Expression, modulation and signalling of IL-17 receptor in fibroblast-like synoviocytes of patients with rheumatoid arthritis (original) (raw)
Abstract
Interleukin-17 (IL-17) has been characterized as a proinflammatory cytokine produced by CD4+ CD45RO+ memory T cells. Overproduction of IL-17 was detected in the synovium of patients with rheumatoid arthritis (RA) compared to patients with osteoarthritis. In contrast to the restricted expression of IL-17, the IL-17 receptor (IL-17R/CDw217) is expressed ubiquitously. Using a real-time RT-PCR assay, we detected similar absolute levels of IL-17R mRNA expression in fibroblast-like synoviocytes (SFC) from patients with RA (mean 9 pg/μg total RNA; ranged from 0·1 pg to 96 pg IL-17R mRNA/μg total RNA) compared to synoviocytes of non-RA patients. Analysis of the IL-17R surface expression confirmed the results obtained for IL-17R mRNA expression. Exposure of SFC to IL-17 led to a mRNA induction of CXC chemokines IL-8, GRO-α and GRO-β. An anti-IL-17 antibody blocked these effects of IL-17. The MAPK p38 appears necessary for the regulation of IL-8, GRO-α and GRO-β expression as shown by inhibition with SB203580. The inhibitors genistein (tyrosine kinase inhibitor) and calphostin C (inhibitor of protein kinase C) reduced significantly the IL-17-stimulated mRNA expression of IL-8, GRO-α and GRO-β in SFC, whereas PD98059 (inhibitor of MEK-1/2) was without effect. Pharmacological drugs used in therapy of RA, such as cyclosporin and methotrexate, induced a fourfold increase of IL-17R mRNA expression and augmented the IL-17-stimulated IL-8 expression. Our results support the hypothesis that IL-17/IL-17R may play a significant role in the pathogenesis of RA contributing to an unbalanced production of cytokines as well as participating in connective tissue remodelling.
Keywords: chemokine, IL-17, IL-17 receptor, rheumatoid arthritis
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by a relapsing and remitting course of joint inflammation. The chronic inflammation process leads to an excessive hyperplasia of the synovium with proliferation of the synovial lining cells, generation of new blood vessels and diffusely scattered or nodular mononuclear cell infiltrates. The proliferation and invasive growth of fibroblast-like cells of the synovium (SFC) results ultimately in the destruction of the joint [1,2]. Cytokines such as interleukin (IL)-1β or TNF-α are known to be involved in the perpetuation of the chronic inflammation in RA [3–6]. Overproduction of the proinflammatory cytokine interleukin-17 (IL-17) was detected in the RA synovium compared to patients with osteoarthritis [7,8]. IL-17 is a 20–30 kDa glycosylated, homodimeric polypeptide secreted by CD4+ activated memory (CD45RO+) T cells [9,10]. IL-17 up-regulates a number of gene products involved in cell activation, growth and proliferation, including IL-1β, IL-6, IL-8, TNF-α, G-CSF and NF-κB[9,11–13,. In the context of arthritis, the effects of IL-17 were associated to joint inflammation and destruction because of the IL-17-stimulated MMP-1 and MMP-9 production, proteoglycan degradation and the IL-17-increased IL-6 and LIF expression in SFC [14–17]. Importantly, IL-17 was found to strengthen the effect of IL-1β and TNF-α, leading to an additive enhancement of cytokine secretion [14].
In contrast to the restricted site of synthesis of IL-17, the IL-17 receptor (IL-17R/CDw217) is expressed ubiquitously [18]. The IL-17R is a type-I membrane protein with no homologue to any of the known family of cytokine receptors. Furthermore, there are no obvious motifs in the receptor’s intracellular domains providing clues how signalling is mediated following binding of IL-17 to its receptor [10,19,20].
To gain knowledge about the expression, modulation and signalling of IL-17R in SFC of patients with RA, we (i) analysed IL-17R expression levels in SFC from patients with RA in comparison to other cells, (ii) studied the modulation of IL-17R mRNA expression in SFC by mediators and (iii) investigated effects of IL-17 binding to its receptor. Our results unravel IL-17 as an additional member of the cytokine network involved in RA.
MATERIALS AND METHODS
Cell culture
SFC were obtained from patients with classical or definite RA undergoing surgical synovectomy by dissociating the minced tissue enzymatically with HBSS containing 0·5 mg/ml collagenase type II (Sigma, Deisenhofen, Germany), 0·15 mg/ml DNase I (Boehringer Mannheim, Germany) and 5 mm Ca++. The cells were cultured in RPMI 1640 containing 10% fetal calf serum (FCS), antibiotics and glutamine as described earlier [21]. Cells were used at confluence at the third to fifth passage. Thyroid carcinoma cell lines (obtained from Dr C. Hoang-Vu, Halle, Germany) and SFC of patients with osteoarthritis (OA; kindly provided by Dr T. Pap, Magdeburg, Germany) were grown in DMEM (Cell Concepts, Umkirch, Germany) with 10% FCS supplemented with penicillin and streptomycin (1%; C.C. Pro, Neustadt/W., Germany). The human monocytic cell line U937 (ATCC) was cultured in RPMI with 10% FCS, antibiotics (1 μl/ml fungizone, 0·75 μl/ml Refobacin) and 50 μmol 2-mercaptoethanol. Synoviocytes from non-RA patients were obtained from patients with meniscectomies (kindly provided by Dr J. Rödel, Jena, Germany) [22] as well as fibroblasts from patients with arthrosis (gift from Dr E. Märker-Herrmann, Mainz, Germany). Chondrocytes from patients with arthrosis were provided by Dr J. Brandt (Halle, Germany). The inhibitors Calphostin C (20 μg/ml; Sigma, Deisenhofen, Germany), PD98059 (10 μm; Alexis, Grünberg, Germany), SB203580 (1 μm; Calbiochem, Schwalbach, Germany) and genistein (10 μg/ml; Gibco BRL, Karlsruhe, Germany) were added to the cultures 30 min prior to IL-17 incubation. Rabbit neutralizing polyclonal anti-hIL-17 antibody (2 μg/ml; Cell Concepts) was co-incubated with rIL-17 for 30 min and added to the cultures.
RNA isolation and cDNA synthesis
Total cellular RNA was isolated according to Chomczynski and Sacchi [23]. The first strand of DNA was synthesized (after a 10-min incubation at 20°C) at 42°C for 50 min using 1 μg total RNA in the presence of dilutions of internal competitive standard RNA in 5·5 μl DEPC water, 2 μl 5 × first strand buffer (250 mm Tris/HCl pH 8·3, 375 mm KCl, 15 mm MgCl2), 0·5 μl dNTP mix (10 mm of each dATP, dCTP, dGTP, dTTP), 1 μl 0·1 m DTT, 0·5 μl (50 pmol) random primer (Boehringer Mannheim, Germany) and 0·5 μl (200 U/μl) SuperscriptTM II-RT (Gibco BRL, Karlsruhe, Germany). For quantitative RT-PCR, IL-17R-standard RNA and total RNA were converted into cDNA in separate tubes.
Construction of RNA standards
The standard for IL-8, IL-17R and IκB-α were constructed by previously described procedures [13,24,25]. For the construction of the internal competitive standard RNA of GRO-α and GRO-β, composite primers were synthesized (see Table 1 for primer sequences). Primer 1 was composed of two specific primers complementary to the coding strand of the respective molecule. Primer 2 contained a sequence for the SP6 RNA polymerase and also one of the specific sequences of primer 1. The purified product of the first PCR amplification with primer 1 and 3 was used as template for the second amplification using primers 2 and 3. The amplified DNA was gel purified (QIA quick Gel Extraction Kit, Qiagen, Hilden, Germany) followed by in vitro transcription by the SP6 promoter using the transcription system of Boehringer. The recombinant RNA was quantified at 260 nm and used as an internal standard in the cDNA synthesis and the competitive PCR reaction.
Table 1.
Sequences of primers used for standard construction and PCR amplification
| Gene | Number | Sequence 5′– 3′ |
|---|---|---|
| GRO-α | 1 | CCAAAGTGTGAACGTGAAGTCCC_GAGGAGGAAGCTGACTGGTG_ |
| 2 | GATTTAGGTGACACTATAGAATACCCAAAGTGTGAACGTGAAGTCCC | |
| 3 | CTCTATCACAGTGGCTGGCA | |
| 4 | CCAAAGTGTGAACGTGAAGTCCC | |
| GRO-β | 1 | CCGAAGTCATAGCCACACTC_GAGAGACACAGCTGCAGAGG_ |
| 2 | GATTTAGGTGACACTATAGAATAC CCGAAGTCATAGCCACACTC | |
| 3 | GGCCATTTTCTTGGATTCCT | |
| 4 | CCGAAGTCATAGCCACACTC | |
| IL-17R | 3 | GTCTGGGCGCAGGTATGTGG |
| 4 | CACCGTGGAGACCCTGGAGGC | |
| 5 | CTGCTCTTCCAGCAGGTC | |
| 6 | ATGAACCAGTACACCCAC | |
| IL-8 | 3 | TCT CAG CCC TCT TCA AAA ACT TCT C |
| 4 | ATG ACT TCC AAG CTG GCC GTG GCT | |
| IκB-α | 3 | ACATCAGCACCCAAGGACAC |
| 4 | GCTGAAGAAGGAGCGGCTAC |
Competitive PCR analysis
cDNA, 1·5 μl, synthesized as described above were diluted in 50 μl of the following solution: 50 mm KCl, 10 mm Tris-HCl pH 9·0, 1·5 mm MgCl2, 0·1% Triton X-100, 0·1 mm of each dNTP, 1 U Taq polymerase (Qiagen) and 50 pmol each of primer 4 and primer 3. PCR was conducted in a thermal cycler (Biometra, Göttingen, Germany) for 30 cycles of 60 s at 94°C, 60 s at 60°C and 60 s at 72°C; 10 μl of each reaction product were run on a 1·5% agarose gel containing 0·1 μg/ml ethidium bromide in TAE buffer. The relative intensities of the bands corresponding to target and standard PCR products were visualized in UV light. The relative amounts of target and standard products were calculated after densitometric analysis using the Scan Pack 3·0 software (Biometra, Göttingen, Germany).
Quantitative PCR analysis
For quantification of IL-17R mRNA content, 1 μl of the reverse transcriptase reaction mixture was added to 25 μl reaction mixture consisting of 1 × reaction buffer, 1·5 U Taq polymerase (Qiagen), 1·8 mm MgCl2, 0·1 × SYBR Green (Biozym, Hess, Oldendorf, Germany), 200 μm of each dNTP and 0·5 μm each of primers 3 and 4. A negative control without template was included. Samples of both, six dilutions of the IL-17R-standard RNA and the cellular RNA, respectively, were run in triplicate in a RG2000 (LTF, Wasserburg, Germany). Initial denaturation at 95°C for 300 s was followed by 40 cycles with denaturation at 95°C for 15 s, annealing at 60°C for 30 s and elongation at 72°C for 20 s. The fluorescence intensity of the double-strand specific SYBR Green, reflecting the amount of formed PCR product, was read after each elongation step at 80°C. IL-17R amounts were determined using the quantification modus of the Rotor-Gene software 4·01.
Western blotting
Cell culture plates were washed twice with ice-cold PBS. Cells were harvested by scraping into ice-cold RIPA buffer (1 × PBS, 1% Nonidet P-40, 0·5% sodium deoxycholate). Inhibitors were added in the following concentrations: 1 mm PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm Na3VO4, 1 mm NaF (Sigma). The cell lysate was transferred to microcentrifuge tubes and incubated on ice for 60 min and was centrifuged at 14 000 r.p.m. for 20 min at 4°C. Protein concentration in the supernatant was quantified by means of the BCA Protein Assay Reagent Kit (KMF, Leipzig, Germany) and 40 μg cell lysate protein was used for Western blot analysis. Proteins were electroblotted from NuPAGE gels (NOVEX, Frankfurt-Hoechst, Germany) onto Hybond ECL membrane (Amersham, Freiburg, Germany). The membrane was blocked with 1% BSA in TBST for 1 h at room temperature. Blots were incubated with the primary antibody (pERK1/2, p-p38, IκB-α, Santa Cruz, Heidelberg, Germany; p-Raf, Biomol, Hamburg, Germany, all 1: 1000 dilution; actin, Sigma, 1: 5000 dilution) in TBST with 1% BSA at room temperature for 2 h. Blots were washed three times and then incubated for 1h with the appropriate secondary antibody (1: 1000 dilution, Dianova, Hamburg, Germany) coupled with horseradish peroxidase. Immunodetection was accomplished using the ECL Western blotting detection reagents (Amersham) for chemiluminescent detection. Immunoreactivity was quantified by scanning densitometry using the Scan Pack 3·0 software (Biometra).
Immunfluorescence staining
For the flow cytometric analysis of IL-17R expression, cells were detached by use of accutase (PAA, Linz, Austria), washed twice with PBS and incubated with the IL-17R MoAb M 203 (10 ng/ml; Immunex, Seattle, WA, USA) at 4°C for 45 min After two washing steps with PBS containing 0·1% sodium acid, the cells were incubated with goat anti-mouse Ig-PE (Dianova) for 30 min at 4°C. Finally, the cells were washed, fixed with 1% paraformaldehyde, and assessed for fluorescence using a FACScan and the Lysis software (Becton Dickinson, Heidelberg, Germany). Results are given as relative mean fluorescence (rFl=mean fluorescence intensity of staining with mAb M203 – mean fluorescence intensity of staining with the secondary antibody/mean fluorescence intensity of staining with the secondary antibody).
Measurement of IL-8 and GRO-α
SFC were cultured in the presence of 20 ng/ml IL-17 for 48 h. Supernatants were collected and assayed for IL-8 using the Chemiluminescence-Enzyme Immunoassay System, Immulite (DPC Biermann, Bad Nauheim, Germany). GRO-α was quantified by means of the Quantikine Human GRO-α Immunoassay (RD Systems, Wiesbaden, Germany).
Statistical analysis
Data are expressed as mean ± s.e.m. The Wilcoxon rank sum test was used to determine whether two experimental values were significantly different, P < 0·05 indicated with *, P < 0·01 with **.
RESULTS
Quantification of IL-17R expression in SFC of patients with RA
To investigate the IL-17R mRNA expression of SFC from patients with RA, we developed a real-time quantitative RT-PCR assay using the specific IL-17R primers 3 and 4 (see Table 1). We detected similar absolute levels of IL-17R mRNA expression in SFC from patients with RA compared to synoviocytes of non-RA patients (Fig. 1a). In SFC of RA patients IL-17R mRNA levels ranged from 0·1 pg to 96 pg IL-17R mRNA/μg total RNA. In SFC of patients with osteoarthritis (OA), we detected 4·4 pg IL-17R mRNA/μg total RNA. The amount of IL-17R mRNA in synoviocytes from patients with meniscectomies was 7·8 pg/μg total RNA, in fibroblasts from patients with arthrosis 2·6 pg/μg total RNA and in tonsillar fibroblasts 4·1 pg/μg total RNA. Chondrocytes of patients with arthrosis expressed 15·2 pg IL-17R mRNA/μg total RNA. Thyroid carcinoma cells expressed between 2·4 pg and 47 pg IL-17R mRNA/μg total RNA. High IL-17R mRNA expression was detected with 88 pg/μg total RNA in U937 cells (early monocytic cell line). Analysis of the IL-17R surface expression using flow cytometry confirmed the results obtained for the IL-17R mRNA expression (Fig. 1b). We measured a relative mean fluorescence intensity (rMFl) of 0·42 for SFC of RA patients, of 0·18 for SFC of OA patients, and of 0·44 for chondrocytes of patients with arthrosis, respectively. The highest IL-17R staining was detected in U937 cells with a rMFl of 10·1.
Fig. 1.
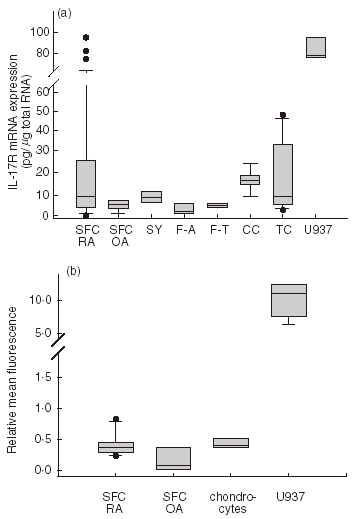
(a) Comparison of the absolute amounts of the IL-17R mRNA in SFC (SFC RA, 15 different patients measured each in duplicate), in synoviocytes of OA patients (SFC OA; five different patients each in duplicate), synoviocytes of patients with meniscectomies (SY; two different patients each in duplicate), fibroblasts of patients with arthrosis (F-A; three different patients each in triplicate experiments), tonsillar fibroblasts (F-T; n =3), in chondrocytes of patients with arthrosis (CC; three different patients each in duplicate), in thyroid carcinoma cells (six different cell lines each in duplicate) and in the monocytic cell line U937 (n =4). Results of quantitative RT-PCR as described in Material and methods. (b) Surface expression of IL-17R on cultured SFC of patients with RA (SFC RA, six different patients) and OA (SFC OA, three different patients), on chondrocytes of patients with arthrosis (CC, three different patients) and U937 cells. Results are given as relative mean fluorescence. Data in (a) and (b) are given as box plots with the median (thin line). The box encompasses the 25th to 75th percentiles. The 5th and 95th percentiles are displayed as error bars; results outside this range are indicated as separate dots.
IL-17 activates signal transduction pathways of NF-κB and MAPKs
We investigated the ability of IL-17 in activating nuclear factor NF-κB. Activation of NF-κB involves the degradation of its cytoplasmatic inhibitor IκB-α which allows the nuclear translocation of NF-κB, and ensures transcriptional activation of genes including IκB-α itself. In Fig. 2a the degradation of IκB-α after IL-17-stimulation for different times is shown. IL-17 treatment leads to a degradation of IκΒ-α within 10 min and resynthesis can be seen after 4 h. Using a competitive RT-PCR, we detected an up-regulated IκB-α mRNA expression in IL-17-stimulated SFC in a time-dependent fashion. This is taken as an additional evidence for NF-κB activation [13]. After 60 min of IL-17 stimulation of SFC, we measured a 2·2-fold increase of IκB-α mRNA expression compared to unstimulated SFC that after 6h already disappeard (Fig. 2b). Furthermore, IL-17 stimulates the phosphorylation of c-Raf and ERK1/2 after 10 min and phosphorylation of p-38 after 30 min (Fig. 3).
Fig. 2.
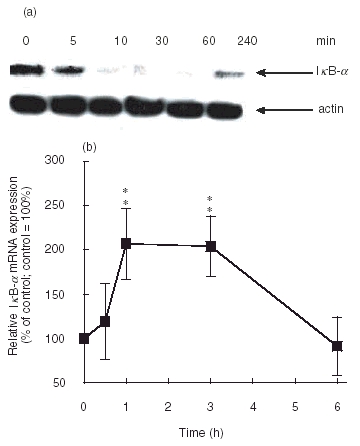
(a) IκB-α degradation in IL-17-stimulated SFC. SFC were treated with 20 ng/ml IL-17 for 5, 10, 30, 60 and 240 min, as indicated. Cytosolic protein was extracted and analysed by immunoblotting using an IκB-α antibody (top panel) or an actin antibody (bottom panel). Results of one representative of three experiments is shown. (b) IL-17 induces expression of IκB-α mRNA in SFC. Results of competitive RT-PCR for IκB-α mRNA expression are given as percentage of the basal control (culture without IL-17=100%; n =221:1011221:1011;4; ** denotes statistically significant differences from control at the P < 0·01 level).
Fig. 3.

Time-dependent effect of IL-17 on phosphorylation of c-Raf, ERK1/2 and p-38. SFC were cultured for different times with IL-17 (20 ng/ml). Cells were lysed, separated, blotted and probed with the appropriate specific antibody as described in Material and methods. Results of one representative of three experiments is shown.
IL-17 increases expression of IL-8, GRO-α and GRO-β in SFC
To determine whether the IL-17 stimulated NF-κB activation resulted in an increase in chemokine expression, we measured the mRNA expression of IL-8, GRO-α and GRO-β in IL-17-stimulated SFC. We detected an up-regulation of mRNA expression of the three chemokines (Fig. 4). The increase measured in mRNA expression was from 1900 fg to 7700 fg/μg total RNA for IL-8, from 55 fg to 195 fg/μ total RNA for GRO-α and from 13 fg to 47 fg/μg total RNA for GRO-β, respectively. Co-incubation with an anti-IL-17 antibody decreased the IL-17-induced mRNA expression of IL-8, GRO-α and GRO-β to 146%, 143% or 126%, respectively, of the unstimulated control (100%; Fig. 5). After 2 days of IL-17-stimulation, the secreted IL-8 amounts increased from 1590 ± 1038 pg/ml to 11 404 ± 4242 pg/ml (n =5). We also found an augmentation of GRO-α protein secretion to 2356 ± 699 pg/ml compared to unstimulated cells secreting 71 ± 35 pg/ml (Fig. 4; n = 5).
Fig. 4.

IL-17 induces expression of IL-8, GRO-α and GRO-β. SFC were cultured in the presence of IL-17 (20 ng/ml). (a) Results of competitive RT-PCR (n =6) as described in Material and Methods. ■, IL-8 mRNA; □, GRO-α mRNA;  , GRO-β mRNA. (b) Culture supernatant were assayed for amounts of IL-8 and GRO-α as described in Material and Methods (n/4; ** denotes statistically significant differences from unstimulated control at the P < 0.01 level). ■, IL-8; □, GRO-α.
, GRO-β mRNA. (b) Culture supernatant were assayed for amounts of IL-8 and GRO-α as described in Material and Methods (n/4; ** denotes statistically significant differences from unstimulated control at the P < 0.01 level). ■, IL-8; □, GRO-α.
Fig. 5.
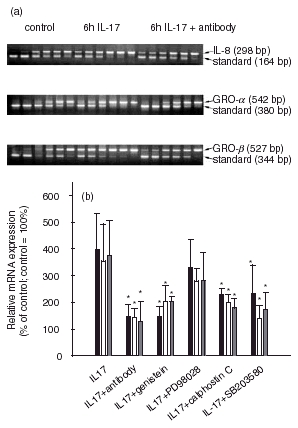
Effects of an anti-IL-17 antibody and protein kinase inhibitors on chemokine mRNA expression. SFC were cultured for 24 h in the presence of IL-17 (20 ng/ml) and the appropriate effector. (a) 0·5μg of total RNA was used for cDNA synthesis in the presence of six dilutions of the appropriate internal competitive standard RNA and random primer in a volume of 10 μl. 1·5 μl of the synthesized cDNA was amplified in the PCR by means of the appropriate primers 4 and 3 as described in Material and methods. 10 μl PCR reaction product was separated on an agarose gel containing ethidium bromide. The relative intensities of the bands of chemokine product and standard product were visualized in UV light. (b) Results of competitive RT-PCR are given as a percentage of the basal control (culture without cytokine = 100%, n = 6, * denotes statistically significant differences from control at the P < 0·05 level). ■, IL-8; □, GRO-α  , GRO-β.
, GRO-β.
To further investigate the intracellular signalling pathways activated by IL-17 and responsible for inducing the chemokine expression, SFC were preincubated with cell-permeable inhibitors of MEK-1/2 (PD98059), p38 (SB203580), PKC (calphostin C), tyrosine kinase (genistein), followed by addition of IL-17, and subsequent chemokine mRNA level analysis. Inhibitors genistein, SB203580 and calphostin C reduced significantly the mRNA expression stimulated by IL-17 of IL-8, GRO-α and GRO-β in SFC. However, with PD98059 we did not measure a significant decrease in the mRNA amount of IL-8, GRO-α and GRO-β (Fig. 5).
Cyclosporin A and methotrexate augment the IL-17R mRNA expression and the IL-8 secretion in SFC
Furthermore, we investigated the influence of antirheumatic drugs on IL-17R mRNA expression. After treatment of SFC with pentoxifylline or indomethacine for 24 h we did not measure significant changes in the IL-17R mRNA level compared to the unstimulated control. However, with cyclosporin A, dexamethasone and methotrexate, we detected an increased IL-17R mRNA expression (Fig. 6). The IL-17R mRNA expression was 2·5-fold higher in dexamethasone-treated SFC, 4·3-fold induced in cyclosporin A-treated SFC, and threefold induced in SFC stimulated with methotrexate. Both antirheumatic drugs, methotrexate and cyclosporin A, are able to increase further the IL-17-stimulated IL-8 secretion whereas dexamethasone inhibited the IL-17-stimulated IL-8 secretion (Fig. 7). After 3 days of methotrexate treatment, the IL-8 secretion stimulated by IL-17 increased from 2240 ± 1008 pg/ml to 18 570 ± 4400 pg/ml. With cyclosporin A we found also an augmentation of the IL-17-stimulated IL-8 secretion to 18 360 ± 4200 pg/ml compared to cells without IL-17 treatment (1085 ± 560 pg/ml). Dexamethasone enhanced the IL-8 amount from 237 ± 120 pg/ml without IL-17 addition to 3813 ± 590 pg/ml in IL-17 stimulated SFC.
Fig. 6.

Effect of pharmacological drugs used for therapy of RA on IL-17R mRNA amount in SFC. SFC were cultured for 24 h in the presence of pentoxifylline (PTX), indomethacine (indo), cyclosporin A (CSA), dexamethasone (DXM) or methotrexate (MTX). Results of quantitative RT-PCR are given as a percentage of the basal control (culture without mediator = 100%, six different patients; * denotes statistically significant differences from control at the P < 0·05 level).
Fig. 7.

Effect of antirheumatic drugs on the IL-8 secretion in SFC. 104 cells were cultured for 48h in the presence of pentoxifylline (PTX), indomethacine (indo), cyclosporin A (CSA), dexamethasone (DXM) or methotrexate (MTX) alone or in combination with IL-17 (20 ng/ml). Culture supernatant were assayed for IL-8 amounts as described in Material and methods (SFC of four different patients; * denotes statistically significant differences from control at the P < 0·05 level). ■, −IL−17; □, +IL−17.
DISCUSSION
The inflamed synovium of RA joints expresses numerous cytokines involved in the complex interactions between lymphocytes and SFC, endothelial cells, synovial macrophages and chondrocytes. IL-17 was found in high levels in the RA synovium and the concentration of this cytokine in synovial fluid of RA patients is elevated [7,8]. Recently, we reported the increased lymphocytic IL-17R mRNA expression after cell–cell contact of lymphocytes with SFC [25]. Here, we investigated the absolute IL-17R mRNA amounts in SFC of patients with RA and compared the data with the expression in fibroblasts from non-RA patients. The median expression level of IL-17R mRNA in SFC of RA patients (median 9 pg/μg total RNA) showed no clearcut difference between SFC of OA patients (4·4 pg/μg total RNA), synoviocytes from patients with meniscectomies (7·8 pg/μg total RNA) or fibroblasts of patients with arthrosis (2·6 pg/μg total RNA). However, the IL-17R mRNA expression ranged from 0·1 pg to 96 pg/μg total RNA in SFC of 15 different RA patients. For comparison, we quantified the expression in chondrocytes of patients with arthrosis, thyroid carcinoma cells, tonsillar fibroblasts and in the monocytic cell line U937, the latter showing the highest expression level with 88 pg/μg total RNA. Our results agree with those of Yao et al. describing the ubiquitous expression of IL-17R with strong Northern blot signals of IL-17R mRNA in lung and liver of mice and weaker concentrations in brain, heart, testes and skeletal muscles [19]. Analysis of IL-17R surface expression using flow cytometry confirmed the results obtained for IL-17R mRNA expression. We measured a rMFl of 0·42 for SFC of RA patients, 0·18 for SFC of OA patients and 0·44 for chondrocytes of patients with arthrosis, respectively. As seen for the IL-17R mRNA levels, the highest IL-17R surface expression was detected in U937 cells with a rMFl of 10·1. Our results of the IL-17R surface expression in SFC agree with the data of Woltman et al., which also used the IL-17R MoAb M 203 from Immunex for staining SFC by means of FACS analysis [26]. Using immunohistochemistry and the IL-17R MoAb M 202 from Immunex, Honorati et al. observed no staining of synoviocytes whereas synovial endothelial cells and chondrocytes from RA patients expressed high levels of IL-17R [27]. We detected similar amounts of IL-17R surface staining in SFC of RA patients and chondrocytes of patients with arthrosis using the IL-17R MoAb M 203.
The present study shows an increased expression of CXC chemokines as IL-8, GRO-α and GRO-β in IL-17-stimulated SFC of patients with RA. In CXC chemokines two conserved cysteines are separated by one unconserved amino acid (http://cytokine.medic.kumamoto-u.ac.jp/). CXC chemokines are potent neutrophil chemoattractants and have been detected in synovial fluids, synovial tissues and sera of RA patients (reviewed in [28]). Using cDNA microarray technology in RA tissue the high chemokine expression could be verified including the CXC chemokines IL-8 and GRO-α[29]. IL-8 induces synovial inflammation as determind by injection of recombinant IL-8 into rabbit knee joint [30]. GRO-α was initially considered an oncogene growth-related peptide, and it has been shown to be identical to melanoma growth-stimulatory activity which expresses autostimulatory activity for human melanoma cell lines [31]. IL-8 and GRO-α may be involved in regulation of collagen turnover in SFC [32] and can stimulate angiogenesis [28]. CXC chemokines such as IL-8 and GRO-α are able to decrease the expression of interstitial collagens by SFC, whereas members of the CC family fail in this respect [32]. Exposure of SFC to IL-17 led to mRNA induction of IL-8, GRO-α and GRO-β. Anti-IL-17 antibody blocked the effects of IL-17. Incubation of cultured SFC with IL-17 for 48 h resulted in a 33-fold increase of GRO-α protein expression and a sevenfold higher IL-8 concentration. In peritoneal mesothelium, IL-17 is capable of selectively recruiting neutrophils via the release of GRO-α[33]. Similar data were obtained for bronchial epithelial cells with the increased expression of IL-8 by IL-17 and the recruitment of neutrophils into the airways [34]. Recently, we reported IL-17-induced IL-8 expression in glial cells as well as the superadditive effects of IL-17 with IL-1β[13[.
In accordance with previous studies, we have found that IL-17 is capable of stimulating the MAPK signalling pathways ERK1/2 and p38 as well as NF-κB [35–37]. In contrast with results of Shalom-Barak et al. in chondrocytes, in human SFC IL-17 did not activate the JNK pathway significantly [37]. The maximum activation of ERK1/2 and p38 was found early after IL-17 stimulation (10–30 min) whereas the resynthesis of IκB-α mRNA peaked between 60 min and 3 h. NF-κΒ regulates the expression of many genes involved in immune as well as in inflammatory responses. This transcription factor acts on genes for proinflammatory cytokines (TNF-α, IL-1β, IL-6), chemokines (IL-8, MIP-1α, GRO), inflammatory enzymes (COX-2), inducible nitric oxide synthase (iNOS), immune receptors (IL-2R, T cell receptor), and for adhesion molecules (ICAM-1, VCAM-1, E-selectin) [38]. Binding of IL-17 to its receptor results in the activation of TRAF-6 and uses NIK and IKK-α in the activation of the NF-κB-dependent CINC promoter which may be the homologue of the human gro gene in the rat system [39]. The inhibitors genistein and calphostin C reduced significantly the effect of IL-17 on the mRNA expression of IL-8, GRO-α and GRO-β in SFC. This means that the induction of these chemokines by IL-17 requires tyrosine-specific protein kinase activities as well as protein kinase C activities for postreceptor signalling processes. However, with PD98059 we did not observe a significant decrease in the mRNA amounts of IL-8, GRO-α and GRO-β. Similar data were obtained for IL-17-induced NO-production in chondrocytes [37]. The MAPK p38 appears necessary for the expression of IL-8 as well as GRO-α and GRO-β, shown by inhibition with SB203580. MAPK p38 is involved in the IL-17-enhanced iNOS production [36], and plays a role in the MMP-9 expression induced by IL-17 [17]. The p38 MAPK pathway regulates a stabilization of IL-8 mRNA through an AU-rich elements-targeted mechanism [40].
Furthermore, we show the effects of antirheumatic drugs as cyclosporin A, dexamethasone and methotrexate on IL-17R mRNA expression. Whereas dexamethasone decreased IL-8 secretion stimulated by IL-17, simultaneous treatment with methotrexate and IL-17 resulted in an increase of IL-17-stimulated IL-8 protein expression. Methotrexate, a folate antagonist, has been shown to exert anti-inflammatory, antiproliferative and immunsuppressive actions that are mediated in part by the release of adenosine [41]. Majumdar and Aggarwal reported the suppression of the TNF-α-induced NF-κB activation by methotrexate [42]. Only when the cells were pretreated for 60 or 120 min with methotrexate was a maximum inhibition of TNF-α-induced NF-κB activation observed. Co-treatment or post-treatment with methotrexate did not inhibit NF-κB activation. This could be an additional explanation for the increased IL-8 levels in the supernatants of SFC co-treated with methotrexate and IL-17. Cyclosporin A showed the strongest effect on IL-17R mRNA expression. This immunosuppressive drug inhibits the enzymatic activity of the serin/threonin phosphatase calcineurin that is required for dephosphorylation of transcription factor NF-AT [43]. Failure to dephosphorylate NF-AT prevents its translocation into the nucleus and results in lack of gene transcription. It has been shown recently that cyclosporin A can block the PMA/ionomycin-induced production of IL-17 as well as IL-17 secretion triggered by IL-15 in PBMC [8]. We observed the augmentation of IL-17R mRNA expression as well as an increase in IL-17-stimulated IL-8 secretion in SFC showing the complex situation of therapeutic interventions.
In summary, our results support the hypothesis that IL-17/IL-17R may play a significant role in the pathogenesis of RA and may contribute to an unbalanced production of cytokines as well as participate in connective tissue remodelling [32]. However, a deeper understanding of the signal transduction pathways of the IL-17/IL-17R complex may be necessary for the understanding of its functions as well as for a development of therapeutic approaches including IL-17/IL-17R as a target.
Acknowledgments
We thank Immunex Corporation, Seattle, WA (USA) for providing the IL-17R MoAb M 203 and Drs T. Pap (Magdeburg, Germany), J. Brandt (Halle, Germany) and E. Märker-Herrmann (Mainz, Germany) for cells. We acknowledge the technical assistance of Sandra Fuhrmann, Michaela Hemp and Grit Helbing. K. Fischer is thanked for support in IL-8 protein quantification.
REFERENCES
- 1.Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis.Passive responders or transformed aggressors? Arthritis Rheum. 1996;39:1781–90. doi: 10.1002/art.1780391103. [DOI] [PubMed] [Google Scholar]
- 2.Weyand CM, Goronzy JJ. The molecular basis of rheumatoid arthritis. J Mol Med. 1997;75:772–85. doi: 10.1007/s001090050167. [DOI] [PubMed] [Google Scholar]
- 3.Breedveld FC. New insights in the pathogenesis of rheumatoid arthritis. J Rheumatol. 1998;53(Suppl):3–7. [PubMed] [Google Scholar]
- 4.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 5.Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–10. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 6.Koch AE, Kunkel SL, Strieter RM. Cytokines in rheumatoid arthritis. J Invest Med. 1995;43:28–38. [PubMed] [Google Scholar]
- 7.Chabaud M, Durand JM, Buchs N, et al. Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 8.Ziolkowska M, Koc A, Luszczykiewicz G, et al. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–8. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]
- 9.Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines [see comments] J Exp Med. 1996;183:2593–603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Z, Painter SL, Fanslow WC, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–6. [PubMed] [Google Scholar]
- 11.Cai XY, Gommoll CP, Jr, Justice L, Narula SK, Fine JS. Regulation of granulocyte colony-stimulating factor gene expression by interleukin-17. Immunol Lett. 1998;62:51–8. doi: 10.1016/s0165-2478(98)00027-3. [DOI] [PubMed] [Google Scholar]
- 12.Jovanovic DV, Di Battista JA, Martel-Pelletier J, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–21. [PubMed] [Google Scholar]
- 13.Kehlen A, Thiele K, Riemann D, Rainov N, Langner J. Interleukin-17 stimulates the expression of IkappaB alpha mRNA and the secretion of IL-6 and IL-8 in glioblastoma cell lines. J Neuroimmunol. 1999;101:1–6. doi: 10.1016/s0165-5728(99)00111-3. [DOI] [PubMed] [Google Scholar]
- 14.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409–14. [PubMed] [Google Scholar]
- 15.Chabaud M, Garnero P, Dayer JM, Guerne PA, Fossiez F, Miossec P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine. 2000;12:1092–9. doi: 10.1006/cyto.2000.0681. [DOI] [PubMed] [Google Scholar]
- 16.Dudler J, Renggli-Zulliger N, Busso N, Lotz M, So A. Effect of interleukin 17 on proteoglycan degradation in murine knee joints. Ann Rheum Dis. 2000;59:529–32. doi: 10.1136/ard.59.7.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jovanovic DV, Martel-Pelletier J, Di Battista JA, et al. Stimulation of 92-kd gelatinase (matrix metalloproteinase 9) production by interleukin-17 in human monocyte/macrophages: a possible role in rheumatoid arthritis. Arthritis Rheum. 2000;43:1134–44. doi: 10.1002/1529-0131(200005)43:5<1134::AID-ANR24>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Spriggs MK. Interleukin-17 and its receptor. J Clin Immunol. 1997;17:366–9. doi: 10.1023/a:1027360106635. [DOI] [PubMed] [Google Scholar]
- 19.Yao Z, Fanslow WC, Seldin MF, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–21. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 20.Yao Z, Spriggs MK, Derry JM, et al. Molecular characterization of the human interleukin (IL)-17 receptor. Cytokine. 1997;9:794–800. doi: 10.1006/cyto.1997.0240. [DOI] [PubMed] [Google Scholar]
- 21.Schwachula A, Riemann D, Kehlen A, Langner J. Characterization of the immunophenotype and functional properties of fibroblast-like synoviocytes in comparison to skin fibroblasts and umbilical vein endothelial cells. Immunobiology. 1994;190:67–92. doi: 10.1016/S0171-2985(11)80284-6. [DOI] [PubMed] [Google Scholar]
- 22.Rodel J, Straube E, Lungershausen W, Roth A, Groh A. Primary cultivation of human synovial cells from nonrheumatic synovial tissue and fluid. Exp Toxicol Pathol. 1996;48:243–7. doi: 10.1016/S0940-2993(96)80006-6. [DOI] [PubMed] [Google Scholar]
- 23.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 24.Navarrete SA, Riemann D, Thiele K, Kehlen A, Navarrete SA, Langner J. Constitutive expression of HLA class II mRNA in synovial fibroblast-like cells from patients with rheumatoid arthritis. Immunol Lett. 1997;58:53–8. doi: 10.1016/s0165-2478(97)02712-0. [DOI] [PubMed] [Google Scholar]
- 25.Thiele K, Riemann D, Navarrete SA, Langner J, Kehlen A. Cell–cell contact of human T cells with fibroblasts changes lymphocytic mRNA expression: increased mRNA expression of interleukin-17 and interleukin-17 receptor. Eur Cytokine Netw. 2000;11:53–8. [PubMed] [Google Scholar]
- 26.Woltman AM, De Haij S, Boonstra JG, Gobin SJ, Daha MR, Kooten CV. Interleukin-17 and CD40-ligand synergistically enhance cytokine and chemokine production by renal epithelial cells. J Am Soc Nephrol. 2000;11:2044–55. doi: 10.1681/ASN.V11112044. [DOI] [PubMed] [Google Scholar]
- 27.Honorati MC, Meliconi R, Pulsatelli L, Cane S, Frizziero L, Facchini A. High in vivo expression of interleukin-17 receptor in synovial endothelial cells and chondrocytes from arthritis patients. Rheumatology (Oxford) 2001;40:522–7. doi: 10.1093/rheumatology/40.5.522. [DOI] [PubMed] [Google Scholar]
- 28.Szekanecz Z, Strieter RM, Kunkel SL, Koch AE. Chemokines in rheumatoid arthritis. Springer Semin Immunopathol. 1998;20:115–32. doi: 10.1007/BF00832002. [DOI] [PubMed] [Google Scholar]
- 29.Heller RA, Schena M, Chai A, et al. Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci U S A. 1997;94:2150–5. doi: 10.1073/pnas.94.6.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Endo H, Akahoshi T, Takagishi K, Kashiwazaki S, Matsushima K. Elevation of interleukin-8 (IL-8) levels in joint fluids of patients with rheumatoid arthritis and the induction by IL-8 of leukocyte infiltration and synovitis in rabbit joints. Lymphokine Cytokine Res. 1991;10:245–52. [PubMed] [Google Scholar]
- 31.Richmond A, Thomas HG. Melanoma growth stimulatory activity. isolation from human melanoma tumors and characterization of tissue distribution. J Cell Biochem. 19848;36:185–98. doi: 10.1002/jcb.240360209. [DOI] [PubMed] [Google Scholar]
- 32.Unemori EN, Amento EP, Bauer EA, Horuk R. Melanoma growth-stimulatory activity/GRO decreases collagen expression by human fibroblasts. Regulation by C-X-C but not C-C cytokines. J Biol Chem. 1993;268:1338–42. [PubMed] [Google Scholar]
- 33.Witowski J, Pawlaczyk K, Breborowicz A, et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. 2000;165:5814–21. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- 34.Laan M, Cui ZH, Hoshino H, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–52. [PubMed] [Google Scholar]
- 35.Attur MG, Patel RN, Abramson SB, Amin AR. Interleukin-17 up-regulation of nitric oxide production in human osteoarthritis cartilage. Arthritis Rheum. 1997;40:1050–3. doi: 10.1002/art.1780400609. [DOI] [PubMed] [Google Scholar]
- 36.Martel-Pelletier J, Mineau F, Jovanovic D, Di Battista JA, Pelletier JP. Mitogen-activated protein kinase and nuclear factor kappa B together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: possible role of transactivating factor mitogen-activated protein kinase-activated proten kinase (MAPKAPK) Arthritis Rheum. 1999;42:2399–409. doi: 10.1002/1529-0131(199911)42:11<2399::AID-ANR19>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 37.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappa B. J Biol Chem. 1998;273:27467–73. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 38.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 39.Awane M, Andres PG, Li DJ, Reinecker HC. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol. 1999;162:5337–44. [PubMed] [Google Scholar]
- 40.Winzen R, Kracht M, Ritter B, et al. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999;18:4969–80. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morabito L, Montesinos MC, Schreibman DM, et al. Methotrexate and sulfasalazine promote adenosine release by a mechanism that requires ecto-5′-nucleotidase-mediated conversion of adenine nucleotides. J Clin Invest. 1998;101:295–300. doi: 10.1172/JCI1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majumdar S, Aggarwal BB. Methotrexate suppresses nf-kappab activation through inhibition of ikappabalpha phosphorylation and degradation. J Immunol. 2001;167:2911–20. doi: 10.4049/jimmunol.167.5.2911. [DOI] [PubMed] [Google Scholar]
- 43.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–42. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]