in ovarian carcinoma vessels, with no difference between VEGF-positive and -negative tumors. Flt-1 expression was also similar between the two groups. Moreover, because vessel count was significantly associated with VEGF expression we analyzed whether ET-1 correlated with VEGF. In our series of 48 ovarian carcinomas, we found a highly significant correlation between ET-1 staining and VEGF immunoreactivity (r = 0.673; P < 0.001).
Figure 5.

Relationship between microvessel count and ET-1 expression in human ovarian carcinomas (n = 48; r = 0.60; P < 0.0002). Solid line indicates mean value and dashed lines SD.
Table 2.
Correlation between Expression of VEGF, Vessel Count, and VEGF Receptors in Human Ovarian Carcinomas
| VEGF expression | Vessel count | Receptors for VEGF | |
|---|---|---|---|
| KDR* | flt-1* | ||
| Positive (n = 38) | 53 ± 33.2 P < 0.0001 | 37 (97%) | 33 (87%) |
| Negative (n = 10) | 23.8 ± 15.1 | 10 (100%) | 9 (90%) |
Ascitic Concentration of ET-1 and VEGF
Because ovarian cancer growth is mainly confined to the peritoneal cavity with the production of ascitic fluid, we evaluated whether advanced ovarian carcinoma would be associated with elevated ascitic concentrations of VEGF and ET-1. In human studies, high levels of VEGF (>10 pmol/L) were measured in malignant ascites. 30 Taking 10 pmol/L as the threshold value for the detection of malignancy, 17 of the 20 samples of ascitic fluid obtained from patients with ovarian carcinoma were VEGF-positive (>10 pmol/L). VEGF levels ranged between 25 and 325 pmol/L in ascitic fluids. High concentrations of ET-1 (≥10 pmol/L) were measured in the same 17 VEGF-positive ascites of the 20 effusions tested (85%). ET-1 levels in the ascitic fluids ranged between 10.4 and 78 pmol/L, whereas only very low or undetectable levels of ET-1 were found in the five patients whose effusions were without cytological evidence of malignancy (<2 pmol/L). Moreover, there was a strong correlation (r = 0.717; P < 0.0026) between ET-1 and VEGF ascitic concentrations (Figure 6) ▶ suggesting that ET-1 and VEGF characterize the advanced disease.
Figure 6.

Relationship between ascitic levels of ET-1 and VEGF in a group of patients (n = 20) with advanced ovarian carcinoma (r = 0.717; P < 0.0026). Solid line indicates mean value and dashed lines SD.
Effect of ET-1 on VEGF Secretion in OVCA 433 Cells
We investigated whether ET-1 might regulate the production of VEGF as an important mechanism by which ET-1 could modulate angiogenesis. ET-1 stimulated VEGF production, with detectable production as early as 1 hour, and maximum stimulation (2.0-fold above control levels) at 24 hours (P < 0.005, Figure 7A ▶ ). The ability of ET-1 to stimulate VEGF production was dose-dependent, reaching maximal stimulation at a concentration of 100 nmol/L (Figure 7B) ▶ . After 24 hour of incubation, VEGF-stimulation by ET-1 (100 nmol/L) was comparable in magnitude to that induced by hypoxia, a recognized potent and important stimulus of VEGF (Figure 8) ▶ . We then investigated the ET-1 receptor subtype involved in the regulation of VEGF secretion. The stimulation by ET-1 (100 nmol/L) was completely blocked by the ETAR antagonist BQ 123, indicating that ET-1 stimulated VEGF production after binding to ETAR in ovarian carcinoma cells.
Figure 7.
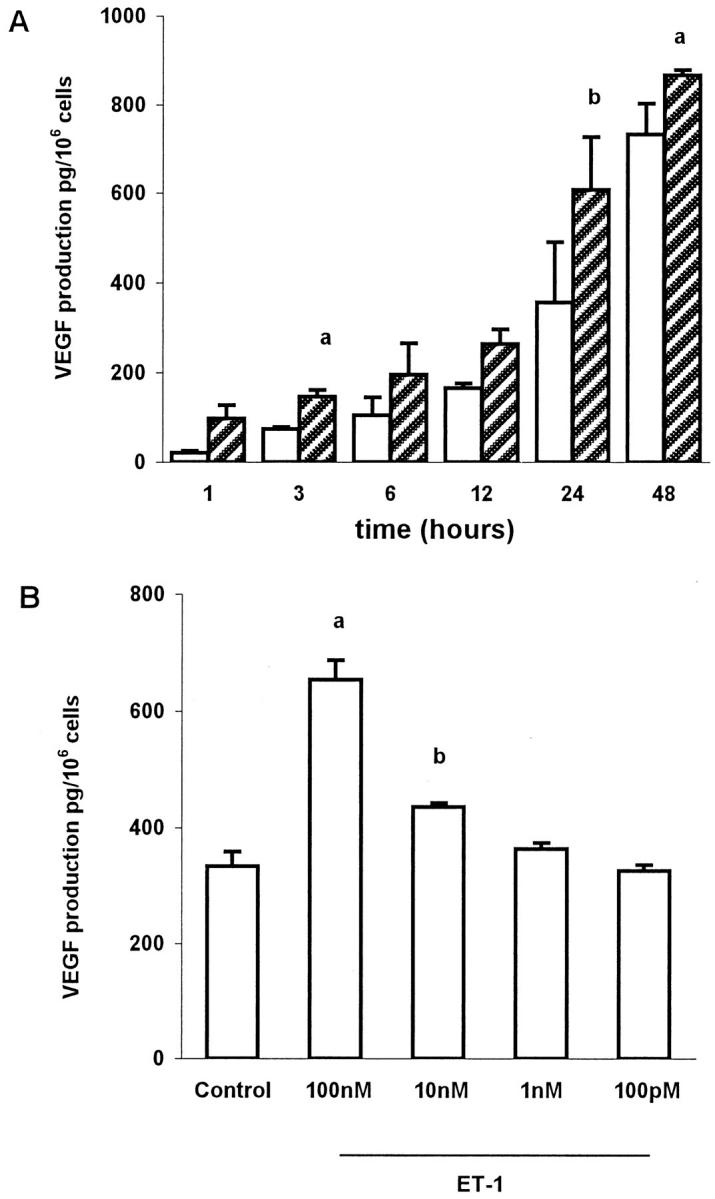
A: Time course of production of VEGF in conditioned media of OVCA 433 cells by ET-1. Open bar, control; hatched bar, ET-1 100 nmol/L. Data expressed are means of results from three experiments each performed in triplicate. Bars, ±SD. a: P ≤ 0.02 compared to control. b: P ≤ 0.005 compared to control. B: Dose response for the actions of ET-1 on VEGF production at 24 hours. Data expressed are means of results from three experiments each performed in triplicate. Bars, ±SD. a: P ≤ 0.005 compared to control. b: P ≤ 0.02 compared to control.
Figure 8.

Inhibition of ET-1-stimulated VEGF production by a ETAR antagonist. Stimulation of VEGF protein in conditioned media of the OVCA 433 cells by ET-1 with ETAR antagonist (100 nmol/L) completely inhibits the stimulation induced by ET-1. Data expressed are means of results from three experiments each performed in triplicate. Bars, ±SD. a: P ≤ 0.001 compared to control. b: P ≤ 0.01 compared to ET-1.
Endothelial Cell Migration Induced by ET-1 Released by Ovarian Carcinoma Cells
To test whether ET-1 released from ovarian carcinoma cells could affect endothelial cell mobility, HUVECs were incubated in a chemotaxis chamber with ascitic fluids obtained from patients with ovarian carcinoma (Figure 9A) ▶ and with conditioned medium of OVCA 433 cells (Figure 9B) ▶ . The ascitic fluid and the OVCA 433 medium contained high levels of ET-1 (38 ± 3.2 pg/ml and 62.5 ± 5.3 pg/ml, respectively) as measured by enzyme-linked immunosorbent assay. The ascites and the spent media (used at a dilution of 1:2) caused a strong increase in endothelial cell migration above serum-free Dulbecco’s modified Eagle’s medium used as control. These stimulatory effects were inhibited by ∼45% after preincubation of conditioned medium and ascitic fluid for 2 hours with an excess of specific antibody to VEGF, which bounds the VEGF secreted. Preincubation with BQ123 induced a partial decrease (∼25%), whereas the addition of BQ 788 determined an higher inhibition (55%) on endothelial cell migration induced by both ascites and conditioned media. Interestingly, co-incubation of VEGF Ab and BQ788 with OVCA 433-conditioned medium reduced by 76% HUVEC migration. Endothelial cell migration induced by 10 nmol/L of basic fibroblast growth factor was not inhibited by the addition of 1 μmol/L M BQ 788 or BQ 123, indicating that the inhibitory effect induced by the ETB and ETA receptor antagonist was specific and was not because of cytotoxicity (data not shown). This in vitro model analyzing the potential paracrine interactions between ovarian tumor cells and endothelial cells indicates that ET-1 produced by tumor cells, together with VEGF, was primarily responsible for the increased HUVEC migration.
Figure 9.

Effects of ET-1 released from ovarian carcinoma cells on endothelial cell migration. A: Stimulation of HUVEC migration by ascitic fluid. Incubation with VEGF antibody (Ab) and ETAR (BQ123) and ETBR (BQ 788) antagonists (1 μmol/L) identifies ET-1 released from tumor cells as an important stimulus for HUVEC mobilization. Data expressed as the number of migrated cells in 10 high-power fields and are means of results from two experiments performed in triplicate. Bars, ± SD. a: P ≤ 0.001 compared to control. b: P ≤ 0.001 compared to ascitic fluid. B: Stimulation of HUVEC migration by conditioned media (CM) from ovarian cancer cell line OVCA 433. Data expressed as migrated cells in 10 high-power fields and are means of results from four experiments each performed in triplicate. Bars, ± SD. a: P ≤ 0.001 compared to control. b: P ≤ 0.01 compared to CM. c: P ≤ 0.006 compared to CM. d: P ≤ 0.02 compared to VEGF Ab. e: P ≤ 0.05 compared to BQ 788.
Discussion
Although the degree of intratumoral stromal vascularization and VEGF expression has been found to correlate with prognosis in patients with early-stage and advanced ovarian cancer, the mechanisms of the angiogenic events in this malignancy are still not well-defined. 3,31-33 Because this knowledge is becoming clinically relevant in view of the emerging therapeutic role of anti-angiogenic agents, we examined the role of ET-1 in the neovascularization of primary and metastatic ovarian carcinomas.
In the present study, elevated ET-1 expression was documented by immunohistochemistry in primary and metastatic ovarian carcinoma. ETAR expression, as detected by in situ hybridization and immunohistochemistry, was localized in carcinoma cells as well as in blood vessels, whereas ETBR was confined to vascular endothelial cells. Autoradiographic binding studies confirmed that ovarian carcinoma cells functionally expressed ETAR but not the ETBR, and that blood vessels co-expressed ETAR and ETBR. The vascular smooth muscle cells predominantly expressed ETAR, whereas endothelial cells expressed ETBR. The localization of the ET receptors in blood vessels adjacent to transformed epithelium suggests that ET-1 might exert angiogenic effects providing an ideal microenvironment for tumor growth. VEGF was expressed in the cytoplasm of tumor cells where it is synthesized and stored, whereas VEGF receptors were expressed by endothelial cells. Because VEGF plays a pivotal role in ascites formation both as a potent inducer of vascular permeability and as an angiogenetic factor, 34 we hypothesized that advanced ovarian carcinoma may be associated with elevated ascitic concentrations of the angiogenic factors VEGF and ET-1. Our findings indeed demonstrate that in the majority of ovarian carcinomas examined, ascitic concentrations of both factors are concordantly elevated. The unprecedented demonstration of high ET-1 levels in a sizeable percentage (85%) of cases implicates this molecule as relevant in the increased neovascularization and vascular permeability of ovarian carcinoma.
Although microvessel count and VEGF expression have been shown to correlate in several tumors, 35-37 to date no such correlation has been found in ovarian carcinoma. 33,38 Our results found a strong correlation of VEGF and vessel density in a significant number of cases. In view of the above results, we extended the same analysis to ET-1 demonstrating a significant correlation between ET-1 expression and microvessel density. ET-1 is a potent mitogen for vascular smooth muscle cells as well as for endothelial cells. Previous observations demonstrated that endothelial cells express ETBR and respond to ET-1, which may exert an angiogenic activity. 5-8 Thus, we have recently demonstrated that ET-1 induces a pro-angiogenic phenotype in human endothelial cells. 39 This phenotype includes both early (ie, increase in cell proliferation, migration, invasion, and MMP-2 production) and late angiogenic events (differentiation into vascular cords). Moreover ET-1 in association with VEGF has a clear angiogenic activity in the Matrigel in vivo assay, comparable to that promoted by bFGF.
Recent studies demonstrated that in vascular smooth muscle cells ET-1, predominantly through ETAR, enhances VEGF secretion and stimulates the VEGF-induced endothelial cell proliferation and invasion. 17
Because the regulation of VEGF production is a critical event in tumor angiogenesis, one can envision that in pathological conditions such as cancer, ET-1 may be up-regulated by various stimuli including hypoxia, growth factors, and inflammatory cytokine, 40 and that ET-1, in turn, might exert angiogenic effect increasing VEGF production and thereby modifying VEGF-related angiogenic responses. In these studies, we demonstrated that ET-1 stimulates VEGF production in ovarian carcinoma cells and is equipotent to hypoxia, a recognized potent and important stimulus of VEGF production. We also showed that the actions of ET-1 were mediated through the ETAR, because the specific antagonist BQ 123 reversed the stimulation of VEGF production.
Furthermore we showed that elevated levels of ET-1 are released by ovarian carcinoma in ascitic fluids and that this growth factor is primarily responsible for endothelial cell migration, acting through ETBR, as demonstrated by the inhibition induced by ETBR antagonists. The significant inhibition obtained by co-incubating HUVECs with the ETBR antagonist and with VEGF Ab, strongly indicates that ET-1, together with VEGF, play a complementary and coordinated role during neovascularization in ovarian carcinoma. Thus, our hypothesis is that during neovascularization, endothelial cells could be initially stimulated by the ET-1/ETBR interaction to migrate, proliferate, and invade surrounding tissue. Thereafter, vessel maturation could be mediated by ET-1/ETAR binding in part through the stimulation of VEGF in the existing tumor or vasculature, resulting in angiogenesis. This working hypothesis links ET-1 to the early as well as to the late stages of angiogenesis involving both ET-1 receptors.
In conclusion, the present study demonstrates that ET-1 and its receptors are expressed by tumor cells as well as by tumor vessels in ovarian carcinoma. The tumor-promoting activity of ET-1 may occur through an autocrine pathway that stimulates tumor cell proliferation and through a paracrine pathway involving direct angiogenic effects on endothelial cells and in part through the stimulation of VEGF in ovarian carcinoma cells. Among the array of factors that concur to ovarian tumor vascularization, we identified ET-1 and its receptors as angiogenic regulators that could represent novel targets for anti-angiogenic therapies. New therapeutic strategies using specific antagonists provide an additional approach to the treatment of ovarian carcinoma in which ET-1 antagonists would play their anti-tumor role as both anti-angiogenic and anti-mitogenic agents. 41,42
Acknowledgments
We thank Marco Varmi and Antonella Mangoni for technical assistance; Dr. Irene Venturo for providing ascitic effusions; Dr. Antonella Stoppacciaro for assistance with the in situ hybridization study; Dr. K. J. Catt for his critical review; and Paula Franke for the formal revision of the manuscript.
Footnotes
Address reprint requests to Anna Bagnato, Laboratory of Molecular Pathology and Ultrastructure, Regina Elena Cancer Institute, Via delle Messi d’Oro 156, 00158 Rome, Italy. E-mail: bagnato@ifo.it.
This work was supported by grants from the Associazione Italiana Ricerca sul Cancro, the Ministero della Sanità, and fellowships from the Fondazione Italiana Ricerca sul Cancro (to D. S. and L. R.).
References
- 1.Folkman J: How is blood vessel growth regulated in normal and neoplastic tissue? Cancer Res 1986, 46:467-473 [PubMed] [Google Scholar]
- 2.Gasparini G, Bonoldi E, Viale G, Verderio P, Boracchi P, Panizzoni GA, Radaelli V, Di Bacco A, Guglielmi RB, Bevilacqua P: Prognostic and predictive value of tumor angiogenesis in ovarian carcinomas. Int J Cancer 1996, 69:205-211 [DOI] [PubMed] [Google Scholar]
- 3.Levin ER: Endothelins. N Engl J Med 1995, 333:356-363 [DOI] [PubMed] [Google Scholar]
- 4.Rubanji GM, Polokoff MA: Endothelins: molecular biology, biochemistry, pharmacology, physiology and pathophysiology. Pharmacol Rev 1994, 46:325-415 [PubMed] [Google Scholar]
- 5.Morbidelli L, Orlando C, Maggi CA, Ledda F, Ziche M: Proliferation and migration of endothelial cells is promoted by endothelins via activation of ETB receptors. Am J Physiol 1995, 269:4686-4695 [DOI] [PubMed] [Google Scholar]
- 6.Wren AD, Hiley CR, Fan TD: Endothelin-3 mediated proliferation in wounded human umbilical vein endothelial cells. Biochem Biophys Res Comm 1993, 196:369-375 [DOI] [PubMed] [Google Scholar]
- 7.Noiri E, HuY, Bahou WF, Keese CR, Giaever I, Goligorsky MS: Permissive role of nitric oxide in endothelin-induced migration of endothelial cells. J Biol Chem 1997, 272:1747-1752 [DOI] [PubMed] [Google Scholar]
- 8.Goligorsky MS, Budzikowski AS, Tsukahara H, Noiri E: Co-operation between endothelin and nitric oxide in promoting endothelial cell migration and angiogenesis. Clin Exp Pharmacol Physiol 1999, 26:269-271 [DOI] [PubMed] [Google Scholar]
- 9.Alberts GF, Peifley KA, Johns A, Kleha JF, Winkles JA: Constitutive endothelin-1 overexpression promotes smooth muscle cell proliferation via an external autocrine loop. J Biol Chem 1994, 269:10112-10118 [PubMed] [Google Scholar]
- 10.Bagnato A, Catt KJ: Endothelins as autocrine regulators of tumor cell growth. Trends Endocrinol Metab 1998, 9:378-383 [DOI] [PubMed] [Google Scholar]
- 11.Bagnato A, Tecce R, Moretti C, Di Castro V, Spergel DJ, Catt KJ: Autocrine actions of endothelin-1 as a growth factor in human ovarian carcinoma cells. Clin Cancer Res 1995, 1:1059-1066 [PubMed] [Google Scholar]
- 12.Bagnato A, Salani D, Di Castro V, Wu-Wong JR, Tecce R, Nicotra MR, Venuti A, Natali PG: Expression of endothelin-1 and endothelin A receptor in ovarian carcinoma: evidence for an autocrine role in tumor growth. Cancer Res 1999, 59:1-8 [PubMed] [Google Scholar]
- 13.Bagnato A, Tecce R, Di Castro V, Catt KJ: Activation of mitogenic signaling by endothelin-1 in ovarian carcinoma cells. Cancer Res 1997, 57:1306-1311 [PubMed] [Google Scholar]
- 14.Stiles JD, Ostrow PT, Balos LL, Greenberg SJ, Plunkett R, Grand W, Heffner RR: Correlation of endothelin-1 and transforming growth factor β1 with malignancy and vascularity in human gliomas. J Neuropathol Exp Neurol 1997, 56:435-439 [DOI] [PubMed] [Google Scholar]
- 15.Shankar A, Loizidou M, Aliev G, Fredericks S, Holt D, Boulos PB, Burnstock G, Taylor I: Raised endothelin-1 levels in patients with colorectal liver metastases. Br J Surg 1998, 85:502-506 [DOI] [PubMed] [Google Scholar]
- 16.Zaho Y, Spingall D, Hamid P, Levene M, Polak J: Localization and characterization of endothelin-1 receptor binding in the blood vessels of human pulmonary tumors. J Cardiovasc Pharmacol 1995, 26(Suppl 3):S9341-S345 [PubMed] [Google Scholar]
- 17.Pedram A, Rasandi M, Hu R-M, Levin ER: Vasoactive peptides modulate vascular endothelial cell growth factor production and endothelial cell proliferation and invasion. J Biol Chem 1997, 272:17097-17103 [DOI] [PubMed] [Google Scholar]
- 18.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF: Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219:983-985 [DOI] [PubMed] [Google Scholar]
- 19.Leung DW, Cachiane G, Kuang WJ, Goeddal DV, Ferrara N: Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246:1306-1309 [DOI] [PubMed] [Google Scholar]
- 20.Dvorak HF, Brown LF, Detmar M, Dvorak AM: Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995, 146:1029-1039 [PMC free article] [PubMed] [Google Scholar]
- 21.Perez RP, Godwin AK, Hamilton TC, Ozols RF: Ovarian cancer biology. Semin Oncol 1991, 18:186-204 [PubMed] [Google Scholar]
- 22.Mesiano S, Ferrara N, Jaffe RB: Role of vascular endothelial growth factor in ovarian cancer: inhibition of ascites formation by immunoneutralization. Am J Pathol 1998, 153:1249-1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Vries C, Escobedo JA, Ulno H, Houck K, Ferrara N, Williams LT: The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255:989-991 [DOI] [PubMed] [Google Scholar]
- 24.Terman BI, Carrion ME, Kovacs E, Rossmussan BA, Eddy RL, Shows TB: Identification of a new endothelial growth factor receptor tyrosine kinase. Oncogene 1991, 6:1677-1683 [PubMed] [Google Scholar]
- 25.Boocock CA, Chornock-Jones S, Sharkey AM, McLaren J, Baker PJ, Wright KA, Twentyman PR, Smith SK: Expression of vascular endothelial growth factor and its receptors flt and KDR in ovarian carcinoma. J Natl Cancer Inst 1995, 87:506-516 [DOI] [PubMed] [Google Scholar]
- 26.Weidner N, Semple JP, Welch WR, Folkman J: Tumor angiogenesis and metastasis-correlation in invasive breast carcinoma. N Engl J Med 1991, 324:1-8 [DOI] [PubMed] [Google Scholar]
- 27.Millan N, Aguilera G, Wynn PC, Mendelsohn FAO, Catt KJ: Autoradiographic localization of brain receptors for peptide hormones: angiotensin II, corticotropin-releasing factor, and gonadotropin-releasing hormone. Methods Enzymol 1986, 124:590-606 [DOI] [PubMed] [Google Scholar]
- 28.Orre M, Rogers PAW: VEGF, VEGF-1, VEGF-2, microvessel density and endothelial cell proliferation in tumours of the ovary. Int J Cancer 1999, 84:101-108 [DOI] [PubMed] [Google Scholar]
- 29.Abu-Jawdeh GM, Faix JD, Niloff J, Tognazzi K, Mauseam E, Dvorak HF, Brown LF: Strong expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in ovarian borderline and malignant neoplasms. Lab Invest 1996, 74:1105-1115 [PubMed] [Google Scholar]
- 30.Yeo K-T, Wang HH, Nagy JA, Sioussat TM, Ledbetter SR, Hoogewerf AJ, Zhou Y, Masse EM, Senger DR, Dvorak HF, Yeo T-K: Vascular permeability factor (vascular endothelial growth factor) in guinea pig and human tumor and inflammatory effusions. Cancer Res 1993, 53:2912-2918 [PubMed] [Google Scholar]
- 31.Alvarez AA, Krigman HR, Whitaker RS, Dodge RK, Rodriguez GC: The prognostic significance of angiogenesis in epithelial ovarian carcinoma. Clin Cancer Res 1999, 5:587-591 [PubMed] [Google Scholar]
- 32.Paley PJ, Staskus KA, Gebhard K, Mohanraj D, Twiggs LB, Carson LF, Ramakrishnan S: Vascular endothelial growth factor expression in early stage ovarian carcinoma. Cancer 1997, 80:98-106 [DOI] [PubMed] [Google Scholar]
- 33.Holligsworth HC, Kdrun EC, Steinberg SM, Rothenberg ML, Merino MJ: Tumor angiogenesis in advanced stage ovarian carcinoma. Am J Pathol 1995, 147:33-41 [PMC free article] [PubMed] [Google Scholar]
- 34.Olson TA, Moharnraj D, Carson LF, Ramakrishnan S: Vascular permeability factor gene expression in normal and neoplastic human ovaries. Cancer Res 1994, 54:276-280 [PubMed] [Google Scholar]
- 35.Takahashi Y, Cleary KR, Mai M, Kitadai Y, Bucana CD, Ellis LM: Significance of vessel count and vascular endothelial growth factor and its receptor (KDR) in intestinal-type. Clin Cancer Res 1996, 2:1679-1684 [PubMed] [Google Scholar]
- 36.Fontanini G, Vignati S, Boldrini L, Chiné S, Silvestri V, Lucchi M, Mussi A, Angeletti CA, Bevilacqua G: Vascular endothelial growth factor is associated with neovascularization and influences progression of non-small cell lung carcinoma. Clin Cancer Res 1997, 3:861-865 [PubMed] [Google Scholar]
- 37.Kitadai Y, Haruma K, Tokutomi T, Tanaka S, Sumii K, Carvalho M, Kuwabara M, Yoshida K, Hirai T, Kajiyama G, Tahara E: Significance of vessel count and vascular endothelial growth factor in human esophageal carcinomas. Clin Cancer Res 1998, 4:2195-2200 [PubMed] [Google Scholar]
- 38.Hartenbach EM, Olson TA, Goswitz JJ, Mohanraj D, Twiggs LB, Carson LF, Ramakrishnan S: Vascular endothelial growth factor (VEGF) expression and survival in human epithelial ovarian carcinomas. Cancer Lett 1997, 121:169-175 [DOI] [PubMed] [Google Scholar]
- 39.Salani D, Taraboletti G, Rosano L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A: Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol 2000, 157:1703-1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kourembanas S, Maraden PA, Mcquillan LP, Faller DV: Hypoxia induces endothelin gene expression and secretion in cultured endothelium. J Clin Invest 1991, 88:1054-1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lahav R, Heffner G, Patterson PH: An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci USA 1999, 96:11496-11500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davos K, Loizidou M, Shankar A, Ali H, Taylor I: Angiogenesis in cancer: role of endothelin-1. Ann R Coll Surg Engl 1999, 81:306-310 [PMC free article] [PubMed] [Google Scholar]