Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker (original) (raw)
. Author manuscript; available in PMC: 2008 Apr 13.
Summary
Hsp70 chaperones assist in protein folding, disaggregation and membrane translocation by binding to substrate proteins with an ATP-regulated affinity that relies on allosteric coupling between ATP-binding and substrate-binding domains. We have studied single and two-domain versions of the E. coli Hsp70, DnaK, to explore the mechanism of interdomain communication. We show that the interdomain linker controls ATPase activity by binding to a hydrophobic cleft between subdomains IA and IIA. Furthermore, the domains of DnaK dock only when ATP binds and behave independently when ADP is bound. Major conformational changes in both domains accompany ATP-induced docking: Of particular importance, some regions of the substrate-binding domain are stabilized, while those near the substrate-binding site become destabilized. Thus, the energy of ATP binding is used to form a stable interface between the nucleotide and substrate-binding domains, which results in destabilization of regions of the latter domain, and consequent weaker substrate binding.
Introduction
Hsp70 molecular chaperones play diverse roles in cells, including chaperoning nascent protein chains, assisting in import of proteins into organelles, enabling survival under stress conditions such as heat shock, and dissociation of macromolecular complexes and aggregates (Bukau et al., 2006; Mayer and Bukau, 2005; Young et al., 2004). All such functions are mediated by interaction of extended, hydrophobic regions of substrate proteins with the Hsp70 C-terminal substrate-binding domain (SBD). The affinity and kinetics of this interaction are modulated by nucleotide binding to the N-terminal ATPase domain (ATPase); in the ADP-bound state, substrate binds with high affinity, but upon exchange for ATP, substrate affinity is substantially decreased with faster on/off rates. Substrate binding in turn increases the catalytic activity of the ATPase domain. The two Hsp70 domains are connected by a short, highly conserved and hydrophobic linker that is important for interdomain allosteric communication (Han and Christen, 2001; Laufen et al., 1999; Mayer et al., 1999; Vogel et al., 2006).
How ligand binding in one Hsp70 domain transmits an allosteric signal to the other domain remains a mystery, although a considerable body of evidence supports a significant ligand-induced conformational change. ATP binding has been shown to decrease the radius of gyration of Hsc70 (Wilbanks et al., 1995), and the sole intrinsic tryptophan in the ATPase domain of DnaK (W102) becomes sequestered from solvent by packing against the helical lid of the SBD when ATP binds (Buchberger et al., 1995; Moro et al., 2003; Palleros et al., 1992; Sivendran, 2004). Both hydrogen exchange and limited proteolytic digestion of DnaK demonstrate that ATP binding stabilizes the ATPase domain and protects the interdomain linker while simultaneously destabilizing the SBD (Buchberger et al., 1995; Rist et al., 2006). This ATP-dependent conformational change in the SBD involves loss of electrostatic stabilizing interactions between the β and helical subdomains (Fernandez-Saiz et al., 2006; Moro et al., 2006). Several residues from each domain and the linker participate in the allosteric mechanism (Montgomery et al., 1999; Vogel et al., 2006 and references therein).
Generally speaking, one might propose several models for the interaction of the two domains of Hsp70 during the cycle of ATP binding and hydrolysis. For instance, the two domains could remain docked throughout the cycle, with ligands modulating the interdomain packing interaction. Alternatively, the two domains might act independently in some ligand-bound states, and come together to form a stable tertiary packing arrangement in others. A third possibility is that the two domains never interact strongly, but that signaling from one domain to the other occurs via binding and release of some switch element. Indeed, one might envision the interdomain linker playing such a role in the Hsp70 family.
Structure determination of a full-length Hsp70 protein would greatly help in understanding interdomain communication, but has remained a challenge, preventing a full exploration of the ligand-induced conformational changes. Structures of the individual domains have been available (for review, see Mayer and Bukau, 2005), and recently a two-domain crystal structure of bovine Hsc70 was reported (Jiang et al., 2005). This nucleotide-free structure shows a domain packing arrangement that leaves both the interdomain linker and the residue homologous to W102 (in DnaK) exposed to solvent, indicating that this structure does not reflect the allosterically active ATP-bound state. In fact, the SBD helix that makes up half the interdomain interface in the crystal structure can be removed without disabling allosteric interdomain communication in DnaK (Pellecchia et al., 2000). Further conflicting data comes from our solution NMR study, which showed the two domains of nucleotide-free DnaK to behave independently with no stable interaction formed (Swain et al., 2006). Finally, a different domain arrangement has been proposed on the basis of NMR residual dipolar couplings and chemical shift perturbation using T. thermophilus DnaK bound to ADP (Revington et al., 2005). Thus, the allosterically active interdomain packing arrangement in Hsp70s remains unclear (Swain and Gierasch, 2006).
In order to explore the full allosteric cycle in Hsp70s, we have deployed a combination of NMR and other biophysical methods to study both the isolated domains and the two-domain DnaK protein from E. coli. We propose a global model for Hsp70 allosteric regulation based on our results and substantiated by previous literature: ATP binding causes docking of the SBD onto the ATPase domain with concomitant major structural changes in both domains; the primary role of substrate binding is to uncouple the two domains, and at the same time to enable the interdomain linker to act as a switch to enhance the ATP hydrolysis rate of the ATPase domain.
Results
The Hsp70 interdomain linker acts as a switch to stimulate the ATPase activity
Previous mutagenesis experiments have demonstrated the importance of the linker to the DnaK allosteric mechanism (Han and Christen, 2001; Laufen et al., 1999). Therefore, we created two versions of the isolated ATPase domain of DnaK, corresponding to residues 1-388 and 1-392, the latter of which retains the conserved hydrophobic VLLL sequence of the interdomain linker. As seen earlier with DnaK(1-385) (Moro et al., 2003), the ATPase activity of DnaK(1-388) (0.06 ± 0.02 min−1) was similar to that of full-length DnaK (0.09 ± 0.04 min−1), but to our surprise, inclusion of the VLLL sequence resulted in a 13-fold ATPase stimulation (0.78 ± 0.01 min−1), similar to the stimulation caused by addition of the substrate peptide p5 (CLLLSAPRR) to full-length DnaK (0.56 ± 0.03 min−1, Fig. 1A (Dinler et al., 2004)). A similar effect of the linker has also recently been reported by others (Vogel et al., 2006). Replacement of the two central leucines in the VLLL sequence of DnaK(1-392) with aspartates, which causes an allosteric defect in the context of full-length DnaK (Laufen et al., 1999), reduced the ATPase activity four-fold, to a level (0.18 ± 0.04 min−1) only 2–3 fold stimulated over DnaK(1-388) or full-length DnaK (Fig. 1A).
Figure 1.
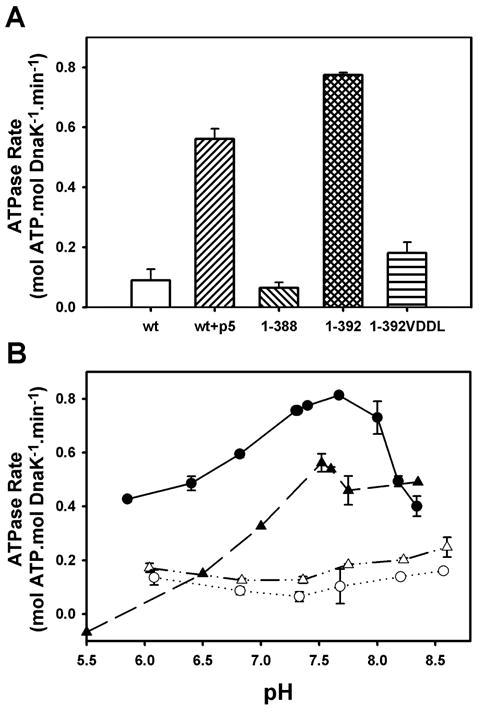
The linker stimulates the ATPase activity of the isolated ATPase domain, similar to the effect of substrate on full-length DnaK. (A) Steady-state ATPase rates measured at pH 7.6. (B) pH-dependence of steady-state ATPase activities for full-length wild-type DnaK in the absence (△) and presence (▲) of p5 peptide, DnaK(1-388) (○), and DnaK(1-392) (●). Error bars represent standard deviation from ≥3 experiments.
Based on previous work that showed the substrate-stimulated ATPase activity of DnaK to be pH-dependent (Sehorn et al., 2002), we explored the pH-activity profiles for these proteins as well. Like the substrate-stimulated ATPase activity of full-length DnaK, the activity of DnaK(1-392) was exceptionally pH-dependent, with maximal activity between pH 7.5 and 8.0 (Fig. 1B). In contrast, the ATPase rates of unstimulated full-length DnaK and DnaK(1-388) were relatively insensitive to pH (Fig. 1B). The similarity of DnaK(1-392) and the substrate-stimulated full-length protein in terms of both activity levels and pH dependencies, coupled with the fact that mutation of the VLLL sequence of the linker in full-length DnaK blocks substrate activation of the ATPase activity (Laufen et al., 1999), leads us to the conclusion that the linker is both necessary and sufficient for allosteric control of the ATPase domain by the SBD. We then used this simplified model system (i.e., DnaK(1-388) vs. DnaK(1-392)) to explore the thermodynamic and conformational differences between the unstimulated and stimulated states of the DnaK ATPase domain.
The interdomain linker induces a closed state of the ATPase domain
CD thermal melts of DnaK(1-388) and DnaK(1-392) showed two transitions (Fig. 2A). The transition observed at higher temperature was not reversible and thus we have not focused on it further. On the other hand, the first transition for both proteins was reversible and corresponded to a 30% loss in ellipticity at 222 nm, reflecting a substantial loss of secondary structure. This transition was also accompanied by a decrease in W102 fluorescence and loss of most TROSY NMR peaks, indicating a shift to a molten-globule-like state lacking stable tertiary structure (data not shown). Significantly, for DnaK(1-392), the first transition occurred 3.5 degrees higher than in DnaK(1-388) (50.5°C vs. 47°C, Fig. 2A), indicating that the linker confers increased stability on the ATPase domain.
Figure 2.
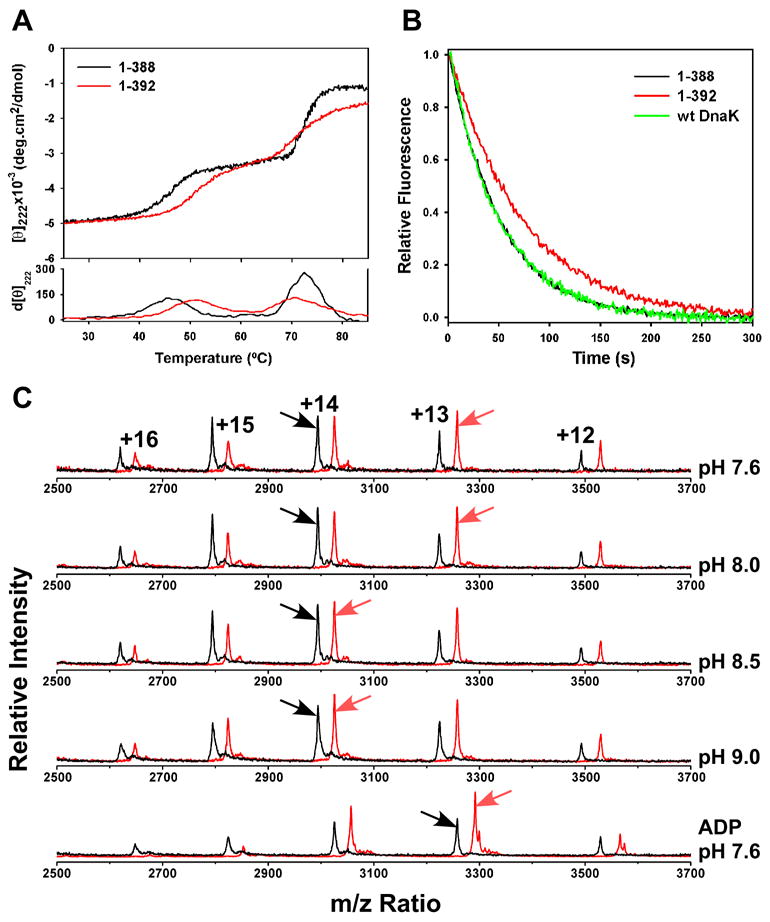
Presence of the linker on the isolated ATPase domain confers enhanced stability and domain closure. (A) Thermal melts of nucleotide-free DnaK(1-388) in black and DnaK(1-392) in red monitored by CD at 222 nm. First derivative of the melting curves is shown at the bottom. (B) MABA-ADP release was monitored by fluorescence after addition of excess ADP. Full-length wild-type DnaK is shown in green, DnaK(1-388) in black, and DnaK(1-392) in red. (C) ESI-MS charge state distributions of DnaK(1-388) in black and DnaK(1-392) in red, with the most abundant charge state indicated by an arrow. The top four spectra were collected on the nucleotide-free state at the indicated pH, and the bottom spectrum on the ADP-bound state at pH 7.6.
The linker also shifted the charge-state distributions from electrospray ionization mass spectrometry (ESI-MS), which have been shown to be a sensitive measure of protein conformation: more compact conformations hold fewer charges (Chowdhury et al., 1990; Kaltashov and Eyles, 2002). At pH 7.6, the most abundant charge state of DnaK(1-392) was +13, whereas the +14 charge state was most abundant for DnaK(1-388) (Fig. 2C, top trace). At more alkaline pH, where the activity level of DnaK(1-392) approaches that of DnaK(1-388), the charge state distribution of DnaK(1-392) increased to +14, whereas that for DnaK(1-388) remained +14 up to pH 11.5 (Fig. 2C and data not shown). Thus the linker compacts and activates the ATPase domain at neutral pH; at higher pH the effect of the linker is reduced.
It has been proposed that the cleft of the Hsp70 ATPase domain opens and closes during the cycle of nucleotide binding and hydrolysis (Flaherty et al., 1990). Indeed, crystal structures of nucleotide-free Hsp70 ATPase domains (co-crystallized with nucleotide exchange factors) show a more open cleft than those bound to nucleotides (Flaherty et al., 1994; Harrison et al., 1997; Sondermann et al., 2001). Using CD thermal melts, we found that ADP binding increased the melting temperature of the ATPase domain fragments (to 54°C from 47°C and 50.5°C for DnaK(1-388) and DnaK(1-392), respectively; data not shown), and ESI-MS analysis showed that the charge state distribution shifted to the more compact +13 state upon ADP binding (Fig. 2C, bottom trace). Since binding of either the linker or ADP stabilize and compact the ATPase domain, we propose that, like ADP binding, linker binding closes the cleft between the lobes. In order to test this, we determined the dissociation rate for ADP, which should be slower from a closed conformation of the ATPase domain. For simplicity, we measured the off-rate of MABA-ADP, which is equivalent to the ADP dissociation rate in full-length DnaK (Theyssen et al., 1996). Indeed, the off-rate of MABA-ADP was slower for DnaK(1-392) (0.0138 ± 0.0002 s−1, Fig. 2B) than for either DnaK(1-388) (0.0201 ± 0.0004 s−1) or full-length DnaK (0.0190 ± 0.0011 s−1), consistent with the hypothesis that the linker favors a closed conformation of the ATPase domain.
The linker binds to a hydrophobic cleft between subdomains IA and IIA on the ATPase domain
TROSY NMR spectra of the ADP-bound ATPase domain with and without the linker showed that the mobility of the C-terminus is constrained by the presence of the VLLL sequence. In a stack plot representation, one can clearly distinguish four narrow and intense resonances in DnaK(1-388) (indicated by arrows in Fig. 3A), which we have assigned to the highly dynamic extreme C-terminus based on the fact that residues 384-388 lacked electron density in the crystal structure of this construct (Harrison et al., 1997). In TROSY NMR spectra of DnaK(1-392), there are no such narrow intense peaks (Fig. 3A). Chemical shift analysis revealed interaction of the linker sequence with a specific site on the ATPase domain, as the presence of the linker induces significant shifts in 40–50 peaks (Fig. 3B). Selective labeling of both ATPase domain constructs with 15N-leucine enabled assignment of L390-L392 in DnaK(1-392) (Fig. 3C), and linewidth analysis of both C-termini revealed significant mobility differences. While the resonances corresponding to L390-L392 in DnaK(1-392) are as broad or broader than most other resonances in the ATPase domain (31.5–33.1 Hz compared to a range of values between ca. 28 and 31 Hz), the C-terminal residues of DnaK(1-388) are extremely narrow, indicating high mobility (21–27 Hz). Thus, interaction of the VLLL segment with a specific site on the ATPase domain constrains the mobility of the entire linker.
Figure 3.
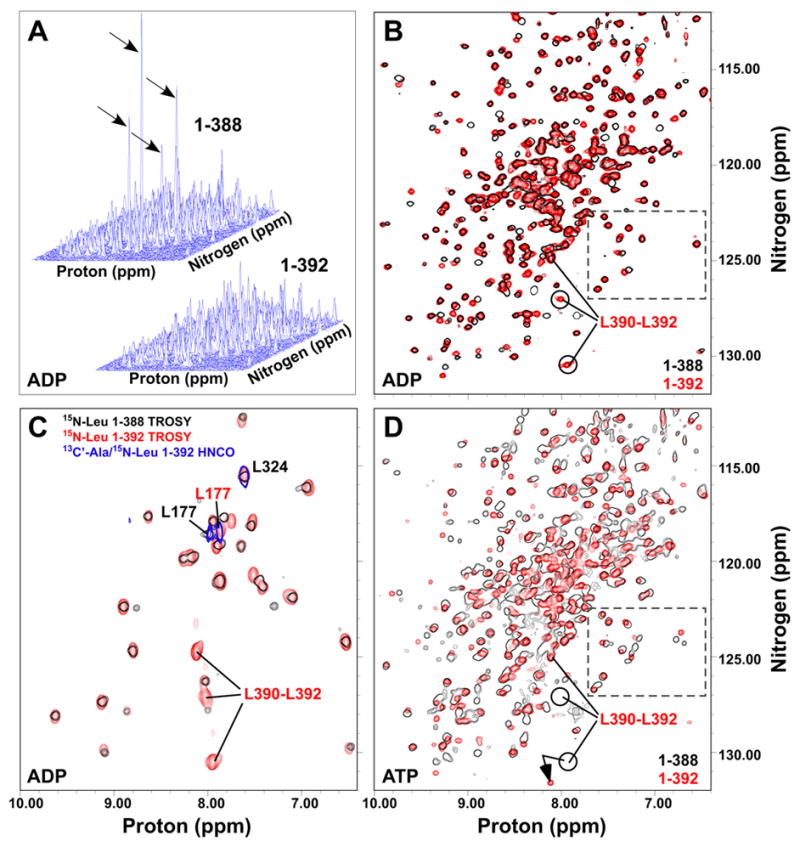
The VLLL sequence of the linker docks onto the ATPase domain. (A) Stack plots of TROSYs on [15N]DnaK(1-388)·ADP (upper left) and [15N]DnaK(1-392)·ADP (lower right) . Intense peaks in DnaK(1-388) are indicated by arrows. (B) Same spectra as in (A), but shown overlaid in a contour map with 1-388 in black outlines and 1-392 in red. (C) Overlay of TROSY spectra for [15N]leucine-labeled DnaK(1-388)·ADP (black outlines) and DnaK(1-392)·ADP (red) with a 2D HNCO of [13C’]alanine/[15N]leucine-labeled DnaK(1-392)·ADP (blue outlines). (D) Overlay of TROSY spectra for [15N]DnaK(1-388)T199A·ATP (black) and [15N]DnaK(1-392)T199A·ATP (red). In B, C and D, the ADP-bound positions of C-terminal leucines of DnaK(1-392) are indicated. Resonances within the dashed box (panels B and D) provide an example of conformational heterogeneity in ATP-bound DnaK(1-388).
In order to localize the site of linker binding on the ATPase domain, we surveyed the surface of this domain for conserved hydrophobic patches within reach of the VLLL segment of the linker. We then pursued a directed assignment strategy in which residue pairs that occur in these patches and that are rare overall in the protein sequence were selectively labeled with 13C on the carbonyl of the i-1 residue and 15N in the i residue amide, followed by detection with a 2D HNCO experiment (see Supplementary Experimental Procedures for details of assignment procedure). By labeling of AL pairs (Fig. 3C), we determined that L177, located in an exposed hydrophobic cleft between subdomains IA and IIA (Mayer and Bukau, 2005), was highly sensitive to linker binding (Δδav = 0.13 ppm). In addition to L177, the structurally adjacent and solvent-exposed residue I373 undergoes a large shift upon linker binding (Δδav = 0.08 ppm), and both I18 and A144 experience modest shifts consistent with an indirect effect of linker binding (Δδav = 0.05 and 0.04 ppm, respectively).
The linker binding site is reciprocally coupled to the nucleotide-binding site
Intriguingly, the resonances of the linker bound in this site are sensitive to nucleotide binding. Two peaks corresponding to linker leucine residues undergo large chemical shift changes between the ADP- and ATP-bound states (Fig. 3B,D; a well-characterized hydrolysis-defective mutant, T199A, was used for ATP-bound spectra (Buchberger et al., 1995; Palleros et al., 1993)). These striking effects clearly demonstrate two-way allostery between the linker-binding site and the nucleotide-binding site: Not only does linker binding stimulate the rate of ATP hydrolysis, but conversely, nucleotide binding is sensed by the bound linker. It is thus tantalizing to propose that this linker-binding site is part of the interdomain interface that mediates allostery between the ATPase and SBD domains.
Conformational equilibrium of the ATPase domain is modulated by linker binding
Since the presence of the linker speeds the catalytic step of the ATPase cycle (Vogel et al., 2006), one might expect the largest influence of the linker on the state that occurs just pre-catalysis, i.e. the ATP-bound state. We find that ATP binding substantially stabilizes the DnaK ATPase domain, with an 8° increase in the CD thermal melting transition relative to the ADP-bound state (to 62°C, data not shown), and also causes chemical shift changes and some doubling of TROSY NMR resonances (Fig. S1 and 3D, boxed region). These doubled resonances do not result from incomplete saturation with nucleotide ligand (Fig. S1); mass spectrometric analysis also ruled out proteolysis as the cause (data not shown). Therefore, we conclude that the 1-388 ATPase domain undergoes slow conformational exchange between at least two states when ATP is bound. Linker binding shifted more than 100 resonances in the ATP-bound state, and also reduced the doubled resonances to singlets (Fig. 3D). The latter effect could mean either that the rate of conformational exchange between multiple states is faster in DnaK(1-392)T199A·ATP, or that only one conformation is sampled (Fig. 3D). We therefore propose that linker binding either accelerates a slow conformational transition to the catalytically active state or increases the population of this state, with the end result in either case that the slow basal ATPase activity is increased. In summary, our analysis has shown that the presence of the linker on the isolated ATPase domain mimics the substrate-stimulated state of this domain: the linker binds to an allosteric site between the “crossing helices” of subdomains IA and IIA, leading to lobe closure and more efficient catalysis.
The two domains of ADP-bound DnaK behave independently
To gain a fuller understanding of the allosteric coupling between domains in Hsp70 proteins, we turned to a two-domain construct that we have previously shown to be functional both in vivo and in vitro and that is well-behaved for NMR studies (Swain et al., 2006). This allosterically competent protein, DnaK(1-552)ye, features a C-terminal truncation and two mutations in the C-terminal tail that abrogate self-binding of the C-terminus into the substrate-binding pocket. Similar to what we previously observed with the nucleotide-free protein (Swain et al., 2006), when we compared TROSY NMR spectra of this two-domain ADP-bound protein with those of the isolated SBD (H6DnaK(387-552)ye) and the isolated ADP-bound ATPase domain (DnaK(1-388)·ADP), we found a nearly perfect correlation: the sum of the spectra of the two individual domains replicates the spectrum of the two-domain protein (Fig. 4A). The fact that the chemical shift changes are so limited when the isolated domains are put into a two-domain context means that the two domains do not interact significantly in either the ADP-bound or nucleotide-free states (Swain et al., 2006).
Figure 4.
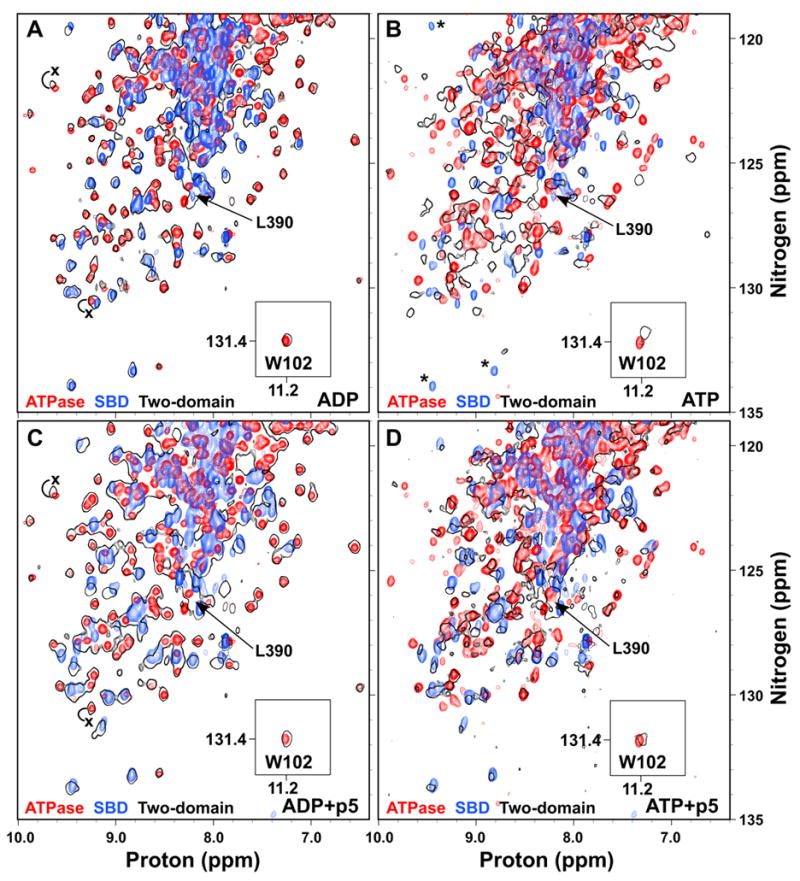
ATP binding to the two-domain DnaK protein induces domain docking that is largely reversed by substrate binding. Overlay of TROSY NMR spectra of the 15N-labeled ATPase domain (DnaK(1-388); red), SBD (H6DnaK(387-552)ye; blue) and two-domain protein (DnaK(1-552)ye; black outlines) bound to ADP (A), ATP (B), ADP and p5 peptide (C), or ATP and p5 peptide (D). The T199A mutant was used for observation of ATP states. The W102 side chain amide is shown in an inset. The ADP-bound position of L390 in the two-domain protein is indicated in all panels by an arrow. In (B), SBD residues that apparently disappear upon ATP binding to the two domain protein are indicated by asterisks. In (A) and (C), the positions of selected ATPase domain resonances that shift upon linker binding are labeled with an x at their position in DnaK(1-392).
Given the key role of the linker in interdomain allostery in DnaK, we assigned the leucine and valine resonances of the interdomain linker (V386, V389-L392) by uniform 15N-labeling of these residue types, in order to follow them in the context of the two-domain protein. Consistent with a role for the linker as a flexible tether between independent domains, the peaks corresponding to these linker residues were narrow and intense. Furthermore, the two-domain spectrum overlays well with DnaK(1-388), but not with DnaK(1-392) (Figs. 3B and 4A); therefore the linker is not bound to the ATPase domain in the ADP-bound two-domain protein. These data paint a picture of the two domains of ADP-bound DnaK behaving largely independently of one another, and connected by a short flexible linker, and are consistent with the proteolytic lability of the linker in this state (Buchberger et al., 1995).
ATP binding to the two-domain protein causes domain packing and gross conformational change in both domains
Using a similar strategy, we investigated the ATP-bound state by taking advantage of the same catalytically-defective T199A mutant used for the ATPase domain studies. Based on previous reports that described a decrease in radius of gyration and sequestration of W102 between the domains when ATP was bound (Moro et al., 2003; Sivendran, 2004; Wilbanks et al., 1995), we expected chemical shift changes relative to the isolated domain spectra for residues involved in this interface, as well as further chemical shift changes within the SBD reflecting conformational change to the low affinity state. Strikingly, global chemical shift changes occur relative to the isolated SBD and ATPase domains, indicating major conformational changes in both domains upon ATP-induced domain docking (Fig. 4B). Notably, the spectrum of the two-domain protein in Fig. 4B is overlaid with that of the same state of the isolated ATPase domain (i.e. T199A and ATP-bound), so the chemical shift changes within the domain due to ATP binding or the T199A mutation are already accounted for. The observed peak shifts in both domains in the context of the two-domain protein relative to the isolated domains thus reflect major conformational changes throughout the protein as a result of domain docking.
Intriguingly, several well-dispersed SBD peaks disappear upon ATP binding (Fig. 4B, asterisks), indicating conformational exchange broadening in the low substrate affinity state, which would be entirely consistent with increased trypsinolysis and hydrogen exchange of the SBD in this state (Buchberger et al., 1995; Rist et al., 2006). The valine and leucine resonances of the interdomain linker are shifted away from their ADP-bound positions (indicated for L390 in Fig. 4B), which is consistent with protection of the linker from trypsinolysis and hydrogen exchange in the ATP-bound state (Buchberger et al., 1995; Rist et al., 2006).
Chemical shift changes for the W102 side chain amide upon ATP binding accurately mirror the well-described fluorescence effects at this tryptophan (Buchberger et al., 1995; Moro et al., 2003; Palleros et al., 1992). Whereas ATP binding to the isolated ATPase domain has no effect on either the chemical shift or the fluorescence of W102, ATP binding to the two-domain protein causes a significant NMR peak shift, and a fluorescence blue shift and quench (Fig. 4A,B) (Buchberger et al., 1995). The clear parallel between the W102 chemical shift behavior observed in this work and the well-established W102 fluorescence changes reveals that both are reporting on the same domain docking phenomenon.
Hydrogen-deuterium exchange reveals a potential docking site for the ATP-bound ATPase domain on the SBD
The ATP-bound state of the two-domain protein is very different in both structure and dynamics from any high resolution Hsp70 structures currently available. In order to gain a better understanding of how the underlying conformational change enables the DnaK molecular machine to release bound substrates, we used hydrogen exchange to follow the conformational differences in the SBD in the ATP and ADP-bound states. In this experimental protocol (Fig. 5A), the effect of ATP on the SBD is read out on the known ADP/peptide-bound assignments. Briefly, hydrogen exchange was initiated in matched samples of DnaK(1-552)ye bound to ATP or ADP, and after an hour, both samples were converted to the ADP/p5-bound state and observed by TROSY NMR (see Experimental Procedures). The ratio of individual SBD peak intensities between ATP- and ADP-pulsed samples serves as an indicator of the relative amount of hydrogen exchange during the pulse of ATP versus ADP.
Figure 5.
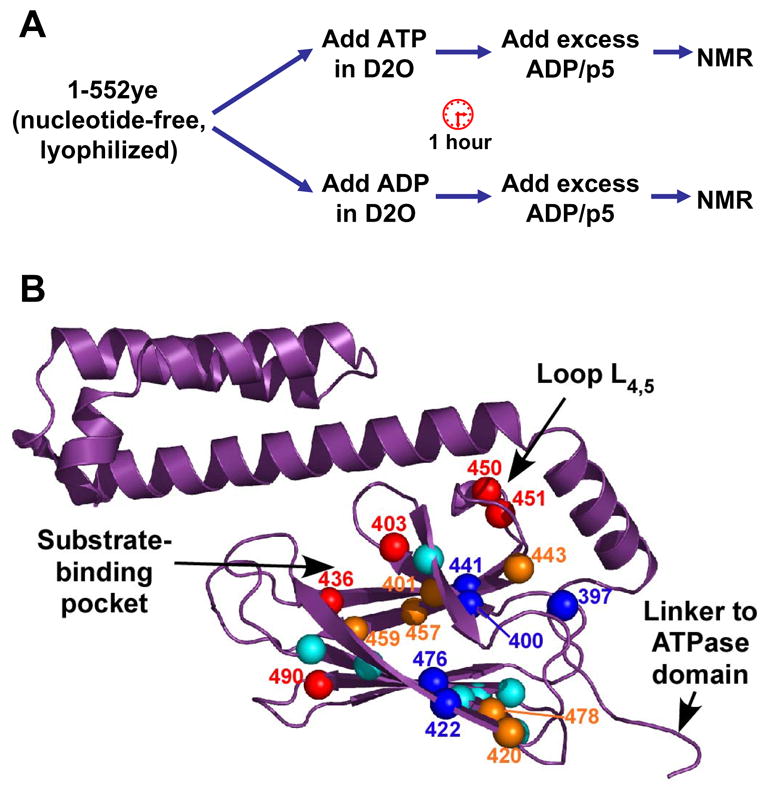
Hydrogen-deuterium exchange (HDX) reveals differential effects of ATP binding in the SBD. (A) Outline of HDX experiment. (B) Peak intensity ratios (IATP/IADP) for the 23 residues that could be quantified are represented on the SBD structure (PDB code 1DKZ). Residues undergoing faster exchange in ATP are indicated by red (IATP/IADP <0.5) and orange balls (IATP/IADP between 0.5 and 0.9). Residues that are more protected in ATP relative to ADP are shown as dark blue balls (IATP/IADP>1.1), and relatively unaffected residues are shown as cyan balls. Figure prepared using PyMOL (http://www.pymol.org).
Note that due to the experimental design, we quantify only SBD resonances that are well-protected and well-dispersed in the ADP/p5 state; 23 resonances fit these criteria (Fig. 5B, Table S1). Of these 23, 11 were less intense in the ATP-pulsed sample, arguing for the expected destabilization in the ATP-bound state (IATP/IADP <0.9; 401, 403, 420, 436, 443, 450, 451, 457, 459, 478, and 490; indicated by red and orange balls in Fig. 5B). These residues primarily cluster near the substrate-binding pocket and in the interior loop L4,5, and are consistent with loss of substrate affinity in this state. Strikingly, however, five resonances were actually more intense after a pulse of ATP versus a pulse with ADP, arguing that these sites were stabilized in the ATP state (IATP/IADP >1.1; 397, 400, 422, 441, and 476; dark blue balls in Fig. 5B). Two pairs of ATP-protected residues form reciprocal hydrogen bonds between the central strands β1 and β4 in the upper sheet and the edge strands β3 and β6 of the lower sheet (Fig. 5B). The clustering of these residues adjacent to the site of attachment of the interdomain linker suggests a model in which this region of the SBD is stabilized due to formation of an interface with the ATPase domain.
Substrate binding reverses the domain packing induced by ATP
We have previously shown that substrate binding to the nucleotide-free state of the two-domain protein causes only intradomain effects within the SBD (Swain et al., 2006). This is also the case for the ADP-bound state, as shown in Fig. 4C, where the overlay of the two-domain protein bound to both ADP and p5 peptide with the isolated domains bound to their respective ligands shows virtually no chemical shift changes in the ATPase domain. Furthermore, the valine and leucine resonances of the interdomain linker are not shifted by p5 binding in the ADP state (shown for L390 in Fig. 4C). Thus there is no interdomain signal upon substrate binding to the ADP-bound state; substrate binding only affects the SBD resonances.
We hypothesized that an interface must be established with the ATPase domain in order for substrate to exert its effects on the ATPase domain, resulting in faster ATP catalysis. Strikingly, binding of p5 peptide to the domain-docked ATP-bound state apparently renders the two domains more independent (Fig. 4D). As substrate is titrated into the ATP-bound state of DnaK(1-552)yeT199A, resonances begin to appear at the positions of the isolated p5-bound SBD. Similarly, peaks also appear or shift to positions of the isolated ATP-bound ATPase domain. Intriguingly, the W102 side chain amide is shifted back to its position in the ADP-bound two-domain protein, consistent with the manner in which its fluorescence emission maximum shifts to the ADP-bound wavelength when substrate is added to the ATP-bound state (Slepenkov and Witt, 1998). As expected for a functional two-domain Hsp70, substrate affinity is substantially reduced in the ATP-bound state, so saturation of the substrate-binding site was more difficult and required at least seven-fold excess of substrate over protein, compared to the stoichiometric binding observed for the ADP-bound state. It is interesting to note, however, that not all resonances re-appear at their isolated domain positions at the end point of the titration, suggesting that some remnant of a binding interface may remain between the domains. These data show that substrate binding affects the ATPase domain only when ATP is bound, with the result that the ATP-induced interdomain coupling is largely reversed.
Discussion
We have been able to follow the complete functional cycle of a two-domain Hsp70 protein by NMR through the use of a well-behaved and fully-functional two-domain DnaK construct. These findings are summarized in the model presented in Figure 6A. Our study has demonstrated that the two domains of DnaK only interact when ATP is bound. The ADP-bound state, like the nucleotide-free state (Swain et al., 2006), consists of two independently-acting domains connected by a short flexible linker. The interaction of the two domains in the ATP-bound state leads to global chemical shift changes in both domains, suggesting major conformational changes not only in the SBD but also in the ATPase domain upon docking. Substrate binding disrupts this domain packing arrangement in the ATP-bound state, leading to ATPase stimulation.
Figure 6.
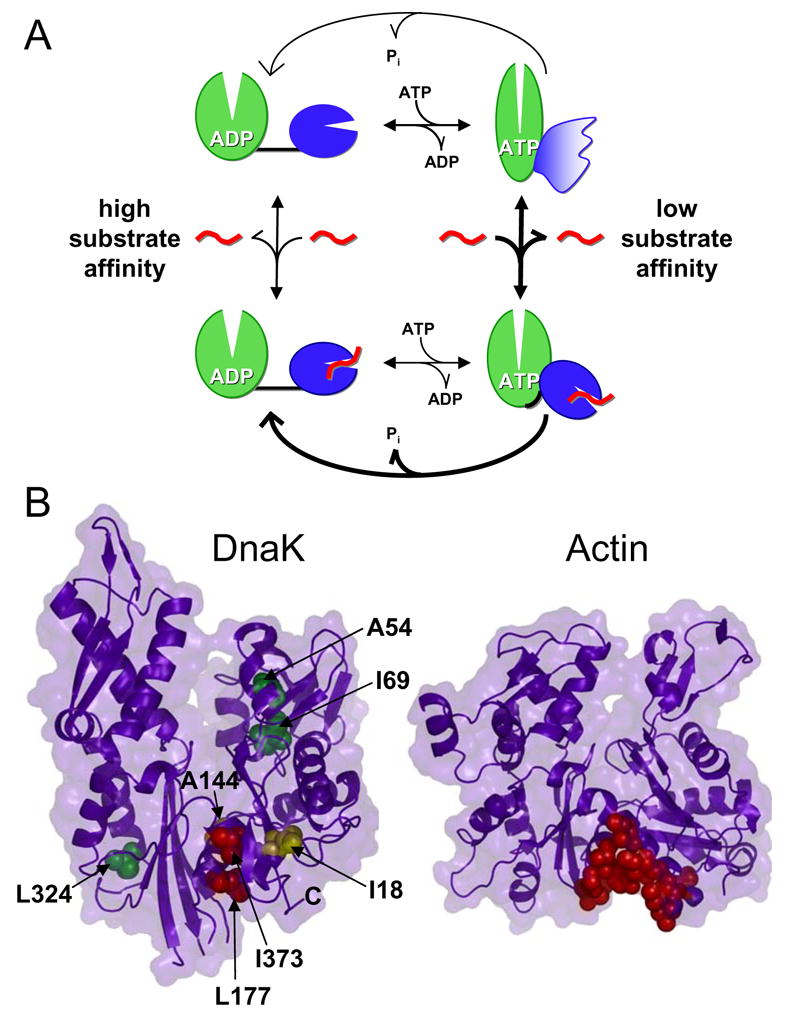
A model for interdomain coupling in Hsp70 proteins. (A) In the ADP-bound state, the two domains are independent and connected by a flexible linker. When ATP binds, domain docking is accompanied by large conformational changes in both domains, and the linker is sequestered from solvent. In this docked state, portions of the SBD are stabilized while regions forming the substrate-binding pocket become dynamic. When both substrate (indicated in red) and ATP are bound, the domains are less intimately associated and the interdomain linker binds to the ATPase domain, stimulating ATP hydrolysis rates. (B) Determination of the linker binding site on the ATPase domain. Left panel, assigned ATPase domain resonances are shown as spheres colored according to their degree of shift upon linker binding (red, Δδav > 0.08 ppm; yellow, Δδav = 0.04–0.08 ppm; green, Δδav < 0.04 ppm; PDB code 1DKG). Right panel, an actin hot spot for protein interactions is indicated by red spheres (PDB code 1ATN) (Dominguez, 2004). Figure prepared using PyMOL (http://www.pymol.org).
Our TROSY NMR spectra of the ATP-bound state of DnaK displayed loss of SBD peaks, indicating conformational exchange-broadening within the SBD. This result is consistent with prior limited trypsinolysis, hydrogen exchange and difference infrared spectroscopy of DnaK, all of which have shown ATP-dependent conformational changes in the SBD, albeit at low resolution, and were interpreted as a domain-wide destabilization (Buchberger et al., 1995; Moro et al., 2006; Rist et al., 2006). However, the site-specific resolution afforded by hydrogen exchange NMR allowed us to see differential effects of ATP binding across the substrate-binding domain, with some regions close to the site of entry of the interdomain linker protected from exchange, and other more distal regions surrounding the substrate-binding pocket destabilized, leading to lowered substrate affinity. The regions protected from exchange in ATP are in fact stabilized by formation of an interface with the ATPase domain. These data are supported by our previous identification of an allosterically defective mutation at K414, which is directly adjacent to the ATP-protected residue L397 (Montgomery et al., 1999). This location for the interdomain interface is also consistent with the results of an evolutionary statistical coupling analysis of 739 members of the Hsp70 family (R. Smock, J. Swain, W. Russ, R. Ranganathan and L. Gierasch, manuscript in preparation).
The present work also provides the first evidence of a major conformational change in the ATPase domain upon docking with the SBD, as evidenced by global peak shifts and substantial broadening relative to the isolated ATP-bound ATPase domain. Previous to this work, the only indications of a docking-induced effect on the ATPase domain were from changes in susceptibility to proteolysis and in the fluorescence signature of W102 (Buchberger et al., 1995; Moro et al., 2003; Palleros et al., 1992). The W102 fluorescence effects are satisfyingly mirrored in our data by chemical shift changes of the W102 side chain amide.
Allostery in the reverse direction, wherein substrate binding regulates ATPase activity, has never been as well-characterized as ATP-induced substrate release. Our data clearly show that substrate binding is only communicated to the ATPase domain when ATP is bound. This is consistent with the fact that every prior experiment that has shown substrate binding to affect the properties of the ATPase domain has, of necessity, been conducted in the presence of ATP (Jordan and McMacken, 1995; Slepenkov and Witt, 1998; Slepenkov and Witt, 2003). Our data show that binding of substrate peptide apparently causes disruption of the packing between the domains, as many chemical shifts return to those expected for the isolated domains. However, if substrate binding to the ATP-bound state were to render the domains completely independent and simultaneously catalyze ATP hydrolysis, then one would expect the isolated ATPase domain (i.e., DnaK(1-388)) to have high ATPase activity, which is clearly not the case (Fig. 1) (Moro et al., 2003). We therefore propose that substrate binding to the ATP-bound state results in a less intimate packing arrangement, but with some element remaining in contact with the ATPase domain. Based on our demonstration that the interdomain linker binds to the isolated ATPase domain and causes faster catalysis, we propose that the interdomain linker plays a major role in this state.
Our studies on the isolated DnaK ATPase domain with and without the interdomain linker sequence show that the linker binds to a discrete site on the ATPase domain, causing compaction and closure of the lobes, and resulting in enhanced ATPase activity. Interestingly, the linker modulates the conformational heterogeneity observed in the ATP-bound state of the ATPase domain, either by increasing the rate of conversion to the catalytically active conformation or by increasing the population of this state. We also observed that the linker causes faster overall ATPase cycling but slower ADP release. This apparent contradiction can be resolved if the linker causes a substantial increase in the rate-limiting catalytic step, which is indeed the case (Vogel et al., 2006). Therefore we propose that the linker favors a closed conformation of the ATPase domain lobes, with the catalytic residues optimally organized for catalysis; this state has modestly slower product release, but the end result is a faster ATPase cycle.
Intriguingly, the binding site of the linker on the ATPase domain corresponds to a “hot spot” for interaction of the structurally homologous protein actin with regulatory binding partners and marine toxins (Fig. 6B) (Dominguez, 2004). One such interaction partner, the WH2 motif, has been shown to reduce actin nucleotide release by rigidifying actin against opening/closing motions in a manner completely analogous to that proposed here for DnaK (Chereau et al., 2005). These regulatory proteins typically bind actin by presenting hydrophobic residues along one face of a helix. In the case of the DnaK linker, however, the hydrophobic residues presented are consecutive, so the binding mode is likely to differ somewhat, but nevertheless the analogy is illuminating. The same cleft in the more structurally distant protein glucokinase also forms an allosteric activating site that has been the target of drug discovery efforts for type 2 diabetes (Kamata et al., 2004). These disparate studies all point to a common mechanism of regulation in these proteins dictated by their shared fold. In DnaK, we have been able to show that the bound linker senses ATP binding, showing a direct two-way allosteric coupling between this cleft and the catalytic machinery.
Notably, the linker binding site is included in an interdomain coupling network identified by our statistical coupling analysis of the Hsp70 family (R. Smock, J. Swain, W. Russ, R. Ranganathan and L. Gierasch, manuscript in preparation). In particular, residues V389 and L390 of the linker display high mutational covariance with residues L177 and L382, respectively, of the crossing helices.
Since the linker is both necessary and sufficient to stimulate the activity of the ATPase domain, we propose that the linker acts as a switch in the context of the full-length protein: normally the linker is prevented from interacting with the ATPase domain, but it becomes available upon substrate binding to stimulate the ATPase activity of the ATPase domain. In the context of the two-domain protein, the linker is clearly not bound to the ATPase domain in the ADP- and/or peptide-bound states. This is in contrast to our results on the isolated ATPase domain, where the linker was able to bind to the ADP-bound domain. Presumably, the presence of the SBD in the two-domain protein prevents the linker from binding to the ATPase domain, either directly, by attracting it to a hydrophobic surface on the SBD, or indirectly, by the geometric constraints dictated by two large independent domains connected by a short linker. However, spectral quality and complexity make it difficult to demonstrate that the linker is bound to the ATPase domain in the substrate/ATP-bound state of the two-domain protein, as our model would predict. Of the ATPase domain resonances that are diagnostic for linker binding in the isolated domain, in the ATP/substrate-bound state of the two-domain protein some lie closer to their position in DnaK(1-392) and some are closer to their position in DnaK(1-388). Presumably, other residues from the SBD beside the linker are also in contact with the ATPase domain, causing additional chemical shift changes relative to the isolated domain.
Our model for interdomain interactions in Hsp70 family members differs from the two recently published structural models for bovine Hsc70 (Jiang et al., 2005) and T. thermophilus DnaK (Jiang et al., 2005; Revington et al., 2005). In the former, the two domains of Hsc70 were observed to interact in the nucleotide-free state, burying ~900 Å of solvent-accessible surface area between them (Jiang et al., 2005). It is somewhat troubling that two mutations (D213A/E214A), which were necessary to generate well-diffracting crystals, lie at the interdomain interface, raising the possibility that the mutations themselves may have favored a non-physiological interdomain crystal packing interaction. In contrast, the two-domain model of T. thermophilus DnaK from NMR defined a different interface and orientation of the two interacting domains, in this case in the ADP-bound form (Revington et al., 2005; Swain and Gierasch, 2006). However, the protein used carried several mutations and deletions, and was not assayed for overall function. Indeed, less than a handful of resonances in the SBD were shifted by ATP binding. Given the limitations of these studies, we reconcile them with our data by suggesting that in those studies a non-physiological domain interaction was favored. Our two-domain NMR construct is functional in vivo, allosterically competent, and can be monitored in solution (Swain et al., 2006); our results conclusively demonstrate that the two domains of E. coli DnaK do not interact in either the ADP or nucleotide-free states, but only dock when ATP binds. This docked state is thus the allosterically active state. Ongoing research will focus on assigning this allosterically active state for a complete structural characterization.
Experimental Procedures
Plasmid constructs
Plasmids encoding DnaK(1-552)ye and H6DnaK(387-552)ye were obtained as previously described (Swain et al., 2006). Plasmids pMS-DnaK(1-388) and pMS-DnaK(1-392) were created from pMSK (Montgomery et al., 1999) by PCR. Mutations were incorporated by QuikChange mutagenesis.
Protein/substrate synthesis and purification
Proteins were expressed in E. coli and purified as described (Montgomery et al., 1999; Swain et al., 2006). Details of isotopic labeling are given in Supplementary Experimental Procedures. Peptide p5 (CLLLSAPRR) was synthesized and purified as described (Swain et al., 2006).
ATPase activity assay
Steady-state ATPase activity measurements were performed at 30 °C as described (Montgomery et al., 1999) except that 5 mM DTT was included. For substrate-stimulated activity, 75 μM p5 peptide was pre-incubated with 1 μM protein for 15 minutes at room temperature prior to measurement.
Circular Dichroism (CD)
CD temperature scans were recorded on a Jasco J-715 spectropolarimeter at 30°C/hr with a 0.1°C step size, 2 sec response time and 1.0 nm bandwidth. Protein samples (10–15 μM) were prepared in HMK buffer (20 mM HEPES, 5 mM MgCl2 and 100 mM KCl, pH 7.6) with 5 mM ADP and ATP added where indicated. The transition midpoints were determined from the first derivative of the melting curve.
Native-state electrospray ionization mass spectrometry (ESI-MS)
Proteins (3–3.5 mg/ml) were dialyzed against 20 mM NH4HCO3 buffer at the pH indicated, and then diluted 20-fold using the same buffer and pH. Sample pH was corrected when necessary. Samples were injected at a rate of 3 μL/min into an MStation mass spectrometer coupled to a standard ESI source (JEOL, Tokyo, Japan), using identical ESI source conditions (orifice potential, 80 V; ring lens potential, 180 V; orifice temperature, 150°C; and desolvating plate temperature, 80°C). The instrument was calibrated and tuned with tridecafluoroheptanoic acid in acetonitrile and sodium bicarbonate. Approximately 40 scans (m/z range of 1000 to 5100, magnet scanning rate of 5 s/decade) were averaged for each spectrum. Experiments were repeated three times to confirm reproducibility.
ADP-bound samples were prepared from ATP-bound proteins by exhaustive dialysis against 4 L HMK at pH 7.6 prior to ammonium bicarbonate dialysis as above. The mass measured in ESI-MS confirmed that the protein was ADP-bound.
ADP Dissociation Rate Measurements
Equimolar MABA-ATP (N8-(4-N’-methylanthraniloylaminobutyl)-8 amino-adenosine 5’-triphosphate, Jena Bioscience, Germany) was preincubated with 0.64 μM protein in HMK buffer, pH 7.6, overnight at 30°C in order to hydrolyze MABA-ATP to MABA-ADP. Dissociation of the fluorescently-labeled nucleotide by addition of 125 μM unlabelled ADP was monitored on an Alpha Scan Fluorometer (Photon Technology International, Birmingham, NJ, USA) with excitation at 360 nm and emission at 440 nm. Rates were calculated by fitting the data to a single exponential using SigmaPlot (Systat Software Inc., Point Richmond, CA). Reported results represent the average of three independent measurements.
NMR
Details of protein preparations are given in Supplementary Experimental Procedures. TROSY data were collected on a 600 MHz Bruker Avance spectrometer using a TXI conventional probe or TCI cryoprobe at 30°C, and processed and analyzed using Felix 2000 (Accelrys, San Diego, CA) and XEasy (Bartels et al., 1995). Weighted average chemical shift changes (Δδav) were calculated as in Radhakrishnan et al.
Hydrogen-Deuterium Exchange Experiments
Matched nucleotide-free samples of 15N-DnaK(1-552)ye (0.3 mM in NMR buffer) were lyophilized and resuspended in D2O containing either 2 mM ATP or ADP, followed by a 1 h incubation at 30°C. Saturating ADP and p5 substrate peptide were added to 10 mM and 0.45 mM, respectively, and single TROSY NMR spectra (duration 20 min.) were begun after 30 minutes incubation at 30°C. Given a basal single-turnover ATPase rate of 0.046 min−1 (Swain et al., 2006), 40% of the ATP should be hydrolyzed before addition of peptide. A three-fold rate acceleration upon peptide addition would be sufficient to hydrolyze all ATP by the start of the NMR experiment; the actual stimulation by peptide is likely 5–10 fold (Swain et al., 2006).
Supplementary Material
01
Acknowledgments
This work was supported by NIH grant GM027616 to LMG. We thank Rob Smock, Steve Eyles and Hwa-Ping (Ed) Feng for critical reading of the manuscript. Mass spectral data were obtained at the University of Massachusetts Mass Spectrometry Facility, which is supported in part by the National Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR. 1995;5:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- Buchberger A, Theyssen H, Schroder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J Biol Chem. 1995;270:16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Chereau D, Kerff F, Graceffa P, Grabarek Z, Langsetmo K, Dominguez R. Actin-bound structures of Wiskott-Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc Natl Acad Sci U S A. 2005;102:16644–16649. doi: 10.1073/pnas.0507021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury SK, Katta V, Chait BT. Probing conformational changes in proteins by mass spectrometry. J Amer Chem Soc. 1990;112:9012–9013. [Google Scholar]
- Dinler G, Montgomery DL, Sivendran R, Gierasch LM. Mapping the interaction surface of the E. coli Hsp70 family molecular chaperone, DnaK. Biophys J. 2004;86:90a. [Google Scholar]
- Dominguez R. Actin-binding proteins--a unifying hypothesis. Trends Biochem Sci. 2004;29:572–578. doi: 10.1016/j.tibs.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Fernandez-Saiz V, Moro F, Arizmendi JM, Acebron SP, Muga A. Ionic contacts at DnaK substrate binding domain involved in the allosteric regulation of lid dynamics. J Biol Chem. 2006;281:7479–7488. doi: 10.1074/jbc.M512744200. [DOI] [PubMed] [Google Scholar]
- Flaherty KM, Deluca-Flaherty C, McKay DB. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature. 1990;346:623–628. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- Flaherty KM, Wilbanks SM, DeLuca-Flaherty C, McKay DB. Structural basis of the 70-kilodalton heat shock cognate protein ATP hydrolytic activity. J Biol Chem. 1994;269:12899–12907. [PubMed] [Google Scholar]
- Han W, Christen P. Mutations in the interdomain linker region of DnaK abolish the chaperone action of the DnaK/DnaJ/GrpE system. FEBS Lett. 2001;497:55–58. doi: 10.1016/s0014-5793(01)02435-8. [DOI] [PubMed] [Google Scholar]
- Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl FU, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science. 1997;276:431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- Jiang J, Prasad K, Lafer EM, Sousa R. Structural basis of interdomain communication in the Hsc70 chaperone. Mol Cell. 2005;20:513–524. doi: 10.1016/j.molcel.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R, McMacken R. Modulation of the ATPase activity of the molecular chaperone DnaK by peptides and the DnaJ and GrpE heat shock proteins. J Biol Chem. 1995;270:4563–4569. doi: 10.1074/jbc.270.9.4563. [DOI] [PubMed] [Google Scholar]
- Kaltashov IA, Eyles SJ. Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spectrom Rev. 2002;21:37–71. doi: 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure (Camb) 2004;12:429–438. doi: 10.1016/j.str.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A. 1999;96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Laufen T, Paal K, McCarty JS, Bukau B. Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance spectroscopy. J Mol Biol. 1999;289:1131–1144. doi: 10.1006/jmbi.1999.2844. [DOI] [PubMed] [Google Scholar]
- Montgomery DL, Morimoto RI, Gierasch LM. Mutations in the substrate binding domain of the Escherichia coli 70 kDa molecular chaperone, DnaK, which alter substrate affinity or interdomain coupling. J Mol Biol. 1999;286:915–932. doi: 10.1006/jmbi.1998.2514. [DOI] [PubMed] [Google Scholar]
- Moro F, Fernández V, Muga A. Interdomain interaction through helices A and B of DnaK peptide binding domain. FEBS Letters. 2003;533:119–123. doi: 10.1016/s0014-5793(02)03752-3. [DOI] [PubMed] [Google Scholar]
- Moro F, Fernandez-Saiz V, Muga A. The allosteric transition in DnaK probed by infrared difference spectroscopy. Concerted ATP-induced rearrangement of the substrate binding domain. Protein Sci. 2006;15:223–233. doi: 10.1110/ps.051732706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palleros DR, Reid KL, McCarty JS, Walker GC, Fink AL. DnaK, hsp73, and Their Molten Globules - Two Different Ways Heat Shock Proteins Respond to Heat. J Biol Chem. 1992;267:5279–5285. [PubMed] [Google Scholar]
- Palleros DR, Reid KL, Shi L, Welch WJ, Fink AL. ATP-Induced Protein Hsp70 Complex Dissociation Requires K+ But Not ATP Hydrolysis. Nature. 1993;365:664–666. doi: 10.1038/365664a0. [DOI] [PubMed] [Google Scholar]
- Pellecchia M, Montgomery DL, Stevens SY, Vander Kooi CW, Feng HP, Gierasch LM, Zuiderweg ERP. Structural insights into substrate binding by the molecular chaperone DnaK. Nat Struct Biol. 2000;7:298–303. doi: 10.1038/74062. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan I, Pérez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Structural analyses of the CREB-CBP transcriptional activator-coactivator complexes by NMR spectroscopy: implications for mapping the boundaries of structural domains. J Mol Biol. 1999;287:859–865. doi: 10.1006/jmbi.1999.2658. [DOI] [PubMed] [Google Scholar]
- Revington M, Zhang Y, Yip GN, Kurochkin AV, Zuiderweg ERP. NMR investigations of allosteric processes in a two-domain Thermus thermophilus Hsp70 molecular chaperone. J Mol Biol. 2005;349:163–183. doi: 10.1016/j.jmb.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in Hsp70 chaperones important for allosteric regulation. J Biol Chem. 2006;281:16493–501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- Sehorn MG, Slepenkov SV, Witt SN. Characterization of two partially unfolded intermediates of the molecular chaperone DnaK at low pH. Biochemistry. 2002;41:8499–8507. doi: 10.1021/bi025810x. [DOI] [PubMed] [Google Scholar]
- Sivendran R. PhD, University of Massachusetts; Amherst: 2004. [Google Scholar]
- Slepenkov SV, Witt SN. Peptide-induced conformational changes in the molecular chaperone DnaK. Biochemistry. 1998;37:16749–16756. doi: 10.1021/bi981738k. [DOI] [PubMed] [Google Scholar]
- Slepenkov SV, Witt SN. Detection of a concerted conformational change in the ATPase domain of DnaK triggered by peptide binding. FEBS Lett. 2003;539:100–104. doi: 10.1016/s0014-5793(03)00207-2. [DOI] [PubMed] [Google Scholar]
- Sondermann H, Scheufler C, Schneider C, Hohfeld J, Hartl FU, Moarefi I. Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science. 2001;291:1553–1557. doi: 10.1126/science.1057268. [DOI] [PubMed] [Google Scholar]
- Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Swain JF, Schulz EG, Gierasch LM. Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states. J Biol Chem. 2006;281:1605–1611. doi: 10.1074/jbc.M509356200. [DOI] [PubMed] [Google Scholar]
- Theyssen H, Schuster HP, Packschies L, Bukau B, Reinstein J. The second step of ATP binding to DnaK induces peptide release. J Mol Biol. 1996;263:657–670. doi: 10.1006/jmbi.1996.0606. [DOI] [PubMed] [Google Scholar]
- Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006;281:38705–38711. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- Wilbanks SM, Chen L, Tsuruta H, Hodgson KO, McKay DB. Solution small-angle X-ray scattering study of the molecular chaperone hsc70 and its subfragments. Biochemistry. 1995;34:12095–12106. doi: 10.1021/bi00038a002. [DOI] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01