Toll-like Receptors Induce a Phagocytic Gene Program through p38 (original) (raw)
Abstract
Toll-like receptor (TLR) signaling and phagocytosis are hallmarks of macrophage-mediated innate immune responses to bacterial infection. However, the relationship between these two processes is not well established. Our data indicate that TLR ligands specifically promote bacterial phagocytosis, in both murine and human cells, through induction of a phagocytic gene program. Importantly, TLR-induced phagocytosis of bacteria was found to be reliant on myeloid differentiation factor 88–dependent signaling through interleukin-1 receptor–associated kinase-4 and p38 leading to the up-regulation of scavenger receptors. Interestingly, individual TLRs promote phagocytosis to varying degrees with TLR9 being the strongest and TLR3 being the weakest inducer of this process. We also demonstrate that TLR ligands not only amplify the percentage of phagocytes uptaking Escherichia coli, but also increase the number of bacteria phagocytosed by individual macrophages. Taken together, our data describe an evolutionarily conserved mechanism by which TLRs can specifically promote phagocytic clearance of bacteria during infection.
Keywords: macrophage, phagocytosis, toll-like receptor, scavenger receptor, p38
Introduction
Bacterial infection is a major cause of illness and morbidity in multicellular organisms. In the innate immune system, macrophages play a key role in combating infection. Macrophages rely heavily on phagocytosis and subsequent degradation of microbes in order to help clear the body of invading pathogens (1). Pathogens are detected by macrophages through a variety of pattern recognition receptors (PRRs; reference 2). This, in turn, leads to receptor-mediated endocytosis, digestion of the pathogen, and presentation of antigens in the context of MHC.
In addition to direct pathogen uptake, other classes of PRRs, such and toll-like receptors (TLRs), play essential roles in activating signal transduction pathways leading to the killing and clearance of pathogens. TLRs recognize highly conserved, pathogen-coded molecular structures termed pathogen-associated molecular patterns (PAMPs; reference 3). A great deal has been learned in the past 10 years regarding specific recognition of microbial PAMPs by individual TLRs (4). For example, TLR4 recognizes LPS from gram-negative bacteria whereas TLR9 recognizes bacterial hypomethylated CpG DNA motifs. After ligand binding, all TLRs sequentially recruit the adaptor molecules myeloid differentiation factor 88 (MyD88), IL-1 receptor–associated kinase (IRAK), and tumor necrosis factor receptor–associated factor 6 (TRAF6). These adaptors in turn mediate the activation of the jun NH2-terminal kinase (JNK), nuclear factor (NF)-κB, p38, extracellular signal–related kinase 1/2 (ERK 1/2), and phosphoinositide 3–kinase signaling pathways leading to activation of inflammatory target genes (5).
Significant progress has been also made in describing the inflammatory and antimicrobial responses induced by individual TLRs (6–8). Inflammation is promoted by TLRs through the production of cytokines such as TNF-α and IL-Iβ (9). TLR activation also promotes recruitment of inflammatory cells (10), and has been shown to mediate direct antimicrobial activity against intracellular bacteria (11). Moreover, a few studies have suggested that certain TLR ligands may play a role in modulating the ability of macrophages to bind extracellular bacteria (12–14). However, the ability of various TLRs to directly modulate macrophage phagocytosis remains unclear, and no molecular mechanism for such a process has been described.
In our current study, we demonstrate that numerous TLR ligands specifically enhance phagocytosis of bacteria such as Escherichia coli and Staphylococcus aureus, while exhibiting minimal effects on nonbacterial targets such as latex beads. Various TLRs differentially promote phagocytosis through induction of a phagocytic gene program, with TLR9 being the strongest mediator of this process and TLR3 being the weakest. In addition, we show that TLR-mediated induction of scavenger receptors (SRs) occurs through MyD88, IRAK4, and p38, and that activation of this pathway is essential for TLR promotion of phagocytosis. This mechanism appears to be evolutionarily conserved between mice and humans, suggesting that this pathway is a critical mediator of bacterial clearance by mammals after infection.
Materials and Methods
Cell Culture and Reagents.
Murine bone marrow–derived macrophages (BMMs) were differentiated from marrow as described previously (6). A129 (IFNAR-1−/−; reference 15) and B6129SF2/J wild-type control mice were obtained from B&K Universal Ltd. and Jackson ImmunoReasearch Laboratories, respectively. Mice deficient in MyD88 and littermate controls were obtained from Shizuo Akira (University of Osaka, Osaka, Japan). C57/B6 mice were used for all experiments not involving the A129 or MyD88-deficient mice (Jackson ImmunoReasearch Laboratories). RAW 264.7 murine macrophage cells were cultured in DMEM media supplemented with 10% fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin. THP-1 human monocytes were cultured in RPMI media supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Peripheral blood mononuclear cells were isolated from human whole blood using Ficoll-Paque (Amersham Biosciences) density gradient centrifugation following the manufacturer's recommended protocol. Monocytes were enriched using Percoll (Amersham Biosciences) density gradient centrifugation and then adhered onto plastic plates for 2 h in RPMI containing 1% fetal bovine serum (Hyclone). The nonadherent cells are washed off with PBS and the adherent cells are cultured in complete RPMI supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin, and treated with stimuli. Specific TLR activation was achieved using F-583 (Rd mutant) E. coli lipid A for TLR4 (Sigma-Aldrich), CpG oligonucleotides for TLR9 (Invitrogen), purified peptidoglycan (PGN) from Staphylococcus aureus (Sigma-Aldrich) for TLR2, polyinosinic acid (poly I):C for TLR3 (Amersham Biosciences), and imiquimod for TLR7 (Sequoia Research). ERK 1/2 and p38 were both inhibited using 10 μM uo126 or sb202190, respectively (Calbiochem). Poly I was obtained from Sigma-Aldrich and added to cells 1 h before bacterial challenge. Macrophage colony-stimulating factor (M-CSF)–containing media was obtained by growing L929 cells 4 d past confluency and then harvesting the conditioned media.
RNA Quantitation.
For microarray studies, BMMs were treated with media or 1 ng/ml lipid A for 4 h or 1 μg/ml poly I:C or 100 nM CpG for 4 or 12 h. After stimulation, total RNA was extracted and labeled cRNA synthesized as described previously (6). The labeled cRNA was used to hybridize to Affymetrix Mu11K chip sets and data analyzed using Affymetrix Microarray Suite 4.0 data mining software. To identify PAMP-induced genes, comparisons were performed using the media-only–treated sample as a baseline. Genes were considered induced if they displayed (a) a threefold change relative to baseline, (b) they had a poststimulation average difference (a value representing absolute expression level) of at least 500, and (c) they had an average difference change relative to baseline of at least 600 (roughly twice chip background). Average difference change values for genes involved in phagocytosis were processed using Cluster and data presented as a dendrogram using the Treeview program (www.rana.lbl.gov/EisenSoftware.htm). For quantitative realtime PCR (Q-PCR), total RNA was isolated and cDNA synthesized as described previously (6). PCR was then performed using the iCycler thermocycler (Bio-Rad). _I_κ_B_α, L32, and intercellular adhesion molecule-1(ICAM-1) primers were the same as those described previously (6, 16). The following primers were synthesized by Invitrogen: macrophage SR with collagenous structure (MARCO) forward ATCCTGCTCACGGCAGGTACT, and reverse GCACATCTCTAGCATCTGGAGCT, scavenger receptor-A (SR-A1) forward TCCTTGCAGAGTCTGAATATGACACT, and reverse CCTCCTGTTGCTTTGCTGTAGATT, lectin-like oxidized LDL receptor (LOX-1) forward GTGGACACAATTACGCCAGGTA, and reverse GCCCTTCCAGGATACGATCC, and _IL-1_β forward GAGCTGAAAGCTCTCCACCTCA, and reverse TCGTTGCTTGGCTCCTTGTAC. It is of note that we refer to SR-A1 mRNA as SR-A throughout this manuscript.
EMSA, Western Blotting, and GST Pulldown Assays.
For EMSA, BMMs were stimulated with TLR ligands and nuclear extracts were prepared as described previously (17). For p38 and phosphorylated p38 immunoblotting, cells were lysed in modified RIPA buffer (50 mM Tris-Cl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate containing 2 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 100 μg/ml PMSF) containing protease and phosphatase inhibitors and 40 _μ_g of protein was loaded per lane and separated by SDS-PAGE on 10% gels. After transfer, nitrocellulose filters were immunoblotted using anti-p38 and antiactivated p38 antibodies from Cell Signaling Technologies as per manufacturer's recommendations. A human pEFBOS.TLR9 construct was provided by Shizuo Akira. ESTs containing the intracellular domain of hTLR3 and full-length hMyD88 were obtained from Research Genetics. The cloning of the intracellular domains of TLR3 and TLR9 into expression vectors and binding assays were performed as described previously (16).
Phagocytosis Assays.
For each experiment, the green fluorescence protein (GFP)–expressing E. coli (a gift from Dr. Jeff F. Miller, UCLA, Los Angeles, CA) were grown up from a single colony in Luria-Bertani broth containing 100 μg/ml ampicillin. The culture was then subcultured and grown until log phase growth was obtained. Bacteria were then washed in PBS two times and then resuspended at the appropriate concentration in either DMEM or RPMI media without serum or antibiotics. Before infection, macrophages were washed two times in media without serum or antibiotics before bacterial challenge. BMMs and RAW264.7 cells were infected with GFP–E. coli at an multiplicity of infection (MOI) of 5 or 25, respectively. The optimal time point for measurement of phagocytosis was determined to be at 45 min after infection. Macrophage cells were then washed with cold PBS in order to stop additional bacterial uptake or destruction of bacteria in the phagolysosome. Cells were washed a total of three times in cold PBS before harvesting in cold PBS containing 5 mM EDTA. Cells were then fixed using paraformaldehyde (1% final concentration) and then subjected to FACS® analysis. Experiments using acetylated low-density lipoproteins (Molecular Probes), fluorescently labeled latex beads (Sigma-Aldrich), or BODIPY®–conjugated S. aureus (Wood strain without protein A) BioParticles® (Molecular Probes) were performed in a similar fashion at 5 μg/ml, at an MOI of 1 or 10, respectively.
Flow Cytometry, Laser Scanning Cytometry, and Fluorescence Microscopy.
MARCO and SR-A protein expression on the macrophage cell surface was detected by using an anti-MARCO–FITC antibody (clone ED31; 1 μg/ml, along with IgG1 isotype control), or anti–SR-A–FITC antibody (clone 2F8, which recognizes both SR-AI and SR-AII isoforms; 1 μg/ml, along with IgG2b isotype control), respectively. These antibodies, along with unconjugated anti–SR-A–blocking antibody (clone 2F8), were obtained from Serotec, Inc. An anti-CD11c antibody conjugated to PE was obtained from Becton Dickinson. All staining antibodies were diluted in FACS® buffer (phosphate-buffered saline, 1% bovine serum albumin, 1% fetal calf serum, 0.1% sodium azide) containing 1% normal mouse serum. FACS® analysis was conducted using a FACSCaliber® (Becton Dickinson) machine and CellQuest software (Becton Dickinson). To assess changes in phagocytic efficiency, RAW 264.7 cells were plated in eight-well chamber slides (Nalge NUNC) and treated as indicated. Cells were fixed with 4% paraformaldehyde for 30 min and analyzed with a laser scanning cytometer (CompuCyte). Cells were counterstained with 6′-diamidino-2-phenylindole (DAPI; Molecular Probes) at a concentration of 10 μg/ml to identify DNA and to distinguish cell cycle stage. The UV laser was used to contour on DAPI using a minimum cell area of 20 μm. The argon laser was used to measure fluorescence of GFP–E. coli shown as percent of cells that have been phagocytosed by macrophages. The number of GFP–E. coli per cell was measured by laser scanning microscopy using ultraviolet light to excite DAPI and the argon laser was used to measure GFP fluorescence within a minimum of 2 pixels in area. LSC-based fluorescence in situ hybridization (FISH) analysis software was used to show the total number of GFP–E. coli per macrophage. The LSC was used to photograph the GFP–E. coli with a CCD camera and colored with CompuCyte color software (Laser Scanning Cytometry). For fluorescence microscopy a Leica DM IRBE fluorescent microscope using 10× and 63× objectives (Leica Microsystems) and Openlab software (Improvision) was used for image acquisition. Cells were counter stained with DAPI and then stained with rhodamine–phalloidin (Molecular Probes) as per the manufacturer's protocol to label cytoskeletal F-actin.
Results
TLR Ligands Specifically Enhance Phagocytosis of Bacteria.
To determine the role of TLRs in promoting phagocytosis, we pretreated RAW 264.7 murine macrophage cells with TLR ligands and measured the uptake of the GFP-labeled Topp10 strain of E. coli. We chose to perform these experiments using the RAW264.7 cell line because we found that these cells have a low basal level of phagocytosis as determined by FACS® (Fig. 1 A). As shown in Fig. 1 A, pretreatment of these cells with various TLR ligands markedly enhanced the level of phagocytosis of the gram-negative bacterium E. coli. We also found that TLR activation leads to increased uptake of BioParticles® derived from the gram-positive bacterium S. aureus (Fig. 1 C). Different TLRs promoted phagocytosis to different extents, with TLR9 being the strongest, followed by TLR2 and TLR7 (data not shown) and TLR4 and TLR3 being the weakest inducers of this process.
Figure 1.

TLR ligands specifically increase macrophage phagocytosis of bacteria. (A) RAW 264.7 macrophage cells were pretreated with media, CpG (100 nM), PGN (20 μg/ml), lipid A (50 ng/ml), or poly I:C (10 μg/ml) for 24 h and then challenged with GFP–E. coli at an MOI of 25. (B) RAW 264.7 cells pretreated with 100 nM CpG or media alone were treated with fluorescently labeled latex beads at an MOI of 1. (C) BODIPY®-conjugated S. aureus BioParticles® at an MOI of 10. (D) THP-1 monocytes were treated with media or PGN (20 μg/ml) for 36 h. Cells were then challenged with GFP–E. coli at an MOI of 5, and analyzed for the presence of phagocytosed bacteria. (E) Primary human monocytes were collected from four independent human donors (donor 1, square; donor 2, circle; donor 3, diamond; donor 4, triangle; mean, line) and subjected to the same phagocytosis experiments as in D. After microbial or particle challenge, cells were washed and subjected to FACS® analysis. All FACS® data are represented as percent positive cells. Data are representative of at least two independent experiments.
To determine if TLR-induced phagocytosis is specific for bacteria, or whether it represents an uptake pathway that can target various products, we performed phagocytosis experiments using fluorescently labeled latex beads or acetylated low-density lipoproteins. Fig. 1 B shows that CpG pretreatment results in only a mild increase in the phagocytosis of latex beads by macrophage cells as compared with media-pretreated samples. Uptake of acetylated low-density lipoproteins was also only minimally increased after TLR activation (data not shown). These findings suggest that TLR ligands induce a phagocytic phenotype that can specifically target both gram-negative and -positive bacteria. Moreover, TLRs seem to have diverged functionally with individual TLRs being able to promote phagocytosis to varying degrees.
To determine if TLR-induced phagocytosis is an evolutionarily conserved process, we first tested the human monocyte cell line THP-1. Because human monocytes are unresponsive to CpG, we used the TLR2 ligand PGN as it was also a relatively strong inducer of phagocytosis in our murine studies and was also able to induce IL-12 p40 production (data not shown). Fig. 1 D demonstrates that PGN pretreatment significantly increased the ability of THP-1 cells to ingest E. coli. We next wanted to test if primary human monocytes also have increased phagocytic capabilities after TLR activation. PGN treatment was also able to significantly up-regulate phagocytosis in primary human monocytes, causing a roughly twofold increase in the percent of cells containing bacteria (Fig. 1 E). In addition, we noticed a correlation between the degree of IL-12 p40 induction (a control for efficient TLR2 stimulation and basal activation state of the monocytes used) and the level of increase in phagocytosis after PGN treatment (data not shown).
TLRs Induce Genes Involved in Phagocytosis.
To define the molecular mechanisms underlying the TLR-induced phagocytic phenotype observed in Fig. 1, we analyzed TLR-regulated target genes on a global basis. Murine bone marrow–derived macrophage cells (BMMs) were treated with CpG, lipid A, and poly I:C for 4 and 12 h, and then subjected to a microarray analysis to assess target gene expression changes (Fig. 2 A). We selected the concentrations of TLR ligands used as they gave relatively equal activation of NF-κB by electrophoretic mobility shift assay (EMSA; Fig. 2 B, top), as well as equal up-regulation of _I_κ_B_α mRNA, suggesting a similar strength of signal (Fig. 1 B, top). Upon analyzing the microarray results, we identified a number of genes that were up-regulated by TLR9, TLR4, and TLR3 that are involved in phagocytosis (Fig. 2 A). Genes such as CD36, thrombospondin, and the apoptotic cell clearance receptor are known to be involved in the clearance of apoptotic cells (18–20). In addition, genes involved in opsonin-dependent phagocytosis (Fc and complement receptor genes as well as Lyn and Syk) were found to be up-regulated by TLRs (21, 22). We also noticed a number of genes involved in opsonin-independent phagocytosis of bacteria. Among these genes were a subset of SRs, including MARCO and SR-A (23). Genes coding for certain signaling molecules (Protein Kinase C δ and Lyn) that are thought to act downstream of SRs were also up-regulated (24).
Figure 2.

TLR ligands induce expression of genes involved in phagocytosis. Murine bone marrow–derived macrophage cells were stimulated with media (m), CpG (100 nM), lipid A (1 ng/ml), or poly I:C (1 μg/ml) for the indicated times. (A) Total RNA was collected after 4 or 12 h of TLR ligand stimulation and subjected to a microarray analysis as described in Materials and Methods. A subset of genes involved in phagocytosis is depicted as a dendrogram. Red represents up-regulation of the gene, black represents no change in expression, and green represents down-regulation of the gene. (B) Nuclear extract from each sample (2 μg) was used to assay inducible DNA binding of NF-κB by EMSA (top), or RNA was collected, converted to cDNA, and subjected to Q-PCR analysis to assay _I_κ_B_α gene induction by 1 h (bottom). (C) Total RNA was collected from BMMs treated with indicated PAMPs, converted to cDNA, and Q-PCR was used to verify expression of MARCO, SR-A, and LOX-1 using sequence specific primers. (D) BMMs were stimulated with TLR ligands for 24 h and cells were stained with anti-MARCO–FITC antibody to detect MARCO via FACS®. The data are represented as percent-positive cells. Q-PCR data are represented in relative expression units and all values have been normalized to L32. All data, except the microarray, are representative of at least three independent experiments.
Although TLR3, TLR4, and TLR9 all induced complement, Fc, and SRs, we chose to focus on SRs due to their ability to directly interact with microbes under nonopsonized conditions (same conditions used in Fig. 1). Using Q-PCR analysis, we confirmed the inducible expression of SR-A and MARCO by the different TLR ligands (Fig. 2 C). We also tested the expression of other SRs and found that LOX-1 was also induced by TLR ligands at early time points (Fig. 2 C). Similar to the pattern of induction obtained in our phagocytosis assays, we found that TLR9 induced the expression of these genes to the highest degree, whereas TLR4-mediated induction was more intermediate and TLR3 was the weakest inducer of the SRs tested. In addition, whereas both TLR9 and TLR4 were able to up-regulate all three of these genes, TLR3 was only able to induce LOX-1 and SR-A, but not MARCO. To determine if induction of SR transcript correlated with increased levels of protein we performed flow cytometry using antibodies specific to SR-A and MARCO and found that TLRs can induce expression of these proteins (Fig. 2 D and Fig. 3, B and C) . These data suggest that TLRs promote phagocytosis though the up-regulation of a phagocytosis gene program that includes SRs.
Figure 3.

TLRs use a MyD88_–_IRAK4_–_p38 signaling pathway in order to induce expression of the SRs MARCO, LOX-1, and SR-A. (A) BMMs from MyD88−/−, IRAK4−/− mice, or wild-type BMMs pretreated with either sb202190 (sb, 10 μM) or uo126 (10 μM) were stimulated with media (m) or CpG (100 nM) for 4 or 12 h. Total RNA was collected, converted to cDNA, and then Q-PCR was used to assay the inducible expression of SR-A, LOX-1, and MARCO under the different conditions. To control for specificity of p38 and ERK 1/2 inhibition, induction of ICAM-1 and IL-1β were also measured. Q-PCR data are represented in relative expression units and normalized to L32. (B) Wild-type, MyD88−/−, IRAK4−/−, and sb202190 (10 μM) pretreated wild-type BMMs were stained for MARCO surface expression after 24 h of media, CpG (100 nM), lipid A (1 ng/ml), or poly I:C (1 μg/ml) treatment and FACS® data are represented as the mean fluorescent intensity (MFI) of each cell population. (C) RAW 264.7 cells were stimulated with CpG (100 nM) or PGN (20 μg/ml) for 24 h, either in the presence of absence or 10 μM sb202190, stained with a FITC-labeled αSR-A antibody, and then analyzed by FACS®. (D) BMMs from wild-type and IRAK4−/− mice were stimulated with media or CpG (100 nM) for the indicated times. Whole cell extract was then subjected to immunoblotting to detect phosphorylated (activated) p38 or total p38. (E) 293T cells were transfected with a MyD88 expression vector. Extract was collected and incubated with glutathione beads containing either TLR9 (intracellular domain)–GST, TLR3 (intracellular domain)–GST, or GST alone. Bound MyD88 was eluted by boiling and visualized by immunoblotting using an anti-MyD88 polyclonal antibody. The dashed line represents a cropping border where irrelevant lanes have been removed. Data are representative of at least two independent experiments.
TLRs Use MyD88, IRAK4, and p38 to Induce SRs.
The signal transduction pathways activated by different TLRs are known to differ. Some TLRs preferentially activate the MyD88-dependent pathway whereas others also activate the MyD88-independent pathway (25, 26). Due to this fact, we wanted to clarify which TLR signaling arm was responsible for the inducible expression of these SRs. Using mice deficient in MyD88, we show in Fig. 3 A that MyD88 is required for the inducible expression of MARCO, LOX-1, and SR-A at the mRNA level in response to TLR9 stimulation with CpG. This dependency was also verified for MARCO at the protein level (Fig. 3 B). In addition, we found that autocrine/paracrine induction of type 1 interferon does not contribute to TLR9- or TLR4-induced LOX-1 or MARCO expression, indicating that the MyD88-independent pathway does not play a role in the up-regulation of the SRs tested (data not shown; reference 6).
Recently, the serine–threonine kinase IRAK4 has been shown to be an important component of the MyD88-dependent pathway (27). To determine if the TLR–MyD88 signal that is responsible for SR up-regulation is mediated through IRAK4, we stimulated BMMs deficient in IRAK4 with CpG and assessed the up-regulation of MARCO and LOX-1. Identical to MyD88 knockout BMMs, the IRAK4 deficiency ablated up-regulation of these TLR target genes (Fig. 3 A). This IRAK4 requirement was also verified at the protein level for MARCO (Fig. 3 B).
In an effort to determine which signaling pathway downstream from IRAK4 is required for SR gene expression, we used different pharmacological inhibitors to block known TLR signaling pathways including p38 (sb202190) and ERK 1/2 (uo126). We used a concentration of 10 μM for each of these inhibitors because we found that this concentration led only to inhibition of the intended pathway and did not nonspecifically affect other signaling pathways (unpublished results). Supporting this claim, ICAM-1 induction (whose transcription is NF-κB dependent) is not effected by treatment with sb202190 (Fig. 3 A). However, inhibition of p38 activity, but not ERK 1/2 activity, blocked TLR-induced expression of SR transcripts in response to CpG (Fig. 3 A). FACS® analysis shows that TLRs also potently up-regulate MARCO and SR-A at the protein level and that inhibition of p38 activity completely blocks this induction (Fig. 3, B and C). The effectiveness of uo126 is exemplified by the fact that TLR-induced IL-1β transcription is greatly reduced after blockade of ERK 1/2 with this compound (Fig. 3 A).
In IRAK4-deficient mice, p38 activation is completely abolished after IL-1β treatment (27). Additionally, it has been shown that CpG-mediated p38 activation is completely abolished in the absence of MyD88 (28). To determine if IRAK4 is also required for p38 activation downstream from TLR9, we treated IRAK4−/− BMMs with CpG and assessed p38 activation by conducting Western blotting using an antibody that specifically recognizes phosphorylated p38. We found that in the absence of IRAK4, p38 was no longer activated by TLR9 signaling (Fig. 3 D).
As previously mentioned, we had noticed that TLR9 was overall the strongest inducer of SR expression, whereas TLR3 was the weakest (Fig. 2, A and C). Because the signaling events leading to SR expression appear to be dependent on MyD88, we tested the relative affinities of the cytoplasmic domains of TLR3 and TLR9 for MyD88. We show in Fig. 3 E that the GST–TLR9 intracellular domain has a higher affinity for MyD88 than does GST–TLR3 intracellular domain using pulldown assays with overexpressed MyD88. Collectively, these data suggest that TLRs differentially up-regulate a subset of bacterial binding SRs via a MyD88–IRAK4–p38-dependent pathway. In addition, the strength of signaling activated by various TLRs may be determined by the relative affinity of these different TLRs for MyD88.
TLR-induced Bacterial Phagocytosis Is MyD88, p38, and SR Dependent.
To determine if TLR-induced phagocytosis of bacteria is dependent on MyD88–IRAK4–p38-mediated SR up-regulation, we first compared TLR-induced phagocytosis of bacteria by BMMs derived from wild-type and MyD88 knockout mice (Fig. 4 A). Although BMMs have a high basal level of phagocytosis relative to RAW 264.7 cells, which is likely due to up-regulation of SR-A by M-CSF in the culture media (data not shown; and references 29 and 30), CpG pretreatment still significantly increases bacterial uptake by BMMs in a MyD88-dependent manner. Interestingly, the basal level of BMM phagocytosis was significantly decreased in MyD88−/− macrophage cells compared with wild-type controls.
Figure 4.

TLR-induced phagocytosis of bacteria is MyD88, p38, and SR dependent_._ (A) BMMs derived from either wild-type or MyD88-deficient mice were stimulated with media or 100 nM CpG for 24 h followed by infection with GFP-expressing E. coli at an MOI of 10 for 45 min. Cells were washed twice with cold PBS and subjected to FACS® analysis. (B) RAW 264.7 macrophage cells were stimulated with media, CpG (100 nM), or PGN (20 μg/ml) for 24 h. Cells were cotreated with 10 μM sb202190 for the entire duration of the media, CpG or PGN pretreatment, or just for the final 1 h. Cells were then challenged with GFP–E. coli for 45 min at an MOI of 25, washed, and subjected to FACS® analysis. (C) RAW 264.7 cells were pretreated with either media, CpG (100 nM), or PGN (20 μg/ml) for 24 h. For the final 1 h, the cells were treated with poly I (100 μg/ml), anti–SR-A (3 μg/ml), or IgG2b isotype control (3 μg/ml) and phagocytosis assays were performed. (D) THP-1 monocytes were treated with media or PGN (20 μg/ml) for 36 h. The same conditions were also used along with sb202190 (10 μM) for 36 h or poly I (100 μg/ml) for the final 1 h of TLR ligand pretreatment. Cells were then challenged with GFP–E. coli at an MOI of 5, washed, and subjected to FACS® analysis. Data represent at least two independent experiments.
We next tested whether or not inhibiting the p38 signaling pathway could prevent TLR-induced phagocytosis. In agreement with our data showing that p38 is required for the TLR-mediated up-regulation of SRs such as MARCO and SR-A (Fig. 3, A–C), the TLR-dependent increase in phagocytosis of E. coli was completely blocked by the p38 inhibitor (Fig. 4 B). However, if the RAW 264.7 macrophage cells were pretreated with TLR ligands and given the p38 inhibitor for only 1 h before bacterial challenge, TLR-induced phagocytosis was relatively unaffected (Fig. 4 B). These data suggest that TLR-induced augmentation of phagocytosis requires p38 for the up-regulation of phagocytic mediators such as SRs, but is p38 independent once these proteins are in place on the cell surface.
SR-A has been shown to play an important role in bacterial phagocytosis (29, 31). Furthermore, mice with a targeted disruption of the SR-A gene have defects in bacterial clearance (31). Thus, we sought to determine if TLR-inducible phagocytosis specifically involved SR-A. Fig. 4 C reveals that a blocking antibody against SR-A, but not the antibody isotype control, significantly reduced TLR-induced phagocytosis. Because the block was not complete, it implied that other SRs may be involved in the uptake of bacteria. To test if this is the case, we performed phagocytosis assays in the presence of the SR inhibitor, poly I. These experiments showed that TLR-inducible phagocytosis was almost completely inhibited by poly I and indicated that multiple SRs are, in fact, involved in the TLR-mediated potentiation of bacterial phagocytosis (Fig. 4 C). The involvement of multiple SRs in TLR9- and TLR2-induced bacterial phagocytosis correlates with a p38-dependent increase in expression of not only SR-A but also MARCO and LOX-1 after CpG treatment (Fig. 3, A–C). These data imply that TLR-inducible macrophage phagocytosis of bacteria is through a MyD88–IRAK4–p38-dependent up-regulation of multiple SRs that, together, contribute to an overall increase in bacterial binding and phagocytosis.
TLR Activation Causes an Increase in Phagocytic Efficiency.
The broad shift in mean fluorescence intensity present in the FACS® data (Fig. 1 A) from our phagocytosis assays suggested that TLR ligand treatment may not only cause an increased number of macrophages able to participate in bacterial phagocytosis, but they also promote an increase in the amount of bacteria uptake by individual macrophage cells. Fluorescence microscopy confirmed this, showing that TLR ligand treatment of RAW 264.7 cells not only causes an increase in the number of cells engaged in phagocytosis, but also an enhancement in the number of E. coli taken up per individual macrophage cell (Fig. 5 A). We used LSC to quantitate the observed increase in the number of bacteria localized within individual macrophages (32). We found LSC to be comparable to FACS® in determining the percentage of TLR-induced phagocytic cells (data not shown). We performed an LSC-based FISH analysis, which enabled us to determine the topographic distribution of E. coli, and therefore allowed us to measure the number of bacteria bound or phagocytosed by each macrophage cell. These experiments showed quantitatively that CpG pretreatment of macrophages causes a great increase in the number of E. coli phagocytosed per individual macrophage, as compared with the media control (Fig. 5 B). In addition, treatment with sb202190 (sb) showed that this increase in phagocytic efficiency was found to be dependent on p38. Similar results were also obtained for cells pretreated with PGN. LSC-based FISH analysis revealed that human monocytes also take up a larger number of bacteria after TLR2 activation (data not shown). Our data suggest that all TLR ligands tested can induce macrophage phagocytosis of E. coli to varying degrees. Interestingly, TLR2 and TLR9 caused the greatest increase in bacterial uptake, with TLR9 being the strongest potentiator out of all of the TLRs tested. Furthermore, we found that TLR-induced nonopsonized phagocytosis can increase both the number of phagocytic cells in the population as well as the number of E. coli taken up on a per cell basis.
Figure 5.
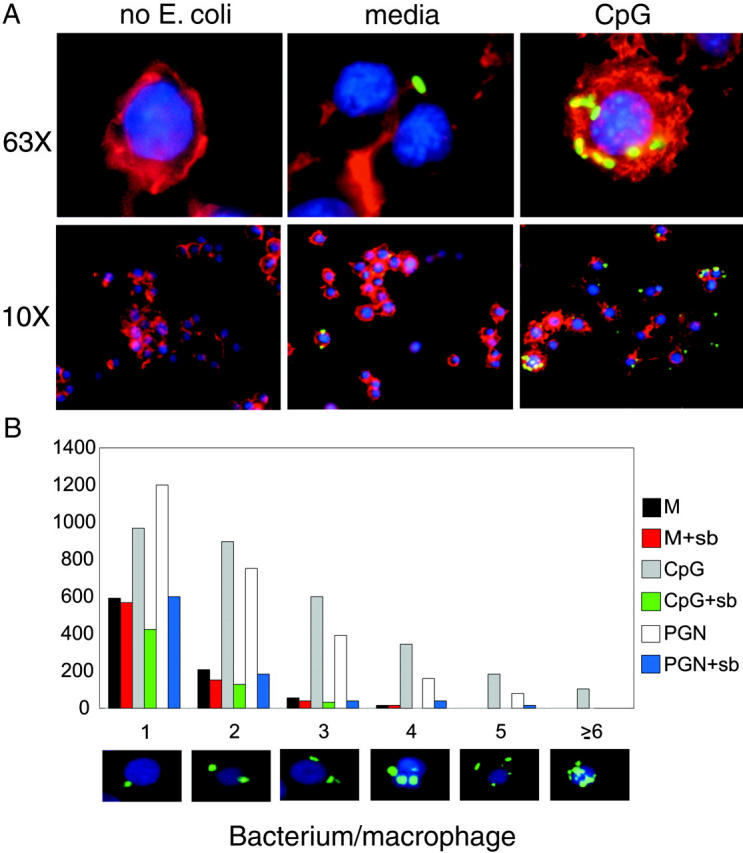
TLR activation results in an increase in the number of bacteria per individual macrophage. (A) RAW 264.7 cells were pretreated with media or CpG (100 nM) for 24 h, phagocytosis assays were performed using GFP–E. coli (green), and cells were fixed, stained with DAPI (blue) to stain chromosomal DNA and rhodamine-phalloidin (red) to stain cytoplasmic F-actin, and subjected to fluorescence microscopy at both 10× and 63× resolution. (B) The same cells as seen in A were subjected to LSC-based FISH analysis using the LSC to determine the number of GFP signals on a per cell basis. Data are represented as the number of macrophage cells (out of 5,000 scanned) that had either 1, 2, 3, 4, 5, or ≥6 GFP signals per cell. Pictures along the x axis are representative of macrophage cells in each grouping that were counted by the LSC. Data represent at least two independent experiments.
Discussion
Phagocytosis, a hallmark function of macrophage cells in innate immune responses, involves a series of coordinated events including recognition of pathogens by cell surface receptors, receptor-mediated endocytosis, fusion of the phagosome with the endosome, digestion of pathogens, as well as presentation of antigens through MHC. The regulation of these individual events during host infection by pathogen, however, is complex and not well understood. TLR family members represent a major class of innate immune receptors abundant on the surface of macrophage cells. A great deal has been learned in the past 10 years regarding the recognition of microbial PAMPs by individual TLRs, and the inflammatory response genes induced by these TLRs. Whereas certain TLRs such as TLR3 and TLR4 have been shown to have antiviral activity through the production of IFNβ (6), relatively less is known about the role of TLRs in mediating direct antibacterial responses. Our current study characterizes a molecular mechanism that clearly links TLRs to macrophage-mediated phagocytosis of bacteria.
We have identified a TLR-inducible gene subset that is indicative of enhanced macrophage bacterial binding and uptake. Among this subset of genes are SRs including SR-A, LOX-1, and MARCO. The role of SR-A in TLR-induced phagocytosis was demonstrated by using a blocking antibody specific for SR-A. Indeed, mice with targeted disruption of the SR-A gene have defects in bacterial clearance (31). The fact that the general SR inhibitor poly I can further inhibit TLR-induced phagocytosis in both human and mouse cells suggests that other SRs must assist SR-A in binding and phagocytosing of bacteria. In addition to SRs, TLRs mediate the induction of other genes involved in the recognition of pathogens and apoptotic cells including the mannose-6-phosphate receptor, ABC1, complement receptors (33–38). The microarray results also demonstrate that each TLR tested can induce both common and specific genes involved in phagocytosis, which suggests that different TLRs can induce unique combinations of phagocytic mediators that may differentially target pathogens.
Our data support a model whereby TLRs transduce signals through MyD88, IRAK4, and p38 resulting in the up-regulation of SRs, leading to a significant increase in macrophage/monocyte phagocytosis of gram-negative and -positive bacteria. The TLR-mediated increase in phagocytosis appears to be specific for bacteria since only a slight increase in uptake of either latex beads or acetylated low-density lipoproteins was observed. These data are especially interesting since uptake of acetylated low-density lipoproteins is also mediated by SRs and suggest that TLRs may be able to differentially regulate the ability of SRs to mediate uptake of different ligands.
Our gene expression data reveal that although TLR3, TLR4, and TLR9 all induce expression of SRs, TLR9 appears to be the strongest inducer of SR-A, MARCO, and LOX-1, as well as of phagocytosis. Because the induction of these genes and phagocytosis of E. coli is dependent on MyD88, it is not surprising that TLR9 has a high relative affinity for this adaptor (Fig. 3 E). MARCO, LOX-1, and SR-A were analyzed specifically in this manuscript because they were all TLR induced, had steady-state mRNA, and protein levels that were dependent on the same signaling cascade (MyD88–IRAK4–p38), and have all been shown to be able to bind bacteria, including E. coli and S. aureus (23). Despite these similarities, each of these SR genes displayed differential induction kinetics, thus further studies will be required to understand the additional mechanisms controlling their TLR-induced expression. Regardless, it is evident that SRs are critical players in TLR-induced macrophage phagocytosis of bacteria, and that they work in a concerted manner. Besides TLR ligands, other stimuli may also induce the surface expression of SRs. Previous and current studies revealed that the SR levels are also up-regulated in response to M-CSF, which explains why bone marrow–derived murine macrophages (cultured in M-CSF) have relatively high levels of SR expression compared with RAW 264.7 cells (cultured without M-CSF [data not shown]; references 28 and 29). There is even a possible relationship between TLRs and M-CSF, as our gene chip data indicated that all TLRs tested can induce expression of M-CSF (data not shown). It is important to emphasize that, besides the up-regulation of SRs, TLRs may likely up-regulate the expression or activity of other molecules involved in phagocytosis, which may not be able to be induced by other stimuli such as M-CSF.
A number of reports have shown that both murine and human TLRs play a vital role in host antibacterial responses. Systemic administration of CpG to bacterially infected wild-type mice results in the suppression of bacterial load (data not shown; and reference 39). Additionally, mice deficient in either MyD88 or IRAK4 have shown increased susceptibility to bacterial infection (27, 40). Recent studies have also revealed that a mutation in the human IRAK4 gene is linked to the inability to clear bacterial infections (41), and specific polymorphisms in human TLR2 result in hyporesponsiveness to bacterial ligands and impaired bacterial clearance by the host (42–44). It is well known that TLRs and the MyD88 signaling pathway play critical roles in pro-inflammatory cytokine production, which is essential for antimicrobial responses. The data presented in this manuscript provide an additional explanation—that TLRs play an important role in promoting clearance of bacteria through the up-regulation of macrophage phagocytic activity through MyD88, IRAK4, and p38.
Acknowledgments
We would like to thank Dr. Stephen Smale at UCLA for donating the use of his flow cytometer and Dr. Shizuo Akira at Osaka University for the MyD88−/− and MyD88−/− mice.
R. O'Connell was supported by the United States Public Health Service National Research Service Award GM07185. S.A. Vaidya was supported by a UCLA Medical Scientist Training Program training grant (GM 08042). E.K. Chow was supported by the UCLA ACCESS Graduate Program. G. Cheng is a Research Scholar supported by the Leukemia and Lymphoma Society of America. W.-C. Yeh was supported by an operating grant from Canadian Institutes of Health Research (MOP-57734). T. Lane was supported by the National Institutes of Health research grant S10 RR15862-01 and Stop Cancer II, and G. Miranda was supported by the Department of Defense grant OC 98-0006. Part of this work was also supported by National Institutes of Health research grants R01 CA87924 and R37 AI47868.
Sean E. Doyle and Ryan M. O'Connell contributed equally to this work.
Abbreviations used in this paper: BMM, bone marrow–derived macrophage; DAPI, 6′-diamidino-2-phenylindole; ERK 1/2, extracellular signal–related kinase 1/2; FISH, fluorescence in situ hybridization; GFP, green fluorescence protein; ICAM-1, intercellular adhesion molecule-1; IRAK, IL-1 receptor–associated kinase; LOX-1, lectin-like oxidized LDL receptor; MARCO, macrophage scavenger receptor with collagenous structure; M-CSF, macrophage colony-stimulating factor; MOI, multiplicity of infection; MyD88, myeloid differentiation factor 88; NF, nuclear factor; PGN, peptidoglycan; poly I, polyinosinic acid; PRR, pattern recognition receptor; Q-PCR, quantitative realtime PCR; SR, scavenger receptor; SR-A, scavenger receptor-A; TLR, toll-like receptor; TRAF6, tumor necrosis factor receptor–associated factor 6.
References
- 1.Sansonetti, P. 2001. Phagocytosis of bacterial pathogens: implications in the host response. Semin. Immunol. 13:381–390. [DOI] [PubMed] [Google Scholar]
- 2.Gordon, S. 2002. Pattern recognition receptors: doubling up for the innate immune response. Cell. 111:927–930. [DOI] [PubMed] [Google Scholar]
- 3.Janeway, C.A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. [DOI] [PubMed] [Google Scholar]
- 4.Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2:675–680. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi, O., and S. Akira. 2001. Toll-like receptors: their physiological role and signal transduction system. Int. Immunopharmacol. 1:625–635. [DOI] [PubMed] [Google Scholar]
- 6.Doyle, S., S. Vaidya, R. O'Connell, H. Dadgostar, P. Dempsey, T. Wu, G. Rao, R. Sun, M. Haberland, R. Modlin, and G. Cheng. 2002. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 17:251–263. [DOI] [PubMed] [Google Scholar]
- 7.Gao, J.J., V. Diesl, T. Wittmann, D.C. Morrison, J.L. Ryan, S.N. Vogel, and M.T. Follettie. 2002. Regulation of gene expression in mouse macrophages stimulated with bacterial CpG-DNA and lipopolysaccharide. J. Leukoc. Biol. 72:1234–1245. [PubMed] [Google Scholar]
- 8.Suzuki, T., S. Hashimoto, N. Toyoda, S. Nagai, N. Yamazaki, H.-Y. Dong, J. Sakai, T. Yamashita, T. Nukiwa, and K. Matsushima. 2000. Comprehensive gene expression profile of LPS-stimulated human monocytes by SAGE. Blood. 96:2584–2591. [PubMed] [Google Scholar]
- 9.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 11:115–122. [DOI] [PubMed] [Google Scholar]
- 10.Netea, M.G., C.A. Van Der Graaf, A.G. Vonk, I. Verschueren, J.W. Van Der Meer, and B.J. Kullberg. 2002. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J. Infect. Dis. 185:1483–1489. [DOI] [PubMed] [Google Scholar]
- 11.Thoma-Uszynski, S., S. Stenger, O. Takeuchi, M.T. Ochoa, M. Engele, P.A. Sieling, P.F. Barnes, M. Rollinghoff, P.L. Bolcskei, M. Wagner, et al. 2001. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 291:1544–1547. [DOI] [PubMed] [Google Scholar]
- 12.Utaisincharoen, P., W. Kespichayawattana, N. Anuntagool, P. Chaisuriya, S. Pichyangkul, A.M. Krieg, and S. Sirisinha. 2003. CpG ODN enhances uptake of bacteria by mouse macrophages. Clin. Exp. Immunol. 132:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalpke, A.H., M.K. Schafer, M. Frey, S. Zimmermann, J. Tebbe, E. Weihe, and K. Heeg. 2002. Immunostimulatory CpG-DNA activates murine microglia. J. Immunol. 168:4854–4863. [DOI] [PubMed] [Google Scholar]
- 14.Souza, M.C., M. Correa, S.R. Almeida, J.D. Lopes, and Z.P. Camargo. 2001. Immunostimulatory DNA from Paracoccidioides brasiliensis acts as T-helper 1 promoter in susceptible mice. Scand. J. Immunol. 54:348–356. [DOI] [PubMed] [Google Scholar]
- 15.Muller, U., U. Steinhoff, L.F. Reis, S. Hemmi, J. Pavlovic, R.M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science. 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 16.Doyle, S.E., R. O'Connell, S.A. Vaidya, E.K. Chow, K. Yee, and G. Cheng. 2003. Toll-like receptor 3 mediates a more potent antiviral response than toll-like receptor 4. J. Immunol. 170:3565–3571. [DOI] [PubMed] [Google Scholar]
- 17.Lee, H., H. Dadgostar, Q. Cheng, J. Shu, and G. Cheng. 1999. NF-kappaB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc. Natl. Acad. Sci. USA. 96:9136–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown, E.J., and W.A. Frazier. 2001. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 11:130–135. [DOI] [PubMed] [Google Scholar]
- 19.Krieser, R.J., and K. White. 2002. Engulfment mechanism of apoptotic cells. Curr. Opin. Cell Biol. 14:734–738. [DOI] [PubMed] [Google Scholar]
- 20.Febbraio, M., D.P. Hajjar, and R.L. Silverstein. 2001. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Invest. 108:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Underhill, D.M., and A. Ozinsky. 2002. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 20:825–852. [DOI] [PubMed] [Google Scholar]
- 22.García-García, E., and C. Rosales. 2002. Signal transduction during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 72:1092–1108. [PubMed] [Google Scholar]
- 23.Peiser, L., S. Mukhopadhyay, and S. Gordon. 2002. Scavenger receptors in innate immunity. Curr. Opin. Immunol. 14:123–128. [DOI] [PubMed] [Google Scholar]
- 24.Peiser, L., and S. Gordon. 2001. The function of scavenger receptors expressed by macrophages and their role in the regulation of inflammation. Microbes Infect. 3:149–159. [DOI] [PubMed] [Google Scholar]
- 25.Dempsey, P.W., S.A. Vaidya, and G. Cheng. 2003. The art of war: innate and adaptive immune responses. Cell. Mol. Life Sci. 60:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeda, K., and S. Akira. 2003. Toll receptors and pathogen resistance. Cell. Microbiol. 5:143–153. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki, N., S. Suzuki, G.S. Duncan, D.G. Millar, T. Wada, C. Mirtsos, H. Takada, A. Wakeham, A. Itie, S. Li, et al. 2002. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 416:750–756. [DOI] [PubMed] [Google Scholar]
- 28.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 29.Peiser, L., P.J. Gough, T. Kodama, and S. Gordon. 2000. Macrophage class A scavenger receptor-mediated phagocytosis of Escherichia coli: role of cell heterogeneity, microbial strain, and culture conditions in vitro. Infect. Immun. 68:1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Villiers, W.J., I.P. Fraser, D.A. Hughes, A.G. Doyle, and S. Gordon. 1994. Macrophage-colony-stimulating factor selectively enhances macrophage scavenger receptor expression and function. J. Exp. Med. 180:705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas, C.A., Y. Li, T. Kodama, H. Suzuki, S.C. Silverstein, and J. El Khoury. 2000. Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J. Exp. Med. 191:147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darzynkiewicz, Z., E. Bedner, X. Li, W. Gorczyca, and M.R. Melamed. 1999. Laser-scanning cytometry: a new instrumentation with many applications. Exp. Cell Res. 249:1–12. [DOI] [PubMed] [Google Scholar]
- 33.Hamon, Y., C. Broccardo, O. Chambenoit, M.F. Luciani, F. Toti, S. Chaslin, J.M. Freyssinet, P.F. Devaux, J. McNeish, D. Marguet, and G. Chimini. 2000. ABC1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat. Cell Biol. 2:399–406. [DOI] [PubMed] [Google Scholar]
- 34.Le Cabec, V., S. Carreno, A. Moisand, C. Bordier, and I. Maridonneau-Parini. 2002. Complement receptor 3 (CD11b/ CD18) mediates type I and type II phagocytosis during nonopsonic and opsonic phagocytosis, respectively. J. Immunol. 169:2003–2009. [DOI] [PubMed] [Google Scholar]
- 35.Takizawa, F., S. Tsuji, and S. Nagasawa. 1996. Enhancement of macrophage phagocytosis upon iC3b deposition on apoptotic cells. FEBS Lett. 397:269–272. [DOI] [PubMed] [Google Scholar]
- 36.van der Laan, L.J., S.R. Ruuls, K.S. Weber, I.J. Lodder, E.A. Dopp, and C.D. Dijkstra. 1996. Macrophage phagocytosis of myelin in vitro determined by flow cytometry: phagocytosis is mediated by CR3 and induces production of tumor necrosis factor-alpha and nitric oxide. J. Neuroimmunol. 70:145–152. [DOI] [PubMed] [Google Scholar]
- 37.Saraiva, E.M., A.F. Andrade, and W. de Souza. 1987. Involvement of the macrophage mannose-6-phosphate receptor in the recognition of Leishmania mexicana amazonensis. Parasitol. Res. 73:411–416. [DOI] [PubMed] [Google Scholar]
- 38.Syme, R.M., J.C. Spurrell, E.K. Amankwah, F.H. Green, and C.H. Mody. 2002. Primary dendritic cells phagocytose Cryptococcus neoformans via mannose receptors and Fcgamma receptor II for presentation to T lymphocytes. Infect. Immun. 70:5972–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weighardt, H., C. Feterowski, M. Veit, M. Rump, H. Wagner, and B. Holzmann. 2000. Increased resistance against acute polymicrobial sepsis in mice challenged with immunostimulatory CpG oligodeoxynucleotides is related to an enhanced innate effector cell response. J. Immunol. 165:4537–4543. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi, O., K. Hoshino, and S. Akira. 2000. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 165:5392–5396. [DOI] [PubMed] [Google Scholar]
- 41.Picard, C., A. Puel, M. Bonnet, C.L. Ku, J. Bustamante, K. Yang, C. Soudais, S. Dupuis, J. Feinberg, C. Fieschi, et al. 2003. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 299:2076–2079. [DOI] [PubMed] [Google Scholar]
- 42.Bochud, P.Y., T.R. Hawn, and A. Aderem. 2003. Cutting edge: a toll-like receptor 2 polymorphism that is associated with lepromatous leprosy is unable to mediate mycobacterial signaling. J. Immunol. 170:3451–3454. [DOI] [PubMed] [Google Scholar]
- 43.Lorenz, E., J.P. Mira, K.L. Cornish, N.C. Arbour, and D.A. Schwartz. 2000. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect. Immun. 68:6398–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang, T.J., and G.T. Chae. 2001. Detection of toll-like receptor 2 (TLR2) mutation in the lepromatous leprosy patients. FEMS Immunol. Med. Microbiol. 31:53–58. [DOI] [PubMed] [Google Scholar]