Heterogeneous Vascular Dependence of Tumor Cell Populations (original) (raw)
Abstract
Cells within a tumor are highly heterogeneous with respect to a wide range of genotypic and phenotypic characteristics. The latter include such properties as growth, survival, invasion, and metastasis. We asked whether the degree to which individual tumor cells rely on a tumor’s vasculature might also be heterogeneous. By adapting an intravital Hoechst 33342 staining technique, we labeled and isolated tumor cells based on their relative proximity to perfused vessels. Because tumor regions distal to the vasculature are likely hypoxic, we examined cells deficient for hypoxia-inducible factor-1α (HIF-1α), a transcription factor that has been shown to mediate hypoxia-induced responses, including apoptosis. Despite reduced vascularization in HIF-1α−/− embryonic stem cell-derived tumors, their growth in vivo was found to be accelerated relative to HIF-1α+/+ tumor counterparts. We hypothesized that this paradoxical observation is because of decreased apoptotic rate, resulting in diminished vascular dependence of HIF-1α−/− cells. Analysis of heterogeneous tumors established from mixtures of HIF-1α+/+ with HIF-1α−/− cells revealed that the proportion of cells expressing wild-type HIF-1α was increased in perivascular areas and decreased in distal tumor regions. Thus, cells expressing HIF-1α were found to be highly dependent on proximity to blood vessels for their growth and survival in vivo, whereas cells that had lost HIF-1α expression were much less so. Heterogeneity in angiogenesis dependence was also observed among cell subpopulations isolated from human melanoma xenografts. This potential for selection of less vascular-dependent tumor cell variants throughout the course of disease progression may have important implications for the long-term efficacy of anti-angiogenic therapy.
A significant potential advantage of treating tumors using anti-angiogenic therapy is the possibility of circumventing the problem of acquired drug resistance. 1-3 This is based on the relative genetic stability of the cellular target of such therapy—the normal host endothelial cell of newly formed tumor neovasculature. 1 In contrast, malignant tumor cells have a far greater ability to mutate rapidly and give rise to variants having heritable drug-resistant phenotypes. 1,2 An interesting clinical example of the possibility of anti-angiogenic resistance-free therapy is the chronic treatment of life-threatening hemangiomas 4 or giant cell tumors of the mandible 5 using daily low-dose interferon α; such treatment can result in very gradual but eventually complete regression of such tumors, without any evidence of relapse. Similarly, chronic, intermittent treatment of various transplantable mouse tumors with endostatin is not compromised by manifestation of acquired resistance to this direct-acting anti-angiogenic drug. 3
There are reasons, however, to anticipate that certain types of anti-angiogenic therapy might be associated with some degree of acquired resistance. 6 For example, given the redundancy of pro-angiogenic growth factors produced by tumor cells, targeting only one such growth factor should, in theory, lead to variants that can sustain an angiogenic response by producing alternative angiogenesis stimulators. This may explain why long-term treatment of transplanted tumors in mice with antibodies to the flk-1 vascular endothelial growth factor (VEGF) (type 2) receptor can lead to eventual relapse after a period of dormant growth. 7 It is possible that variants having low-grade resistance to direct-acting angiogenesis inhibitors may emerge with chronic treatment as well. Thus, continuous treatment using low doses of a chemotherapeutic drug used as an anti-angiogenic agent, eg, cylophosphamide 8 or vinblastine 7 can result in eventual tumor relapse. Such relapses can be indefinitely delayed by the use of more potent combination anti-angiogenic therapy using an additional anti-angiogenic drug such as TNP-470 8 or monoclonal flk-1 antibodies along with the chemotherapeutic drug. 7 Resistance to the single treatment may be because of epigenetic changes induced in the endothelial cells or, conceivably, to the selection of tumor cell variants that have a reduced vascular dependence for survival. The latter may occur as a consequence of tumor cell heterogeneity, the subject of this study.
It is well known that cells varying in such properties as growth, invasiveness, metastatic potential, and drug resistance can be isolated from single tumors. 9-14 In theory, it is clearly possible that this tumor cell heterogeneity may apply to angiogenesis, ie, variant populations may also differ in their expression of angiogenic factors, and consequently in their ability to induce angiogenesis. If this were the case, variability in blood vessel density within different regions of a tumor might be expected. 15 Indeed, such areas of high vessel density, (so called “vascular hot spots”) are characteristic of most tumors. 16,17 Although it is clear that acquisition of the angiogenic phenotype is an important step in tumor progression, 18 what has not generally been considered is the possibility that the relative dependence of tumor cells on angiogenesis itself may also become subject to the same types of selective pressures. Throughout time, the outgrowth of subpopulations of less vascular-dependent or angiogenesis-dependent malignant cells could potentially occur, particularly in the context of certain types of long-term anti-angiogenic therapy, 6 with selection of such traits as an increased capacity to survive in nutrient- or oxygen-deprived areas of a tumor. Indeed, there is evidence that hypoxia provides a selection mechanism for cells with diminished susceptibility to apoptosis, as it has been shown that small numbers of cells harboring mutations in the p53 tumor suppressor gene overtake wild-type cells under hypoxic growth conditions. 19
An important component of the hypoxic response is the activation of gene expression mediated by hypoxia-inducible factor (HIF-1). Inactivation of the HIF-1α gene in embryonic stem cells by Carmeliet and colleagues 20 has revealed a new role for this transcription factor in the hypoxic regulation of cell growth and apoptosis. Although apoptosis was increased in HIF-1α+/+ cells on exposure to hypoxia, HIF-1α−/− cells were found to be relatively hypoxia-resistant. Striking differences were found in tumors derived on injection of these ES cells into mice. Compared to HIF-1α+/+ tumors, HIF-1α−/− tumors were poorly vascularized, deficient in large vessels, and contained more hypoxic areas. Despite these angiogenic defects, the growth of HIF-1α−/− tumors was actually accelerated relative to the HIF-1α+/+ tumors, because of a decrease in hypoxia-induced tumor cell apoptosis. One possible explanation for this paradoxical coupling of increased tumor growth rate with reduced vascularization in HIF-1α−/− tumors might be that the loss of HIF-1α in these cells renders them less vascular-dependent.
To investigate whether HIF-1α loss might decrease vascular dependence in these cells, and by extension allow us to study the possibility of variable vascular dependence of tumor cell subpopulations in a more general way, we established tumors from mixtures of HIF-1α+/+ and HIF-1α−/− cells, and examined the distribution of each population relative to perfused blood vessels. Our results demonstrated that HIF-1α+/+ cells predominantly and preferentially localized to areas immediately surrounding perfused blood vessels whereas HIF-1α−/− cells were located more distal to such vessels. We also studied heterogeneity in vascular dependence of human tumors by selecting human melanoma variants from xenografts based on their relative proximity to perfused vessels. Our results suggest that cells within a single tumor may be heterogeneous with respect to angiogenesis or blood vessel dependence, and these differences may arise from genetic changes occurring during malignant tumor progression.
Materials and Methods
Cell Lines and Culture Conditions
The HIF-1α−/− embryonic stem (ES) cells generated by homologous recombination, and HIF-1α+/+ ES cells harboring a randomly integrated targeting vector have been previously characterized. 20 The yellow fluorescent protein (YFP) and _lacZ_-tagged HIF-1 α+/+ ES cells (YC5 and C16, respectively) were derived by one of us. The C16 cells are functionally normal, whereas harboring a heterozygous deletion of the FGFR2 gene. All ES cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 15% fetal bovine serum (HyClone, Logan, UT), 0.1 mmol/L nonessential amino acids, 1 mmol/L sodium pyruvate, 2 mmol/L l-glutamine, 50 μg/ml each penicillin and streptomycin (all Life Technologies, Inc., Rockville, MD), 100 μmol/L β-mercaptoethanol (Sigma Chemical Co., St. Louis, MO) and leukemia inhibitory factor. Human melanoma cells were grown in RPMI 1640 medium (Life Technologies, Inc.) with 5% fetal bovine serum, and bovine aortic endothelial cells were cultured in Dulbecco’s modified Eagle’s medium (Life Technologies, Inc.) with 5% fetal bovine serum.
Establishment of Heterogeneous HIF-1α−/− and HIF-1α+/+ Tumors
ES cell-derived tumors (teratomas) were produced by subcutaneous injection of 5 × 10 6 ES cells into athymic nude (nu/nu) mice (Charles River, Wilmington, MA). For heterogeneous tumors, equal numbers of HIF-1α−/− and HIF-1α+/+ (untagged, YFP-tagged, or _lacZ_-tagged) ES cells, totaling 5 × 10 6 cells, were mixed before injection. Growth of tumors was monitored until an estimated volume of at least 1,000 mm 3 was reached, as calculated using the standard formula (length × width 2 × 0.5).
Labeling of Tumor Cells as a Function of Distance from Vasculature
Tumor-bearing mice were injected via the tail vein with 200 μl of 10 mg/kg Hoechst 33342 dye (Sigma Chemical Co.), which was allowed to circulate and diffuse for 5 minutes before tumor removal. Under these conditions, a reproducible perivascular tumor cell-labeling gradient was achieved, as described elsewhere. 21,22 Tumor tissue was disaggregated with an enzyme cocktail containing collagenase type III (Worthington, Lakewood, NJ), hyaluronidase (Sigma Chemical Co.), and collagenase type IV (Sigma Chemical Co.), washed several times, and resuspended in phosphate-buffered saline (PBS) to produce a single cell suspension suitable for fluorescence-activated cell sorting (FACS) analysis.
Analysis of HIF-1α Genotype in Proximal and Distal Tumor Cell Subpopulations
For HIF-1α−/− and untagged HIF-1α+/+ mixed tumors, the tumor cell suspension was sorted based on Hoechst 33342 fluorescence intensity on an EPICS Elite V flow cytometer (Coulter Electronics, Hialeah, FL). Gates were set to collect the 5% most brightly stained cells, designated “proximal,” and the 5% least brightly stained cells, designated “distal,” based on their relative proximity to tumor blood vessels. Genomic DNA was isolated from the two cell subpopulations, and the composition of each, with respect to HIF-1α genotype, was determined by polymerase chain reaction using _Eco_RI-digested DNA and primers flanking the deletion site. The following primers were used: HIF700, 5′-CAAGCATTCTTAAATGTGGAGC-TATCT-3′; HIF960, 5′-TTGTGTTGGGGCAGTACTGGAAAGATG-3′; NEO187, 5′-CGAAGGGGCCACCAAAGAACGGAGCCG-3′. Amplification of the wild-type allele using HIF960 and HIF700 yielded a product of 270 bp, whereas a 340-bp product was obtained by amplification of the HIF-1α−/− allele with primers HIF960 and NEO187. Primers flanking the neo sequence were used as an internal control, producing a product of 426 bp, and the sequences were as follows: H1, 5′-TCCACCATGATATTCGGCAA-3′; H4, 5′-TGAATGAACTGCAGGACGAG-3′.
For mixed tumors composed of HIF-1α−/− and YFP-labeled HIF-1α+/+ ES cells, the HIF-1α genotype was determined directly by flow cytometric analysis of the tumor cell suspension. Using a FACStar Plus flow cytometer (Becton Dickinson, Mountain View, CA) equipped with a dual laser, cells could be analyzed simultaneously for Hoechst and YFP fluorescence intensity. The percentage of YFP-positive HIF-1α+/+ cells among the 5% most brightly (proximal) and 5% most dimly (distal) Hoechst-stained cells could be ascertained from the data.
In Vivo Selection of Tumor Cell Subpopulations for Varying Vascular Dependence
Tumors were established from the WM239A human melanoma cell line by intradermal injection of 10 6 cells into athymic nude mice. After tumor growth, mice were injected with Hoechst 33342 dye, as before, followed by euthanasia and dissection of the tumor under sterile conditions. Pieces of tumor were saved for histological assessment, and the remaining tissue digested as described. The cell suspension was sorted on an EPICS V flow cytometer (Coulter Electronics), equipped with a UV laser. Windows were set to collect the 15% most and least brightly stained cells (proximal and distal cells, respectively). Sorted cells were plated and grown in vitro, then re-injected into mice. This sequence of tumor growth, cell sorting, and establishment of sorted cells in culture constituted one round of in vivo vascular selection. The procedure was repeated for a total of four rounds to evolve the proximal and distal cell lines, respectively, selected to be close to, or distant from perfused tumor vasculature.
Histology and Immunostaining
For lacZ staining of ES cell-derived tumor tissues, specimens were fixed in lacZ fixative (0.2% glutaraldehyde, 50 mmol/L EGTA, pH 7.3, 100 mmol/L MgCl2 in 0.1 mol/L sodium phosphate, pH 7.3) for 4 hours on ice with shaking. Samples were washed in PBS, and cryoprotected in 15% sucrose in PBS followed by 30% sucrose in PBS at 4°C. Tissue was placed in Tissue-Tek OCT (Sakura) at 4°C for at least 1 hour before freezing over dry ice. Cryosections of 10 μm were cut and placed on silanized slides.
Slides were fixed in cold PBS containing 0.2% glutaraldehyde, then washed in lacZ wash buffer (2 mmol/L MgCl2, 0.01% sodium deoxycholate, 0.02% Nonidet-P40 in 0.1 mol/L sodium phosphate, pH 7.3). Staining was performed in lacZ stain solution (0.5 mg/ml X-gal, 5 mmol/L potassium ferrocyanide, and 5 mmol/L potassium ferricyanide in lacZ wash buffer) at 37°C for 4 hours, protected from light. To identify blood vessels, slides were washed in PBS after lacZ staining, and stained with a rat anti-CD31 antibody (Pharmingen, La Jolla, CA) used at a dilution of 1:200. Immunoreactivity was visualized by incubation with 3-amino-9-ethylcarbazole (Zymed, South San Francisco, CA).
For the vascular selection experiments, tumors produced by cells from the final round of vascular selection, and tumors produced by the original unsorted WM239A cells were characterized as follows: tumor tissue was fixed in modified Carnoy’s (60% ethanol, 30% chloroform, 10% acetic acid) or 3% paraformaldehyde. Cryostat sections (6 to 8 μm) were cut and viewed under UV epifluorescence to visualize Hoechst staining. Paraffin sections (6 μm) were stained with an antibody to Von Willebrand Factor (DAKO, Carpinteria, CA) to detect vascular endothelium. Immunocomplexes were visualized by incubation with diaminobenzidine tetrahydrochloride (Boehringer Mannheim, Indianapolis, IN) or 3-amino-9-ethylcarbazole, and slides were lightly counterstained with hematoxylin.
Analysis of WM239 Melanoma Cell Lines and Tumors
Vascular density was assessed in tumor sections immunostained for von Willebrand Factor according to established methods. 23 To assess tumor cell apoptosis, we quantified the number of apoptotic bodies in 10 nonnecrotic oil immersion fields of hematoxylin and eosin stained slides. Apoptotic bodies were characterized as highly pkynotic nuclear fragments, associated with shrunken cells.
The angiogenic activity of WM239 parent, proximal-4 (P-4), and distal-4 (D-4) cells were quantified by examining the effects of medium conditioned by these cells for 24 hours, and control medium, on bovine aortic endothelial cell proliferation in vivo. Briefly, 2.5 × 10 4 bovine aortic endothelial cells were plated into 12-well plates in complete media, and after 16 hours replaced with test media. Cells were incubated for an additional 72 hours, followed by cell counting.
Results
In Vivo Labeling of Perivascular Tumor Cells with Hoechst 33342
To distinguish cells located in perivascular areas from cells located in more hypoxic regions of a tumor, we adapted a technique previously used to analyze drug response of cells within solid tumors and spheroids. 21,24 The nontoxic, fluorescent, DNA-binding dye Hoechst 33342 was injected intravenously into tumor-bearing mice at nonsaturating concentrations. By allowing the dye to circulate for a short time before tumor removal, cells were fluorescently labeled based on their relative distance from perfused blood vessels (Figure 1) ▶ . Cells immediately surrounding tumor vasculature were highly fluorescent, whereas cells located farther away did not fluoresce, or had a much lower level of fluorescence intensity. After disaggregation of the tumor cells into a single cell suspension, the sample was analyzed for Hoechst fluorescence by flow cytometry and the cells showing the 5% highest and lowest Hoechst fluorescence intensities were designated proximal or distal relative to perfused vasculature.
Figure 1.

Identification of perivascular tumor cells by intravital Hoechst 33342 staining and flow cytometry. A: Cells were labeled based on blood vessel proximity by intravenous injection and circulation of nonsaturating concentrations of the DNA-binding fluorescent dye. B: Cryosection of a WM239 melanoma tumor shows Hoechst-stained islands of cells (top) clustered around CD31-positive blood vessels (bottom), with nuclear brightness diminishing in cells located distal from these vessels. The cells showing the 5% highest and lowest fluorescence intensities were designated “proximal” or “distal” relative to perfused vasculature, and could be sorted by FACS or analyzed directly for YFP fluorescence.
Differential Perivascular Distribution of HIF-1α+/+ and HIF-1α−/− Cells in Mixed Tumors
Once the proximal and distal cell populations in the tumor were identified, the proportion of HIF-1α+/+ and HIF-1α−/− cells in these populations was determined. First, tumors generated by injecting a mixture of YFP-labeled wild-type ES cells with untagged HIF-1α−/− cells were analyzed. By studying the tumor cell suspensions simultaneously for Hoechst and YFP fluorescence, the proportion of HIF-1α+/+ YFP-fluorescing cells in the proximal and distal population was determined directly. In every tumor examined, it was found that on removal ∼4 weeks after injection of cells, the overall proportion of HIF-1α+/+ cells was decreased. Although the original mixture injected was composed of wild-type and HIF-1α−/− cells in equal numbers, the final percentage of HIF-1α+/+ cells was diminished to an average of only 12%, and in some tumors was as low as 4 to 5%. As expected, analysis of control tumors derived from YFP-tagged HIF-1α+/+ cells alone showed some loss of YFP expression because of the tumor cell isolation protocol. At completion of the experiment, the percentage of YFP-positive cells was decreased from 100 to 76%. However, even if a similar reduction in YFP expression in mixed tumors is taken into account, the overall decrease in YFP-tagged HIF-1α+/+ cells in mixed tumors from 50 to 12% (more than fourfold) remains statistically significant (_t_-test, P < 0.005).
There were also dramatic differences in the distribution of HIF-1α+/+ and HIF-1α−/− cells relative to blood vessels. HIF-1α+/+ cells accounted for only 4% of distal cells, compared to 29% of the proximal cell population (Figure 2A) ▶ . In contrast, YFP-positive cells in control tumors were not preferentially localized to the perivascular tumor regions: whereas the perivascular tumor cell population was 77.6% YFP-positive, the distal population was 74.8% YFP-positive, a difference that is not statistically significant. The significant sevenfold increase (P < 0.002) of HIF-1α+/+ cells in proximal regions of mixed tumors indicates that HIF-1α+/+ cells localize primarily around perfused vessels, and suggests that throughout time, these cells might be dependent on, and/or selected by blood vessel proximity. In contrast, cells lacking HIF-1α seem to be much less dependent on the vasculature.
Figure 2.

The proportion of HIF-1α+/+ cells is decreased in tumor regions distal from perfused blood vessels. A: HIF-1α+/+ cells were identified by flow cytometric analysis of YFP-fluorescence in mixed tumors generated from YFP-tagged wild-type and untagged HIF-1α−/− ES cells. Although YFP-positive (HIF-1α+/+ cells) constituted 29% of the proximal cell population, this was decreased to only 4% in distal tumor areas (*, P < 0.002) In contrast, YFP-positive cells in control tumors derived from YFP-tagged HIF-1α+/+ cells alone showed no significant differences between proximal and distal regions (**, not significant). Results are displayed as mean values ± SD. B: Polymerase chain reaction analysis of HIF-1a status in tumor cell populations. Amplification of genomic DNA yields distinct products for HIF-1α+/+ and HIF-1α−/− cells (lanes 1 and 2). Lanes 3 to 5 show the presence of both HIF-1α+/+ and HIF-1α−/− cells in mixed tumors before sorting. Lanes 6 to 9 show the composition of distal and proximal cell populations of two tumors after sorting by Hoechst fluorescence. HIF-1α+/+ cells are localized proximal to perfused blood vessels but are rare in more distal tumor regions. As a control, neomycin resistance gene sequences were co-amplified in each reaction.
Disappearance of HIF-1α+/+ Genotype in Tumor Regions Distant from the Vasculature
A more direct analysis of the predominant genotype within proximal and distal cell populations was undertaken, using polymerase chain reaction to distinguish between the wild-type and disrupted HIF-1α gene. This analysis could be expected to be unaffected by fluctuations in gene expression and cell viability during tumor growth and experimental procedures. As shown in Figure 2B ▶ , amplification of the wild-type gene yields a product of 270 bp whereas a larger product of 340 bp is amplified from HIF-1α−/− DNA. As expected, both bands were detected in DNA isolated from unsorted mixed tumor cells, indicating the presence of both HIF-1α+/+ and HIF-1α−/− cells. However, on sorting of these cells into proximal and distal populations based on Hoechst fluorescence, it was found that whereas both bands were detected in proximal cell DNA, the HIF-1α+/+-specific band was absent or much weaker in the distal cell DNA. These results again demonstrate that HIF-1α+/+ cells localize predominantly to regions surrounding perfused blood vessels and thus seem to be more vessel-dependent than HIF-1α−/− cells, which were prevalent in both cell populations.
To directly visualize the differential perivascular distribution of HIF-1α+/+ and HIF-1α−/− cells in mixed tumors in a Hoechst-independent manner, _lacZ_-tagged HIF-1α+/+ cells were used. The localization of these wild-type cells relative to tumor blood vessels was determined by histochemical staining for lacZ expression accompanied by immunostaining for CD31 as a marker of the tumor vasculature. As shown in Figure 3 ▶ , blue _lacZ_-positive HIF-1α+/+ cells were observed scattered throughout the tumor section, with decreased numbers relative to unstained HIF-1α−/− cells. This is expected, as a reduction of their overall contribution to only 12% of tumor cells was demonstrated in earlier experiments using the YFP-tagged HIF-1α+/+ cells. Furthermore, although present throughout the tumor, the HIF-1α+/+ cells showed a tendency to cluster around vascular structures, directly confirming the results that were obtained previously with the differentially Hoechst-labeled disaggregated tumor cells.
Figure 3.
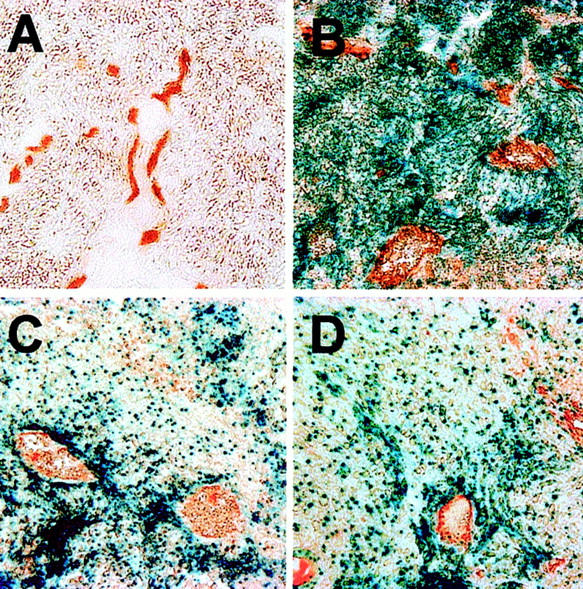
Histochemical β-gal staining and CD31 immunostaining. Tumor sections were stained for lacZ expression to identify location of _lacZ_-tagged HIF-1α+/+ cells relative to CD31-positive blood vessels. A: HIF-1α−/− tumor. B: _LacZ_-positive HIF-1α+/+ tumor. C and D: Staining of mixed tumors shows clustering of blue _lacZ_-positive HIF-1α+/+ cells around tumor vasculature.
Thus the observation that HIF-1α+/+ cells localize to areas proximal to the vasculature strongly suggests that these cells, which are more hypoxia-sensitive, seem to have an increased dependence on blood vessels for survival. Conversely, cells that have lost HIF-1α, and which are known to exhibit a decreased susceptibility to hypoxia-induced growth inhibition and apoptosis, 20 seem to be much less vascular-dependent.
Vascular Selection of Human Melanoma Xenografts
To examine whether heterogeneity in vascular dependence might exist in the more relevant context of a human tumor, a reverse experiment was performed. An unbiased vascular selection strategy was devised to segregate cells in human tumor xenografts based on their proximity to perfused microvasculature. The human melanoma cell line WM239A was chosen for this selection, and proximal and distal cell populations were sorted from xenografted tumors based on differential Hoechst 33342 fluorescence as described earlier. However, the sorted cells were passaged in culture, followed by re-injection into mice to generate tumors, whereupon tumors were once again removed and sorted. To establish the proximal cell line, only sorted proximal cells were retained and propagated, and to derive the distal cell line, only distal cells were used. This sequence of tumor growth, cell sorting, and establishment of sorted cells in culture was repeated for a total of four rounds, to produce the final proximal and distal cell lines (Figure 4) ▶ . This process allowed progressive enrichment for tumor cell subpopulations derived from perivascular or hypoxic areas of the tumor.
Figure 4.

Schematic of vascular selection protocol. Human melanoma tumor xenografts were established in mice, then cells selected based on differential Hoechst fluorescence. Sorted cells were expanded in vitro, and re-injected for a total of four rounds of vascular selection from the original parent WM239A.
When the resulting cell lines were re-injected into mice, dramatic differences in tumor take and growth were observed (Figure 5, A and B) ▶ . Distal cell populations showed enhanced tumor growth relative to the parental WM239A cells after two rounds of vascular selection, which did not increase with subsequent iterations. These cells showed no differences in tumor take compared to parental cells. In contrast, proximal cells showed greatly reduced growth rates in vivo. By three rounds of selection, this reduced growth was significantly different from parental cells, and continued to decrease with subsequent selection. Tumor take also decreased; by the fourth round of selection, only one of five mice had tumors after 2 weeks, and two of five mice did not develop detectable tumors even after 20 weeks. There was no difference in growth rates of any of these cell lines under monolayer culture conditions.
Figure 5.
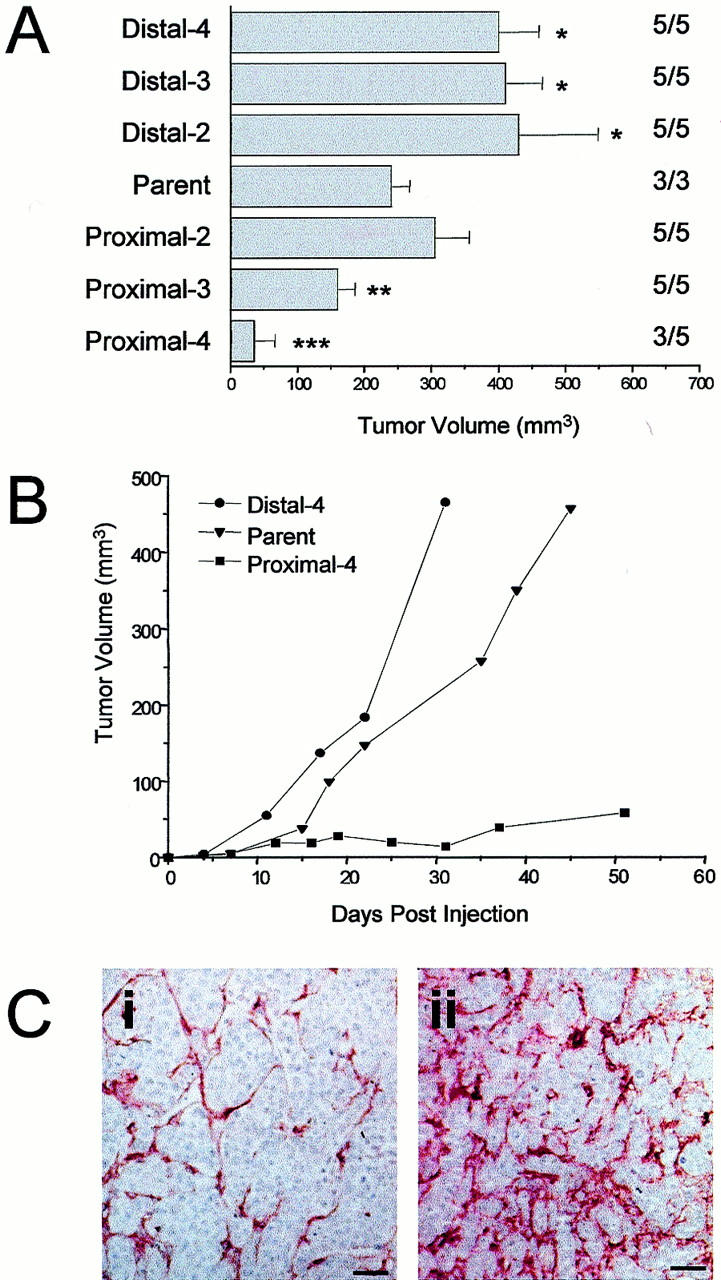
In vivo growth of WM239A melanoma cell lines. A: Tumor volume was measured 28 days after intradermal injection of 10 6 cells. Values to the right indicate number of mice with tumors/number of mice injected. After two rounds of vascular selection, distal cells produced tumors that grew significantly faster in vivo than parental cells (*, P < 0.05). By three rounds of selection, the growth of tumors from proximal-3 (P-3) cells was significantly reduced from parental (**, P < 0.05). An additional round of vascular selections resulted in proximal-4 (P-4) cells with reduced tumor take (60%) and significant slowest growth (***, P < 0.05). B: Growth of tumors produced by injection of parental WM239A, P-4, and D-4 cells. C: Tumor sections immunostained for von Willebrand Factor. Blood vessels are relatively sparse in D-4 tumors (i), compared to P-4 tumors (ii), where an extensive vascular network is visible. Scale bar, 50 μm.
Staining for the endothelial cell marker, von Willebrand Factor, revealed striking differences in the vascular patterns between tumors originating from proximal (P-4) and distal (D-4) tumor cell populations (Figure 5C) ▶ . Quantification of blood vessels unexpectedly indicated significantly higher numbers per hot spot in tumors derived from the less tumorigenic P-4 cells than in more aggressive tumors from D-4 cells (Table 1) ▶ . Tumors from parental WM239 cells had intermediate vascular densities. These differences in blood vessel density may be reflective of either increase in angiogenic proficiency of P-4 cells or their inability to proliferate or survive distant from blood vessels. Our results support the latter interpretation, as analysis of apoptosis in these tumors showed significantly more apoptotic cells in P-4 tumor sections than in D-4 sections (P < 0.05, Table 1 ▶ ). Furthermore, we examined the effects of medium conditioned by WM239 parent, P-4, and D-4 cells on the proliferation of bovine aortic endothelial cells in vitro, and we observed no significant differences between the cell lines (Table 1) ▶ . There was also no significant difference in VEGF protein secretion by these cells under either normoxic or hypoxic conditions as measured by enzyme-linked immunosorbent assay (data not shown). As P-4 cells were therefore not found to be more angiogenic than their D-4 counterparts, we interpret the differences in vascular density as an indirect measure of differential vascular dependence between the tumor cell lines.
Table 1.
Vessel Density Is Higher in P-4 Tumors than Parent WM239 or D-4 Tumors; However, P-4 Tumors Are More Apoptotic, and P-4 Cells Are Not More Angiogenic in Vitro
| Cell line | Tumor vascular density | Tumor apoptotic index | BAEC proliferation in response to serum-free CM (mean cell number × 105) |
|---|---|---|---|
| Parent WM239A | 52 ± 10.5 | 0.67 ± 0.58 | 1.69 ± 0.52 |
| D-4 | 37 ± 8.1* | 0.73 ± 0.25 | 1.62 ± 0.48 |
| P-4 | 69 ± 9.5† | 1.97 ± 0.46† | 1.34 ± 0.37 |
Discussion
Genetic and Epigenetic Determinants of Heterogeneous Vascular Dependence
Tumor cells require blood vessels to supply them with metabolites such as oxygen and glucose. Hypoxic regions are a common and significant feature of tumors, because the disorganized vasculature is often insufficient to meet the needs of a rapidly proliferating tumor mass. Hypoxia occurs in tumor tissue that is >150 to 200 μm away from a functional vessel. 25 In some tumors, this blood vessel dependence may be manifested in the formation of cuffs of viable tumor cells tightly surrounding a single central blood vessel, with areas of necrosis occurring beyond the critical oxygen diffusion distance. However, some of the genetic and epigenetic changes in tumor cells may result from adaptive and selective processes induced by protracted focal ischemia. Hypoxia induces apoptosis in oncogenically transformed cells through a p53-dependent pathway, 26 and thereby selects for tumor cells that have lost functional p53, resulting in increased survival capacity. Furthermore, p53 influences the hypoxic response of cells through regulation of HIF-1α stability. 27 Oncogenic transformation itself may also facilitate adaptation to hypoxia, for instance Ras-transformation and hypoxia synergize to induce VEGF expression. 28 The concept of vascular dependence therefore potentially encompasses a broad range of cellular functions, in particular cell proliferation and cell survival under conditions of metabolic stress such as hypoxia and hypoglycemia, as well as responses to endothelial cell-derived paracrine signals. 19,22
An Experimental Approach to Studying Heterogeneous Vascular Dependence of Tumor Cells and a Possible Genetic Basis of the Phenomenon
It has been previously shown that ES cells genetically disrupted in the HIF-1α locus are relatively resistant to hypoxic stress, indicating a new role for HIF-1α in mediating hypoxia-induced apoptosis, beyond functioning in the homeostatic response to cellular hypoxia. 20 Paradoxically, although tumors derived from HIF-1α−/− cells were found to express low levels of VEGF along with significantly reduced vascular density and function, they nevertheless exhibited an accelerated rate of tumor growth relative to wild-type HIF-1α+/+ cells because of a reduced rate of hypoxia-induced growth arrest and apoptosis. As mentioned above, certain other naturally occurring genetic changes during tumor progression may result in operationally similar outcomes of decreased reliance on oxygen and growth factors, and therefore diminished relative vascular dependence. 6
To examine the possibility that hypoxia response may influence cellular selection within heterogeneous tumor cell populations, tumors were established from mixtures of hypoxia-sensitive HIF-1α+/+ and hypoxia-resistant HIF-1α−/− cells. Tumor cells were then separated as a function of distance from blood vessels by intravenous injection of Hoechst 33342 dye. This procedure yields brightly fluorescent cells proximal to perfused vasculature, and a lower fluorescence intensity in more distal tumor regions. 21,24 These proximal and distal cells were distinguished by FACS, and analyzed to determine the proportions of HIF-1α+/+ and HIF-1α−/− cells in each cell population. Assuming one subpopulation of tumor cells differed from another in terms of vascular/angiogenesis dependence, one would expect to find a nonrandom distribution of these cells within the tumor. In particular, the more vascular-dependent cell type would likely be located to a greater extent in areas surrounding blood vessels than in the more hypoxic and distal regions simply because such cells would have a greater chance to die of hypoxia-induced apoptosis. It should be noted that oxygenation in tumor vessels has been shown to be heterogeneous 29 and hence this argument would apply only to areas surrounding well-perfused capillaries. This is also the most significant limitation of the Hoechst technique, in that only vessels that are perfused at the time of injection are accessible to the dye. Consequently, fluctuations in vessel perfusion throughout time or space might theoretically result in some of the proximal, vascular-dependent cells being caught in the hypoxic zones of the tumor.
Despite these reservations, in the HIF-1α model system, the HIF-1α+/+ (hypoxia-sensitive) cells did indeed show increased perivascular localization compared to the relatively hypoxia-resistant HIF-1α−/− cells. This was shown by three independent methods. First, FACS analysis showed that the proportion of YFP-tagged wild-type (HIF-1α+/+) cells was increased by sevenfold in areas close to perfused vasculature. Analysis of proximal and distal cell DNA by polymerase chain reaction to determine HIF-1α genotype confirmed these results. Although HIF-1α−/− cells were found in both the proximal and distal cell populations, HIF-1α+/+ cells were detected in regions proximal to vessels, but were scarce or even absent in the more distal regions. Second, the nonrandom localization of wild-type cells was also visualized directly by histochemical staining of _lacZ_-tagged HIF-1α+/+ cells in tumor sections, which clearly showed clustering of these cells around the vasculature. Finally, HIF-1α−/− cells showed an overall selective growth advantage in mixed tumors, with their proportion increasing from 50% at the time of cell injection, to almost 90% at the time of tumor removal, consistent with their previously described increased malignant properties. 20 An analogous trend has also been observed in a series of Ras-transformed cell lines differing in malignant potential. 22 Similarly, a more aggressive subline was derived by a vascular selection procedure from WM239 melanoma xenografts, which also contain a less aggressive, blood vessel-associated (ie, dependent) cell population. All of the approaches consistently support the notion of greater malignancy of cells that are able to survive within hypoxic regions of a tumor. Such cells might be considered less angiogenesis-dependent.
The results obtained with the murine ES cell-derived teratomas should not be interpreted as a suggestion that HIF-1α loss itself may be favorable for tumor progression. HIF-1α−/− cells merely represent a phenocopy of hypoxia-resistant tumor cells. In other words, loss of HIF-1α may not be a favored pathway leading to hypoxia-resistant cellular phenotype in human tumors. Indeed, there is growing evidence that the opposite seems to be true, as a recent study showed that HIF-1α is overexpressed in many common human cancers and their metastases. 30 Because HIF-1α seems to be important for the vascularization of tumors, it is likely that its activity may be maintained at a level sufficient for expression of genes such as VEGF, but without induction of apoptosis. However, our experiments demonstrate that in principle, cells harboring certain defined genetic defects that promote their survival under stressful conditions such as hypoxia, show differences in angiogenesis dependence, as indicated by their nonrandom localization relative to tumor blood vessels. The HIF-1α status of the sublines derived from WM239 melanoma was investigated by Western blotting, and no difference in protein expression was detected between P-4 and D-4 cells when cultured in the presence of cobalt chloride to mimic hypoxia (data not shown). However, other elements of HIF-1α signaling (DNA binding, gene transcription) have not been investigated in detail and the role of this mechanism in vascular selection cannot be definitively ruled out. It is also possible that this selection may be driven by more complex sets of mechanisms including negative pressures such as acidification, glucose, or growth factor deprivation in conjunction with hypoxia.
Implications for Anti-Angiogenic Therapy of Tumors
Our results clearly have potentially important implications for anti-angiogenic therapy. First, it might be predicted that anti-angiogenic drugs would preferentially affect tumor cells immediately adjacent to tumor vessels rather than those located in hypoxic regions more distal to such vessels. Indeed, this is exactly what was reported by Bergers and colleagues. 31 Thus, treatment of islet cell pancreatic tumors arising in transgenic oncomice with a combination of angiostatin and endostatin, or TNP-470, also resulted in tumor regressions that were accompanied by apoptosis of tumor cells found in close apposition to tumor capillaries. As noted by Bergers and colleagues, 31 this observation is counterintuitive as one might have expected preferential apoptosis of the hypoxic cells after such a therapeutic intervention. Our results help explain such findings. Second, although tumors will likely never become entirely angiogenesis-independent, the degree to which they rely on blood vessels could conceivably decrease throughout time. Therefore, over the course of long-term anti-angiogenic therapy, tumors may become less responsive to treatment with certain anti-angiogenic drugs, especially when used as single agents. 6 Hence, it may be necessary to increase the intensity of anti-angiogenic treatments, for example, by using combination treatment regimens. We successfully used such a strategy by combining continuous low-dose vinblastine chemotherapy (as an anti-vascular targeting strategy) with anti-flk-1 antibodies. 7 This drug combination did not result in acquired drug resistance, ie, the emergence of angiogenesis-independent tumor cell variants, despite a very long term (eg, 6 months) of continuous treatment of human neuroblastoma xenografts or mouse tumors. 7,8 Similarly, a combination of angiostatin and endostatin is clearly superior to either drug used alone as an anti-angiogenic treatment strategy. 31,32
The success of such combinations as continuous low-dose chemotherapy and anti-flk-1 antibodies may be related to the necessity for neutralizing varioius resistance factors for activated endothelial cells of new blood vessels. Indeed, VEGF itself may be one such resistance factor, 33 eg, by up-regulating anti-apoptosis genes such as bcl-2, A1, XIAP, and survivin in endothelial cells. 34-37
In summary, this study suggests that tumor cell heterogeneity may apply to angiogenesis, ie, cells within human and rodent tumors may differ in their vascular dependence, and such differences could, in principle, arise from genetic alterations occurring during cancer progression, eg, changes that affect the apoptotic response of a tumor cell to hypoxia. Contrary to what is often believed, high blood vessel density may not always be indicative of high angiogenic capacity of a given tumor, but may also, at least in some cases, signify the inability of less malignant tumor cells to thrive beyond certain short distances from their capillaries. A relative loss of vascular dependency among tumor cells may therefore constitute a significant determinant in promoting malignant progression, with possible implications for metastatic potential, as well as having important consequences for the long-term efficacy of certain types of anti-angiogenic therapy.
Acknowledgments
We thank Ms. Gisele Knowles (Hospital for Sick Children, Toronto) and Ms. Juliet Sheldon (Ontario Cancer Institute, Toronto) for their expert technical assistance in flow cytometry; Ms. Shan Man for her assistance with animal work; Ms. Luba Roncari and Mrs. Kanwal Minhas for their assistance with immunostaining; and Ms. Lynda Woodcock and Mrs. Cassandra Cheng for their excellent secretarial assistance.
Footnotes
Address reprint requests to Robert S. Kerbel, Ph.D., Sunnybrook Health Science Centre, Division of Cancer Research, Rm. S-218, Reichmann Research Building, 2075 Bayview Ave., Toronto, Ontario, Canada M4N 3M5. E-mail: robert.kerbel@swchsc.on.ca.
Supported by grants from the Medical Research Council of Canada (Canadian Institutes of Health Research), the National Institutes of Health, United States (CA-41233 to R. S. K.), and the National Cancer Institute of Canada (11162 to B. L. C.). J. L. Y. is the recipient of a Medical Research Council of Canada doctoral research award.
References
- 1.Kerbel RS: Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. BioEssays 1991, 13:31-36 [DOI] [PubMed] [Google Scholar]
- 2.Kerbel RS: Acquired drug resistance driven by tumor cell genetic instability: circumvention by direct acting anti-angiogenic vascular targeting agents. Ehrlich M eds. DNA Alterations in Cancer: Genetic and Epigenetic Changes. 2000, :pp 489-501 Biotechniques Books, Natick [Google Scholar]
- 3.Boehm T, Folkman J, Browder T, O’Reilly MS: Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390:404-407 [DOI] [PubMed] [Google Scholar]
- 4.Ezekowitz RA, Mulliken JB, Folkman J: Interferon alfa-2a therapy for life-threatening hemangiomas of infancy. N Engl J Med 1992, 326:1456-1463 [DOI] [PubMed] [Google Scholar]
- 5.Kaban LB, Mulliken JB, Ezekowitz RA, Phil D, Ebb D, Smith PS, Folkman J: Antiangiogenic therapy of a recurrent giant cell tumor of the mandible with interferon alpha-2a. Pediatrics 1999, 103:1145-1149 [DOI] [PubMed] [Google Scholar]
- 6.Rak J, Kerbel RS: Treating cancer by inhibiting angiogenesis: new hopes and potential pitfalls. Cancer Metastasis Rev 1996, 15:231-236 [DOI] [PubMed] [Google Scholar]
- 7.Klement G, Baruchel S, Rak J, Man S, Clark C, Hicklin D, Bohlen P, Kerbel RS: Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest 2000, 105:R15-R24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Browder T, Butterfield CE, Kraling BM, Marshall B, O’Reilly MS, Folkman J: Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 2000, 60:1878-1886 [PubMed] [Google Scholar]
- 9.Dexter DL, Calabresi P: Intraneoplastic diversity. Biochim Biophys Acta 1982, 695:97-112 [DOI] [PubMed] [Google Scholar]
- 10.Fidler IJ, Hart IR: Biological diversity in metastatic neoplasms: origins and implications. Science 1982, 217:998-1003 [DOI] [PubMed] [Google Scholar]
- 11.Poste G, Greig R: On the genesis and regulation of cellular heterogeneity in malignant tumors. Invasion Metastasis 1982, 2:137-176 [PubMed] [Google Scholar]
- 12.Heppner GH, Miller BE: Tumor heterogeneity: biological implications and therapeutic consequences. Cancer Metastasis Rev 1983, 2:5-23 [DOI] [PubMed] [Google Scholar]
- 13.Heppner GH: Tumor heterogeneity. Cancer Res 1984, 44:2259-2265 [PubMed] [Google Scholar]
- 14.Heppner GH, Dexter DL, DeNucci T, Miller FR, Calabresi P: Heterogeneity in drug sensitivity among tumor cell subpopulations of a single mammary tumor. Cancer Res 1978, 38:3758-3763 [PubMed] [Google Scholar]
- 15.Rak J, St. Croix B, Kerbel RS: Consequences of angiogenesis for tumor progression, metastasis and cancer therapy. Anti-Cancer Drugs 1995, 6:3-18 [DOI] [PubMed] [Google Scholar]
- 16.Weidner N, Semple JP, Welch WR, Folkman J: Tumor angiogenesis and metastasis—correlation in invasive breast carcinoma. N Engl J Med 1991, 324:1-8 [DOI] [PubMed] [Google Scholar]
- 17.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J: Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol 1993, 143:401-409 [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86:353-364 [DOI] [PubMed] [Google Scholar]
- 19.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ: Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379:88-91 [DOI] [PubMed] [Google Scholar]
- 20.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E: Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394:485-490 [DOI] [PubMed] [Google Scholar]
- 21.Chaplin DJ, Durand RE, Olive PL: Cell selection from a murine tumour using the fluorescent probe Hoechst 33342. Br J Cancer 1985, 51:569-572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rak J, Filmus J, Kerbel RS: Reciprocal paracrine interactions between tumor cells and endothelial cells. The “angiogenesis progression” hypothesis. Eur J Cancer 1996, 32A:2438-2450 [DOI] [PubMed] [Google Scholar]
- 23.Weidner N: Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat 1995, 36:169-180 [DOI] [PubMed] [Google Scholar]
- 24.Olive PL, Chaplin DJ, Durand RE: Pharmacokinetics, binding and distribution of Hoechst 33342 in spheroids and murine tumours. Br J Cancer 1985, 52:739-746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomlinson RH, Gray LH: The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer 1955, 9:539-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graeber TG, Peterson JF, Tsai M, Monica K, Fornace AJJ, Giaccia AJ: Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol Cell Biol 1994, 14:6264-6277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A: Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1-alpha. Genes Dev 2000, 14:34-44 [PMC free article] [PubMed] [Google Scholar]
- 28.Mazure NM, Chen EY, Yeh P, Laderoute KR, Giaccia AJ: Oncogenic transformation and hypoxia synergistically act to modulate vascular endothelial growth factor expression. Cancer Res 1996, 56:3436-3440 [PubMed] [Google Scholar]
- 29.Helmlinger G, Yuan F, Dellian M, Jain RK: Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med 1997, 3:177-182 [DOI] [PubMed] [Google Scholar]
- 30.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW: Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 1999, 59:5830-5835 [PubMed] [Google Scholar]
- 31.Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D: Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 1999, 284:808-812 [DOI] [PubMed] [Google Scholar]
- 32.Yokoyama Y, Dhanabal M, Griffioen AW, Sukhatme VP, Ramakrishnan S: Synergy between angiostatin and endostatin: inhibition of ovarian cancer growth. Cancer Res 2000, 60:2190-2196 [PubMed] [Google Scholar]
- 33.Castilla MA, Caramelo C, Gazapo RM, Martin O, Gonzalez-Pacheco FR, Tejedor A, Bragado R, Arroyo MV: Role of vascular endothelial growth factor (VEGF) in endothelial cell protection against cytotoxic agents. Life Sci 2000, 67:1003-1013 [DOI] [PubMed] [Google Scholar]
- 34.Nor JE, Christensen J, Mooney DJ, Polverini PJ: Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol 1999, 154:375-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerber HP, Dixit V, Ferrara N: Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem 1998, 273:13313-13316 [DOI] [PubMed] [Google Scholar]
- 36.Tran J, Rak J, Sheehan C, Saibil SD, LaCasse E, Korneluk RG, Kerbel RS: Marked induction of the IAP family anti-apoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem Biophys Res Commun 1999, 264:781-788 [DOI] [PubMed] [Google Scholar]
- 37.O’Conner DS, Schechner JS, Adida C, Mesri M, Rothermel AL, Li F, Nath AK, Pober JS, Altieri DC: Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol 2000, 156:393-398 [DOI] [PMC free article] [PubMed] [Google Scholar]