Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-α) production acting on leucocytes resembling immature dendritic cells (original) (raw)
Abstract
Patients with active SLE often have an ongoing production of IFN-α. We therefore searched for an endogenous IFN-α-inducing factor (IIF) in SLE patients and found that their sera frequently induced production of IFN-α in cultures of peripheral blood mononuclear cells (PBMC) from healthy blood donors, especially when the PBMC were costimulated with the cytokines IFN-α2b and granulocyte-macrophage colony-stimulating factor (GM-CSF). The phenotype of the IFN-α-producing cells (IPC) as determined by flow cytometry corresponded to that of the natural IPC, resembling immature dendritic cells. The IIF activity in SLE sera was sometimes as high as that of a virus and was present especially in patients with active disease and with measurable IFN-α levels in serum. The IIF had an apparent molecular weight of 300–1000 kD and appeared to consist of both immunoglobulin and DNA, possibly being immune complexes. This endogenous IFN-α inducer may be of pathogenic significance, since a reported occasional adverse effect of IFN-α therapy in patients with non-autoimmune disorders is development of anti-dsDNA antibodies and SLE.
Keywords: systemic lupus erythematosus, interferon-alpha, dendritic cells, interferon inducer
INTRODUCTION
SLE is a multisystem autoimmune disease characterized by multiple immunological abnormalities including production of antibodies reactive with many different autoantigens. The pathogenesis is largely unknown, although several factors of importance for the development of the disease have been reported [1]. In naturally occurring SLE both circulating IFN-α and increased levels of IFN-α-inducible proteins have been reported [2–8]. Because long-term IFN-α therapy of patients with non-autoimmune disorders can induce antinuclear antibodies, antibodies to native DNA and also an SLE-like syndrome [9–11], the endogenous production of IFN-α in SLE could play a role in the development of this autoimmune disease. That possibility is also supported by findings that IFN-α, beside its antiviral activity, also has several immunoregulatory functions [12–17].
The ongoing production of IFN-α in many SLE patients suggests the presence of an IFN-α inducer, because IFN-α/β gene expression usually must be triggered by, for example, microorganisms or their constituents [18]. In the present investigation we therefore searched for an endogenous IFN-α-inducing factor (IIF) by co-culturing sera from SLE patients with peripheral blood mononuclear cells (PBMC) of normal blood donors. We found that such sera often induced IFN-α production, especially when the PBMC were costimulated by recombinant IFN-α2b and granulocyte-macrophage colony-stimulating factor (GM-CSF) and when sera were from patients with active disease. We therefore characterized the IFN-α-inducing factor (SLE-IIF) and the type of PBMC responsible for the IFN-α production.
PATIENTS, MATERIALS AND METHODS
Patients and controls
Consecutive patients entering the rheumatology clinic who fulfilled the ACR classification criteria for SLE [19] were included. The 34 SLE patients (27 female and seven male) with a median age of 47 years (range 21–79 years) had a mean duration of disease of 12.9 years (1–36 years). The mean ACR index for the patients was 4.9 (range 4–8). Seven patients were untreated, 21 patients were treated with prednisolone (mean 8.8 mg/day) and eight individuals also received azathioprine (mean 100 mg/day). Disease activity was assessed by a modified SLE Disease Activity Index (SLEDAI) score where complement levels and titre of anti-DNA antibodies were excluded [20]. A total of 18 staff members (14 female and four male) with a median age of 47 years (range 28–58 years) served as controls. Sera were collected and stored in aliquots at −80°C. The study protocol was approved by the Committee of Ethics, Faculty of Medicine, Uppsala University.
Preparation of PBMC
The PBMC were prepared by Ficoll–Hypaque (Pharmacia, Uppsala, Sweden) density gradient centrifugation of buffy coats from normal blood donors. The cells were washed in PBS, and either frozen as described [21] or immediately used in the IFN-α induction cultures (see below). Before use, the cells were suspended in RPMI 1640 medium (ICN Biomedical, Costa Mesa, CA) with penicillin 60 μg/ml, streptomycin 100 μg/ml, l-glutamine 2 mm, 20 mm HEPES and 0.1% (v/v) human serum albumin (HSA; Kabi-Pharmacia, Stockholm, Sweden).
Herpes simplex virus
The herpes simplex virus (HSV) type 1 was propagated in human WISH cells, grown in Dulbecco's modified Eagle's medium (DMEM; ICN Biomedical) with penicillin 60 μg/ml, streptomycin 100 μg/ml, l-glutamine 2 mm, 20 mm HEPES and 5% fetal calf serum (FCS; Gibco BRL, Life Technologies, Paisley, UK). The HSV (2 × 107 plaque-forming units (PFU)/ml) was used either infectious (f-HSV) or UV-inactivated (1 J at 254 nm; UV-HSV), at optimal concentrations for the induction of IFN-α.
IFN-α induction cultures
The PBMC were cultured at a final concentration of 8 × 106 (flow cytometry) or 2 × 106 (all other experiments) cells per ml in presence of 10–25% SLE serum (as indicated), the indicated volumes of fractionated or enzyme-treated sera, or HSV. Cultures were in triplicates using final volumes of 0.1 ml/well in 96-well flat-bottomed plates (Nunclon; Nunc, Roskilde, Denmark), or for subsequent in situ hybridization or flow cytometry 1 ml volumes per well in 24-well plates (Nunc). When indicated, cultures were supplemented with 500 U/ml recombinant IFN-α2b (Intron-A; Schering-Plough, Bloomfield, NJ), 1 ng/ml GM-CSF (Leucomax; Schering-Plough), or 100 μg/ml cycloheximide (Boehringer Mannheim, Mannheim, Germany). The combination IFN-α2b and GM-CSF was chosen because these cytokines enhance HSV-induced production of IFN-α in PBMC [22]. The PBMC cultures were incubated for 5 h (in situ hybridization, flow cytometry) or 24 h (all other experiments) at 37°C and 7% CO2.
Immunoassays for IFN-α
The levels of IFN-α in 24-h culture supernatants were determined by a previously described dissociation-enhanced lanthanide fluoroimmunoassay (DELFIA) [23], which detects the majority of IFN-α subtypes but not the IFN-α2b used to supplement the IFN-α induction cultures. Serum levels of IFN-α were determined by a modified, more sensitive DELFIA [24]. Lower detection levels were 1 and 0.5 U/ml, respectively. The IFN-α immunoassay standard as well as the bioassay standard (see below) were calibrated against the NIH reference leucocyte IFN-α GA-23-902-530.
IFN bioassay
Supernatants were assayed for biologic IFN activity using Madin Darby bovine kidney (MDBK) cells as indicator cells and vesicular stomatitis virus for challenge [25]. For neutralization, supernatants were preincubated for 1 h at 37°C with 2 × 103 neutralizing U/ml of a sheep antiserum to IFN-α (batch 435; titre 4 × 105 neutralizing U/ml) produced in our laboratory using as antigen immunosorbent purified IFN-α derived from Sendai virus-stimulated blood leucocytes.
In situ hybridization
Cells stimulated by SLE serum or f-HSV were suspended, washed in PBS and fixed overnight in paraformaldehyde (PFA) [22]. In situ hybridization for the detection of cells containing mRNA for IFN-α was then performed with a 35S-labelled cRNA probe to human IFN-α2b [26] as described [27]. Slides were evaluated in the light microscope as described [26], cells with more than five grains were scored as positive and the approximate numbers of grains per positive cell were recorded.
Analysis of cells by flow cytometry
The PBMC stimulated by SLE sera or UV-HSV were fixed by PFA, stained and analysed by flow cytometry (FCM) for the simultaneous expression of cell surface antigen and intracellular IFN-α as described [28]. In short, fixed PBMC were incubated with MoAb reactive with cell surface antigen (see below) and then with FITC-conjugated goat anti-mouse immunoglobulin antibody (Dako A/S, Glostrup, Denmark) prior to permeabilization with 0.05% Tween 20. Subsequently, the PBMC were incubated with a biotinylated MoAb reactive with IFN-α, and finally with PE-conjugated streptavidin (Jackson ImmunoReseach, West Grove, PA).
The MoAb to cell surface antigen included OKT3 (CD3; Ortho Diagnostic systems, Raritan, NJ), MT310, SS2/36, HD37, 1F8, BIRMA-K3 (CD4, CD10, CD19, CD21 and CD34, respectively; Dakopatts A/S, Glostrup, Denmark), clone 44 (CD11b; British Biotechnology, Abingdon, UK), S-HCL-3, L48, L307.4, L243 (CD11c, CD45RA, CD80, and HLA-DR, respectively; Becton Dickinson, San Jose, CA), IV:414 (CD14; produced in our laboratory [23]), 2E1, FA6-152, MAB 89 (CD32, CD36, CD40; Immunotech SA, Marseille, France), HB15a (CD83; kindly provided by Dr T. F. Tedder, Department of Immunology, Duke University Medical Center, Durham, NC [29]) and IT2.2 (CD86; PharMingen, San Diego, CA). The muIgG1 isotype control was from Dakopatts.
Ultra filtration
Sera were prefiltered using 0.45-μm filters (Acrodisc; Gelman Sciences, Ann Arbor, MI), diluted to 25% in RPMI 1640 medium or when indicated in the mixture 33% RPMI 1640 and 67% 100 mm Tris–HCl pH 7.5. Ultrafiltration by centrifugation in Filtron Microsep units (Filtron Technology Corp., Northborough, MA) was done as recommended by the manufacturer. The retentate volumes (20–45 μl) were adjusted to the original 0.4 ml with RPMI 1640 medium or with RPMI and 50% of the Tris buffer as indicated above. The fractions were tested for their IFN-α-inducing capacity as described above in cytokine-supplemented cultures using 50-μl volumes of each fraction per well.
Treatment with enzymes
Volumes (25 μl) of sera or HSV diluted in RPMI 1640 medium were mixed with an equal volume of either the nuclease Benzonase (final concentration 800 U/ml; Nycomed Pharma A/S, Copenhagen, Denmark), DNase I 20 U/ml, RNase A 1 U/ml or RNase T1 100 U/ml (Boehringer Mannheim, Mannheim, Germany). After 1 h at 37°C the mixtures were added to PBMC in RPMI 1640 medium with IFN-α2b and GM-CSF supplement.
Separation on protein G columns
The sera were 0.45 μm filtered and diluted with equal volumes of RPMI 1640 medium. They were separated on protein G Sepharose (Pharmacia) as recommended by the manufacturer. The pH of the eluates was adjusted to pH 7 by the addition of 1 m Tris–HCl pH 9.0, and dialysed against PBS. Serum, effluent and eluate were tested alone or combined for ability to induce IFN-α production by adding 25 μl volumes per well to cytokine-supplemented PBMC cultures (see above).
Statistical analysis
The significance of differences between groups was determined by χ2 test or by Mann–Whitney _U_-test, as indicated. The IFN-α-inducing capacity of an SLE serum was considered significant (P < 0.05) when the IFN-α level was > 2 s.d. above the mean of IFN-α levels induced by the control sera.
RESULTS
Induction of IFN-α production in PBMC by SLE sera
Serum from between three and seven (depending on the PBMC donor) of 34 SLE patients induced production of up to 220 U/ml of IFN-α in cultures of PBMC from normal individuals (Fig. 1). In contrast, no IFN-α was induced by sera from healthy controls (n = 18). When such PBMC cultures were supplemented with IFN-α2b and GM-CSF, all sera from both patients and controls induced production of IFN-α (Fig. 1). The levels of IFN-α were, however, low with the control sera (range 1–25 U/ml, median 16 U/ml for the PBMC donor in Fig. 1), while as much as 103 U IFN-α/ml (range 3–1030 U/ml, median 41 U/ml in Fig. 1) was observed with sera from SLE patients. Depending on the PBMC donor used, sera from 14 and 21 of the 34 SLE patients induced significantly (P < 0.05) higher levels of IFN-α than control sera. The SLE sera which were IFN-α inducers in the absence of costimulatory IFN-α2b and GM-CSF caused 5–100-fold higher IFN-α production in PBMC cultures supplemented with these cytokines. Similar results as in Fig. 1 were obtained with heparinized or defibrinized plasma (data not shown).
Fig. 1.
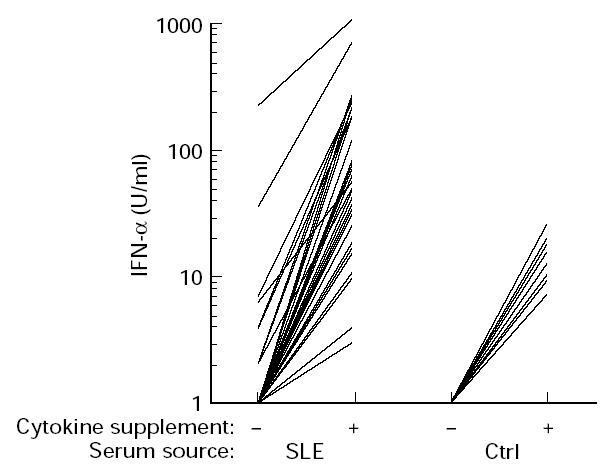
IFN-α production of normal peripheral blood mononuclear cells (PBMC) (previously frozen) in response to stimulation by sera (20%) from SLE patients (SLE; n = 34), or healthy controls (ctrl; n = 18). The PBMC cultures were either supplemented (+) or not (–) with 500 U/ml of IFN-α2b and 1 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF). Levels of produced IFN-α were determined by a specific immunoassay, not detecting the added IFN-α2b. The median IFN-α production occurring in cytokine-supplemented cultures without serum was 1 U/ml (range < 1–4 U/ml) and < 1 U/ml in unsupplemented cultures. Results from one of four representative PBMC donors with similar results. See Patients, Materials and Methods for further details.
The protein synthesis inhibitor cycloheximide prevented IFN-α production induced by SLE sera in the PBMC cultures, proving it is due to new synthesis of IFN-α (data not shown). The IFN-α detected by immunoassay was biologically active, because the antiviral activity induced in PBMC by two selected SLE sera in the absence of costimulatory cytokines was 500 and 400 U/ml in a cytopathic effect inhibition bioassay using vesicular stomatitis virus and target MDBK cells, was fully neutralized (< 2 U/ml) by anti-IFN-α antibodies, and agreed with immunoassay results (380 and 510 U/ml).
Relation between the IFN-α-inducing capacity of SLE sera, presence of IFN-α in serum and disease activity
As shown in Table 1, both the proportion of patients with IFN-α-inducing sera and the levels of serum-induced IFN-α production in PBMC cultures without costimulatory cytokines were significantly higher in SLE patients with active disease (SLEDAI score ≥ 2) than in patients in remission (SLEDAI = 0). The difference between the two groups of patients was less pronounced with IFN-α2b/GM-CSF costimulated PBMC.
Table 1.
Relation between the disease activity in SLE patients and the IFN-α-inducing capacity in serum or presence of IFN-α in serum†
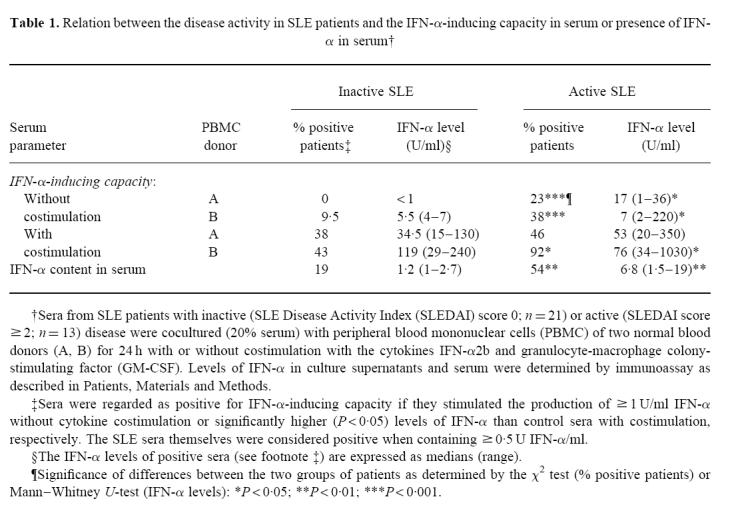
Eleven of 34 SLE patients had measurable serum levels of IFN-α (≥ 0.5 U/ml; Table 1). Both the proportion of IFN-α-positive patients and their serum levels of IFN-α were significantly higher in patients with active than inactive SLE.
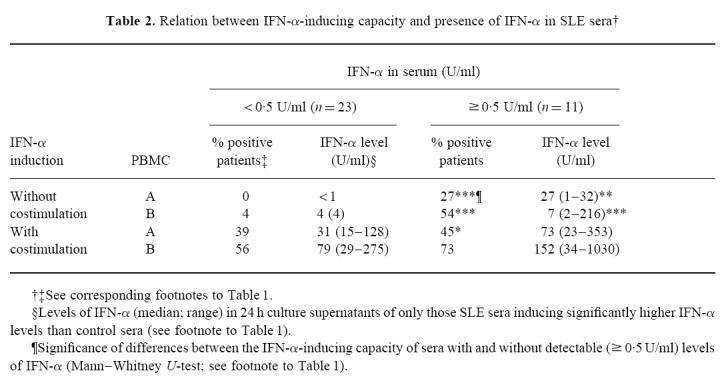
Furthermore, SLE sera with detectable levels of IFN-α induced IFN-α production in PBMC more frequently and at higher levels than sera without detectable IFN-α levels (Table 2). This difference was not seen when the PBMC cultures were costimulated with IFN-α2b and GM-CSF.
Table 2.
Relation between IFN-α-inducing capacity and presence of IFN-α in SLE sera†
Identification of the IFN-α-producing cells
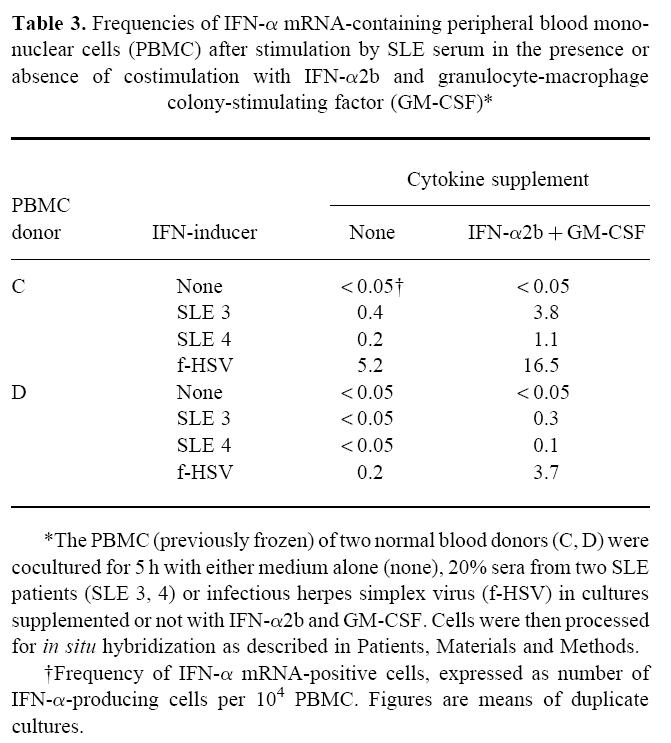
Infrequent but heavily labelled IFN-α mRNA-containing IFN-α-producing cells (IPC) were identified by means of in situ hybridization using a 35S-labelled cRNA probe among PBMC exposed in vitro to SLE serum (Fig. 2). Their labelling intensity was comparable to the IFN-α mRNA levels in cells induced by HSV (not shown), but the frequency was considerably lower (Table 3) and correlated to the IFN-α levels in the culture medium measured by immunoassay (results not shown). Similar to the effects on the protein level, the presence of costimulatory IFN-α2b and GM-CSF during the induction of the IFN-α responses increased the IPC frequencies for both SLE serum and HSV stimulations. The IPC frequencies varied both with the PBMC donor and the SLE serum used.
Fig. 2.
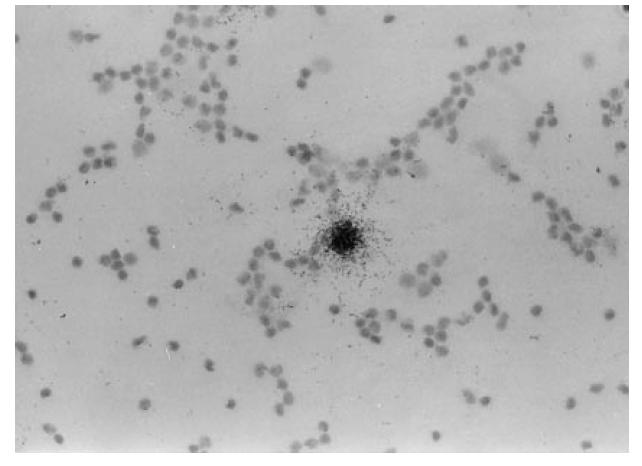
Infrequent but strongly IFN-α mRNA-positive peripheral blood mononuclear cells (PBMC) detected by in situ hybridization. The PBMC were stimulated by 20% of an SLE serum (donor C and serum 3 in Table 3) for 5 h, fixed and processed for in situ hybridization as described in Patients, Materials and Methods. (Mag. × 400.)
Table 3.
Frequencies of IFN-α mRNA-containing peripheral blood mononuclear cells (PBMC) after stimulation by SLE serum in the presence or absence of costimulation with IFN-α2b and granulocyte-macrophage colony-stimulating factor (GM-CSF)*
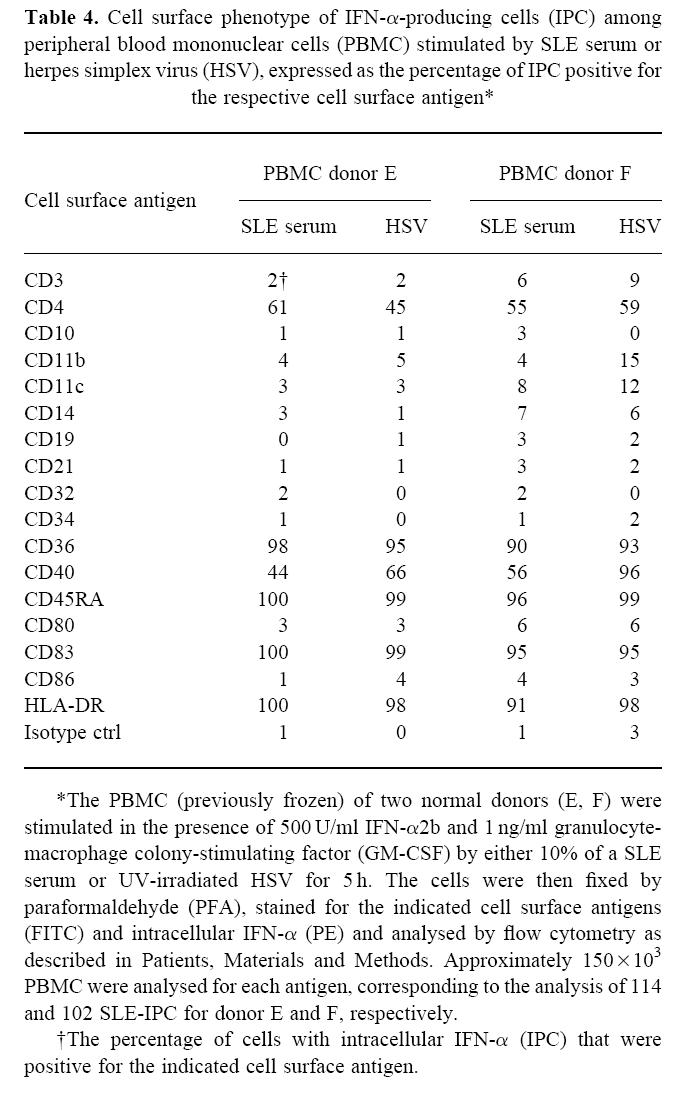
The phenotype of IPC induced by SLE serum and by HSV were clearly similar, as determined by FCM with analysis of cell surface antigens on cells stained for intracellular IFN-α (Table 4). Both types of IPC clearly expressed CD36, CD45RA, CD83 and HLA-DR as well as lower and more variable levels of CD4 and CD40. They did not express the costimulatory molecules CD80 and CD86, and several antigens normally present on T or B cells or monocytes/macrophages.
Table 4.
Cell surface phenotype of IFN-α-producing cells (IPC) among peripheral blood mononuclear cells (PBMC) stimulated by SLE serum or herpes simplex virus (HSV), expressed as the percentage of IPC positive for the respective cell surface antigen*
Characterization of the IFN-α-inducing activity
Separation of the IFN-α-inducing factor in SLE serum (SLE-IIF) by gel filtration on Sephacryl S-400 or Sepharose 4B columns revealed no significant IFN-α-inducing activity in any of the fractions (results not shown). However, ultrafiltration showed that most of the SLE-IIF was retained by 100- and 300-kD cut-off filters, but not by a 1000-kD filter (Table 5). Therefore most of the SLE-IIF was between 300 and 1000 kD in size.
Table 5.
Characterization of the IFN-α-inducing activity in SLE and control serum by means of ultrafiltration, adsorption to protein G Sepharose or treatment with nucleases*
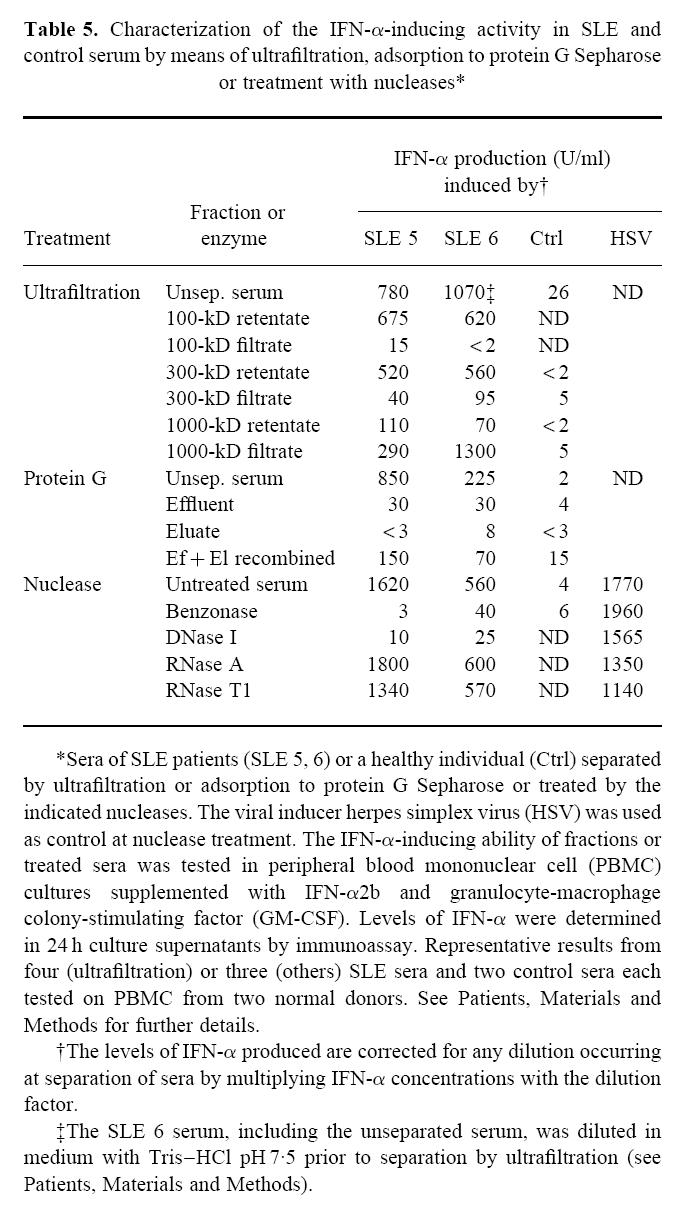
An obvious possibility was that the SLE-IIF consisted of small antigen–antibody complexes. In support of this, the IFN-α- inducing activity was eliminated by passage of SLE serum over a protein G Sepharose column, i.e. no activity was found in the effluent (Table 5). While no IFN-α-inducing activity was eluted from the column, combining effluent and eluate partially restored the IFN-α-inducing activity. The low IFN-α-inducing activity of a control serum was not clearly decreased in the protein G Sepharose effluent, but a moderate increase was seen in the combined effluent and eluate. When SLE serum was passed over a control unconjugated Sepharose CL-4B column, all IFN-α-inducing activity was recovered in the effluent (results not shown).
Both the nuclease Benzonase and DNase I, but not RNase A or T1, largely destroyed the IFN-α-inducing activity (Table 5). Pretreatment of only the PBMC with these enzymes and subsequent washing had no effect on their IFN-α-producing ability (results not shown). Furthermore, treatment of HSV with enzymes did not affect the IFN-α production induced by this virus (Table 5). Finally, Benzonase treatment of sera from healthy control individuals did not decrease their already low IFN-α-inducing capacity.
DISCUSSION
Serum (as well as plasma) from as many as 20% of the SLE patients in our study triggered new synthesis of bioactive IFN-α in cultures of PBMC from normal individuals, and some SLE sera were as potent IFN-α inducers as the virus HSV. Obviously, such a high IFN-α-inducing activity in the blood could have profound effects in patients. It could well explain the previously reported activation of the type I IFN system in SLE [2–8], a possibility further suggested by our finding of a significant association between the presence of IFN-α-inducing activity and of IFN-α in patient sera.
We found that IFN-α production of normal PBMC cocultured with SLE sera was enhanced 5–100 times when the cells were also exposed to IFN-α2b and GM-CSF. Such a stimulatory effect of these cytokines has been seen also for HSV-induced IFN-α production [22]. Interestingly, SLE sera which did not induce IFN-α production alone did so in the presence of IFN-α2b and GM-CSF. Because these or other cytokines are produced in SLE patients during active disease [30,31], they could also sustain a high IFN-α production by costimulatory effects in vivo.
The actual IFN-α-producing cells (IPC) among PBMC exposed to SLE serum in vitro were infrequent, with an apparently high copy number of IFN-α mRNA, and therefore probably producing large quantities of IFN-α on a per cell basis. They had the phenotype of natural IPC (NIPC) as determined by flow cytometry and thus appear identical to the NIPC activated by many different viruses and bacteria, including HSV [23,28,32]. Because the NIPC resemble circulating immature dendritic cells [28], the IFN-α inducer in SLE may act on cells which are considered central in immune responses.
We have previously shown a 70-fold decrease in the number of NIPC in the blood of SLE patients, but no cells with an ongoing secretion of IFN-α [24]. The endogenous SLE-IIF might therefore possibly recruit IPC and activate them in tissues. In fact, activated NIPC-like cells in mice and pigs injected by viral IFN-α inducers are preferentially localized in lymphoid tissues [17,33,34]. Assuming a similar location in SLE patients, the produced IFN-α could very efficiently influence the immune system.
The first characterization of the SLE-IIF revealed a relatively high molecular weight (300–1000 kD), and probable size heterogeneity at gel filtration. Although protein G column separation indicated immunoglobulin content, a component essential for inducing IFN-α synthesis was sensitive to DNase I and the general nuclease Benzonase, but not to RNases. Consequently, we suggest that the SLE-IIF consists of antigen–antibody complexes containing DNA. Such antigen–antibody complexes probably dissociate at the protein G column separations and destruction or limited reassociation of complex components could explain why their IFN-α-inducing activity was only partially restored. Work in progress will determine the nature of the DNA involved, e.g. whether it is present as nucleosomes, the specificity of the antibodies and whether additional components such as complement factors are required. At present we speculate that the DNA is related to immunostimulatory DNA containing palindromes with unmethylated CpG [35]. The presence of such DNA in sera of SLE patients has been described [36–38] and has also been suggested to be important in the pathogenesis of SLE (reviewed in [39]).
We observed that also sera from normal individuals caused low IFN-α production in PBMC, although only in PBMC costimulated by IFN-α2b and GM-CSF. Besides always requiring such costimulation, this inducing activity in normal serum differed from that in SLE serum in being weaker, as well as not being retained by protein G or destroyed by Benzonase.
We noted an association between serum IFN-α inducer and disease activity and confirmed the previously described correlation between serum IFN-α levels and disease activity [2,5,6]. Although it cannot be excluded that the activation of the type I IFN system is a secondary phenomenon in SLE, the fact remains that the type I IFNs have many immunoregulatory activities [12–17]. In addition, various forms of autoimmune manifestations are frequently seen in patients on IFN-α therapy [10,40], including antinuclear and anti-DNA antibodies as well as occasionally SLE-like syndromes [9–11]. Our hypothesis, in part based on our present findings, is therefore that IFN-α is an important pathogenic factor which precipitates and sustains the naturally occurring SLE in genetically predisposed individuals. The production of such IFN-α can at least initially be caused by viral infections and be maintained by the identified endogenous IFN-α inducer. The further elucidation of the role of the type I IFN system in SLE may consequently provide new insights into the aetiology and pathogenesis of this disease.
Acknowledgments
We thank Mrs Anne Riesenfeld, Mr Anders Perers, Mrs Lotta Karnell and Mrs Inger Ohlsson for excellent technical assistance. This work was supported by grants from the Swedish Rheumatism Association, the Tore Nilson Foundation, the 80 years Foundation of King Gustav V, the Emil and Wera Cornell Foundation and the Swedish Medical Research Council.
REFERENCES
- 1.Wallace DJ, Hahn B. Dubois' lupus erythematosus. 5. Baltimore: Williams and Wilkins; 1997. [Google Scholar]
- 2.Kim T, Kanayama Y, Negoro N, Okamura M, Takeda T, Inoue T. Serum levels of interferons in patients with systemic lupus erythematosus. Clin Exp Immunol. 1987;70:562–9. [PMC free article] [PubMed] [Google Scholar]
- 3.Klippel JH, Carette S, Preble OT, Friedman RM, Grimley PM. Serum alpha interferon and lymphocyte inclusions in systemic lupus erythematosus. Ann Rheum Dis. 1985;44:104–8. doi: 10.1136/ard.44.2.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Preble OT, Rothko K, Klippel JH, Friedman RM, Johnston MI. Interferon-induced 2′-5′ adenylate synthetase in vivo and interferon production in vitro by lymphocytes from systemic lupus erythematosus patients with and without circulating interferon. J Exp Med. 1983;157:2140–6. doi: 10.1084/jem.157.6.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strannegård Ö, Hermodsson S, Westberg G. Interferon and natural killer cells in systemic lupus erythematosus. Clin Exp Immunol. 1982;50:246–52. [PMC free article] [PubMed] [Google Scholar]
- 6.Ytterberg SR, Schnitzer TJ. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum. 1982;25:401–6. doi: 10.1002/art.1780250407. [DOI] [PubMed] [Google Scholar]
- 7.von Wussow P, Jakschies D, Hartung K, Deicher H. Presence of interferon and anti-interferon in patients with systemic lupus erythematosus. Rheumatol Int. 1988;8:225–30. doi: 10.1007/BF00269199. [DOI] [PubMed] [Google Scholar]
- 8.von Wussow P, Jakschies D, Hochkeppel H, Horisberger M, Hartung K, Deicher H. MX homologous protein in mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1989;32:914–8. [PubMed] [Google Scholar]
- 9.Rönnblom LE, Alm GV, Öberg KE. Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990;227:207–10. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 10.Rönnblom LE, Alm GV, Öberg KE. Autoimmunity after alpha- interferon therapy for malignant carcinoid tumors. Ann Intern Med. 1991;115:178–83. doi: 10.7326/0003-4819-115-3-178. [DOI] [PubMed] [Google Scholar]
- 11.Fritzler MJ. Drugs recently associated with lupus syndromes. Lupus. 1994;3:455–9. doi: 10.1177/096120339400300605. [DOI] [PubMed] [Google Scholar]
- 12.Belardelli F. Role of interferons and other cytokines in the regulation of the immune response. APMIS. 1995;103:161–79. doi: 10.1111/j.1699-0463.1995.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 13.D'Cunha J, Ramanujam S, Wagner RJ, Witt PL, Knight E, Jr, Borden EC. In vitro and in vivo secretion of human ISG15, an IFN-induced immunomodulatory cytokine. J Immunol. 1996;157:4100–8. [PubMed] [Google Scholar]
- 14.Rogge L, Barberis-Maino L, Biffi M, et al. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J Exp Med. 1997;5:825–31. doi: 10.1084/jem.185.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray D. Viral immunity: interferons jog old T-cell memories. Curr Biol. 1996;6:1254–5. doi: 10.1016/s0960-9822(02)70710-0. [DOI] [PubMed] [Google Scholar]
- 16.Belardelli F, Gresser I. The neglected role of type I interferon in the T-cell response: implications for its clinical use. Immunol Today. 1996;17:369–71. doi: 10.1016/0167-5699(96)10027-X. [DOI] [PubMed] [Google Scholar]
- 17.Riffault S, Eloranta M-L, Carrat C, Sandberg K, Charley B, Alm G. Herpes simplex virus induced appearance of IFN-α/β producing cells and partially IFN-α/β dependent accumulation of leukocytes in murine regional lymph nodes. J Interferon Cytokine Res. 1996;16:1007–14. doi: 10.1089/jir.1996.16.1007. [DOI] [PubMed] [Google Scholar]
- 18.De Maeyer E, De Maeyer-Guignard J. Interferons and other regulatory cytokines. New York: John Wiley & Sons; 1988. [Google Scholar]
- 19.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 20.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35:630–40. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 21.Cederblad B, Riesenfeld T, Alm GV. Deficient herpes simplex virus-induced interferon-alpha production by blood leukocytes of preterm and term newborn infants. Pediatr Res. 1990;27:7–10. doi: 10.1203/00006450-199001000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Cederblad B, Alm GV. Interferons and the colony-stimulating factors IL-3 and GM-CSF enhance the IFN-alpha response in human blood leucocytes induced by herpes simplex virus. Scand J Immunol. 1991;34:549–55. doi: 10.1111/j.1365-3083.1991.tb01578.x. [DOI] [PubMed] [Google Scholar]
- 23.Svensson H, Cederblad B, Lindahl M, Alm GV. Stimulation of natural interferon-α/β producing cells by Staphylococcus aureus. J Interferon Res. 1996;16:7–16. doi: 10.1089/jir.1996.16.7. [DOI] [PubMed] [Google Scholar]
- 24.Cederblad B, Blomberg S, Vallin H, Perers A, Alm GV, Rönnblom L. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon-α producing cells. J Autoimmun. 1998;11:465–70. doi: 10.1006/jaut.1998.0215. [DOI] [PubMed] [Google Scholar]
- 25.Cederblad B, Alm GV. Infrequent but efficient interferon-alpha- producing human mononuclear leukocytes induced by herpes simplex virus in vitro studied by immuno-plaque and limiting dilution assays. J Interferon Res. 1990;10:65–73. doi: 10.1089/jir.1990.10.65. [DOI] [PubMed] [Google Scholar]
- 26.Gobl AE, Funa K, Alm GV. Different induction patterns of mRNA for IFN-alpha and -beta in human mononuclear leukocytes after in vitro stimulation with herpes simplex virus-infected fibroblasts and Sendai virus. J Immunol. 1988;140:3605–9. [PubMed] [Google Scholar]
- 27.Sandberg K, Eloranta M-L, Campbell IL. Expression of alpha/beta interferons (IFN-α/β) and their relationship to IFN-α/β-induced genes in lymphocytic choriomeningitis. J Virol. 1994;68:7358–66. doi: 10.1128/jvi.68.11.7358-7366.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svensson H, Johannisson A, Nikkilä T, Alm GV, Cederblad B. The cell surface phenotype of human natural interferon-α producing cells as determined by flow cytometry. Scand J Immunol. 1996;44:164–72. doi: 10.1046/j.1365-3083.1996.d01-289.x. [DOI] [PubMed] [Google Scholar]
- 29.Zhou LJ, Schwarting R, Smith HM, Tedder TF. A novel cell-surface molecule expressed by human interdigitating reticulum cells, Langerhans cells, and activated lymphocytes is a new member of the Ig superfamily. J Immunol. 1992;149:735–42. [PubMed] [Google Scholar]
- 30.al Janadi M, al Balla S, al Dalaan A, Raziuddin S. Cytokine profile in systemic lupus erythematosus, rheumatoid arthritis, and other rheumatic diseases. J Clin Immunol. 1993;13:58–67. doi: 10.1007/BF00920636. [DOI] [PubMed] [Google Scholar]
- 31.Horwitz DA, Wang H, Gray JD. Cytokine gene profile in circulating blood mononuclear cells from patients with systemic lupus erythematosus: increased interleukin-2 but not interleukin-4 mRNA. Lupus. 1994;3:423–8. doi: 10.1177/096120339400300511. [DOI] [PubMed] [Google Scholar]
- 32.Fitzgerald-Bocarsly P. Human natural interferon-α producing cells. Pharmac Ther. 1993;60:39–62. doi: 10.1016/0163-7258(93)90021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artursson K, Lindersson M, Varela N, Scheynius A, Alm GV. Interferon-alpha production and tissue localization of interferon-alpha/beta producing cells after intradermal administration of Aujeszky's disease virus-infected cells in pigs. Scand J Immunol. 1995;41:121–9. doi: 10.1111/j.1365-3083.1995.tb03543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eloranta M-L, Sandberg K, Alm GV. The interferon-α/β responses of mice to herpes simplex virus studied at the blood and tissue level in vitro and vivo. Scand J Immunol. 1996;43:355–60. doi: 10.1046/j.1365-3083.1996.d01-62.x. [DOI] [PubMed] [Google Scholar]
- 35.Pisetsky D. Immune activation by bacterial DNA. A new genetic code. Immunity. 1996;5:303–10. doi: 10.1016/s1074-7613(00)80256-3. [DOI] [PubMed] [Google Scholar]
- 36.Krapf F, Herrmann M, Leitmann W, Kalden J. Antibody binding of macromolecular DNA and RNA in the plasma of SLE patients. Clin Exp Immunol. 1989;75:336–42. [PMC free article] [PubMed] [Google Scholar]
- 37.Sano H, Takai O, Harata N, Yoshinaga K, Kodama-Kamada I, Sasaki T. Binding properties of human anti-DNA antibodies to cloned human DNA fragments. Scand J Immunol. 1989;30:51–63. doi: 10.1111/j.1365-3083.1989.tb01188.x. [DOI] [PubMed] [Google Scholar]
- 38.Miyata M, Kanno T, Ishida H, et al. CpG motif in DNA from immune complexes of SLE patients augments expression of intercellular adhesion molecule-1 on endothelial cells. Jpn J Clin Pathol. 1996;44:1125–31. [PubMed] [Google Scholar]
- 39.Krieg A. CpG, DNA: a pathogenic factor in systemic lupus erythematosus. J Clin Immunol. 1995;15:284–92. doi: 10.1007/BF01541318. [DOI] [PubMed] [Google Scholar]
- 40.Strander H. Toxicities of interferons. In: Stuart-Harris R, Penny R, editors. Clinical applications of the interferons. London: Chapman & Hall; 1997. pp. 331–63. [Google Scholar]