Constitutively Active Type I Insulin-Like Growth Factor Receptor Causes Transformation and Xenograft Growth of Immortalized Mammary Epithelial Cells and Is Accompanied by an Epithelial-to-Mesenchymal Transition Mediated by NF-κB and Snail (original) (raw)
Abstract
Type I insulin-like growth factor receptor (IGF-IR) can transform mouse fibroblasts; however, little is known about the transforming potential of IGF-IR in human fibroblasts or epithelial cells. We found that overexpression of a constitutively activated IGF-IR (CD8-IGF-IR) was sufficient to cause transformation of immortalized human mammary epithelial cells and growth in immunocompromised mice. Furthermore, CD8-IGF-IR caused cells to undergo an epithelial-to-mesenchymal transition (EMT) which was associated with dramatically increased migration and invasion. The EMT was mediated by the induction of the transcriptional repressor Snail and downregulation of E-cadherin. NF-κB was highly active in CD8-IGF-IR-MCF10A cells, and both increased levels of Snail and the EMT were partially reversed by blocking NF-κB or IGF-IR activity. This study places IGF-IR among a small group of oncogenes that, when overexpressed alone, can confer in vivo tumorigenic growth of MCF10A cells and indicates the hierarchy in the mechanism of IGF-IR-induced EMT.
Numerous studies have shown the importance of type I insulin-like growth factor receptor (IGF-IR) signaling in mouse mammary gland development and cancer. IGF-IR-null mice have markedly reduced mammary gland development (6), and overexpression of a constitutively active IGF-IR in the mammary gland caused rapid mammary tumorigenesis (8). Similar mammary tumorigenesis was recently shown using inducible overexpression of wild-type IGF-IR (25). Interestingly, transgenic overexpression of the IGF-IR downstream signaling intermediates insulin receptor substrate 1 (IRS1) and IRS2 also caused mammary tumorigenesis (10), and targeted deletion of IRS2 blocked metastasis (32).
IGF-IR is elevated and autophosphorylated in human breast cancer (41). Several strategies have been reported that block IGF-IR activity and inhibit breast cancer growth and metastasis (38), and numerous pharmaceutical companies have agents that target IGF-IR in clinical trials (15). Although IGF-IR signaling is implicated in breast cancer, the molecular mechanisms of IGF-IR-mediated tumorigenesis and metastasis remain unclear.
The role of IGF-IR in malignant transformation has been addressed mainly by studies of mouse fibroblasts. In this setting, IGF-IR acts as a classic oncogene, with overexpression causing transformation (26). Conversely, mouse embryo fibroblasts with a targeted disruption of the IGF-IR gene are resistant to transformation by a variety of viral and cellular oncogenes (35). Reintroduction of IGF-IR renders these cells susceptible to transformation. These results have led to the concept that IGF-IR is itself not only an oncogene, but also “quasi-necessary” for transformation (5). However, recent studies have highlighted important differences between transformation of mouse and human cells and, more importantly, between fibroblasts and epithelial cells (14). To this point, few data exist on IGF-IR-mediated transformation of human epithelial cells.
Two recent studies have used the human MCF10A immortalized mammary epithelial cell line to examine the effect of elevated IGF-IR levels on mammary acinus formation in three-dimensional (3D) culture (24, 47). Both groups found that overexpressed IGF-IR remained ligand dependent, but when stimulated by IGF-I, caused hyperproliferation, decreased apoptosis, and altered polarity, resulting in large, complex, disrupted acini. Blockade of phosphatidylinositol 3-kinase or extracellular signal-regulated kinase 1/2 (ERK1/2) blocked the formation of disrupted acini by MCF10A-IGF-IR cells (47).
Epithelial-to-mesenchymal transition (EMT) has been recognized as a cellular mechanism in normal development as well as, recently, in tumorigenesis (7, 21, 42), and reports strongly indicate that both invasion and metastasis may be dependent on the acquisition of EMT features by primary cancer cells (22, 46). Several transcription factors are central to EMT, including Snail, Slug, Twist, and Zeb1 (4, 22, 46).
IGF-I stimulation of breast cancer cells overexpressing IGF-IR has been shown to cause depolarization and a mesenchyme-like transition (20). Irie et al. noted that MCF10A cells overexpressing IGF-IR showed a subtle conversion, from a cuboidal epithelial morphology to a more spindle-shaped morphology (24), and Yanochko and Eckhart noted that MCF10A-IGF-IR cells showed altered E-cadherin localization (47).
The transcription factor nuclear factor κB (NF-κB) was recently shown to be essential for EMT and metastasis in a breast cancer cell model (22). NF-κB has recently been implicated in cell proliferation and various cancers, including breast cancer (9, 33). High levels of nuclear NF-κB were found in the majority of primary human and rodent breast tumor tissue samples, breast cancer cell lines, and carcinogen-transformed mammary epithelial cells (9, 27). IGF-IR can activate the NF-κB signaling pathway (29). IGF-IR can modulate glycogen synthase kinase 3β (GSK-3β) activity via Akt; GSK-3β regulates Snail via NF-κB (1); and NF-κB binds Snail promoter and increases its activity (3), suggesting a potential IGF-IR-GS3Kβ-NF-κB-Snail signaling pathway in EMT in MCF10A cells.
In this study, we established MCF10A human mammary epithelial cells that overexpress a constitutively active IGF-IR (CD8-IGF-IR-MCF10A). CD8-IGF-IR-MCF10A cells showed numerous features of transformation, including growth factor-independent proliferation, lack of contact inhibition, anchorage-independent growth, invasion, and tumorigenesis in vivo. This study is significant, as relatively few oncogenes, when expressed alone, have been shown to cause in vivo tumorigenic potential in MCF10A cells. In addition, CD8-IGF-IR-MCF10A cells showed an EMT which was associated with the downregulation of multiple epithelial cell markers and concomitant upregulation of many mesenchymal markers. Transcriptional repressor Snail mRNA levels were dramatically induced in CD8-IGF-IR-MCF10A cells, despite no change in Slug, Twist, or Zeb1. NF-κB was highly activated in CD8-IGF-IR-MCF10A cells, and the EMT and upregulation of Snail gene expression were reversed by the blockade of NF-κB or IGF-IR signaling. Therefore, constitutively activated IGF-IR activates NF-κB to induce Snail and cause EMT in MCF10A cells.
MATERIALS AND METHODS
Plasmid cloning.
Construction of the CD8-IGF-IR chimera has been described previously (8). For this study, we isolated CD8-IGF-IR from pcDNA3.1 using NheI and linearized pBabe-puro with BamHI. After the 5′ overhangs of each plasmid were filled in with Klenow enzyme (Promega, Madison, WI), they were ligated with T4 DNA ligase (Promega). Positive clones were identified by restriction digestion and confirmed by sequencing. For the cloning of the dominant negative human Snail (SnaDN), the human Snail gene including zinc finger domains and flanking regions was amplified with a forward primer (NM_005985.2; 5′-ccggatcctctgaggccaaggatctccag-3′) and reverse primer of the T7 promoter region of pCMV-Tag 2B. _Flag_-_Snail_-pCMV-Tag 2B was used as the template for PCR. A 459-bp PCR fragment was digested with BamHI and XhoI, resulting in a 380-bp fragment, and ligated with linearized pCMV-Tag 2B vector. Then, _Flag_-SnaDN was excised with BstXI and XhoI and blunted with Klenow enzyme. pLNCX2 vector was digested with HindIII and then blunted with Klenow enzyme. They were ligated with T4 DNA ligase and positive clones were confirmed by sequencing.
Cell culture, retrovirus production, and establishment of stable cell lines.
The immortalized human breast epithelial cell line MCF10A was cultured in Dulbecco's modified Eagle medium (DMEM)-F-12 (Invitrogen) supplemented with 5% horse serum (Sigma, St. Louis, MO), 20 ng/ml epidermal growth factor (Sigma), 10 μg/ml insulin (Sigma), 0.5 μg/ml hydrocortisone (Sigma), 100 ng/ml cholera toxin (Sigma), and 100 units/ml penicillin-streptomycin (Invitrogen, Carlsbad, CA). The retrovirus was generated by transfection of CD8-IGF-IR-pBabe-puro or pBabe-puro plasmid into PT67 packaging cells (BD Biosciences, San Jose, CA) using Lipofectamine reagent (Invitrogen). The supernatant was harvested 48 h after transfection and MCF10A cells were plated on 60-mm dishes the day before infection and were infected for 48 h with 1 ml of the supernatant containing retrovirus encoding CD8-IGF-IR or empty vector (pBabe-puro) as a control. Infected cells were selected with 5 μg/ml puromycin (Sigma) 48 h after infection, and individual clones were isolated 10 days later. The overexpression of CD8-IGF-IR was confirmed by immunoblot analysis with anti-IGF-IRβ antibody. Twelve individual positive clones were pooled together to rule out clonal artifacts and were named the CD8-IGF-IR-MCF10A pool. For the control clones, isolated individual clones (n = 5) were pooled together (vector-MCF10A). For CD8-IGF-IR-MCF10A cells overexpressing SnaDN, retrovirus was made by the same method as described above and used to infect CD8-IGF-IR-MCF10A cells. Infected cells were selected with 1 mg/ml G418 (Invitrogen), and positive clones were detected with anti-Flag antibody (Sigma).
Monolayer growth assay.
For growth curve experiments, cells were plated in 12-well plates at a concentration of 25,000 cells/well and the next day after cells were plated was designated day 0. For measuring cell numbers in serum-free conditions, cells were placed in serum-free media at day 0. Cell numbers were counted every 2 days using a Beckman Coulter Z series counter (Beckman Coulter, Fullerton, CA).
Anchorage-independent growth assay.
For anchorage-independent colony formation, 1 × 104 cells were suspended in their growth medium containing 0.35% SeaPlaque agarose (Cambrex Bio Science, Rockland, ME) and plated in 6-well plates over a base layer of complete medium containing 0.7% agarose. Colonies were stained with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (5 mg/ml in phosphate-buffered saline [PBS]) by incubation for 4 h at 37°C. Cells were photographed using an Alpha Imager 7000 (Alpha Innotech, San Leandro, CA).
3D Matrigel culture.
3D Matrigel culture was performed as described previously (12). Briefly, cells were treated with trypsin and first resuspended in DMEM-F-12 medium supplemented with 20% horse serum. The cells were centrifuged and resuspended in assay medium (DMEM-F-12 supplemented with 2% horse serum, 10 μg/ml insulin, 1 ng/ml cholera toxin, 100 μg/ml hydrocortisone, 50 U/ml penicillin, and 50 μg/ml streptomycin). The cells were counted and diluted in assay medium to achieve a final concentration of 25,000 cells/ml. Eight-chambered glass slides were coated with 60 μl Matrigel (BD Biosciences) per well and left to solidify for 30 min. The cells were mixed 1:1 with assay medium containing 4% Matrigel and 10 ng/ml epidermal growth factor (EGF), and 400 μl was added to each chamber of the Matrigel-coated slide. This corresponds to a final overlay solution of 5,000 cells/well in medium containing 2% Matrigel and 5 ng/ml EGF. Assay medium containing 5 ng/ml EGF and 2% Matrigel was replaced every 4 days. Immunostaining of acinar structures with antibodies to Ki-67 (Zymed) or laminin V (Chemicon) was performed as described previously (12). For nuclear staining, the acinar structures were fixed in 2% paraformaldehyde at room temperature for 20 min. Fixed structures were washed three times in PBS for 10 min each time. Nuclear staining was performed by incubation for 15 min with PBS containing 5 μM TOPRO-3 (Invitrogen) before cells were mounted with the antifade agent VECTASHIELD (Vector Laboratories, Burlingame, CA). Confocal analyses were performed with a Nikon Eclipse E1000 confocal microscopy system.
Migration and invasion assay.
The migration of cells was analyzed both through an in vitro wound-healing assay and a transwell chamber assay. After overnight serum starvation, monolayers of confluent cultures were lightly scratched with a pipette tip, washed with serum-free medium to remove detached cells, and then incubated with serum-free or serum-containing medium. Cultures were observed regularly for up to 24 h. For the transwell chamber assay, the polycarbonate membranes (8-μm pore size) (Becton Dickinson Labware) of the upper compartments of transwell culture chambers were coated with 10 μg/μl fibronectin. For the invasion assay, a BD Matrigel invasion chamber (BD biosciences) was used. A total of 3 × 104 cells were resuspended in 500 μl serum-free medium and placed in the upper compartment, and the lower compartment was immediately filled with 500 μl growth medium. After 24 h, the cells on the upper surface of the transwell were carefully removed with a cotton swab, the membrane was stained using a Hema 3 Stat pack (Fisher), and cells that passed through the membrane to the lower surface were counted in four different fields at ×10 magnification.
Xenograft studies.
All procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the IACUC of Baylor College of Medicine. Mice were maintained on a 12-h light, 12-h dark schedule with ad libitum access to laboratory chow (Picolab rodent diet 20 and lab diet 5053; PMI Nutrition International Inc., Brentwood, MO) and water. Vector-MCF10A and CD8-IGF-IR-MCF10A cells were harvested and 5 × 106 cells were injected orthotopically into the number 3 mammary fat pads of athymic nu/nu or scid/beige mice (Charles River, Wilmington, MA). The tumor sizes were measured with digital calipers twice a week, and tumor volumes were estimated according to the formula for an ellipse, (short dimension)2 × (long dimension)/2.
Immunoblot analysis.
Forty micrograms of total protein was resuspended in denaturing sample loading buffer (3% dithiothreitol, 0.1 M Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate, 0.2% bromophenol blue, 20% glycerol), separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and electrophoretically transferred to a nitrocellulose membrane overnight at 4°C. The remaining steps were all performed at room temperature. The membrane was blocked with PBS plus 0.05% Tween 20 (PBST) containing 5% nonfat milk (blocking solution) for 1 h followed by incubation with anti-pY1162/1163-IGF-IR (no. 44-804G; Biosource, Camarillo, CA), anti-IGF-IR beta (no. sc-713; Santa Cruz Biotechnology, Santa Cruz, CA), anti-N-cadherin (no. 610920; BD Transduction Laboratories), anti-vimentin (no. 550513; BD Transduction Laboratories), anti-beta-catenin (no. 610154; BD Transduction Laboratories), anti-alpha-catenin (no. sc-7894; Santa Cruz Biotechnology), anti-E-cadherin (no. sc-8426; Santa Cruz Biotechnology), anti-fibronectin (no. sc-9068; Santa Cruz Biotechnology), and anti-β-actin (no. A1978; Sigma) antibodies in blocking solution overnight at 4°C. Subsequently, the membrane was washed three times for 5 min each with PBST and then incubated with a horseradish peroxidase-linked secondary antibody (Amersham Pharmacia Biotech, Piscataway, NJ) at a dilution of 1:4,000 in blocking solution. After the membrane was washed three times for 5 min each with PBST, bands were visualized by enhanced chemiluminescence according to the manufacturer's protocol (Pierce Biotechnology, Rockford, IL) and captured using an Alpha Innotech 7000 imaging system (Alpha Innotech, San Leandro, CA).
Immunofluoresence.
1 × 105 cells were seeded onto two chamber slides (Falcon), and 2 days later, the medium was changed, with or without 1 μM BMS-536924 (45), and the incubation was continued for an additional 24 h. The cells were washed with PBS, fixed for 20 min in 2% paraformaldehyde, and then washed three times with PBS. The cells were permeabilized with 0.5% Triton X-100 in PBS for 20 min, washed three times with PBS, and then blocked with 10% normal goat serum (Jackson Immunoresearch Laboratories Inc., West Grove, PA) in PBST. The cells were stained using monoclonal anti-E-cadherin antibody (BD Transduction Laboratories) and anti-NF-κB antibody (Santa Cruz Biotechnology) at 1:200 dilution in blocking solution overnight at 4°C, and then washed three times for 10 min in PBST buffer. Cells were then stained with goat anti-mouse Alexa Fluor 488 (Invitrogen) at 1:200 dilution in blocking solution, followed by three washes of 10 min in PBST. The slides were mounted using mounting medium (Vectorshield; Vector Laboratories, Burlingame, CA).
Immunohistochemistry.
Vector-MCF10A and CD8-IGF-IR-MCF10A cells were cultured in maintenance media and then placed in serum-free medium (SFM) for 24 h. Cells were trypsinized, fixed briefly with 4% paraformaldehyde, and processed into paraffin. Slides were cut at 3 to 4 microns, baked overnight at 58°C, and deparaffinized using a Shandon-Lipshaw Varistain (program 2). Heat-induced antigen retrieval was performed in 1 mM EDTA (pH 8.0) (pY-IGF-IR) or in 0.1 M Tris-HCl (pH 9.0) (total IGF-IR) for 5 min. All further incubations were performed at room temperature, and all washing was performed with TBST (0.15 M NaCl, 0.01 M Tris-HCl [pH 7.4], 0.05% Tween 20) unless otherwise stated. Endogenous peroxidase activity was blocked by incubation in 3% hydrogen peroxide solution for 5 min. The slides were then incubated with anti-pY1131-IGF-IR (catalogue no. 3021; Cell Signaling Technology) or anti-IGF-IR beta (catalogue no. sc-713; Santa Cruz) for 1 h, biotinylated secondary antibody (1:200) for 30 min, and then horseradish peroxidase-labeled avidin (1:200) for 30 min. As a negative control, the slides were incubated with purified rabbit immunoglobulin (Jackson Laboratories). Detection was achieved by incubation with diaminobenzidine (Dako) for 15 min, followed by enhancement with 0.2% osmium tetroxide for 30 s. The slides were counterstained with hematoxylin, dehydrated, and mounted using cytoseal.
RNA isolation and quantitative real-time PCR.
Total RNA was prepared with an RNeasy mini kit (QIAGEN, CA) according to the instruction manual. Total RNA was reverse transcribed in a final volume of 100 μl using 0.5 μg of total RNA, 0.5 μl random primers, and 25 mM deoxynucleoside triphosphate. After the samples were heated for 5 min at 65°C, 20 μl of 5× first-strand buffer (Invitrogen), 5 μl 0.1 M dithiothreitol, and 0.5 μl of Superscript II RNase H reverse transcriptase (Invitrogen) were added. Reverse transcription was performed on a 9600 GeneAmp PCR system at 25 °C for 5 min, 48°C for 30 min, and 70°C for 10 min. The cDNA was stored at −20°C. Quantitative real-time PCR (Q-PCR) was performed using primers that were already published for human E-cadherin, Twist, and N-cadherin (37); fibronectin (36); Snail and ZEB1 (34); alpha-smooth muscle actin (α-SMA) (44); and Slug (40); and we designed primers for vimentin which consisted of forward 5′-CAACCTGGCCGAGGACAT-3′ and reverse 5′-ACGCATTGTCAACATCCTGTCT-3′. Q-PCR was performed in an ABI PRISM 7700 sequence detector (PE Biosystems) using the power SYBR green PCR master mix (Applied Biosystems). Each reaction mixture contained 5 μl of template cDNA, a final concentration of 0.15 μM of forward and reverse primers, 12.50 μl of 2× SYBR green buffer, and RNase-free H2O to make a final volume of 25 μl. The PCR for each gene consisted of the following: 50°C for 2 min, denaturing at 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 30 s. To determine the primer specificity, three stages (95°C for 15 s, 60°C for 20 s, and 95°C for 15 s, with a ramping time of 20 min) were added to the end of the PCR to obtain dissociation curves for each gene. Analysis of Q-PCR data was performed using Sequence Detector v. 1.7 software (ABI). Relative transcript levels were determined using the 2−ΔΔ_CT_ method and normalized to β-actin (28).
Preparation of nuclear extracts and electrophoretic mobility shift assays.
Nuclear extracts and electrophoretic mobility shift assays were prepared as previously described (43). Nuclear extracts (10 μg) were incubated with 32P-labeled NF-κB or OCt-1 probe (Promega, Madison, WI) for 15 min at room temperature. The mixtures were then subjected to electrophoresis in 6% polyacrylamide gel and autoradiography.
RESULTS
Characterization of immortalized mammary epithelial MCF10A cells expressing a CD8-IGF-IR fusion gene.
We recently reported the generation of a chimeric and constitutively activated IGF-IR that consists of a fusion of the human CD8α extracellular domain with the human IGF-IRβ subunit. CD8-IGF-IR was created to study the effect of IGF-IR in vivo without the complexity of IGF-I ligand and binding protein interactions. Expression of this chimeric receptor in the mammary glands of transgenic mice resulted in rapid mammary tumorigenesis (8) that was associated with an apparent EMT (data not shown). To further study the role of CD8-IGF-IR in transformation and EMT, we utilized MCF10A cells, an immortalized but nontransformed human mammary epithelial cell line (11). These cells exhibit numerous features of normal breast epithelium, including lack of tumorigenicity in nude mice, lack of anchorage-independent growth, and dependence on growth factors and hormones for proliferation and survival. MCF10A cells form polarized functional acini in 3D Matrigel culture and have proven to be a useful model for the study of oncogene disruption of polarity and acinus formation (11). To determine the effect of constitutive receptor activation on MCF10A immortalized human mammary epithelial cells, we generated stable cell lines by retroviral infection. Multiple clones expressing CD8-IGF-IR were identified, and these were combined together to generate a pool (CD8-IGF-IR-MCF10A) for further experiments. Clones expressing empty vector were combined to make a control pool clone (vector-MCF10A). Next we examined whether the overexpressed CD8-IGF-IR was constitutively active in the absence of ligand, IGF-I. Vector-MCF10A cells stimulated with IGF-I showed weak tyrosine phosphorylation of the β-subunit of IGF-IR (P∼IGF-IR) (Fig. 1A). CD8-IGF-IR has a molecular mass of approximately 75 kDa and exists as a doublet below the endogenous IGF-IRβ subunit (95 kDa), and it was constitutively phosphorylated without ligand. The activation was highly dominant because there was no additive or synergistic effect of IGF-I on these cells. Cells were then cultured in serum-free media, placed in paraffin, and immunohistochemistry (IHC) was performed to determine P∼IGF-IR and total IGF-IR levels. CD8-IGF-IR-MCF10A cells had readily detectable P∼IGF-IR staining that was mainly membranous and was completely absent in vector-MCF10A cells (Fig. 1B). IHC for total IGF-IR showed an approximately fivefold increase in their levels, which was consistent with the results of the immunoblotting (Fig. 1A).
FIG. 1.
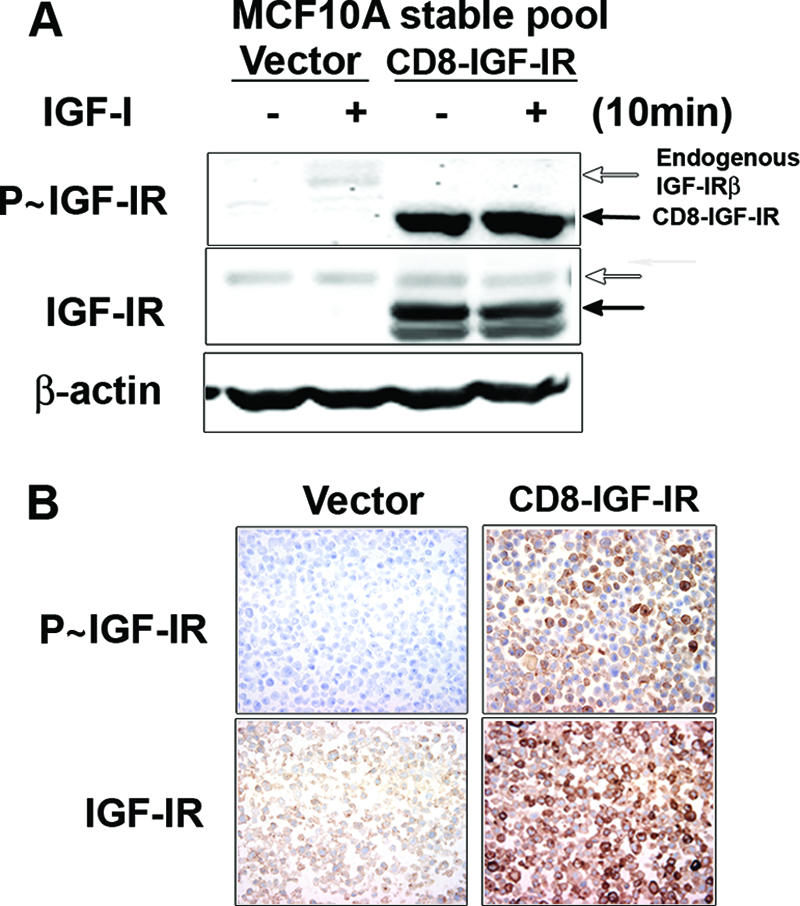
Establishment of MCF10A stable cell lines overexpressing CD8-IGF-IR. (A) Vector-MCF10A and CD8-IGF-IR-MCF10A pools were incubated in SFM for 24 h and then stimulated with IGF-I (10 nM) for 10 min. Forty-microgram amounts of cell lysates were immunoblotted with anti-pY-IGF-IR (P∼IGF-IR) or total IGF-IRβ antibodies. The open and black arrows indicate endogenous IGF-IRβ and CD8-IGF-IR, respectively. β-Actin was used as a loading control. − and + indicate the absence or presence of IGF-I. (B) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were cultured in SFM, harvested, and processed in paraffin. Five-micrometer sections were then processed for IHC using anti-pY-IGF-IR (P∼IGF-IR) or total IGF-IRβ antibodies.
CD8-IGF-IR disrupts normal mammary acinus formation in 3D Matrigel culture.
MCF10A cells grown in 3D Matrigel recapitulate numerous features of breast epithelium in vivo, including the formation of acinus-like spheroids with a hollow lumen, apicobasal polarization, and the basal deposition of basement membrane components (12). Therefore, we examined whether overexpressing CD8-IGF-IR would disrupt the normal mammary acinar morphogenesis of MCF10A cells in 3D Matrigel culture. As shown in Fig. 2A and B, vector-MCF10A cells formed small, well-formed acini, often with empty lumen. In contrast, CD8-IGF-IR-MCF10A acini appeared larger and disrupted, with cells filling the luminal space. The acini sometimes showed protrusions into the Matrigel (Fig. 2A; day 8) that are characteristic of invasive cells (Fig. 2A; arrows). Ki-67, a marker for proliferation, was highly expressed in the multiacinus structure of CD8-IGF-IR-MCF10A cells on day 12, while it was hard to detect Ki-67 staining in the growth-arrested acini of vector-MCF10A cells on the same day (Fig. 2B, upper panels), suggesting that constitutively active IGF-IR signaling induces hyperproliferation of the mammary epithelial cells. Vector-MCF10A acini often had cells in the center of the lumen that were undergoing apoptosis and stained positive for cleaved caspase-3 (Fig. 2B, left middle panel). CD8-IGF-IR cells showed no apoptosis as assessed by cleaved caspase-3 staining, consistent with the observed filling of the lumen (Fig. 2B, right middle panel). The staining of basement membranes of acinar structures with laminin V, a marker of epithelial cell polarity, showed that vector-MCF10A cells formed well-organized acinar structure with apical/basal polarity, while the multiacini of CD8-IGF-IR-MCF10A cells had disrupted polarity, with laminin V staining within the lumen (Fig. 2B, lower panels).
FIG. 2.
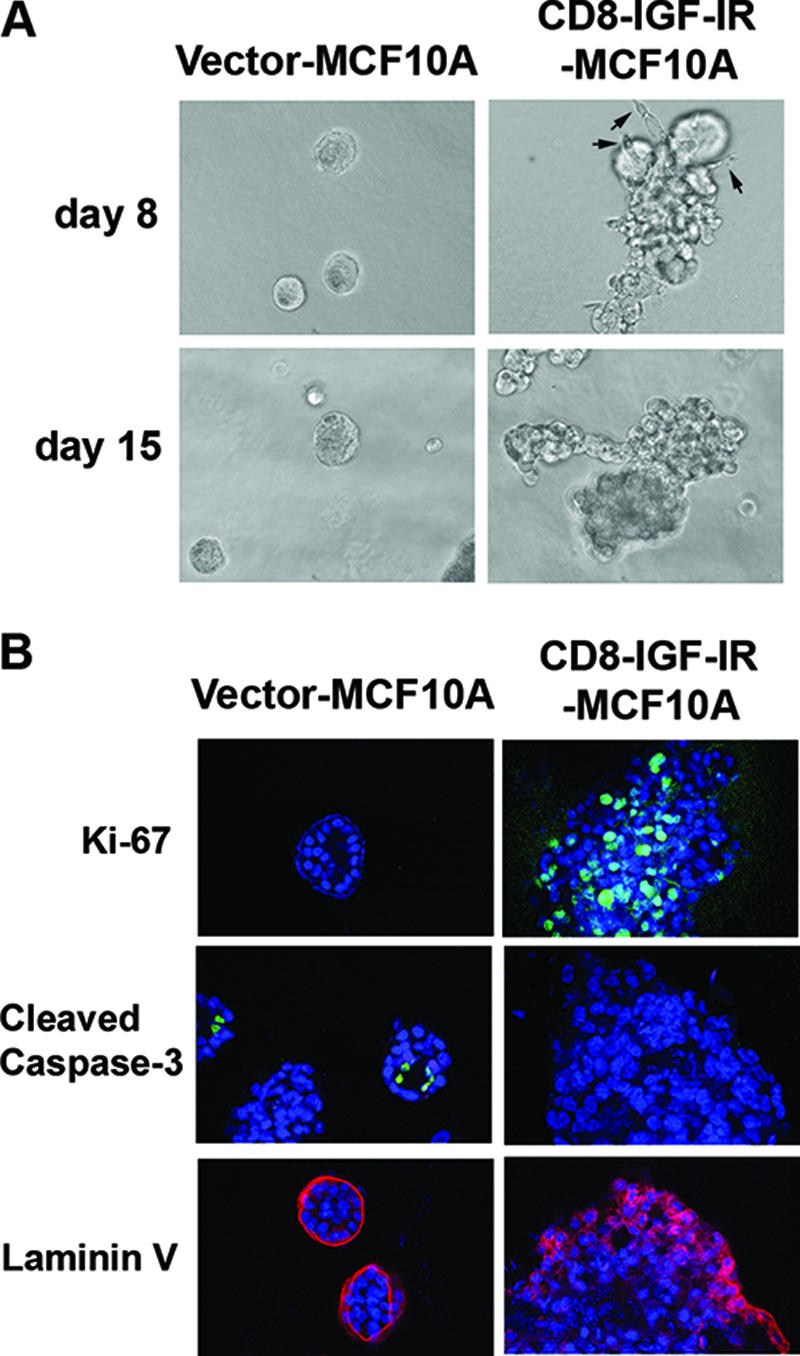
Constitutively active IGF-IR disrupted acinar morphogenesis of MCF10A cells on 3D Matrigel culture. (A) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were plated in Matrigel and cultured for 8 days or 15 days. Phase-contrast images of acini are shown in ×20 magnification. The arrow indicates invasive protrusions. (B) The cells were cultured on Matrigel for 8 to 12 days and then stained with antibodies to Ki-67 (green), cleaved caspase-3 (green) or laminin V (red). The nuclei (blue) were labeled with TOPRO-3 and visualized by confocal microscopy with ×40 magnification.
CD8-IGF-IR causes transformation of MCF10A cells.
To evaluate whether the overexpression of CD8-IGF-IR caused transformation of MCF10A cells, we measured several hallmarks of the transformed cell (Fig. 3). Several experiments repeatedly indicated that the monolayer proliferation of CD8-IGF-IR-MCF10A cells was slightly slower than that of vector-MCF10A cells in normal growth media (Fig. 3A). In contrast, under SFM conditions, vector-MCF10A cells underwent growth arrest, whereas CD8-IGF-IR-MCF10A cells continued to proliferate. Furthermore, CD8-IGF-IR-MCF10A failed to exhibit contact inhibition and formed foci after long-term culture (Fig. 3B). Consistent with their nontransformed phenotype, vector-MCF10A cells failed to grow in soft agar, whereas CD8-IGF-IR-MCF10A cells repeatedly formed numerous colonies (Fig. 3B). Similarly, vector-MCF10A cells showed very poor ability to invade through Matrigel when a modified Boyden chamber was used, whereas CD8-IGF-IR-MCF10A cells were highly invasive (Fig. 3C).
FIG. 3.
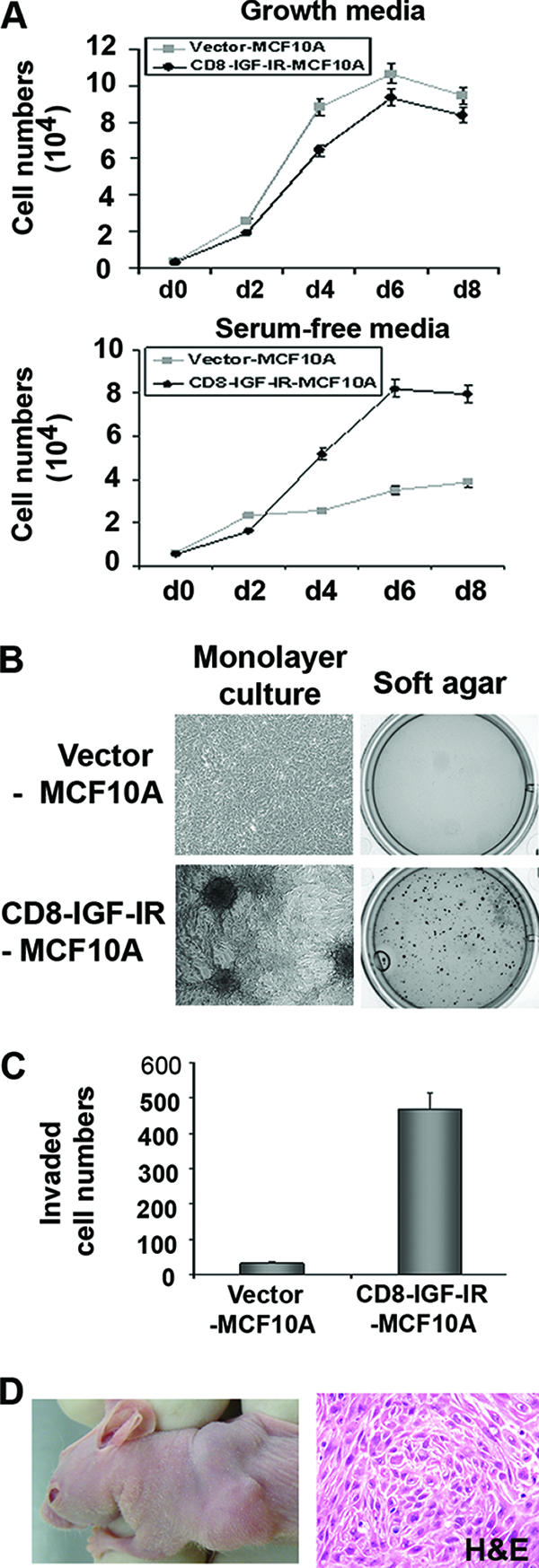
Constitutive IGF-IR activation induced cellular transformation of nontransformed MCF10A cells. (A) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were plated on 12-well plates, and the next day, the medium was changed for serum-free medium to compare the growth rates of these cells in serum-free conditions. The next day after plating was designated day 0 (d0). Cell numbers were counted every 2 days. The error bars represent the standard errors of the means. (B) For monolayer culture, 2 × 105 cells were plated on 6-well plates and cultured for 10 days. Phase-contrast images of focus formation at day 10 are shown in the left panels. For the anchorage-independent growth assay, 1 × 104 cells were suspended in their growth medium containing 0.35% agarose and plated on 6-well plates over a basal layer of complete medium containing 0.7% agarose. Two weeks later, colonies were stained with MTT (right panels). (C) The Matrigel invasion assay showed that the number of CD8-IGF-IR-MCF10A cells that penetrated the membrane was significantly higher than that of vector-MCF10A cells. Cells were counted in four different microscopic fields (×10 magnification). The error bars represent the standard errors of the means. (D) Six-week-old mice were injected with CD8-IGF-IR-MCF10A cells in the number 3 mammary gland. The left panel is a representative photograph of a xenograft at 15 days after injection. The right panel is a representative section of tumor stained with hematoxylin and eosin.
Numerous oncogenes are able to disrupt MCF10A acinus formation and transform MCF10A cells in vitro. However, oncogenes such as ErbB2 disrupt acinus formation (11) but fail to convey in vivo xenograft growth (16). We therefore examined whether CD8-IGF-IR-MCF10A cells could grow in immunocompromised mice. In an initial pilot experiment, we injected vector-MCF10A and CD8-IGF-IR-MCF10A cells with or without Matrigel orthotopically into athymic nude mice. We found that vector-MCF10A cells injected into athymic nude mice were unable to form a palpable mass when injected with (0/3 mice) or without (0/3 mice) Matrigel. In contrast, CD8-IGF-IR-MCF10A cells formed a palpable mass within 7 to 10 days (3/3 mice with Matrigel and 1/3 mice without Matrigel) (Fig. 3D). Histologic analysis of the tumors by hematoxylin and eosin staining revealed them to be undifferentiated carcinomas. Next, we repeated the experiment, and in results similar to those of our pilot experiment, Matrigel caused an increase in tumor take and tumor size after 10 days, with palpable tumors appearing in 7/10 mice with Matrigel and 5/10 without Matrigel. After 3 weeks, xenografts injected with Matrigel had reached a volume of 120.3 ± 3.3 mm3, whereas the xenografts without Matrigel were only 56 ± 21.8 mm3. Taken together, these data indicate that overexpression of a single oncogene (CD8-IGF-IR) in MCF10A cells is sufficient to cause transformation.
CD8-IGF-IR causes EMT in MCF10A cells.
The expression of CD8-IGF-IR in the mammary glands of transgenic mice resulted in rapid mammary tumorigenesis (8) that was associated with an apparent EMT (data not shown). Interestingly, when MCF10A cells were infected with CD8-IGF-IR and were screened to identify those with stable CD8-IGF-IR expression, it was noted that the majority of the puromycin-resistant clones exhibited a morphological change that resembled an EMT. As shown in Fig. 4A, vector-MCF10A cells showed highly organized cell-cell adhesion and cell contact, whereas CD8-IGF-IR-MCF10A cells had an elongated and refractive appearance, with cell scattering and loss of cell-cell contacts. The cobblestone-like morphology of MCF10A cells at confluence was replaced in CD8-IGF-IR-MCF10A cells by a spindle-like fibroblastic morphology. To determine the molecular alterations that occurred in these cells, we examined the expression of the epithelial adherence junction protein E-cadherin and the mesenchymal proteins vimentin and N-cadherin. In 3D culture, vector-MCF10A cells showed high levels of membrane-localized E-cadherin and little or no expression of vimentin or N-cadherin. In stark contrast, CD8-IGF-IR-MCF10A acini showed lower levels of E-cadherin, and the E-cadherin was mislocalized, showing punctate staining. In addition, the CD8-IGF-IR-MCF10A acini showed strong staining of both vimentin and N-cadherin. Immunoblot analysis showed that epithelial adherence junction proteins were dramatically downregulated in CD8-IGF-IR-MCF10A cells compared to their strong expression in vector-MCF10A cells (Fig. 4B). E-cadherin mRNA levels were reduced 10-fold in the CD8-IGF-IR-MCF10A cells compared to their levels in vector-MCF10A cells (Fig. 5A), and this correlated with a twofold reduction in E-cadherin promoter activity (Fig. 4C). The expression of mesenchymal markers, including vimentin, N-cadherin, fibronectin, and α-SMA, were all low or absent in vector-MCF10A cells but highly induced in CD8-IGF-IR-MCF10A cells (Fig. 4B).
FIG. 4.

EMT was induced in CD8-IGF-IR-MCF10A cells. (A) The morphologies of MCF10A cells expressing either the control vector or CD8-IGF-IR were revealed by phase-contrast microscopy (×20 magnification) at day 3. Vector-MCF10A and CD8-IGF-IR-MCF10A cells were cultured on Matrigel for 12 days and then stained with antibodies to E-cadherin (green), vimentin (red), or N-cadherin (red). The nuclei (blue) were labeled with TOPRO-3 and visualized by confocal microscopy with ×40 magnification. (B) Levels of epithelial proteins, including E-cadherin, β-catenin, and α-catenin, and mesenchymal proteins, including N-cadherin, vimentin, fibronectin, and α-SMA, in vector-MCF10A and CD8-IGF-IR-MCF10A cells were examined by immunoblotting. β-Actin was used as a loading control. (C) The activity of a 2-kb fragment of the E-cadherin promoter upstream of a luciferase reporter (E-cad-luc) was compared to that of the control plasmid (pGL2-basic) by transient transfection. The luciferase (luc) activity was normalized to the cotransfected β-galactosidase activity. The error bars represent the standard errors of the means. (D) Results of the transwell migration assay in which vector-MCF10A and CD8-IGF-IR-MCF10A cells were induced to migrate toward growth media. The migrated cells that passed through the membrane to the lower surface were counted in four different microscopic fields at ×10 magnification. (E) Confluent vector-MCF10A and CD8-IGF-IR-MCF10A cells were scratched and photographs were taken immediately (0 h) and 24 h postscratch at ×10 magnification.
FIG. 5.
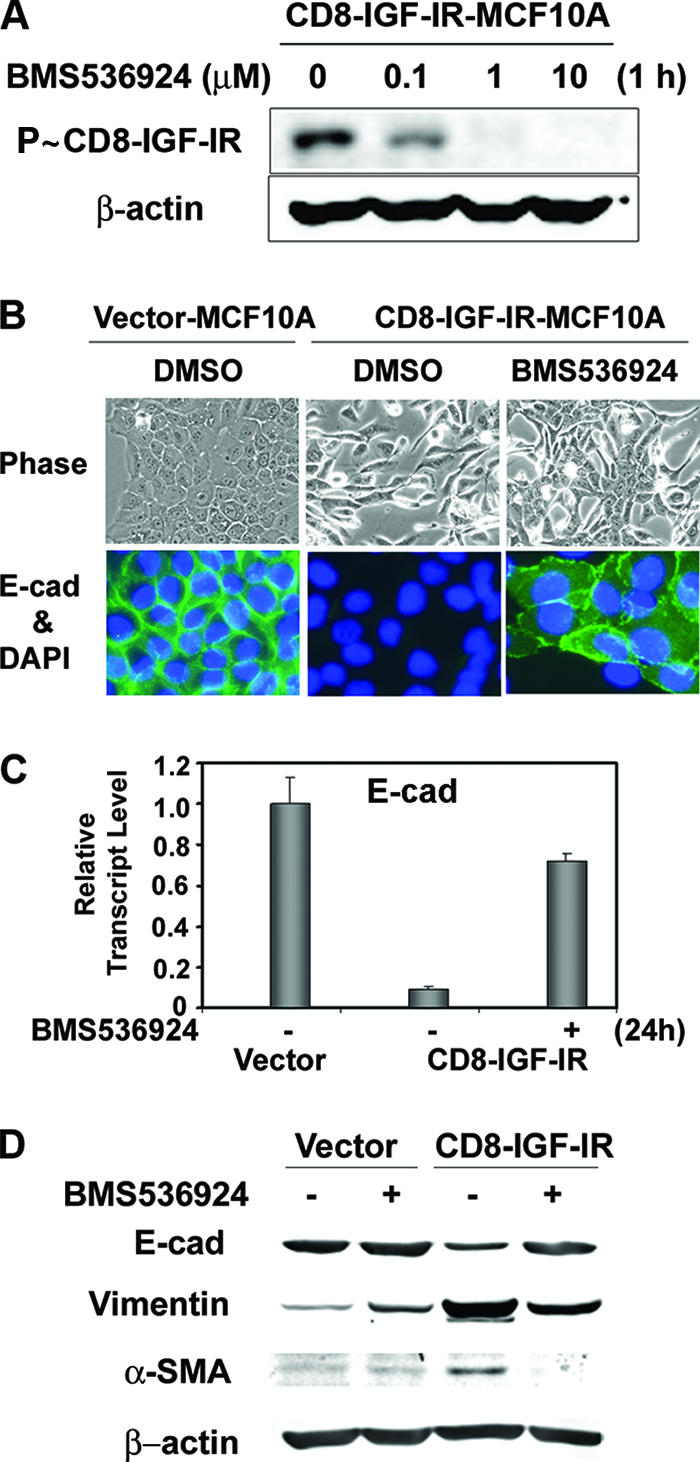
Blocking of constitutive IGF-IR activation by a new small-molecule inhibitor of IGF-IR, BMS-536924, partially reversed the mesenchyme-like morphological changes and the downregulation of E-cadherin levels. (A) CD8-IGF-IR-MCF10A cells were incubated with the indicated concentration of BMS-536924 for 1 h after 16 h of serum starvation, and the phosphorylation of CD8-IGF-IR was analyzed by immunoblotting with anti-P∼IGF-IR antibody. β-Actin was used as a loading control. (B) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were incubated with or without 1 μM BMS-536924 for 24 h and were either visualized by phase-contrast microscopy at ×20 magnification (upper panels) or stained with E-cadherin (E-cad) (×40 magnification; green) (lower panels). The nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI). DMSO, dimethyl sulfoxide. (C) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were incubated with (+) or without (−) 1 μM BMS-536924 for 24 h in growth media. The levels of E-cadherin mRNA were examined by Q-PCR. The results are presented as relative transcript levels compared to vector-MCF10A by using the ΔΔ_CT_ method, with β-actin mRNA levels used as the normalization control. The error bars represent the standard errors of the means. (D) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were incubated with (+) or without (−) BMS-536924 for 24 h and lysed, and the levels of epithelial (E-cadherin) or mesenchymal markers (vimentin and α-SMA) were measured. β-Actin was used as a loading control.
Therefore, both the morphological and molecular changes in the CD8-IGF-IR-MCF10A cells demonstrated that these cells had undergone EMT. The functional consequences of EMT are enhanced cell scattering and migration. Indeed, both the fibronectin-coated transwell chamber assay and the wound-healing assay showed that CD8-IGF-IR-MCF10A cells migrated much faster than vector-MCF10A cells, which were generally unable to migrate (Fig. 4D and E).
EMT and E-cadherin downregulation are reversed by blockade of IGF-IR activity with BMS-536924.
To investigate whether the EMT phenotype of CD8-IGF-IR-MCF10A cells is a specific event mediated by the constitutively active IGF-IR, cells were treated with a small-molecule inhibitor of IGF-IR (BMS-536924) (45). Immunoblot analysis showed that constitutive phosphorylation of CD8-IGF-IR was inhibited in a dose-dependent manner by BMS-536924, with an observed effect at 100 nM of the drug and maximal inhibition at 1 μM (Fig. 5A). CD8-IGF-IR-MCF10A cells treated with BMS-536924 for 24 h showed a partial reversion to a cobblestone-like epithelial morphology, and immunofluorescence results showed that E-cadherin levels were also partially reversed; however, the E-cadherin was not completely properly localized, having some cytoplasmic localization (Fig. 5B). Consistent with a decrease in E-cadherin protein and promoter activity in CD8-IGF-IR-MCF10A cells (Fig. 4C), E-cadherin mRNA levels were reduced 10-fold compared to its levels in vector-MCF10A cells (Fig. 5C). Importantly, this downregulation of E-cadherin mRNA was reversed (by up to 80%) with BMS-536924 (Fig. 5B). We also examined the ability of BMS-536924 to reverse changes in the protein levels of epithelial and mesenchymal markers in CD8-IGF-IR-MCF10A cells; we found that BMS-536924 reversed the reduction of E-cadherin protein levels and also reversed the induction of vimentin and α-SMA (Fig. 5D). It should be noted that, despite the ability of IGF-IR inhibition to reverse the levels of these proteins, the morphological reversion was incomplete, suggesting that there may be irreversible changes that have caused the EMT, or that other pathways, in addition to IGF-IR, need to be inhibited.
Snail transcription factor is an essential regulator of constitutively active IGF-IR-induced EMT.
Since it is known that Snail, Slug, Twist, and Zeb1 are repressors of E-cadherin promoter activity and the subsequent reduction of E-cadherin mRNA and protein levels is a central component of EMT (23), we examined the mRNA expression levels of Snail, Slug, Twist, and Zeb1 by Q-PCR. Snail mRNA was dramatically induced (up to 80-fold) in CD8-IGF-IR-MCF10A cells, whereas no changes were observed for Slug, Twist, or Zeb1, even though mRNA was detected for all three genes (Fig. 6A). Next, we tested whether IGF-I can also induce Snail mRNA in vector-MCF10A cells. Figure 6B shows that IGF-I treatment induced Snail mRNA, even though the induction was not as strong as that in CD8-IGF-IR-MCF10A cells. The induction of Snail mRNA by IGF-I treatment, or in CD8-IGF-IR-MCF10A cells, was mostly reversed by treatment with BMS-536924. However, it should be noted that the inhibitor did not fully reverse the induction by IGF-I or CD8-IGF-IR, suggesting either incomplete blockade of IGF-IR, or that there are other pathways mediating the induction. To show whether Snail was necessary for the downregulation of E-cadherin and the EMT, we generated a SnaDN that consisted of only the zinc finger domains and small flanking regions of wild-type Snail and introduced this construct into CD8-IGF-IR-MCF10A cells by retroviral infection (CD8-IGF-IR/SnaDN-MCF10A cells). Monolayer growth of these cells showed that they had an epithelium-like morphology that was somewhat similar to that of vector-MCF10A cells (Fig. 6C, top panels). The expression of SnaDN in CD8-IGF-IR-MCF10A cells also reversed the abnormal 3D growth of CD8-IGF-IR-MCF10A cells, with acini now appearing smaller and similar to those of vector-MCF10A cells (Fig. 6C). Consistent with this partial morphological reversion, the CD8-IGF-IR/SnaDN-MCF10A cells showed elevated levels of E-cadherin (Fig. 6D).
FIG. 6.
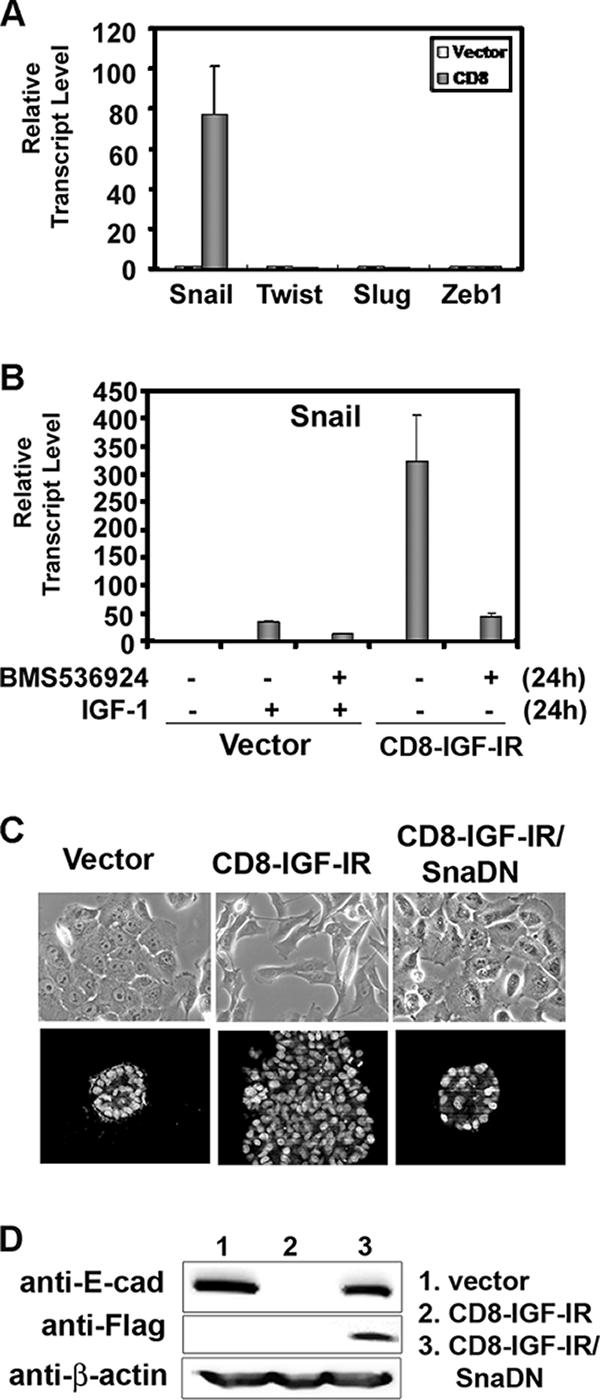
Snail is the essential mediator of the EMT induced by the constitutively active IGF-IR signaling. (A) The expression of Snail, Slug, Twist, and Zeb1 mRNA in vector-MCF10A (open bar) and CD8-IGF-IR-MCF10A (shaded bar) cells was examined by Q-PCR. The results are presented as transcript levels relative to the level in vector-MCF10A cells by using the ΔΔ_CT_ method, with β-actin mRNA levels used as the normalization control. (B) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were incubated with (+) or without (−) 100 ng/ml IGF-I and 1 μM BMS-536924 for 24 h in growth media. The expression of Snail mRNA was examined by Q-PCR as described in Materials and Methods. The values shown are relative to those of vector-MCF10A cells without IGF-I stimulation. The error bars represent the standard errors of the means. (C) Vector-MCF10A, CD8-IGF-IR-MCF10A, and CD8-IGF-IR/SnaDN-MCF10A cells were grown in their growth media for 2 days. The cells were visualized by phase-contrast microscopy at ×20 magnification (upper panels). The cells were cultured on Matrigel for 8 days, and then the nuclei were labeled with TOPRO-3 and visualized by confocal microscopy with ×40 magnification (lower panels). (D) Vector-MCF10A, CD8-IGF-IR-MCF10A, and CD8-IGF-IR/SnaDN-MCF10A cells were immunoblotted for E-cadherin (E-cad) and Flag. β-Actin was used as a loading control.
NF-κB is the upstream regulator of Snail expression in the regulation of CD8-IGF-IR-induced EMT.
We next examined pathways downstream of CD8-IGF-IR, which caused the dramatic elevation of Snail mRNA levels. The most-studied regulator of Snail promoter and mRNA levels is ERK1/2 (13). However, we found that incubation of CD8-IGF-IR-MCF10A cells with the ERK1/2 inhibitor U0126 (10 μM) had absolutely no effect on the 80-fold elevation of Snail mRNA levels in CD8-IGF-IR-MCF10A cells (Fig. 7A). U0126 also had no effect upon the morphology of CD8-IGF-IR-MCF10A cells (Fig. 7E). This is consistent with our observation that ERK1/2 is not active in these cells (data not shown). Recent reports indicated that NF-κB is an essential regulator of EMT and metastasis in a breast cancer cell model (23) and that NF-κB can bind the human Snail promoter and increase its activity (3). It was also reported that IGF-I stimulated sustained activation of NF-κB in human multiple myeloma cells (29). To determine whether the increased NF-κB was involved in the induction of Snail and EMT, we used inhibitors of the NF-κB signaling pathway. Treatment of CD8-IGF-IR-MCF10A cells for 24 h with two different inhibitors of the NF-κB pathway (IκB kinase [IKK] inhibitor II and Helenalin) partially reversed the induction of Snail mRNA in CD8-IGF-IR-MCF10A cells, and this reversion was similar to the results observed with BMS-536924 treatment (Fig. 7B).
FIG. 7.

NF-κB is the upstream regulator of Snail in CD8-IGF-IR-induced EMT. (A) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were incubated with dimethyl sulfoxide (DMSO) (open bars) or 10 μM U0126 (shaded bars) for 24 h, and then Snail mRNA levels were measured by Q-PCR as described in Materials and Methods. Representative levels are compared to those of vector-MCF10A cells incubated with DMSO. The bars represent the averages ± standard errors of the means of three measurements. (B) Vector-MCF10A and CD8-IGF-IR-MCF10A cells were plated and the next day, cells were incubated with or without 1 μM BMS-536924 (BMS) and two NF-κB pathway inhibitors, including 20 μM IKK inhibitor II (IKK) and 20 μM Helenalin (Hele), for 24 h. Snail mRNA levels were measured by Q-PCR as described above for panel A. DMSO was used as the vehicle for the reagents. The transcript levels are relative to the level in vector-MCF10A cells incubated with DMSO. The bars represent the averages ± standard errors of the means of three measurements. (C) The cells were cultured for 2 days and then stained with anti-NF-κB p65 antibody (green). The nuclei (blue) were stained with TOPRO-3 and visualized by confocal microscopy (×60 magnification with oil). (D) The cells were harvested at day 1 or day 2, and nuclear extracts were prepared and incubated with oligonucleotides corresponding to NF-κB or OCt-1 consensus sequences. OCt-1 binding was used as a loading control. (E) Vector-MCF10A (a) and CD8-IGF-IR-MCF10A (b to e) cells were incubated with DMSO (a and b), BMS-536924 (c), 20 μM IKK inhibitor II (d), or 10 μM U0126 (e) for 24 h, and photographs were taken with phase-contrast microscopy at ×20 magnification. (F) Vector-MCF10A (a) and CD8-IGF-IR-MCF10A (b to d) cells were grown in 3D Matrigel and then incubated with DMSO (a and b), 1 μM BMS (c), or 20 μM IKK inhibitor II (d). They were harvested at day 12 and then stained with TOPRO-3 for nuclear staining.
We next examined NF-κB activity in CD8-IGF-IR-MCF10A cells. First, we compared the nuclear localization of the p65 subunit of NF-κB in both vector-MCF10A cells and CD8-IGF-IR-MCF10A cells, because activated NF-κB translocates from the cytoplasm to the nucleus. Immunofluorescence staining of p65 showed exclusive cytoplasmic staining in vector-MCF10A cells; however, p65 was detected in the nuclei of CD8-IGF-IR-MCF10A cells (Fig. 7C). To confirm whether nuclear NF-κB is active, gel mobility shift assays were performed. The results shown in Figure 7D show that NF-κB binding activity was highly induced in CD8-IGF-IR-MCF10A cells compared to the activity in vector-MCF10A cells.
Finally, we examined whether blockade of NF-κB activity could reverse the EMT phenotype of CD8-IGF-IR-MCF10A cells in a manner similar to that observed with blockade with the IGF-IR inhibitor BMS-536924. CD8-IGF-IR-MCF10A cells in monolayer culture were elongated and refractile compared to MCF10A cells, which showed a classic cobblestone epithelial appearance. Treatment of CD8-IGF-IR-MCF10A cells with IKK inhibitor II partially reversed the EMT phenotype, with cells now showing more cell attachment (Fig. 7E). This partial reversion was similar to that seen with BMS-536924 and was not seen with U0126 (Fig. 7E). We also tested the same inhibitors in 3D culture and found that both IKK and BMS-536924 caused marked alterations in the growth of CD8-IGF-IR-MCF10A cells, with smaller acini that resembled those of vector-MCF10A cells (Fig. 7F).
DISCUSSION
IGF-IR is a critical regulator of cell proliferation and survival. Consistent with this, this receptor also has a role in tumorigenesis and metastasis and is an attractive target for cancer therapy (38). We recently reported the generation and characterization of the first transgenic mouse overexpressing IGF-IR (8). In this model, we found that overexpression of a constitutively active IGF-IR chimera (CD8-IGF-IR) in the mouse mammary gland led to rapid mammary tumorigenesis (8), finally resulting in metastasis to the lungs (unpublished data). In this report, we show that overexpression of the same CD8-IGF-IR chimera in MCF10A immortalized mammary epithelial cells caused transformation when cells were grown in immunocompromised mice. Furthermore, the cells underwent EMT, which was associated with increased NF-κB activity, induction of Snail transcriptional repressor, and downregulation of E-cadherin. The EMT was correlated with dramatically enhanced migration and invasion, and the induction of the Snail and EMT phenotypes could be reversed by small-molecule inhibitors of IGF-IR and NF-κB.
MCF10A cells are a useful tool for examining the effects of oncogenes on cell polarity and transformation. Several “classic” oncogenes, such as HER-2, activated Ras, and cyclin D1, have been shown to disrupt cell polarity and cause transformation in vitro; however, all are unable to confer xenograft growth in vivo (11). In contrast to those results, other, less-studied oncogenes, including these for growth hormone (51), ephA2 (49), and αB-crystallin (31), have recently been shown to confer xenograft growth on MCF10A cells. Similarly, we have shown that CD8-IGF-IR alone can cause transformation of MCF10A cells in vitro and that it also confers growth in athymic nude mice when the cells are injected, with or without Matrigel.
During this study, two groups reported on the overexpression of wild-type IGF-IR in MCF10A cells (24, 47). Both groups found that overexpressed IGF-IR remained ligand dependent but, when hyperstimulated by IGF-I, caused increased proliferation, decreased apoptosis, and loss of polarity. This resulted in large, complex, disrupted acini; however, both studies found that cells were noninvasive. We found a similar induction of proliferation, absence of apoptosis, and loss of polarity that resulted in disrupted acinus formation in our 3D cultures of CD8-IGF-IR-MCF10A cells. However, CD8-IGF-IR-MCF10A cells also showed invasive properties in Matrigel culture and, after 15 days of culture, were found to grow together. This invasion in Matrigel was supported by the ability of the cells to invade when the modified Boyden chamber assay was used. This suggests that CD8-IGF-IR may be activating selected pathways to a greater and more sustained level than the hyperstimulated overexpressed wild-type receptor or that CD8-IGF-IR may be activating alternate pathways.
Irie et al. noted a subtle conversion from a cuboidal epithelial morphology to a more spindle-shaped morphology (24). Most intriguingly, downregulation of Akt1, but not Akt2, in IGF-IR-overexpressing clones caused cells to undergo a profound EMT, similar to the results with our CD8-IGF-IR clones. The EMT caused by Akt1 downregulation in the presence of overexpressed IGF-IR was attributed to a subsequent increase in ERK1/2 activity. In contrast to the results of the studies of Irie et al. (24), we found that CD8-IGF-IR caused EMT even though there was little or no activation of ERK1/2 (data not shown). Yanochko and Eckhart (47) did not comment on the morphology of their MCF10A-IGF-IR cells, but did indicate that in 3D culture, the cells showed areas of punctate E-cadherin staining, suggesting changes in cell adhesion. Interestingly, we noted that CD8-IGF-IR-MCF10A cells showed a dramatic change in E-cadherin staining, with a distinct punctate pattern perhaps somewhat similar, but exaggerated, compared to that of Yanochko and Eckhart (47). It is possible that the constitutive IGF-IR we have used is able to cause EMT due to higher and more-prolonged constitutive activation of downstream signaling pathways, or perhaps via the induction of novel signaling pathways not normally activated by the wild-type overexpressed receptor.
IGF-I stimulation of breast cancer cells overexpressing IGF-IR has been shown to cause depolarization and a mesenchyme-like transition (20). Other growth factor receptors, such as fibroblast growth factor receptor (39), epidermal growth factor receptor (17), and transforming growth factor β-receptor (48), are known to induce EMT. Furthermore, there is considerable cross-talk between these receptors and downstream signaling pathways and transcription factors that lead to EMT (19). Several transcription factors have been implicated in the repression of E-cadherin and EMT, including Snail/Slug, Twist, and Zeb1 (23). We found that CD8-IGF-IR-MCF10A cells showed a specific and dramatic elevation of Snail mRNA levels. Recent studies have implicated Snail as a critical regulator of both the EMT process in MCF10A cells (1) and breast cancer recurrence (30).
Snail expression has been shown previously to be regulated by numerous growth factors (13); however, ours is the first study to show that IGF-I and IGF-IR can induce Snail mRNA. The mechanism of Snail induction by receptor tyrosine kinases has generally focused on the role of Ras signaling (13). We found that CD8-IGF-IR caused an increase in Snail mRNA levels; however, this increase was not due to ERK1/2 activity, as levels of ERK1/2 were low in CD8-IGF-IR-MCF10A cells, and blockade of ERK1/2 with U0126 did not lower Snail levels. Phosphatidylinositol 3-kinase and Akt are required for transforming growth factor β-induced EMT (2), and constitutive Akt can induce EMT (18). GSK-3β has also been shown to repress EMT, via repression of Snail mRNA (1) and by phosphorylating Snail on two motifs that cause its proteasome-dependent degradation and also modulate its nuclear localization (50). We have found constitutive P∼Akt and loss of P∼GSK-3β in CD8-IGF-IR-MCF10A cells (data not shown), indicating that this may be a mechanism for the regulation of Snail. However, recent evidence has implicated the IGF-IR signaling cascade in the activation of the NF-κB signaling pathway (29), and inhibition of GSK-3β has been shown to activate NF-κB and increase Snail mRNA levels (1). Indeed, we found that NF-κB was highly active in CD8-IGF-IR-MCF10A cells and that blockade of NF-κB reduced Snail levels in these cells. This is consistent with previous data indicating that NF-κB binds Snail promoter and increases its activity (3) and strongly suggests a signaling scenario whereby IGF-IR leads to activation of Akt, loss of GSK-3β, increased NF-κB activity, increased levels of Snail, loss of E-cadherin, and subsequent EMT (Fig. 8). This model is supported by the numerous other studies described above.
FIG. 8.
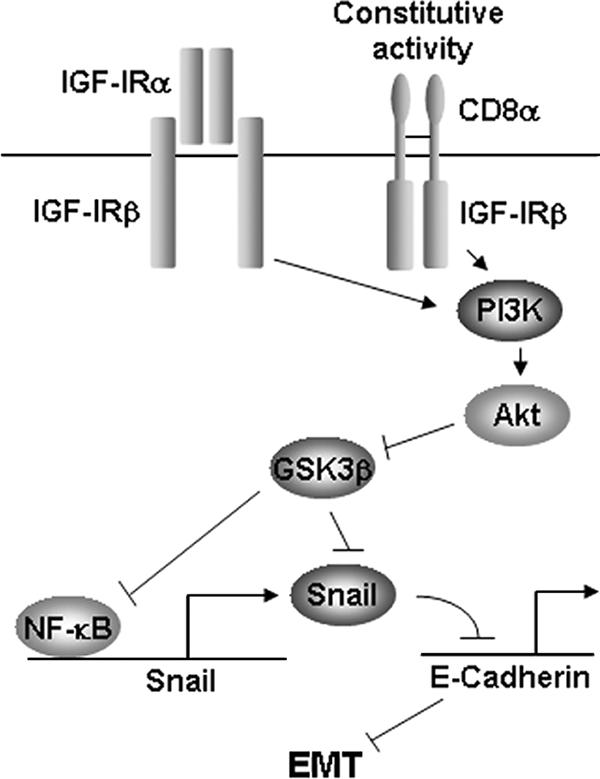
Schematic indicating how CD8-IGF-IR leads to EMT. This model is based upon studies presented in this report indicating that CD8-IGF-IR induces NF-κB, Snail, repression of E-cadherin mRNA and protein levels, and subsequently, EMT. The hierarchy is shown by the reversion of Snail mRNA levels and EMT by IGF-IR or NF-κB inhibitors. The model is supported by studies showing that IGF-IR activates Akt to repress GSK-3β, GSK-3β is a repressor of NF-κB activity and Snail mRNA and protein levels, IGF-IR activates NF-κB, and NF-κB binds the Snail promoter and increases Snail mRNA levels. PI3K, phosphatidylinositol 3-kinase.
In summary, we show that the expression of a constitutively active IGF-IR in human mammary epithelial cells results in tumorigenic conversion that is accompanied by a transition to a mesenchyme-like phenotype. This study is novel in that relatively few single oncogenes have been shown to confer xenograft growth on MCF10A cells and thus provides a new model for examining MCF10A cell growth in vivo. This is also the first study to show that IGF-I and its receptor can modulate Snail levels and cause EMT in mammary epithelial cells.
Acknowledgments
We thank Ora Britton for help with the xenograft studies, Craig Allred and the Pathology core of the Breast Center at Baylor College of Medicine for IHC, and Steffi Oesterreich for critical reading of the manuscript. Flag-Snail-pCMV-Tag 2B was a gift from Mien-Chie Hung at the M. D. Anderson Cancer Center, Houston, TX.
This work was supported in part by Public Health Service grants R01CA94118 (A.V.L.) and P01CA30195 (C.K.O./A.V.L.). A.V.L. is the recipient of a T. T. Chao Scholar award (Department of Medicine, Baylor College of Medicine). X.C. is the recipient of a Career Development Award (P50CA58183).
Footnotes
▿
Published ahead of print on 12 February 2007.
REFERENCES
- 1.Bachelder, R. E., S. O. Yoon, C. Franci, A. G. de Herreros, and A. M. Mercurio. 2005. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J. Cell Biol. 168**:**29-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakin, A. V., A. K. Tomlinson, N. A. Bhowmick, H. L. Moses, and C. L. Arteaga. 2000. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 275**:**36803-36810. [DOI] [PubMed] [Google Scholar]
- 3.Barbera, M. J., I. Puig, D. Dominguez, S. Julien-Grille, S. Guaita-Esteruelas, S. Peiro, J. Baulida, C. Franci, S. Dedhar, L. Larue, and A. Garcia de Herreros. 2004. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 23**:**7345-7354. [DOI] [PubMed] [Google Scholar]
- 4.Barrallo-Gimeno, A., and M. A. Nieto. 2005. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 132**:**3151-3161. [DOI] [PubMed] [Google Scholar]
- 5.Baserga, R. 1999. The IGF-I receptor in cancer research. Exp. Cell Res. 253**:**1-6. [DOI] [PubMed] [Google Scholar]
- 6.Bonnette, S. G., and D. L. Hadsell. 2001. Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology 142**:**4937-4945. [DOI] [PubMed] [Google Scholar]
- 7.Boyer, B., A. M. Valles, and N. Edme. 2000. Induction and regulation of epithelial-mesenchymal transitions. Biochem. Pharmacol. 60**:**1091-1099. [DOI] [PubMed] [Google Scholar]
- 8.Carboni, J. M., A. V. Lee, D. L. Hadsell, B. R. Rowley, F. Y. Lee, D. K. Bol, A. E. Camuso, M. Gottardis, A. F. Greer, C. P. Ho, W. Hurlburt, A. Li, M. Saulnier, U. Velaparthi, C. Wang, M. L. Wen, R. A. Westhouse, M. Wittman, K. Zimmermann, B. A. Rupnow, and T. W. Wong. 2005. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res. 65**:**3781-3787. [DOI] [PubMed] [Google Scholar]
- 9.Cogswell, P. C., D. C. Guttridge, W. K. Funkhouser, and A. S. Baldwin, Jr. 2000. Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene 19**:**1123-1131. [DOI] [PubMed] [Google Scholar]
- 10.Dearth, R. K., X. Cui, H. J. Kim, I. Kuiatse, N. A. Lawrence, X. Zhang, J. Divisova, O. L. Britton, S. Mohsin, D. C. Allred, D. L. Hadsell, and A. V. Lee. 2006. Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2. Mol. Cell. Biol. 26**:**9302-9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debnath, J., and J. S. Brugge. 2005. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 5**:**675-688. [DOI] [PubMed] [Google Scholar]
- 12.Debnath, J., S. K. Muthuswamy, and J. S. Brugge. 2003. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30**:**256-268. [DOI] [PubMed] [Google Scholar]
- 13.De Craene, B., F. van Roy, and G. Berx. 2005. Unraveling signalling cascades for the Snail family of transcription factors. Cell Signal. 17**:**535-547. [DOI] [PubMed] [Google Scholar]
- 14.Dimri, G., H. Band, and V. Band. 2005. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res. 7**:**171-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garber, K. 2005. IGF-1: old growth factor shines as new drug target. J. Natl. Cancer Inst. 97**:**790-792. [DOI] [PubMed] [Google Scholar]
- 16.Giunciuglio, D., M. Culty, G. Fassina, L. Masiello, A. Melchiori, G. Paglialunga, G. Arand, F. Ciardiello, F. Basolo, E. W. Thompson, et al. 1995. Invasive phenotype of MCF10A cells overexpressing c-Ha-ras and c-erbB-2 oncogenes. Int. J. Cancer 63**:**815-822. [DOI] [PubMed] [Google Scholar]
- 17.Grande, M., A. Franzen, J. O. Karlsson, L. E. Ericson, N. E. Heldin, and M. Nilsson. 2002. Transforming growth factor-beta and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J. Cell Sci. 115**:**4227-4236. [DOI] [PubMed] [Google Scholar]
- 18.Grille, S. J., A. Bellacosa, J. Upson, A. J. Klein-Szanto, F. van Roy, W. Lee-Kwon, M. Donowitz, P. N. Tsichlis, and L. Larue. 2003. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 63**:**2172-2178. [PubMed] [Google Scholar]
- 19.Grunert, S., M. Jechlinger, and H. Beug. 2003. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell. Biol. 4**:**657-665. [DOI] [PubMed] [Google Scholar]
- 20.Guvakova, M. A., and E. Surmacz. 1999. The activated insulin-like growth factor I receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp. Cell Res. 251**:**244-255. [DOI] [PubMed] [Google Scholar]
- 21.Hay, E. D. 1995. An overview of epithelio-mesenchymal transformation. Acta Anat. 154**:**8-20. [DOI] [PubMed] [Google Scholar]
- 22.Huber, M. A., N. Azoitei, B. Baumann, S. Grunert, A. Sommer, H. Pehamberger, N. Kraut, H. Beug, and T. Wirth. 2004. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 114**:**569-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber, M. A., N. Kraut, and H. Beug. 2005. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 17**:**548-558. [DOI] [PubMed] [Google Scholar]
- 24.Irie, H. Y., R. V. Pearline, D. Grueneberg, M. Hsia, P. Ravichandran, N. Kothari, S. Natesan, and J. S. Brugge. 2005. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 171**:**1023-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones, R. A., C. I. Campbell, E. J. Gunther, L. A. Chodosh, J. J. Petrik, R. Khokha, and R. A. Moorehead. 4 September 2006. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene doi: 10.1038/sj.onc.1209955. [Epub ahead of print.] [DOI] [PubMed]
- 26.Kaleko, M., W. Rutter, and A. Miller. 1990. Overexpression of the human insulin-like growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol. Cell. Biol. 10**:**464-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim, D. W., M. A. Sovak, G. Zanieski, G. Nonet, R. Romieu-Mourez, A. W. Lau, L. J. Hafer, P. Yaswen, M. Stampfer, A. E. Rogers, J. Russo, and G. E. Sonenshein. 2000. Activation of NF-kappaB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis 21**:**871-879. [DOI] [PubMed] [Google Scholar]
- 28.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25**:**402-408. [DOI] [PubMed] [Google Scholar]
- 29.Mitsiades, C. S., N. Mitsiades, V. Poulaki, R. Schlossman, M. Akiyama, D. Chauhan, T. Hideshima, S. P. Treon, N. C. Munshi, P. G. Richardson, and K. C. Anderson. 2002. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene 21**:**5673-5683. [DOI] [PubMed] [Google Scholar]
- 30.Moody, S. E., D. Perez, T. C. Pan, C. J. Sarkisian, C. P. Portocarrero, C. J. Sterner, K. L. Notorfrancesco, R. D. Cardiff, and L. A. Chodosh. 2005. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 8**:**197-209. [DOI] [PubMed] [Google Scholar]
- 31.Moyano, J. V., J. R. Evans, F. Chen, M. Lu, M. E. Werner, F. Yehiely, L. K. Diaz, D. Turbin, G. Karaca, E. Wiley, T. O. Nielsen, C. M. Perou, and V. L. Cryns. 2006. αB-crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J. Clin. Investig. 116**:**261-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagle, J. A., Z. Ma, M. A. Byrne, M. F. White, and L. M. Shaw. 2004. Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol. Cell. Biol. 24**:**9726-9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orlowski, R. Z., and A. S. Baldwin, Jr. 2002. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 8**:**385-389. [DOI] [PubMed] [Google Scholar]
- 34.Pena, C., J. M. Garcia, V. Garcia, J. Silva, G. Dominguez, R. Rodriguez, C. Maximiano, A. Garcia de Herreros, A. Munoz, and F. Bonilla. 2006. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int. J. Cancer 119**:**2098-2104. [DOI] [PubMed] [Google Scholar]
- 35.Resnicoff, M., and R. Baserga. 1998. The role of the insulin-like growth factor I receptor in transformation and apoptosis. Ann. N. Y. Acad. Sci. 842**:**76-81. [DOI] [PubMed] [Google Scholar]
- 36.Rodgers, K., B. McMahon, D. Mitchell, D. Sadlier, and C. Godson. 2005. Lipoxin A4 modifies platelet-derived growth factor-induced pro-fibrotic gene expression in human renal mesangial cells. Am. J. Pathol. 167**:**683-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosivatz, E., I. Becker, K. Specht, E. Fricke, B. Luber, R. Busch, H. Hofler, and K. F. Becker. 2002. Differential expression of the epithelial-mesenchymal transition regulators Snail, SIP1, and twist in gastric cancer. Am. J. Pathol. 161**:**1881-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sachdev, D., and D. Yee. 2006. Inhibitors of insulin-like growth factor signaling: a therapeutic approach for breast cancer. J. Mammary Gland Biol. Neoplasia 11**:**27-39. [DOI] [PubMed] [Google Scholar]
- 39.Strutz, F., M. Zeisberg, F. N. Ziyadeh, C. Q. Yang, R. Kalluri, G. A. Muller, and E. G. Neilson. 2002. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 61**:**1714-1728. [DOI] [PubMed] [Google Scholar]
- 40.Sugimachi, K., S. Tanaka, T. Kameyama, K. Taguchi, S. Aishima, M. Shimada, K. Sugimachi, and M. Tsuneyoshi. 2003. Transcriptional repressor Snail and progression of human hepatocellular carcinoma. Clin. Cancer Res. 9**:**2657-2664. [PubMed] [Google Scholar]
- 41.Surmacz, E. 2000. Function of the IGF-I receptor in breast cancer. J. Mammary Gland Biol. Neoplasia 5**:**95-105. [DOI] [PubMed] [Google Scholar]
- 42.Thiery, J. P. 2003. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 15**:**740-746. [DOI] [PubMed] [Google Scholar]
- 43.Wang, D., Y. You, S. M. Case, L. M. McAllister-Lucas, L. Wang, P. S. DiStefano, G. Nunez, J. Bertin, and X. Lin. 2002. A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat. Immunol. 3**:**830-835. [DOI] [PubMed] [Google Scholar]
- 44.Wang, L., J. Wang, B. E. Wang, P. G. Xiao, Y. J. Qiao, and X. H. Tan. 2004. Effects of herbal compound 861 on human hepatic stellate cell proliferation and activation. World J. Gastroenterol. 10**:**2831-2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wittman, M., J. Carboni, R. Attar, B. Balasubramanian, P. Balimane, P. Brassil, F. Beaulieu, C. Chang, W. Clarke, J. Dell, J. Eummer, D. Frennesson, M. Gottardis, A. Greer, S. Hansel, W. Hurlburt, B. Jacobson, S. Krishnananthan, F. Y. Lee, A. Li, T. A. Lin, P. Liu, C. Ouellet, X. Sang, M. G. Saulnier, K. Stoffan, Y. Sun, U. Velaparthi, H. Wong, Z. Yang, K. Zimmermann, M. Zoeckler, and D. Vyas. 2005. Discovery of a (1H-benzoimidazol-2-yl)-1H-pyridin-2-one (BMS-536924) inhibitor of insulin-like growth factor I receptor kinase with in vivo antitumor activity. J. Med. Chem. 48**:**5639-5643. [DOI] [PubMed] [Google Scholar]
- 46.Yang, J., S. A. Mani, J. L. Donaher, S. Ramaswamy, R. A. Itzykson, C. Come, P. Savagner, I. Gitelman, A. Richardson, and R. A. Weinberg. 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117**:**927-939. [DOI] [PubMed] [Google Scholar]
- 47.Yanochko, G. M., and W. Eckhart. 2006. Type I insulin-like growth factor receptor over-expression induces proliferation and anti-apoptotic signaling in a three-dimensional culture model of breast epithelial cells. Breast Cancer Res. 8**:**R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zavadil, J., and E. P. Bottinger. 2005. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24**:**5764-5774. [DOI] [PubMed] [Google Scholar]
- 49.Zelinski, D. P., N. D. Zantek, J. C. Stewart, A. R. Irizarry, and M. S. Kinch. 2001. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 61**:**2301-2306. [PubMed] [Google Scholar]
- 50.Zhou, B. P., J. Deng, W. Xia, J. Xu, Y. M. Li, M. Gunduz, and M. C. Hung. 2004. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6**:**931-940. [DOI] [PubMed] [Google Scholar]
- 51.Zhu, T., B. Starling-Emerald, X. Zhang, K. O. Lee, P. D. Gluckman, H. C. Mertani, and P. E. Lobie. 2005. Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res. 65**:**317-324. [PubMed] [Google Scholar]