Placenta growth factor (PlGF) induces vascular endothelial growth factor (VEGF) secretion from mononuclear cells and is co-expressed with VEGF in synovial fluid (original) (raw)
Abstract
The aims of this study were (i) to determine whether PlGF, VEGF and PlGF/VEGF heterodimers are detected in synovial fluid (SF) and plasma samples from patients with a range of arthropathies; (ii) to describe whether any correlation exists between SF PlGF, VEGF and PlGF/VEGF heterodimer levels and the total and differential SF leucocyte counts; and (iii) to investigate the regulation of peripheral blood mononuclear cell (PBMC) VEGF secretion by stimuli relevant to inflammatory joints. PlGF, VEGF and PlGF/VEGF heterodimer levels were measured in the SF and plasma of patients with a range of arthropathies and normal controls by ELISA. Western blotting for PlGF was performed on SF from three patients with rheumatoid arthritis (RA) and primary inflammatory arthropathies. VEGF was quantified in cell culture supernatants after stimulation with lipopolysaccharide (LPS), PlGF or cobalt ions of PBMC isolated from RA patients and controls. PlGF and VEGF were detected in all SF samples. PlGF/VEGF heterodimers were detected in 10.2% of SF samples, most frequently in RA samples. Western blotting confirmed the presence of PlGF in RA SF. PlGF was detected in 52% of RA and 31% of control plasma samples, and VEGF was detected in 38% of RA and 38% of control plasma samples. PlGF/VEGF heterodimers were detected in 21% of RA samples and none of the control samples. In primary inflammatory arthropathy patients, SF PlGF and VEGF levels correlated significantly with the SF total leucocyte count and the neutrophil count. PlGF was the most potent inducer of PBMC VEGF production in both RA and control subjects. This is the first report of the detection of PlGF and PlGF/VEGF heterodimers in the SF of patients with inflammatory arthropathies, and we have shown for the first time that PlGF up-regulates PBMC VEGF production. PlGF may therefore play a key role in the production of VEGF in the inflammatory joint.
Keywords: placenta growth factor, vascular endothelial growth factor, arthritis, synovial fluid heterodimers
INTRODUCTION
Early events in the clinical course of rheumatoid arthritis (RA) focus on the changes occurring in the synovial lining. Histologically, these changes are marked by neovascularization and inflammatory cell infiltration within the synovium with associated synoviocyte hyperplasia, which together produce synovitis and the formation of inflamed vascular pannus. The synovitis is responsible for the effusion and the pannus covers and erodes into articular cartilage, eventually leading to joint destruction. Synovial fluid (SF) accumulation associated with synovitis in the inflamed joint is one of the first symptoms of disease activity in RA and can reach the point where synovial blood flow is compromised [1], accentuating the hypoxia and acidosis common in joints of severely affected patients [2].
VEGF is a potent angiogenic factor which stimulates the proliferative phase of endothelial growth [3], induces endothelial expression of plasminogen activators, plasminogen activator inhibitor-1 [4] and interstitial collagenase [5], and is a regulator of endothelial cell integrin expression and chemoattraction [6]. These factors control the extracellular proteolysis which is essential for capillary morphogenesis and the migration of endothelial cells during new capillary formation. VEGF is also one of the most potent modulators of capillary permeability (50 000 times more active than histamine on a molar basis), having been characterized as vascular permeability factor (VPF) [7]. Therefore, the identification of increased levels of VEGF in RA SF [8,9] is highly suggestive that VEGF may be part of the inflammatory mechanism driving the destruction of the joint.
Immunohistochemical studies have localized VEGF protein to inflamed synovial macrophages and microvascular endothelium, though only the former had detectable mRNA by in situ hybridization [8]. VEGF mediates its biological function through binding to two specific tyrosine kinase receptors; flt-1 (fms-like tyrosine kinase) [10] and KDR (kinase insert domain containing receptor) [11], also known as VEGFR1 and VEGFR2. In tissues, these receptors are expressed predominantly on endothelial cells in keeping with the endothelial cell-specific roles for VEGF, though the flt-1 receptor is also present on monocytes and neutrophils and enables these cells to respond to a chemoattractant gradient of VEGF [12]. High levels of VEGF in SF could therefore promote a continual influx of such inflammatory cells into the joint.
PlGF, a member of the same family of polypeptides as VEGF, has also been found to be mitogenic for endothelial cells [13], to stimulate tissue factor production and chemotaxis in monocytes [14] and also bind the VEGF receptor flt-1 [15]. Naturally occurring heterodimers formed between VEGF and PlGF have been identified and have been found to be more active than PlGF homodimers and nearly as potent as VEGF homodimers in assays of mitogenesis [13]. Therefore, some of the biological activities attributed to VEGF homodimers might be mediated by PlGF/VEGF heterodimers and therefore play an important role in the processes involved in RA.
The aims of this study were (i) to determine whether PlGF, VEGF and PlGF/VEGF heterodimers are detected in the SF and plasma samples from patients with a range of arthropathies; (ii) to describe whether any correlation exists between SF VEGF, PlGF and PlGF/VEGF heterodimer levels and the total and differential SF leucocyte counts; and (iii) to investigate the regulation of peripheral blood mononuclear cell (PBMC) VEGF secretion by stimuli relevant to inflammatory joints.
PATIENTS AND METHODS
Patient samples
SF samples were obtained from 88 patients by joint aspiration performed as part of routine clinical management. SF samples were categorized on the basis of pathology alone [16] into the following groups: RA (18 females, 10 males, mean age (range) 61 years (38–85 years)); osteoarthritis (OA) (three females, four males, 62 years (46–79 years)), primary inflammatory arthropathies (seven females, eight males, 46 years (18–75 years)), non-inflammatory arthropathies (three females, 12 males, 50 years (17–78 years)), crystal arthropathies (one female, eight males, 58 years (43–75 years)), juvenile chronic arthritis (five females, one male, 13 years (10–16 years)) and seronegative arthropathies (three females, five males, 43 years (29–52 years)). Full clinical data regarding mean (± s.e.m.) disease duration were available for the patients with RA (12.8 ± 1.9 years), OA (4.6 ± 0.7 years), primary inflammatory arthropathies (6.0 ± 1.8 years), non-inflammatory arthropathies (3.7 ± 0.4 years) and crystal arthropathies (5.4 ± 1.9 years).
EDTA-anti-coagulated whole blood samples were collected from 61 RA patients (51 females, 10 males, mean age (range) 59 years (30–77 years)) diagnosed and categorized according to criteria of the American College of Rheumatology (ACR). Plasma samples from 29 healthy individuals with no history of rheumatological disease acted as controls (20 female, nine male, mean age (range) 34 years (24–56 years)). Plasma and SF samples were stored as multiple aliquots at −70°C prior to measurement. Ethical approval for the study was granted by the Central Manchester Health Authority Ethical Committee.
Reagents
Recombinant (baculovirus-derived) VEGF165 and soluble flt-1 proteins were a gift from Zeneca Pharmaceuticals (Alderley Edge, UK). Recombinant (_Escherichia coli_-derived) PlGF and PlGF/VEGF and antibodies to PlGF were purchased from R&D Systems (Abingdon, UK) and the polyclonal antibody to VEGF was purchased from Serotec (Kidlington, UK). Lipopolysaccharide (LPS) and cobalt chloride were purchased from Quadratech (Epsom, UK) and Merck (Poole, UK), respectively. Heparin Sepharose, desalting columns and molecular weight markers were purchased from Amersham Pharmacia Biotech (St Albans, UK). Chemiluminescent substrate was obtained from Pierce & Warriner (Chester, UK).
PlGF ELISA
SF and plasma PlGF levels were measured by an in-house ELISA using a combination of a monoclonal and a biotinylated polyclonal antibody specific for PlGF. These antibodies were selected as they show no cross-reactivity with VEGF (manufacturer's data sheet). Ninety-six-well Dynatech Immulon 2 plates were coated with 100 μl of 2.5 μg/ml MoAb to PlGF in 0.05 m carbonate buffer pH 9.6, overnight at 4°C. Plates were blocked for a minimum of 1 h at room temperature with 300 μl of 5% sucrose, 1% bovine serum albumin (BSA) in PBS, prior to the addition of samples, controls and standards. A standard curve ranging from 30 pg/ml to 4 ng/ml was produced using rPlGF diluted in ELISA buffer (0.1% BSA, 0.05% Tween 20 in TBS). Positive and negative control samples of rPlGF, rVEGF and rPlGF/VEGF all at 1 ng/ml were added to every plate. The plates were then incubated for 2 h prior to the addition of 100 μl of 150 ng/ml biotinylated polyclonal antibody to PlGF for 2 h. Detection involved 100 μl streptavidin peroxidase diluted 1:6000 for 25 min, then 100 μl of TMB substrate for 10 min. The reaction was stopped by the addition of 50 μl 0.5 m sulphuric acid. Plates were then read at 450 nm. All incubations were carried out at room temperature except coating, and plates were washed six times with 400 μl of 0.1 m PBS with 0.05% Tween after each step. Samples were measured in duplicate and mean values calculated using Softmax software. The intra- and interplate coefficients of variation were 23.5% and 18.0%. The assay was shown to be specific for PlGF, with VEGF and PlGF/VEGF heterodimer positive control samples only recording background levels.
VEGF receptor capture ELISA
SF and plasma VEGF levels were measured using an in-house flt-1 receptor capture ELISA previously described in detail [17]. This assay has been shown to measure free VEGF but not VEGF complexed to flt-1. Intra- and interplate coefficients of variation for this assay were 4.5% and 11.5%, respectively. We have previously established that serum levels of VEGF do not reflect circulating levels due to platelet release of VEGF during clotting [17] and therefore have quantified VEGF levels in plasma.
PlGF/VEGF heterodimer ELISA
Ninety-six-well Dynatech Immulon 2 plates were coated with 100 μl of 2.5 μg/ml polyclonal antibody to VEGF in 0.05 m carbonate buffer pH 9.6, overnight at 4°C. Plates were blocked for a minimum of 1 h at room temperature with 300 μl of 5% sucrose, 1% BSA in PBS, prior to the addition of samples, controls and standards. A standard curve ranging from 30 pg/ml to 4 ng/nl was produced using rPlGF/VEGF diluted in ELISA buffer (0.1% BSA, 0.05% Tween 20 in TBS). Other steps were then identical to those described above for PlGF. Intra- and interplate coefficients of variation were 14.6% and 19.3%. The assay was shown to be specific for PlGF/VEGF heterodimers, with VEGF- and PlGF-positive control samples only recording background levels.
Western blotting for PlGF in SF
Three SF samples from RA patients containing significant levels of PlGF and three SF samples from non-inflammatory arthropathy patients with low levels as determined by ELISA were analysed by SDS–PAGE and Western blotting. Samples (200 μl) of SF (all from knee joints) were diluted 1:5 in 10 mm phosphate buffer and applied to a heparin Sepharose column, washed with buffer and eluted in buffer containing 1.0 m NaCl. Samples were desalted on a desalting column and the eluant was lyophilized. Samples were dissolved in 100 μl of SDS non-reducing sample loading buffer and after boiling for 5 min, 30 μl of each sample were applied to an 8–16% gradient polyacrylamide gel run under non-reducing conditions with molecular weight markers and rPlGF. Proteins were transferred to nitrocellulose by electroblotting for 90 min in Tris–glycine transfer buffer containing 10% methanol. The blot was blocked with 3% milk protein in Tris-buffered saline (blocking solution) for 1 h and then affinity-purified, biotinylated goat anti-PlGF antibody (R&D Systems), at 0.2 μg/ml in blocking solution, was applied for 2 h with shaking at 4°C. Following thorough washing in Tris-buffered saline (three times for 15 min) streptavidin–peroxidase at 1:10 000 in blocking solution was applied for 30 min with shaking at room temperature. Following washing, the blot was incubated with a chemiluminescent substrate for 1 min and exposed to Biomax MR film.
Microscopic analysis of SF
SF total leucocyte counts were performed on all samples using a haemocytometer and leucocyte differential counts were carried out on samples containing sufficient cells. Cells were diluted to 400 cells/mm3 in PBS, then 100 μl was cytospun at 200 g for 30 min. The slides were then air-dried prior to fixation in methanol for at least 5 min. Slides were stained using Jenner–Giemsa stain, then air-dried and mounted in aquamount prior to microscopic analysis.
Regulation of VEGF secretion by PBMC
PBMC were isolated using standard Ficoll density gradient centrifugation [18]. Cells were counted using a haemocytometer and resuspended at 2 × 106/ml in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS).
To investigate the regulation of PBMC VEGF secretion by mechanisms which may occur within the synovial joint, PBMC isolated from 10 RA patients (eight females, two males, 57 years (43–70 years)) and 10 healthy controls (six females, four males, 36 years (26–48 years)) were incubated for 24 h and 72 h at 37°C/5% CO2 in the presence of LPS (10 U/ml), cobalt chloride (50 μm), PlGF (10 ng/ml), LPS and cobalt together and all three agents together. Unstimulated cells cultured under identical conditions acted as controls.
Statistical analysis
Data were analysed using the one-way anova with Bonferroni correction or Pearson correlation and linear regression using SPSS 6.1 for Windows.
RESULTS
SF PlGF, VEGF and PlGF/VEGF heterodimer levels
PlGF and VEGF were detected in all SF samples, with significantly higher levels of PlGF being found in RA, primary inflammatory arthropathy and juvenile chronic arthritis samples compared with non-inflammatory arthropathy and OA samples, and significantly higher levels of VEGF being found in RA, primary inflammatory arthropathy and crystal arthropathy samples compared with non-inflammatory arthropathy samples (P < 0.05, one-way anova with Bonferroni correction) (Fig. 1a,b). PlGF/VEGF heterodimers were only detected in a total of nine (10.2%) SF samples, most frequently in RA samples (5/28) (Fig. 1c). No statistically significant differences existed between heterodimer levels in the different disease groups. No overall correlation existed between VEGF, PlGF and PlGF/VEGF heterodimer levels, but heterodimers were detected only in samples having high levels of PlGF and VEGF.
Fig. 1.
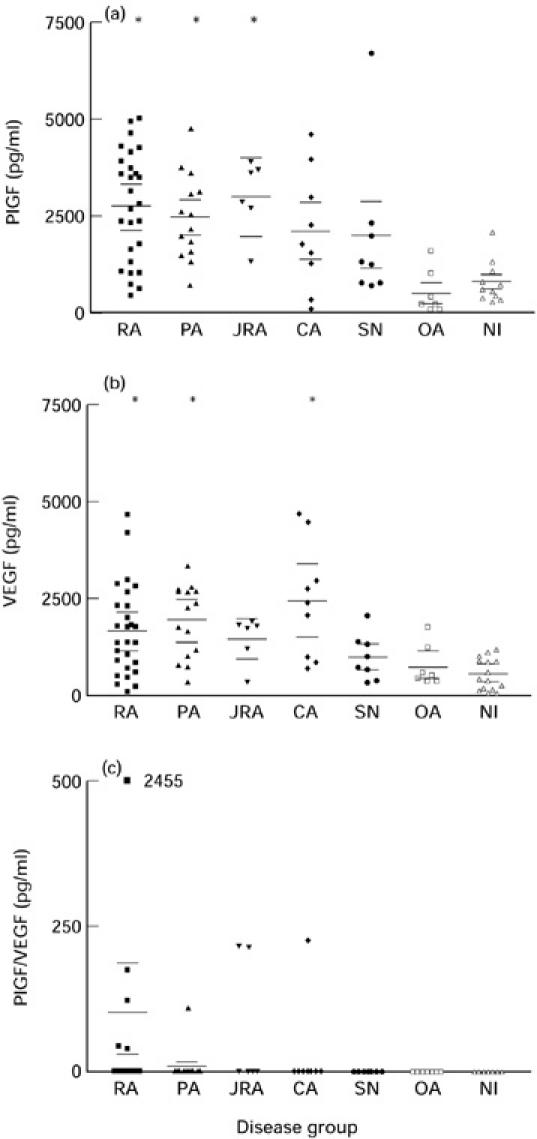
Synovial fluid levels (mean ± s.e.m.) of PlGF (a) (*P < 0.05 compared with non-inflammatory arthropathy (NI) group, one-way anova with Bonferroni correction), VEGF (b) (*P < 0.05 compared with NI and osteoarthritis (OA) groups, one-way anova with Bonferroni correction) and PlGF/VEGF heterodimers (c) (P = NS). RA, Rheumatoid arthritis; PA, primary inflammatory arthropathy; JRA, juvenile rheumatoid arthritis; CA, crystal arthropathy; SN, seronegative arthritis.
Plasma PlGF, VEGF and PlGF/VEGF heterodimer levels
PlGF was detected in nine (31%) of the control plasma samples and 32 (52%) of the RA patient samples; no significant difference was observed in the levels of the positive samples between the two groups' samples (mean ± s.e.m. values 367 ± 45 pg/ml versus 293 ± 96 pg/ml, respectively; P = NS). VEGF was detected in 11 (38%) of the control samples and 23 (38%) of the RA samples. The positive RA samples contained significantly higher VEGF levels compared with controls (216 ± 21 pg/ml versus 133 ± 18 pg/ml; P = 0.005). All control samples were negative for PlGF/VEGF heterodimers, whereas 13 (21%) of the RA samples had detectable levels.
Western blotting for PlGF
Western blotting using the anti-PlGF antibody revealed a significant band at 48–50 kD in the three SF samples from RA patients and a detectable but less prominent band at the same position in the samples from non-inflammatory arthropathy patients (Fig. 2). This is in agreement with the reported molecular size of native PlGF at 50 kD [19], which being N-glycosylated migrates as a higher molecular size species in comparison with the unglycosylated E. coli recombinant PlGF at 45 kD.
Fig. 2.

Western blot of PlGF detected in synovial fluid (SF). Lane 1, rPlGF-positive control; lanes 2–4, SF from three patients with rheumatoid arthritis; lanes 5–7, SF from three patients with primary inflammatory arthropathies.
Microscopic analysis of SF
A comparison was made between PlGF and VEGF levels detected in SF and the absolute and differential leucocyte counts of these fluids determined by microscopic analysis. In the primary inflammatory arthropathy group, SF PlGF and VEGF levels correlated significantly with the total leucocyte count (r = 0.69, P = 0.006 for PlGF, and r = 0.76, P = 0.001 for VEGF) and also the neutrophil count (r = 0.67, P = 0.009, and r = 0.79, P < 0.001, respectively) (Fig. 3a,b). No similar correlations were observed in the RA patient group or the crystal arthropathy group, which had the highest mean SF VEGF level.
Fig. 3.
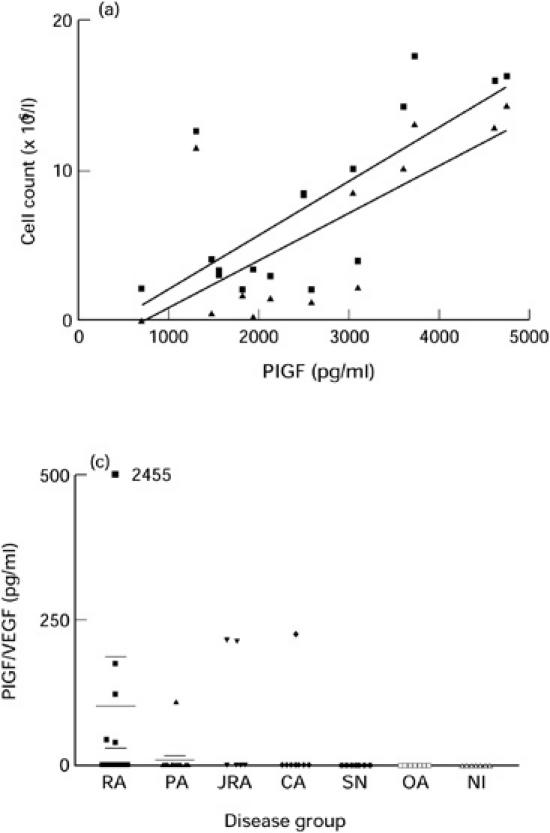
Correlation between synovial fluid (SF) PlGF and total leucocyte and neutrophil counts (a) (total leucocyte count r = 0.69, P = 0.006 and neutrophil count r = 0.67, P = 0.009) and correlation between SF VEGF and total leucocyte and neutrophil counts (b) (total leucocyte count r = 0.76, P = 0.001 and neutrophil count r = 0.79, P < 0.001). ▪, Leucocytes; ▴, neutrophils.
Regulation of VEGF secretion by PBMC
PlGF was the most potent inducer of PBMC VEGF production in RA and control samples at both time points, causing significantly increased VEGF secretion compared with unstimulated, LPS- or cobalt-stimulated cells at 24 h, and unstimulated and cobalt-stimulated cells at 72 h in both groups (P < 0.01, one-way anova with Bonferroni correction) (Fig. 4a,b). LPS was the only single stimulant that produced a late increase in VEGF secretion at 72 h compared with 24 h (P < 0.02, one-way anova with Bonferroni correction). No significant difference in VEGF production existed between RA and control PBMC at any time point. Combinations of the stimuli did not have any significant synergistic effect on VEGF secretion (data not shown).
Fig. 4.
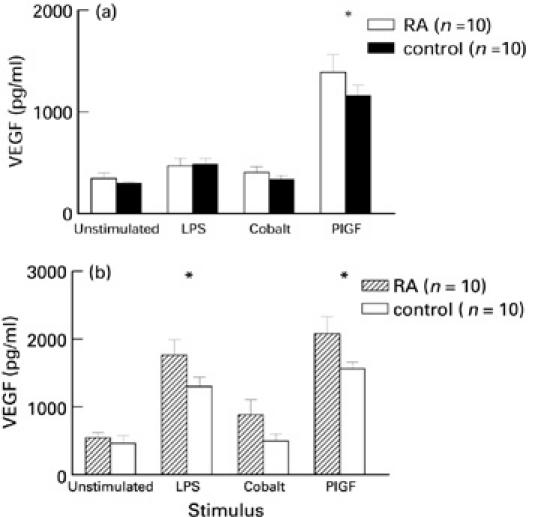
VEGF secretion (mean ± s.e.m.) by lipopolysaccharide (LPS), cobalt and PlGF-stimulated peripheral blood mononuclear cells isolated from 10 rheumatoid arthritis (RA) patients and 10 normal controls after 24 h culture (a) (*P < 0.05 compared with LPS and cobalt, one-way anova with Bonferroni correction) and 72 h culture (b) (*P < 0.05 compared with cobalt, one-way anova with Bonferroni correction).
DISCUSSION
This is the first study describing the specific detection of PlGF in the SF of patients with RA and other inflammatory arthropathies using a sensitive and specific immunoassay, and is confirmed by Western blotting. PlGF is the second described member of the VEGF superfamily which includes VEGF-B [20,21], VEGF-C/VRP [22,23] and VEGF-D/FlGF [24,25]. PlGF has been shown to have minimal activity in assays of vascular endothelial cell growth and permeability, but can potentiate the activity of VEGF in vitro and in vivo [15]. Naturally occurring heterodimers of PlGF and VEGF have been purified and characterized [13,26]; interestingly, these heterodimers induce endothelial cell mitogenesis, whereas PlGF homodimers have minimal activity. Like VEGF, increased levels of PlGF have been detected in diabetic retinopathy [27] and goitres resected from patients with Graves' disease [28].
SF PlGF levels were significantly higher in RA, primary inflammatory arthropathy and juvenile chronic arthritis patients compared with OA and non-inflammatory arthropathy patients. Interestingly, VEGF/PlGF heterodimer was detected only in a small number of SF samples, detection being more likely in samples with high PlGF levels and within the patient groups with the higher mean PlGF levels. PlGF may therefore also be important in driving the pathology of the inflammatory joint, either indirectly by enhancing the effects of VEGF, or directly by being chemoattractant to monocytes [12,14]. It may also however play a regulatory role by forming heterodimers with VEGF to control the differential triggering of VEGF receptors, the resultant heterodimers only binding to flt-1 and not to KDR. Furthermore, recent work has suggested that PlGF may have angiogenic properties and also induce growth and migration of endothelial cells from bovine coronary post-capillary venules and human umbilical veins [29], raising the possibility that PlGF may also be acting as a direct mitogen and angiogenic agent in RA. PlGF mRNA, like VEGF, can exhibit alternative splicing generating three potential protein species, PlGF-1 (131 aa), PlGF-2 (152 aa) or PlGF-3 (203 aa) [30]. Currently, there are no antibodies specific for the individual protein variants, but of these proteins only PlGF-2 contains the heparin binding domain (exon 6). Therefore, the use of heparin columns in this study to extract and concentrate PlGF from SF samples prior to Western blotting confirms the presence of PlGF-2 in SF. PlGF-2 has been shown to compete with VEGF and FGF-2 for heparan sulphate binding sites in extracellular matrix, and it has been suggested that PlGF may promote solubilization of these potent angiogenic factors from sequestered matrix depots [31].
VEGF has previously been detected in SF in a number of inflammatory arthropathies [8,9], but these early studies are confusing as they report widely differing absolute VEGF values. Fava et al. [8] detected mean VEGF levels of 59.1 pm (equivalent to 2.4 ng/ml), whereas Koch et al. [9] reported mean levels of 386 ng/ml. The reasons for this level of discrepancy are not clear, though different anti-VEGF antibodies, standards and assays were employed.
In this study we have used a VEGF receptor capture assay, which has been completely validated in performance against a widely available assay [17]. The VEGF levels detected ranged from 80 to 4000 pg/ml, in keeping with those reported by Fava et al. [8]. We have found that VEGF levels were significantly higher in RA, primary inflammatory and crystal arthropathies compared with non-inflammatory arthropathies. Our findings differ from previous studies in that we have detected raised levels of VEGF in OA SF compared with the non-inflammatory arthropathy group. It has been suggested that the VEGF found within the inflamed joint originates from synovial macrophages [8,9], though we have previously shown that PBMC [32] and neutrophils [33] are able to express VEGF mRNA and secrete VEGF protein upon stimulation, possibly implicating these cells in the pathogenesis of the inflammatory synovium, where large numbers of these cells are seen. Macrophages and neutrophils within SF are also able to secrete VEGF (unpublished observation). Monocytes and neutrophils have recently been shown to express flt-1, which may mediate chemotaxis to VEGF, resulting in recruitment of these cells in arthritic lesions [12]. In the primary inflammatory arthropathy group, the VEGF level correlated strongly with the neutrophil (but not lymphocyte or monocyte) count in the synovial cellular infiltrate, supporting a potential role for these cells in the primary immune response and the initiation of joint damage.
The high levels of SF VEGF detected may be accounted for by a lack of sflt-1 production within the joint. We have previously shown that in the presence of sflt-1, the receptor capture assay and other commercial assays are unable to detect VEGF [17] and it is therefore unlikely that the VEGF detected in SF is in the form of VEGF–flt-1 complex. Neutralizing VEGF with sflt-1 may therefore provide a useful therapeutic tool for controlling VEGF activity. That plasma VEGF levels were relatively low in RA plasma compared with SF samples provides some further evidence to support a local rather than systemic role for VEGF in the disease pathogenesis.
PlGF was the most potent enhancer of PBMC VEGF secretion, resulting in an approximately four-fold increase compared with unstimulated cells in both RA and control samples at 24 h and 72 h. This would provide an alternative explanation of the findings of Park et al., who reported enhanced VEGF bioactivity in the presence of PlGF [15], and emphasizes the potential importance of the detection of high SF PlGF levels in inflammatory joints. PlGF may promote VEGF-induced angiogenesis and changes in vascular permeability and may also enhance monocyte migration into the joint through binding to the flt-1 receptor on monocytes, resulting in chemotaxis [14]. PlGF, like transforming growth factor-beta (TGF-β) and tumour necrosis factor-alpha (TNF-α) may therefore be acting as an indirect angiogenic agent in RA [34,35].
The use of cobalt chloride replicated the stimulatory effect of hypoxia on VEGF expression, mimicking the hypoxic conditions which occur in the acutely inflamed rheumatoid joint [36,37]. Cobalt chloride enhanced PBMC VEGF secretion with a largest mean increase of 57% in RA patients after both 24 h and 72 h. Interestingly, RA PBMC appeared to be more responsive to cobalt compared with normal control PBMC at both time points. This may reflect an increased responsiveness to hypoxia in patients with RA. Cobalt has previously been shown to up-regulate VEGF mRNA in cultured human endothelial cells [38] and human epithelial cells by mediating increases in mRNA stability [39], but the corresponding protein secretion was not measured. Combining the cobalt and LPS stimuli gave no additive effects, with the mean percentage increase in VEGF secretion being equivalent to LPS alone (data not shown). Indeed, when all three stimuli were added the mean increase in secretion was similar to the PlGF-induced response alone.
In conclusion, we have shown for the first time (i) that PlGF and PlGF/VEGF heterodimers are present in the SF and plasma of patients with RA and a number of other arthropathies, and (ii) that PlGF up-regulates PBMC VEGF expression. The presence in SF of PlGF and VEGF, two members of the same family of potent angiogenic growth factors, together with PlGF/VEGF heterodimers, in particular, in patients with RA indicates a role for these growth factors in the pathological angiogenesis that fuels pannus development in the inflamed joint. We have provided evidence for two potential pathways of sustaining VEGF expression in the joint, a hypoxia-driven induction of VEGF from mononuclear cells infiltrating SF, and a direct up-regulation of VEGF by PlGF from these cells. Although pharmacological inhibition of angiogenesis resulting in suppression of arthritis has been reported in experimental models [40], this remains to be confirmed in clinical studies. We suggest that specific modulation of VEGF and PlGF is a rational therapeutic target for inhibition of angiogenesis in the joint. In the immediate future, this could be achieved by direct injection into the joint of neutralizing humanized anti-VEGF and anti-PlGF antibodies, or better still, injection of recombinant soluble flt-1 receptor which would potentially neutralize both growth factors. In the longer term, orally active signal transduction inhibitors that block the down-stream consequences of VEGF and PlGF receptor triggering may prove to be the effective angiogenesis suppressive agents in clinical practice [41].
Acknowledgments
This project was funded by the Arthritis Research Council, UK Grant B0558. The authors would like to thank Angela Thompson for her help in sample collection and Dr Don Ogilvie for the kind gift of the recombinant VEGF165 and sflt-1. Some of this work has previously been presented in poster form at the ACR Annual meeting, 1997 and the BSR Annual meeting, 1998.
REFERENCES
- 1.Jayson MIV, Dixon ASJ. Intra-articular pressure in rheumatoid arthritis of the knee. II Effect of intra-articular pressure on blood circulation to the synovium. Ann Rheum Dis. 1970;29:266–8. doi: 10.1136/ard.29.3.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falchuk KH, Goetzl EJ, Kulka JP. Respiratory gases of synovial fluids. An approach to synovial tissue circulatory–metabolic imbalance in rheumatoid arthritis. Am J Med. 1970;49:223–31. doi: 10.1016/s0002-9343(70)80078-x. [DOI] [PubMed] [Google Scholar]
- 3.Connolly DT, Heuvelman DM, Nelson R, et al. Tumour vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–8. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pepper MS, Ferrara N, Orci L, Montesano R. Vascular endothelial growth factor (VEGF) induces plasminogen activators and plasminogen activator inhibitor-1 in microvascular endothelial cells. Biochem Biophys Res Commun. 1991;181:902–6. doi: 10.1016/0006-291x(91)91276-i. [DOI] [PubMed] [Google Scholar]
- 5.Unemori EN, Ferrara N, Bauer EA, Amento EP. Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J Cell Physiol. 1992;153:557–62. doi: 10.1002/jcp.1041530317. [DOI] [PubMed] [Google Scholar]
- 6.Senger DR, Ledbetter SR, Claffey KP, Papadopoulossergiou A, Perruzzi CA, Detmar M. Stimulation of endothelial cell migration by vascular permeability factor vascular endothelial growth factor through co-operative mechanisms involving the alpha (v) beta (3) integrin, osteopontin and thrombin. Am J Pathol. 1996;149:293–305. [PMC free article] [PubMed] [Google Scholar]
- 7.Connolly DT, Olander JV, Heuvelman DM, et al. Human vascular permeability factor. J Biol Chem. 1989;264:20017–24. [PubMed] [Google Scholar]
- 8.Fava RA, Olsen NJ, Spencer-Green G, et al. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994;180:341–6. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koch AE, Harlow LA, Haines GK, et al. Vascular endothelial growth factor—a cytokine modulating endothelial function in rheumatoid arthritis. J Immunol. 1994;152:4149–56. [PubMed] [Google Scholar]
- 10.de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–91. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 11.Terman BI, Dougher-Vermazen M, Carrion ME, et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial growth factor. BBRC. 1992;187:1579–86. doi: 10.1016/0006-291x(92)90483-2. [DOI] [PubMed] [Google Scholar]
- 12.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–43. [PubMed] [Google Scholar]
- 13.Disalvo J, Bayne ML, Conn G, et al. Purification and characterization of a naturally occurring vascular endothelial growth factor-placenta growth factor heterodimer. J Biol Chem. 1995;270:7717–23. doi: 10.1074/jbc.270.13.7717. [DOI] [PubMed] [Google Scholar]
- 14.Clauss M, Weich H, Breier G, et al. The vascular endothelial growth factor receptor Flt-1 mediates biological activities. Implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. J Biol Chem. 1996;271:17629–34. doi: 10.1074/jbc.271.30.17629. [DOI] [PubMed] [Google Scholar]
- 15.Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor—potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994;269:25646–54. [PubMed] [Google Scholar]
- 16.Freemont AJ, Denton J. Atlas of synovial fluid cytopathology. Vol. 18. Higham, USA: Kluwer Academic Publishers; 1991. [Google Scholar]
- 17.Webb NJA, Bottomley MJ, Watson CJ, Brenchley PEC. VEGF is released from platelets on blood clotting: implications for the measurement of VEGF in clinical disease. Clin Sci. 1998;94:395–405. doi: 10.1042/cs0940395. [DOI] [PubMed] [Google Scholar]
- 18.Boyum A. Separation of white blood cells. Nature. 1964;204:793–4. doi: 10.1038/204793a0. [DOI] [PubMed] [Google Scholar]
- 19.Khaliq A, Li XF, Shams M, et al. Localisation of placenta growth factor (PlGF) in human term placenta. Growth Factors. 1996;13:243–50. doi: 10.3109/08977199609003225. [DOI] [PubMed] [Google Scholar]
- 20.Olofsson B, Pajusola K, Kaipainen A, et al. Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc Natl Acad Sci USA. 1996;93:2576–81. doi: 10.1073/pnas.93.6.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grimmond S, Lagencrantz J, Drinkwater C, et al. Cloning and characterization of a human gene related to vascular endothelial growth factor. Genome Res. 1996;6:124–31. doi: 10.1101/gr.6.2.124. [DOI] [PubMed] [Google Scholar]
- 22.Joukov V, Pajusola K, Kaipainen A, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for Flt-4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:290–8. [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Gray A, Yuan J, Luoh SM, Avraham H, Wood WI. Vascular endothelial growth factor-related protein: a ligand and specific activator of the tyrosine kinase receptor Flt-4. Proc Natl Acad Sci USA. 1996;93:1988–92. doi: 10.1073/pnas.93.5.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamada Y, Nezu J-I, Shimane M, Hirata Y. Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics. 1997;42:483–8. doi: 10.1006/geno.1997.4774. [DOI] [PubMed] [Google Scholar]
- 25.Rocchigiani M, Lestingi M, Luddi A, et al. Human FIGF. Cloning, gene structure, and mapping to chromosome Xp22.1 between PIGA and the GRPR genes. Genomics. 1998;47:207–16. doi: 10.1006/geno.1997.5079. [DOI] [PubMed] [Google Scholar]
- 26.Cao Y, Chen H, Zhou L, et al. Heterodimers of placenta growth factor/ vascular endothelial growth factor. Endothelial activity, tumour cell expression, and high affinity binding to Flk-1/KDR. J Biol Chem. 1996;271:3154–62. doi: 10.1074/jbc.271.6.3154. [DOI] [PubMed] [Google Scholar]
- 27.Khaliq A, Foreman D, Ahmed A, et al. Increased expression of placenta growth factor in proliferative diabetic retinopathy. Lab Invest. 1998;78:109–16. [PubMed] [Google Scholar]
- 28.Viglietto G, Romano A, Manzo G, et al. Upregulation of the angiogenic factors PlGF, VEGF and their receptors (Flt-1, Flk-1/KDR) by TSH in cultured thyrocytes and in the thyroid gland of thiouracil-fed rats suggest a TSH-dependent paracrine mechanism for goiter hypervascularization. Oncogene. 1997;15:2687–98. doi: 10.1038/sj.onc.1201456. [DOI] [PubMed] [Google Scholar]
- 29.Ziche M, Maglione D, Ribatti D, et al. Placenta growth factor-1 is chemotactic, mitogenic, and angiogenic. Lab Invest. 1997;76:517–31. [PubMed] [Google Scholar]
- 30.Barillari G, Albonici L, Franzese O, et al. The basic residues of placenta growth factor type 2 retrieve sequestered angiogenic factors into a soluble form: implications for tumor angiogenesis. Am J Pathol. 1998;152:1161–6. [PMC free article] [PubMed] [Google Scholar]
- 31.Cao Y, Ji WR, Qi P, Rosin A, Cao Y. Placenta growth factor: identification and characterisation of a novel isoform generated by RNA alternative splicing. Biophys Res Commun. 1997;235:493–8. doi: 10.1006/bbrc.1997.6813. [DOI] [PubMed] [Google Scholar]
- 32.Watson CJ, Webb NJA, Bottomley MJ, Brenchley PEC. Peripheral mononuclear cells express VEGF and the VEGF receptor flt-1. JASN. 1996;7:1725. [Google Scholar]
- 33.Webb NJA, Myers CR, Watson CJ, Bottomley MJ, Brenchley PEC. Activated human neutrophils express vascular endothelial growth factor (VEGF) Cytokine. 1998;10:254–7. doi: 10.1006/cyto.1997.0297. [DOI] [PubMed] [Google Scholar]
- 34.Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C. Macrophages and angiogenesis. J Leuk Biol. 1994;55:410–22. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- 35.Bottomley MJ, Webb NJA, Watson CJ, Holt PJL, Freemont AJ, Brenchley PEC. PBMCs from patients with rheumatoid arthritis spontaneously secrete VEGF: specific up-regulation by TNFα in synovial fluid. Clin Exp Immunol. 1999;117:171–6. doi: 10.1046/j.1365-2249.1999.00949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Page-Thomas DP, Dingle JTM. In-vitro studies of rheumatoid synovium. Preliminary metabolic comparison between synovial membrane and villi. Br J Exp Pathol. 1955;36:195–8. [PMC free article] [PubMed] [Google Scholar]
- 37.Minchenko A, Bauer T, Salceda S, Caro J. Hypoxic stimulation of vascular endothelial growth factor expression in vivo and in vitro. Lab Invest. 1994;71:374–9. [PubMed] [Google Scholar]
- 38.Namiki A, Brogi E, Kearney M, et al. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J Biol Chem. 1995;270:31189–95. doi: 10.1074/jbc.270.52.31189. [DOI] [PubMed] [Google Scholar]
- 39.Shima DT, Deutsch U, D'amour PA. Hypoxic induction of vascular endothelial growth factor (VEGF) in human epithelial cells is mediated by increases in mRNA stability. FEBS Letters. 1995;370:203–8. doi: 10.1016/0014-5793(95)00831-s. [DOI] [PubMed] [Google Scholar]
- 40.Oliver SJ, Cheng TP, Banquerigo ML, Brahn E. Suppression of collagen-induced arthritis by an angiogenesis inhibitor, AGM-1470, in combination with cyclosporin: reduction of VEGF. Cell Immunol. 1995;166:196–206. doi: 10.1006/cimm.1995.9978. [DOI] [PubMed] [Google Scholar]
- 41.Brenchley PEC. Antagonising the expression of VEGF in pathological angiogenesis. Expert opinion on therapeutic patents. Exp Opin Ther Patents. 1998;8:1697–708. [Google Scholar]