IL-6 receptor blockage inhibits the onset of autoimmune kidney disease in NZB/W F1 mice (original) (raw)
Abstract
In the present study, we examined the preventive effect of anti-mouse IL-6 receptor (IL-6R) antibody, MR16-1, on the development of autoimmune kidney disease in female NZB/W F1 (BWF1) mice. Immunological tolerance to MR16-1 or isotype-matched control antibody, KH-5, was induced by the simultaneous administration of anti-CD4 MoAb in mice. Thereafter, mice were intraperitoneally given 0.5 mg of MR16-1, 0.5 mg of KH-5 or saline once a week from 13 to 64 weeks of age. MR16-1 treatment dramatically suppressed proteinuria and prolonged the survival time of BWF1 mice. Only one out of 10 mice died with high levels of proteinuria throughout the experiment. MR16-1 almost completely suppressed the production of IgG forms of anti-DNA and anti-TNP antibodies, but not the IgM forms of these antibodies. In particular, all IgG subclasses (IgG1, IgG2a, IgG2b and IgG3) of anti-DNA antibody production were significantly suppressed. Moreover, serum IgG1, IgG2a and IgG3 levels in MR16-1-treated mice were lower than those in saline- and KH-5-treated mice, whereas serum IgM and IgA levels were not influenced. In conclusion, MR16-1 potently suppressed the development of autoimmune disease in BWF1 mice, and this was attributed to its effect of specific suppression of IgG class antibody production.
Keywords: NZB/W F1 mice, anti-IL-6R antibody, autoimmune kidney disease
INTRODUCTION
IL-6 is a multifunctional cytokine and plays essential roles in host defence mechanisms through the immune, haematopoietic and central nervous systems. However, in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), cardiac myxoma and Castleman's disease, hypergammaglobulinaemia and autoantibody production were associated with elevated levels of IL-6 [1–4].
NZB/W F1 (BWF1) mice spontaneously develop autoimmune disease which resembles human SLE, i.e. hypergammaglobulinaemia, autoantibody production including anti-DNA antibody, immune complex-mediated glomerulonephritis and death from renal failure [5]. Although high serum levels of IL-6 have not been found in BWF1 mice [6], we and others previously reported that B cells from BWF1 mice in vitro show hyperresponsiveness to IL-6 and then produce anti-DNA antibody [7–9], suggesting that IL-6 plays an essential role in autoantibody production in BWF1 mice as well as in human patients.
In the present study, we examined the effects of anti-IL-6 receptor (IL-6R) antibody, MR16-1, on antibody production and the course of autoimmune disease in BWF1 mice. MR16-1 specifically binds to IL-6R and blocks IL-6 binding to IL-6R. MR16-1 is reported to inhibit IL-6 and IL-6R complex-induced osteoclast formation in vitro [10] and to prevent muscle atrophy in cancer-bearing mice [11]. Furthermore, MR16-1 inhibits the proliferation of IL-6-dependent cell line MH60 and IL-6-induced immunoglobulin production dose-dependently in vitro and completely inhibits the development of mesangial-proliferative glomerulonephritis in IL-6 transgenic mice (manuscript in preparation). Our results clearly indicate that IL-6 strongly participated in the development of autoimmune kidney disease via IgG class antibody production.
MATERIALS AND METHODS
Animals
NZB nu/+ and NZW nu/+ mice were obtained from the University of California (Davis, CA) and maintained in our Research Laboratories. Female euthymic BWF1 mice were bred in our laboratories. The animals were specific pathogen-free, and were kept in cages in a room maintained at 24 ± 2°C, with 50–60% relative humidity. Each group included 10 mice, except for the saline group (nine mice).
Antibodies
Hybridoma MR16-1 cells, which produce rat anti-mouse IL-6R monoclonal IgG1 and hybridoma KH-5 cells, which produce rat anti-DNP monoclonal IgG1, were produced in our laboratories [10,11]. Briefly, spleen cells obtained from Wister rats which were immunized with soluble mouse IL-6R and dinitrophenyl (DNP)-bovine serum albumin (BSA), respectively, were fused with mouse P3U1 myeloma cells. Hybridoma GK1.5 cells, which produce rat anti-mouse CD4 monoclonal IgG2b, were obtained from the American Type Culture Collection (Rockville, MD). The cells were injected intraperitoneally into BALB/c nu/nu mice pretreated with pristane, 2,6,10,14-tetramethyldecanoic acid (Aldrich Chemical, Milwaukee, WI). Ascites were collected and IgG was obtained by means of a protein G column.
Experimental schedule
Immunological tolerance to either MR16-1 or KH-5 was induced by the methods of Finck et al. [12]. Briefly, female BWF1 mice (13 weeks old) were divided into three groups. All groups were intraperitoneally administered anti-CD4 MoAb for 3 consecutive days (1 mg/day on days −1, 0 and 1); these anti-CD4-treated animals in their respective groups received, by i.p. injection, either saline (saline group), 0.5 mg of KH-5 (KH-5 group) or 0.5 mg of MR16-1 (MR16-1 group) on day 0; thereafter, they received, weekly, their respective i.p. treatments of saline, KH-5 (0.5 mg) and MR16-1 (0.5 mg).
In another experiment, female BWF1 mice (13 weeks old) were divided into four groups. Two of these groups received anti-CD4 MoAb treatment as described above; these anti-CD4-treated animals in their respective groups received either 0.5 mg of KH-5 (KH-5 group) or 0.5 mg of MR16-1 (MR16-1 group) on day 0; thereafter they received, weekly, their respective i.p. treatments of KH-5 (0.5 mg) and MR16-1 (0.5 mg). The other two groups did not receive anti-CD4 antibody, and were intraperitoneally administered either KH-5 (0.5 mg) or MR16-1 (0.5 mg) on day 0; thereafter they received, weekly, their respective i.p. treatments of KH-5 and MR16-1 (0.5 mg).
Serum was collected regularly and proteinuria was measured every 2 weeks from the age of 14 weeks to the end of the experiments.
Proteinuria
Proteinuria was measured semiquantitatively at 2-week intervals using Uristix test strips (Bayer-Sankyo, Tokyo, Japan). Protein concentrations in the urine > 100 mg/dl were considered positive.
Serum anti-DNA and trinitrophenyl antibody levels
Anti-DNA or anti-trinitrophenyl (TNP) antibody levels in serum were measured by ELISA, as previously described [13]. Briefly, diluted serum was added to each well of heat-denatured bovine DNA (Sigma Chemical Co., St Louis, MO)- or TNP mouse serum albumin-coated immunoplate (Nunc-Immunoplate; MaxiSorp, Roskilde, Denmark), and incubated for 2 h at room temperature. After a well was washed, bound immunoglobulin was measured using peroxidase-conjugated antibody to mouse IgG, IgM (Organon Teknika Corp., West Chester, PA), mouse IgG1, IgG2a, IgG2b or IgG3 (Zymed Labs, San Francisco, CA). Substrate was added and the colourimetric reaction was stopped by 6 n H2SO4 solution.
Spectraphotometric measurement was carried out with a microplate reader at 492–600 nm. To convert optical density (OD) values to IgG or IgM concentration, a standard curve was generated for each assay, using pooled 5-month-old MRL/lpr mouse serum defined as 100 U/ml.
Serum immunoglobulins and C3 level
Serum IgG1, IgG2a, IgG3, IgM, IgA and C3 concentrations were measured with single radial immunodiffusion. Immunoglobulin levels were measured by single radial immunodiffusion kit, according to the manufacturer's instructions (The Binding Site, Birmingham, UK). C3 levels were measured as previously reported [14]. Antibody to mouse C3 was purchased from Organon Teknika Corporation. Serum C3 levels were calculated from a standard curve using 12-week-old BWF1 mouse serum.
Serum IL-6 levels
Serum IL-6 concentration was measured by ELISA, according to the manufacturer's instructions (Amersham International plc., Aylesbury, UK).
Statistical analysis
For the statistical analysis, all data except that for proteinuria and survival were changed to common logarithmic values. Statistical significance of the differences was analysed using logarithmic values by Tukey's test, except for IgG anti-TNP antibody, which was analysed by Student's _t_-test. Proteinuria and survival time were analysed by generalized Wilcoxon test. All analyses were performed using a software package (Statistical Analysis System; SAS Institute Japan Ltd, Tokyo, Japan).
RESULTS
Proteinuria
Saline- and KH-5-treated mice began to show proteinuria at 26 and 36 weeks of age, respectively. However, in the MR16-1-treated mice proteinuria was dramatically suppressed. At 64 weeks of age (the end of experiment) the cumulative incidence of proteinuria was 100%, 90% or 10% in the groups of mice, respectively, treated with saline, KH-5 or MR16-1 (Fig. 1).
Fig. 1.
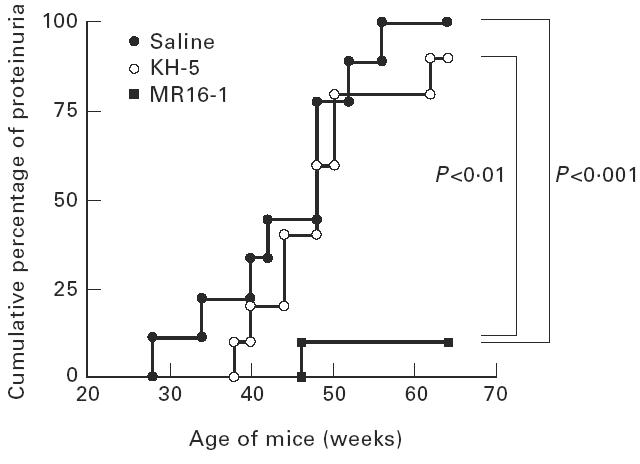
Cumulative percentage of female BWF1 mice treated with anti-IL-6R MoAb with positive proteinuria. BWF1 mice in which tolerance was induced were intraperitoneally given 0.5 mg of KH-5 or MR16-1 once a week, from 13 weeks old to 60 weeks old. Proteinuria over 300 mg/dl was assessed as positive. Each group included 10 mice, except for the saline group (nine mice).
Life span
The 50% mortality times for saline-treated mice and KH-5-treated mice were 50 and 52 weeks of age, respectively, and 80% of mice in both groups died at 64 weeks of age with positive proteinuria. In dramatic contrast, MR16-1 treatment prevented fatal renal disease, and only one out of 10 mice died by 64 weeks of age (Fig. 2).
Fig. 2.
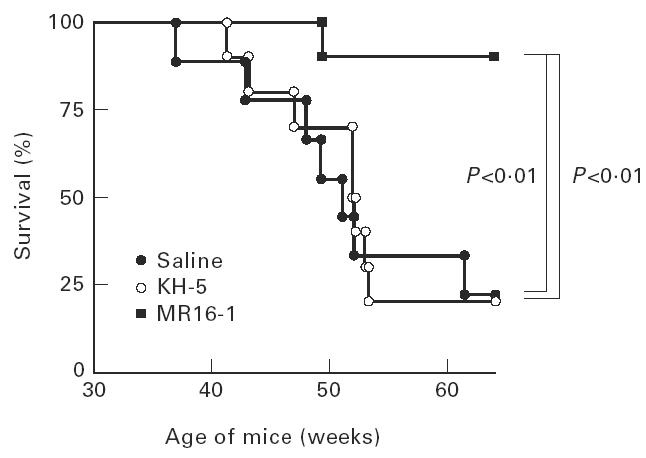
Survival curve of female BWF1 mice treated with anti-IL-6R MoAb. The same animals as in Fig. 1.
Serum anti-DNA and anti-TNP antibody levels
Serum levels of anti-DNA antibody, as an autoantibody, and anti-TNP antibody, as an indicator of polyclonal B cell activation, were measured by ELISA. The production of IgG anti-DNA and anti-TNP antibody in the sera were potently suppressed by MR16-1 treatment (Figs 3 and 4). In addition, MR16-1 potently suppressed the production of all IgG isotypes of anti-DNA antibody (Fig. 5). On the other hand, MR16-1 did not influence the production of IgM anti-DNA or anti-TNP antibody (Figs 3 and 4).
Fig. 3.
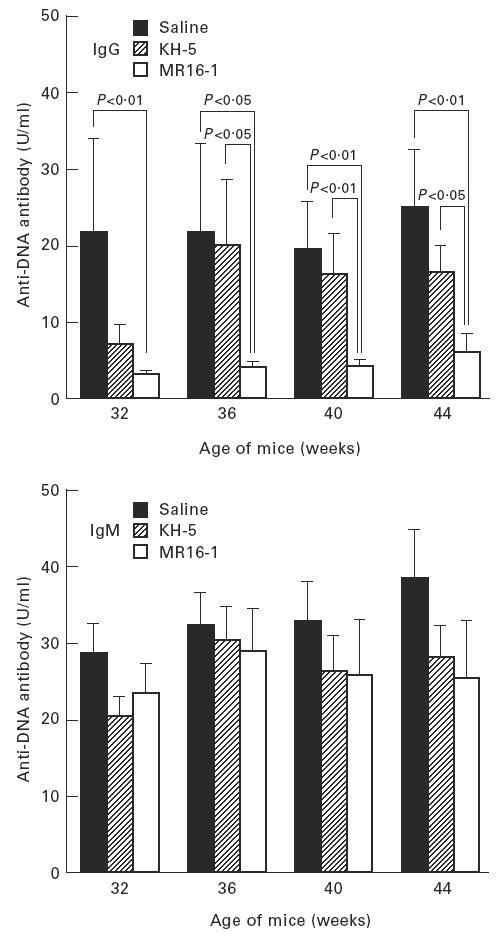
Serum anti-DNA antibody levels. Anti-DNA antibody levels in sera were measured by ELISA. Data are expressed as the mean and s.e.m. of surviving mice at each age. The same batch of mice as in Fig. 1 was used.
Fig. 4.
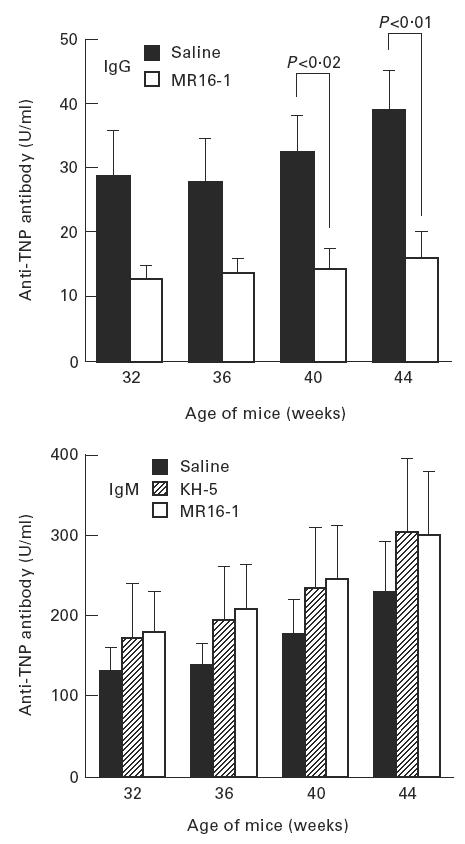
Serum anti-trinitrophenyl (TNP) antibody levels. Anti-TNP antibody levels in sera were measured by ELISA. See legend for Fig. 3.
Fig. 5.
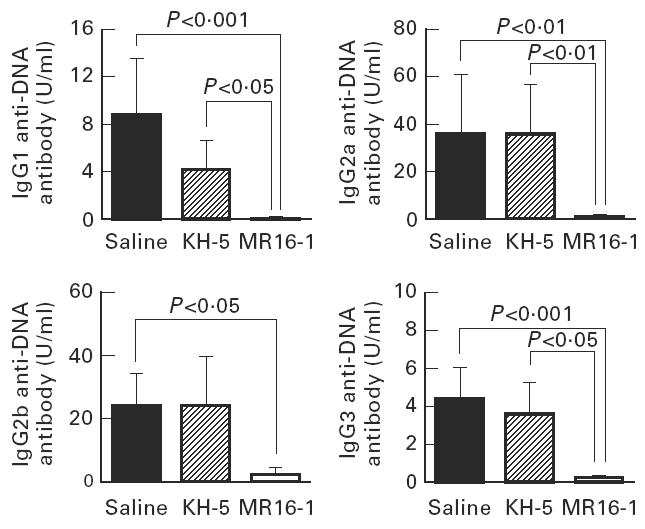
Serum levels of IgG subclass of anti-DNA antibody in 36-week-old BWF1 mice. Data are expressed as the mean and s.e.m. of surviving mice at 36 weeks of age. The same batch of mice as in Fig. 1 was used.
Serum immunoglobulin levels
Serum concentrations of IgG, IgM and IgA at 32 weeks of age were measured. Serum IgG1, IgG2a and IgG3 concentrations in MR16-1-treated mice were lower than in saline- and KH-5-treated mice at 32 weeks of age. However, serum IgM and IgA concentrations were not influenced by the administration of MR16-1 (Fig. 6).
Fig. 6.
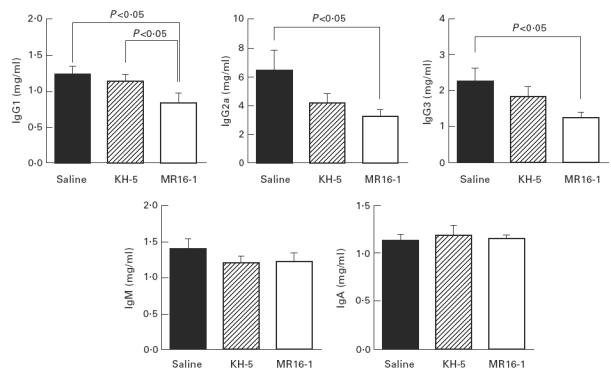
Serum immunoglobulin level at 32 weeks of age. Data are expressed as the mean and s.e.m. of surviving mice at 32 weeks of age. The same batch of mice as in Fig. 1 was used.
Serum IL-6 levels
Serum concentrations of mouse IL-6 at 36 weeks of age were evaluated. IL-6 was not detectable (< 50 pg/ml) in all mice (data not shown).
Serum C3 levels
Serum levels of C3 at 28 and 40 weeks of age were examined. Serum C3 levels of MR16-1-treated mice were similar to both saline- and KH-5-treated mice (data not shown).
Body weight
There was no difference in the body weight gain between the saline and antibody-treated groups (data not shown).
Evaluation of tolerance induction
To evaluate the tolerance induction, female BWF1 mice (13 weeks of age) received MR16-1 (0.5 mg) or KH-5 (0.5 mg) once a week with or without anti-CD4 MoAb treatment in another experiment. Fifty percent of mice (5/10) in the MR16-1 group without anti-CD4 MoAb treatment died within 6 weeks after the first MR16-1 injection. These mice had high levels of IgG class anti-rat IgG1 antibody, 4 weeks after the first MR16-1 injection. In the KH-5-treated group without anti-CD4 MoAb treatment, mice had raised levels of anti-rat IgG1 antibody, but these were lower than those in MR16-1-treated mice and showed a natural survival time. On the other hand, both MR16-1- and KH-5-treated mice with anti-CD4 MoAb treatment did not have anti-rat IgG antibody 4 weeks after first antibody injection.
DISCUSSION
We demonstrate here that long-term administration of anti-IL-6R MoAb prolonged life span and suppressed proteinuria. These results clearly show that IL-6 is an essential cytokine for the development of autoimmune kidney disease in BWF1 mice.
Without any treatment, 50% of BWF1 mice used presented with proteinuria at age 8 months and 50% mortality was observed at 10 months. However, in anti-CD4-treated saline and KH-5 groups, 50% proteinuria and 50% mortality was observed at age 11 months and 12 months, respectively. This delay of autoimmune disease onset may be due to anti-CD4 MoAb treatment.
IgG class antibodies play an important role in the development of kidney disease in lupus mice, i.e. (i) lupus mice have significantly higher polyclonal IgG concentrations at preclinical and clinical stages than normal or young lupus mice [5], (ii) the age-related switching of IgM to IgG anti-DNA antibodies appears to herald the onset of kidney disease [15], (iii) BWF1 nude mice, which do not produce IgG class antibodies, do not develop kidney disease [16]. MR16-1 prevented the increase of serum IgG anti-DNA and anti-TNP antibody levels, indicating that the mechanism of action of MR16-1 is by suppression of production of IgG antibodies.
IL-6 is reported to stimulate mitogen-induced IgG, IgM and IgA production, as well as IL-4-dependent IgE production in vitro [17–19]. However, we showed that IL-6R blockage in vivo decreased only IgG levels, but not completely. This result suggested that IgG production is partially IL-6-dependent, but in vivo IgM and IgA production are not IL-6-dependent. This idea is supported by findings of IL-6 knockout mice studies [20,21].
Finck et al. [12] reported that the administration of anti-IL-6 MoAb showed beneficial effects during development of renal disease in this strain of mice. In their paper, anti-IL-6 MoAb suppressed IgG anti-DNA antibody production without affecting serum IgG isotype levels, suggesting that IL-6 preferentially affects autoantibody-producing B cells. On the other hand, anti-IL-6R MoAb reduced all IgG isotype levels and suppressed the production of IgG anti-TNP antibody as well as anti-DNA antibody, suggesting that IL-6 acts on IgG isotype-producing B cells. Furthermore, the preventive effect of anti-IL-6 MoAb was partial, and the proportion of mice with positive proteinuria gradually increased when the administration was continued. In contrast, anti-IL-6R MoAb completely suppressed the onset of nephritis except in one mouse, which had high level of IgG anti-DNA antibody and in which immunological tolerance to rat IgG1 could not be induced. Although the reason for this difference between anti-IL-6 antibody and anti-IL-6R antibody is obscure, the potency of antibody used for IL-6/IL-6R blockage may be relevant.
Since MR16-1 and KH-5 are rat MoAbs and heterogeneous proteins for mice, long-term administration is impossible. Therefore, we tried to induce tolerance to rat IgG1 using anti-CD4 MoAb, as previously reported [12]. It is reported that simultaneous administration of anti-CD4 MoAb and some antigens induces tolerance to antigen, but this phenomenon is not observed in all cases [22]. However, our findings could support the induction of tolerance: (i) anti-rat IgG1 antibodies were not found in sera from mice receiving simultaneous administration of anti-CD4 MoAb with either MR16-1 or KH-5, (ii) mice without anti-CD4 MoAb presented with high levels of anti-rat IgG1 antibody in long-term administration of MR16-1 and died.
It has been reported that the administration of anti-IL-6 MoAb partially protected the Shwartzman reaction in endotoxin-injected mice and ameliorated clinical symptoms in RA patients with increasing biologically active IL-6 levels in serum [23,24]. IL-6 suppresses IL-1 and tumour necrosis factor-alpha (TNF-α) production [25] and augments the IL-1 receptor antagonist and the soluble TNF receptor [26], indicating that IL-6 is an anti-inflammatory cytokine. Therefore, augmented IL-6 might involve the anti-inflammatory effects of anti-IL-6 MoAb. So we decided to check serum IL-6 levels in MR16-1-treated mice. As it has been reported [6], BWF1 mice did not have detectable levels of IL-6 in sera, and increased production of IL-6 was not found in MR16-1-treated mice.
Calorie-restricted animals lose body weight, and their serum anti-DNA antibodies are significantly decreased [27]. Gain in body weight was unaffected in mice treated with MR16-1. Thus, the beneficial effects of MR16-1 were not due to low calorie intake. Furthermore, since IL-6 up-regulates the biosynthesis of complement components C3 and factor B [28,29], we measured serum C3 levels in MR16-1-treated BWF1 mice at 28 weeks of age (before the onset of nephritis). MR16-1 did not affect serum C3 levels, suggesting that IL-6R blockage could not influence C3 synthesis, and that complement suppression did not involve autoimmune kidney disease suppression. Throughout the experiments, no severe side-effects were observed in MR16-1-treated mice.
In conclusion, IL-6 may be essential for the over-production of IgG class antibodies in BWF1 mice.
References
- 1.Linker-Israeli M, Deans RJ, Wallace DJ, et al. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol. 1991;147:117–23. [PubMed] [Google Scholar]
- 2.Hirano T, Matsuda T, Turner M, et al. Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. 1988;18:1797–801. doi: 10.1002/eji.1830181122. [DOI] [PubMed] [Google Scholar]
- 3.Hirano T, Taga T, Yasukawa K, et al. Human B cell differentiation factor defined by a anti-peptide antibody and its possible role in autoantibody production. Proc Natl Acad Sci USA. 1987;84:228–31. doi: 10.1073/pnas.84.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman's disease. Blood. 1989;74:1360–7. [PubMed] [Google Scholar]
- 5.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–388. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 6.Tang B, Matsuda T, Akira S, et al. Age-associated increase in interleukin 6 in MRL/lpr mice. Int Immunol. 1991;3:273–8. doi: 10.1093/intimm/3.3.273. [DOI] [PubMed] [Google Scholar]
- 7.Mihara M, Fukui H, Koishihara Y, et al. Immunologic abnormality in NZB/W F1 mice. Thymus-independent expansion of B cells responding to interleukin-6. Clin Exp Immunol. 1990;82:533–7. doi: 10.1111/j.1365-2249.1990.tb05485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mihara M, Ohsugi Y. Possible role of IL-6 in pathogenesis of immune complex-mediated glomerulonephritis in NZB/W F1 mice: induction of IgG class anti-DNA autoantibody production. Int Arch Allergy Appl Immunol. 1990;93:89–92. doi: 10.1159/000235285. [DOI] [PubMed] [Google Scholar]
- 9.Alarcon-Riquelme ME, Moller G, Fernandez C. Age-dependent responsiveness to interleukin-6 in B lymphocytes from a systemic lupus erythematosus-prone (NZB X NZW) F1 hybrid. Clin Immunol Immunopathol. 1992;62:264–9. doi: 10.1016/0090-1229(92)90101-s. [DOI] [PubMed] [Google Scholar]
- 10.Tamura T, Udagawa N, Takahashi N, et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci USA. 1993;90:11924–8. doi: 10.1073/pnas.90.24.11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujita J, Tsujinaka T, Yano M, et al. Anti-interleukin-6 receptor antibody prevents muscle atrophy in colon-26 adenocarcinoma-bearing mice with modulation of lysosomal and ATP-ubiquitin-dependent proteolytic pathways. Int J Cancer. 1996;68:637–43. doi: 10.1002/(SICI)1097-0215(19961127)68:5<637::AID-IJC14>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 12.Finck BK, Chan B, Wofsy D. Interleukin 6 promotes murine lupus in NZB/NZW F1 mice. J Clin Invest. 1994;94:585–91. doi: 10.1172/JCI117373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mihara M, Nakano T, Ohsugi Y. An immunomodulating anti- rheumatic drug, lobenzarit disodium (CCA): inhibition of polyclonal B-cell activation and prevention of autoimmune disease in MRL/Mp-lpr/lpr mice. Clin Immunol Immunopathol. 1987;45:366–74. doi: 10.1016/0090-1229(87)90089-4. [DOI] [PubMed] [Google Scholar]
- 14.Mihara M, Ohsugi Y. Autoimmune kidney disease in MRL/Mp-lpr/lpr mice inhibited by OK-432, a streptococcal preparation. Clin Exp Immunol. 1989;78:102–7. [PMC free article] [PubMed] [Google Scholar]
- 15.Steward MW, Hay FC. Changes in immunoglobulin class and subclass of anti-DNA antibodies with increasing age in NZB/W F1 hybrid mice. Clin Exp Immunol. 1976;26:363–70. [PMC free article] [PubMed] [Google Scholar]
- 16.Mihara M, Ohsugi Y, Saito K, et al. Immunologic abnormality in NZB/NZW F1 mice. Thymus-independent occurrence of B cell abnormality and requirement for T cells in the development of autoimmune disease, as evidenced by an analysis of the athymic nude individuals. J Immunol. 1988;141:85–90. [PubMed] [Google Scholar]
- 17.Muraguchi A, Hiranao T, Tang B, et al. The essential role of B cell stimulatory factor 2 (BSF-2/IL-6) for the terminal differentiation of B cells. J Exp Med. 1988;167:332–44. doi: 10.1084/jem.167.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beagley KW, Eldrige JH, Lee F, et al. Interleukins and IgA synthesis: human and murine interleukin 6 induce high rate IgA secretion in IgA-committed B cells. J Exp Med. 1989;169:2133–48. doi: 10.1084/jem.169.6.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vercelli D, Jabara HH, Arai K, et al. Endogenous interleukin-6 plays an obligatory role in interleukin-4 dependent human IgE synthesis. Eur J Immunol. 1989;19:1419–24. doi: 10.1002/eji.1830190811. [DOI] [PubMed] [Google Scholar]
- 20.Kopf M, Baumann H, Freer G, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–42. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 21.Bromander AK, Ekman L, Kopf M, et al. IL-6-deficient mice exhibit normal mucosal IgA responses to local immunizations and Helicobacter felis infection. J Immunol. 1996;156:4290–7. [PubMed] [Google Scholar]
- 22.Gutstein NL, Seaman WE, Scott JH, et al. Induction of immune tolerance by administration of monoclonal antibody to L3T4. J Immunol. 1986;137:1127–32. [PubMed] [Google Scholar]
- 23.Heremans H, Dillen C, Put W, et al. Protective effect of anti-interleukin (IL)-6 antibody against endotoxin, associated with paradoxically increased IL-6 levels. Eur J Immunol. 1992;22:2395–401. doi: 10.1002/eji.1830220932. [DOI] [PubMed] [Google Scholar]
- 24.Wendling D, Racadot E, Wijdenes J. Treatment of severe rheumatoid arthritis by anti-interleukin 6 monoclonal antibody. J Rheumatol. 1993;20:259–62. [PubMed] [Google Scholar]
- 25.Schindler R, Mancilla J, Endres S, et al. Correlations and interactions in the production of interleukin-6 (IL-6), IL-1, and tumor necrosis factor (TNF) in human blood mononuclear cells: IL-6 suppresses IL-1 and TNF. Blood. 1990;75:40–47. [PubMed] [Google Scholar]
- 26.Tilg H, Trehu E, Atkins MB, et al. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood. 1994;83:113–8. [PubMed] [Google Scholar]
- 27.Izui S, Fernandes G, Hara I, et al. Low-calorie diet selectively reduces expression of retroviral envelope glycoprotein gp70 in sera of NZBxNZW F1 hybrid mice. J Exp Med. 1981;154:1116–24. doi: 10.1084/jem.154.4.1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falus A, Rokita H, Walcz E, et al. Hormonal regulation of complement biosynthesis in human cell lines. II. Upregulation of the biosynthesis of complement components C3, factor B and C1 inhibitor by interleukin-6 and interleukin-1 in human hepatoma cell line. Mol Immunol. 1990;27:197–201. doi: 10.1016/0161-5890(90)90115-g. [DOI] [PubMed] [Google Scholar]
- 29.Katz Y, Revel M, Strunk RC. Interleukin 6 stimulates synthesis of complement protein factor B and C3 in human skin fibroblast. Eur J Immunol. 1989;19:983–8. doi: 10.1002/eji.1830190605. [DOI] [PubMed] [Google Scholar]