New Insights into Vitamin D Sterol-VDR Proteolysis, Allostery, Structure-Function from the Perspective of a Conformational Ensemble Model (original) (raw)
. Author manuscript; available in PMC: 2008 Mar 1.
Published in final edited form as: J Steroid Biochem Mol Biol. 2007 Mar;103(3-5):243–262. doi: 10.1016/j.jsbmb.2006.12.004
Abstract
Recently, we have developed a vitamin D sterol (VDS)-VDR conformational ensemble model. This model can be broken down into three individual, yet interlinked parts: a) the conformationally flexible VDS, b) the apo/holo-VDR helix-12 (H12) conformational ensemble, and c) the presence of two VDR ligand binding pockets (LBPs); one thermodynamically favored (the genomic pocket, G-pocket) and the other kinetically favored by VDSs (the alternative pocket, A-pocket). One focus of this study is to use directed VDR mutagenesis to 1) demonstrate H12 is stabilized in the transcriptionally active closed conformation (hVDR-c1) by three salt-bridges that span the length of H12 (cationic residues R154, K264 and R402), 2) to elucidate the VDR trypsin sites [R173 (hVDR-c1), K413 (hVDR-c2) and R402 (hVDR-c3)] and 3) demonstrate the apo-VDR H12 equilibrium can be shifted. The other focus of this study is to apply the model to generate a mechanistic understanding to discrepancies observed in structure-function data obtained with a variety of 1α,25(OH)2-vitamin D3 (1,25D) A-ring and side-chain analogs, and side-chain metabolites. We will demonstrate that these structure-function conundrums can be rationalized, for the most part by focusing on alterations in the VDS conformational flexibility and the elementary interaction between the VDS and the VDR A- and G-pockets, relative to the control, 1,25D.
Keywords: vitamin D receptor; VDR; 1α,25(OH)2-vitamin D3; structure-function; conformational ensemble; induced-fit
INTRODUCTION
The steroid hormone 1α,25(OH)2-vitamin D3 (1,25D, Fig. 1A) and its metabolites collectively form nearly all of the small, lipophilic, molecules that regulate the complex vitamin D endocrine system. The known endocrine functions of vitamin D sterols include regulation of ion homeostasis, bone integrity, apoptosis, and cell growth and differentiation. Vitamin D sterols modulate these processes via regulating multiple tissue specific cellular signaling cascades via binding to the nuclear vitamin D receptor (VDR) and activating genomic and non-genomic cellular responses [1,2].
Fig. 1.

Vitamin D sterols and the VDR
(A) Chemical structures of vitamin D sterols addressed in this study, 1α,25(OH)2-vitamin D3 (1,25D) serves as the reference compound. RCI values are listed under each sterol (see reagents for complete chemical names of 1,25D analogs or natural metabolites). The RCI demonstrates how well the given sterol competes with constant levels of [3H]-1,25D (0.2 pmoles/ml, see methods), in a steroid competition assay. (B) Domain partitioning of the nuclear vitamin D receptor (VDR). All VDR structures displayed herein are composed of aa118-427, Δ165-215 (see methods). Note, the F-domain aa404-427 is the activation function-2 domain (AF2 or Helix-12). (C) The tertiary structure of the holo-VDR molecule. This closed VDR H12 conformation (hVDR-c1) and fold is observed in nearly all other holo-NR x-ray crystallographic results (see NR pdb files). The VDR molecule ligand binding pockets (LBPs) house 1,25D as determined by x-ray crystallography [genomic pocket (G-pocket), green and red ball and stick structure] and molecular modeling [alternative pocket (A-pocket), orange and red ball and stick]. The A-ring domains of these two pockets overlap one another (black circle in figure). When complexed to the G-pocket, 1,25D's side-chain hydroxyl forms H-bonds with H305 and H397, and forms vdW contacts with I268 and V300 (labeled green and blue wireframe structures). In the putative A-pocket, the 1,25D molecule extends towards the H2/ β-sheet region (red ribbons), specifically R158, rather than the activation helices H11/ H12 (rust, violet and pink ribbon). Residues that provide electrostatic stability to H12 (R154, K264 and R402) or are trypsin sites (R402 and K413) are labeled and rendered in wireframe. When 1,25D is bound to the G-pocket a salt-clamp between K246 and E420 (orange dashed line). In this holo-VDR conformation the hydrophobic cleft (blue, transparent Connolly surface) has high affinity for NCoA molecules and low affinity for NCoR molecules.
The VDR, like other nuclear receptors (NRs), is separated into six domains (A-F, Fig. 1B; [3,4,5]). Portions of helices 3, 4 and 5 of domain E (LBD) form the hydrophobic cleft (Fig. 1C). The hydrophobic cleft is the region of the NR/VDR where most transcriptional co-activator (NCoA) and co-repressor (NCoR) molecules compete for binding. The VDR affinity for NCoA versus NCoR is predominantly regulated by hormone binding and the conformation of the VDR C-terminal activation helix (H12 or activation function 2 domain, Fig. 1C) [6,7,8]. NR crystallographic studies demonstrate that under crystallographic conditions nearly all hormones stabilize the transcriptionally active, closed H12 conformation (NR/VDR-c1, Fig. 1C) that facilitates recruitment of NCoA's. It is currently posited that H12 is opened in the absence of ligand (apo-NR) and is induced to close (e.g. activated by induced-fit) upon binding the ligand [9,10].
Crystal structures of a truncated VDR, consisting of portions of domain D and all of domains E and F (Fig. 1B), have shown that agonists occupy the same relative steric space in the VDR LBD, termed the genomic pocket (G-pocket), and stabilize only the closed helix-12 conformation (VDR-c1, Fig. 1C) [11,12,13,14,15]. Analysis of these VDR structures and molecular models with complete AF2-domains, reveal that unlike any other hormonally regulated NR, H12 is held in the closed conformation (c1) by three salt-bridges: 1) K264---E420 2) R154---K413/ R154---L414/ R154---D232 and 3) R402---S427/ S398/ E425 (Fig. 1C). Of these three, the functional importance of only the K264-E420 salt-bridge has been directly tested. The results showed that severing the clamp via site-directed mutagenesis significantly reduced 1,25D activation of a VDR reporter construct [16]. Herein, we will test the functional importance of the other two salt-clamps by designing and functionally evaluating five hVDR mutant constructs (R402E, R402A, R154E, R158E and R154E/R402E).
Currently the most actively used method for assaying VDR H12 conformational heterogeneity in solution, is the partial trypsin digest experiment (protease sensitivity assay, PSA) [17]. Unlike x-ray crystallography, PSA provides insight into solution state H12 dynamics and ultimately yields insight into the population distribution of apo/holo-VDR (H12) isomers under steady state conditions. Previous VDR PSA results indicate that ligands stabilize three different VDR conformations. The observed population distribution (e.g. PSA footprint) of the three VDR conformers varies depending on the chemistry of the vitamin D sterol being assayed. For example, Norman et al. [18] demonstrated that 1,25D and 20-epi-1,25D (i.e. a VDR superagonist [19]) both predominantly stabilize the closed H12 conformation (hVDRwt-c1, ∼34 kDa PSA band), while the two side-chain synthetic analog of 1,25D (KH, Fig. 1A) stabilizes an opened-like, inactive H12 conformation (hVDRwt-c3, ∼30 kDa PSA band). Furthermore, it has been demonstrated a class of VDR genomic antagonists stabilize a significant population of the partially-closed H12 conformation (hVDRwt-c2, ∼32 kDa PSA band) [20]. The trypsin site exposed when H12 is in the closed position (hVDR-c1, Fig. 1C) has been successfully mapped to R173 [21] (insertion loop, Fig. 1B), while the trypsin sites exposed when sterols stabilize hVDRwt-c2 and hVDRwt-c3 remain unidentified. Herein we use the VDR primary structure, the unique KH PSA footprint and the VDR crystal structures to design VDR mutants to elucidate the two presently unmapped trypsin sites, and to demonstrate that some species lack hVDRwt-c2 and hVDRwt-c3 sites.
Like the x-ray crystal structures of VDR superagonists [14] and 1,25D [11], the recently solved KH-VDR (VDR from zebrafish) complex [12] clearly demonstrates that the ligand binding pocket adapts to the KH sterol so it stabilizes the closed VDR H12 conformation (hVDR-c1). The KH-VDR x-ray result does correlate well with its ability to transactivate with similar or greater efficacy relative to 1,25D; however, the structure does not correlate with KH's solution state PSA results or reduced VDR affinity [18]. This is also true for many other analogs of 1,25D [1]. Interestingly, the terminal C23-side chain metabolite of 1,25D (1,23S,25R-lac, Fig. 1A) also preferentially stabilizes the hVDRwt-c3 PSA fragment, but unlike KH is a very poor VDR agonist and considerably weaker VDR binder [22].
These structure-function similarities and differences are central to methods used in designing VDR point mutants to elucidate the unknown VDR trypsin sites and in designing mutants to test the KH-zbVDR crystal structure, and 1,25D, KH and 1,23S,25R-lac hVDR G-pocket molecular models. The comprehensive structure-function results will demonstrate that a) some VDR mutants enhance the intramolecular stability of the apo-VDR molecule; therefore these mutants are capable of maintaining or enhancing VDR potency in the absence of H-bonds formed with the ligands side-chain atoms; and b) the structure-function and computational results are not consistent with a VDR one pocket molecular model.
Finally, it will be demonstrated that most of 1,25D's side-chain metabolites (Fig. 1A; C23 and C24-pathways [23]), share the 1,23S,25R-lac and KH VDR PSA profiles (e.g. abundance of hVDRwt-c3) and have reduced VDR RCI values. Yet functionally the metabolites have been previously shown to have broad ranging VDR agonist potencies [24], often assay or cell specific. In an attempt to mechanistically understand the discrepancies in the structure-function results obtained with 1,25D, 1,23S,25R-lac, KH and the side-chain metabolites, all sterols were docked to the x-ray, hVDR genomic pocket (G-pocket) and putative alternative ligand binding pocket (A-pocket, Fig. 1C) [25]. The VDR A-pocket was discovered using in silico techniques and has been linked to shape and stereospecific induction of nongenomic cellular responses [26], as well as the unique structure-function results obtained for some A-ring and side-chain derivatives of 1,25D [27,28]. An important assumption made in these works is that the VDR A-pocket represents a kinetically favored ligand binding pocket (LBP), while x-ray results clearly indicate the VDR G-pocket is the thermodynamically favored LBP.
We will demonstrate herein that the in silico results suggest that the lack of correlation between these sterols VDR PSA and RCI results and their functional profiles can be understood by application of the vitamin D sterol-VDR conformational ensemble model [25,26,27,28]. We will also demonstrate the putative A-pocket can be used to provide a mechanistic understanding to the structure-function results obtained with 19-nor and 2-CH3 A-ring modifications, a continuation of our previous work with A-ring diastereomers/ metabolites [28]. Thus this study provides novel insights into apo/holo-VDR H12 allostery and suggests VDR cell and/or tissue specific signaling is modulated by vitamin D sterol pools. A putative molecular mechanism that focuses solely on the elementary interaction between the sterol and the VDR and is capable of explaining the comprehensive, often inconsistent, vitamin D sterol-VDR structure-function results is presented.
Materials and Methods
Reagents
1α,25(OH)2-vitamin D3 (1,25D), 21-(3'-Hydroxy-3'-methylbutyl)-1α,25(OH)2-D3 (KH) and 19-nor-KH were gifts from Dr. Milan Uskokovic (Hoffmann La Roche, Nutley NJ). 23S,25R-1α,25(OH)2-D3-26,23-lactone (1,23S,25R-lac), 23R,25S-1α,25(OH)2-D3-26,23-lactone (1,23R,25S-lac), 1α,23S,25(OH)3-D3 (1,23S,25D), 1,24R,25(OH)3-D3 (1,24R,25D), 1,24S,25(OH)3-D3 (1,24S,25D), 24-oxo-1,25(OH)2-D3 (24-oxo-1,25D), 1,24R(OH)2-D3 (1,24R-D) and 1,24S(OH)2-D3 (1,24S-D) were provided by Dr. Seiichi Ishizuka (Teijin Pharma, Ltd., Tokyo Japan).
1,25D and analog genomic transactivation
CV1 cells were transiently transfected with activator 1(-)Nhe1(-)VDR or mutant VDR plasmids (1-427) and an osteocalcin SEAP (secreted alkaline phosphatase) reporter, in accordance with [26] and [29].
Protease Sensitivity Assay (PSA)
[35S]-VDR was generated from 1(-)Nhe1(-)VDR or mutant plasmid (1-427) using the Promega TNT reticulocyte lysate kit. The reaction tubes were incubated for 2 hr at 30°C, then placed on ice. 1,25D or analog (10−5-10−9 M) was added and the tubes were incubated at room temperature for 20 min. Then 15 μg/ ml trypsin was added to the tubes and incubated for 20 min at room temperature followed by a 5 min incubation at 80°C with SDS. Reaction mixtures were then loaded in a 14% SDS-PAGE gel and subjected to electrophoresis. Bands were visualized by autoradiography. Band densities were calculated using the UScanIT software package.
Molecular Modeling
The nuclear vitamin D receptor (VDR) ligand binding domain (LBD) x-ray construct (aa118-427, Δ165-215) complexed to 6-_s_-trans 1,25D (pdb code: 1DB1) was opened in the Biopolymer module of Insight 2000.1 (Accelyrs Inc., San Diego, CA). The missing residues aa375-377 (loop) and aa424-427 (C-terminal portion of helix-12) were added to the protein and their positions optimized while all other atoms of the assembly remained fixed. Hydrogen atoms were then added at pH = 7.4. The assembly generated served as the starting structure for the control minimization experiment [_trans_ 1,25D minimized in the VDR ligand pocket described by the x-ray structure (G-pocket, Fig. 1C] and all subsequent experiments where a prospective ligand was docked in the VDR G-pocket. The final, minimized 1α,25(OH)2-lumisterol3 (JN)/ VDR A-pocket model was used as the starting structure for all VDR alternative pocket (A-pocket, Fig. 1C) docking experiments [26,27,28]. For docking experiments in both pockets, low energy side-chain conformation of the putative ligand from conformational search calculations were used as starting structures (see [26,27,28] for methodology, Fig. 7A). An 8.0 ns MD simulation was performed following assembly minimization to check if the total energy of the complex was conserved [26,28]. For docking KH in the VDR G-pocket, the second side-chain was manually rotated to mimic the orientation observed in the KH-VDR crystal structure [26,27,28]. Steric clashes with L309 were removed by manually rotating L309 out of the G-pocket prior to assembly minimization.
Fig. 7.

1,25D dot map and 19-nor-1,25D C5,C6,C7,C8-dihedral driver experiments
(A) Dot map of the 1,25D CD-ring fragment performed using standard computational methodology [36,37]. The dot map projects the regiospecific placement of the 25-OH H-bonding atom (gray dots) in each of the low energy side-chain conformations (all 4 kcal/mol from lowest energy structure, green wireframe). The dotted red surface provides a projection of the “real space’ occupied by the 25-OH group. All the low energy 1,25D side-chain conformations can be grouped into three C16-C17-C20-C22 dihedral angle populations. Population A is the side-chain orientation preferred by the putative alternative VDR ligand binding pocket (A-pocket, Fig. 1C); Population B is the preferred orientation of the VDR x-ray, G-pocket (Fig. 1C); and Population C is the preferred side-chain orientation observed in the serum vitamin D binding protein (DBP) x-ray structure [54]. The table inset demonstrates that the side-chain metabolites of 1,25D do have reduced conformational mobility (entropy), but oxidation does not alter the global population distribution. (B) Illustration of the C5,6,7,8 dihedral driver experiment performed using PC_Model v9.0 (Serena Software Inc.) for 1,25D (black line) and the 19-nor-1,25D analogs (gray line). The calculated potential energy (kcal/mol) of a given C5,6,7,8 dihedral conformer in one degree increments is plotted. The most energetically favorable conformers (∼ ±110°) show an A-ring geometry where the C5,C6,C10,C19 diene is perpendicular to the C7-C8 double bond.
Potential Interaction Energy (PIE) calculations
PIE values were calculated using the method outlined in [26,27,28] and represent an arbitrary value that reflects the potential non-bond attractive and repulsive interactions formed between the ligand and VDR residues. It does not reflect the total binding energy; therefore, only values obtained when docking an identical ligand in different conformations, epimers and constitutional isomers can be directly compared.
PC_Model seco-B-ring dihedral driver experiments
1,25D and 19-nor-1,25D were energy optimized using the MM2 method. The C5-C6-C7-C8 dihedral angle was then set to 180° and the dihedral rotated in one degree increments to −180°. The single point energy of each conformer was then plotted against the dihedral angle to develop an energy diagram.
RESULTS
Intramolecular electrostatic stabilization of the C-terminal VDR activation helix (H12)
The VDR x-ray crystal structures indicate that the K264 (H5)---E420 (H12) and R154 (H2-loop)---E232 (H3)/K413 (H12)/L414 (H12) electrostatic interactions are integral in stabilizing H12 in the transcriptionally active, closed conformation (hVDRwt-c1, Fig. 1C, yellow circles). Residues N424, E425, I426, and S427 are unresolved in the 1,25D-VDR x-ray structures [11,12,13,14,15], but when added in an α-helical orientation to the x-ray construct (pdb code: 1DB1), prior to assembly minimization, it was observed that R402 (H11) formed Coulombic interactions with either E425 and S398 or S427 and S398 (see methods; Fig. 1C, pink ribbon in yellow circle). In accordance with this in silico observation, mutation of R402 to Glu (E) or Ala (A) increased 1,25D's genomic EC50 ∼10-fold (Table 1, Fig. 2). This is comparable to previous studies that showed fracturing of the K264-E420 salt-bridge (K264A) increased 1,25D's transactivation EC50 ∼10-fold [16].
Table 1.
Summary of 1,25D, KH, 1,23S,25R-lac and MK EC50 results in various hVDR constructs
All data was determined by transiently transfecting CV1 cells with the VDR activator plasmid and an osteocalcin-VDRE driven SEAP-reporter plasmid (see methods; n ≥ 3). If no statistics are shown the experiment was not repeated. A value of >500 indicates no reporter activity was observed up to a 1μM dose of sterol. NS indicates there was no significant induction of reporter relative to basal (EtOH control).
| hVDR Construct | 1,25D EC 50 (nM) | KH EC 50 (nM) | 1,23S,25R-lac EC 50 (nM) | MK EC 50 (nM) |
|---|---|---|---|---|
| hVDRwt | 1.6 ± 1.0 | 0.49 ± 0.36 | > 500 nM | NS |
| R402A | 13.2 | --- | NS | --- |
| R402E | 16.3 ± 4.2 | --- | --- | --- |
| R154E | 147 ± 44 | --- | --- | --- |
| R154E/R402E | 403 ± 125 | --- | --- | --- |
| R158A | 46.2 ± 9.2 | --- | --- | --- |
| R158F | 37.5 ± 7.1 | --- | --- | --- |
| R158E | 40.0 & 42.9 | --- | --- | --- |
| R173Q | 2.0 ± 0.8 | --- | --- | --- |
| R402E/K413Q | 11 ± 2.1 | --- | --- | --- |
| H305A | 25.0 ± 7.5 | 0.96 ± 0.75 | > 500 | --- |
| H305F | 3.2 ± 0.93 | 0.32 ± 0.19 | > 500 | 0.061 ± 0.002 |
| H397F | 9.1± 1.3 | 3.44 ± 1.51 | > 500 | > 500 |
| H305A/H397F | 31.0 ± 10.0 | 3.21 ± 1.36 | > 500 | > 500 |
| H305F/H397F | 1.6 ± 0.13 | 0.95 ± 0.59 | 263 ± 102 | 0.034 ± 0.008 |
| V300Y | 1.02 ± 0.3 μM | 21.4 ± 3.7 | --- | --- |
| I268Y/V300Y | NS | NS | --- | --- |
| H229Y | > 500 | > 500 | --- | --- |
| R158A/H229Y | > 500 | --- | --- | --- |
Fig. 2.
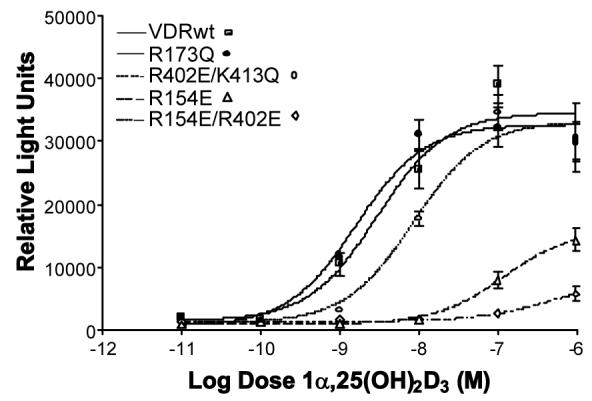
Intramolecular electrostatic stabilization of the VDR AF-2 domain (H12)
Representative 1,25D dose-response curves for hVDRwt and single and double point mutants designed to assess the residues' significance in providing electrostatic stability to the closed, transcriptionally active H12 conformation (Fig. 1C) or serve as tools in elucidation of trypsin sites. The R402E/ K413Q double mutant has the same functional profile as any of the R402 single mutants (see Table 1). Analogs were assayed in CV1 cells transiently transfected with the specified full length hVDR construct and an osteocalcin-VDRE driven SEAP-reporter (see methods). Effective concentrations (EC50) values for all mutants (assayed in triplicate; n ≥ 3) addressed in the text are summarized in table 1. All EC50 values were found to be significantly different (p ≤ 0.05) when compared to hVDRwt with the exception of R173Q.
Mutation of VDR's novel hinge-domain/ AF2-domain (domains D and F, Fig. 1B), R154 salt-bridge (Fig. 1C), caused a 100-fold increase in 1,25D's EC50 (see R154A/ R154E, Fig. 2 and Table 1) and virtually destroyed its potency (see Bmax, Fig. 2). Alternatively, mutation of R158, an Arg residue local to R154 that does not directly contact H12 residues (Fig. 1C) [11], to an A, E or F, only reduced 1,25D's genomic efficacy to ∼20% of WT (Fig. 2, Table 1). Finally, the R154E/R402E double mutant had an additive effect on 1,25D's transactivation efficacy and potency, virtually completely depleting 1,25D's ability to transactivate (Fig. 2, Table 1). These results confirm that the VDR AF2-domain is unique relative to other NRs because H12 is stabilized in the transcriptionally active, closed configuration by three salt-bridges, two of which (R154 and R402 salt-bridges) are unique to the VDR, and the other (K264 salt-bridge), is only observed in TRβ [8,11]. More importantly, the R154 loop residue, forms the most functionally relevant, direct electrostatic non-bond interaction with VDR H12 backbone residues (Fig. 1C). According to bioinformatics sequence alignments (expasy.org) of all known VDR sequences, all species contain R154, K264 and R402 loci.
Elucidation of the three VDR LBD trypsin cleavage sites
The three known VDR tryptic fragments migrate at ∼34 kDa (hVDRwt-c1 = closed H12), ∼32 kDa (hVDRwt-c2 = partially-closed H12 or intermediate), and ∼30 kDa (hVDRwt-c3 = opened H12; Fig. 3A) in a 14% SDS-PAGE gel. R173 is known to be the hVDRwt-c1 trypsin site [21]. Based on the hVDR primary sequence, trypsin cleavage at R173 produces a ∼29 kDa C-terminal fragment (VDR-c1, expasy.org) and a ∼21 kDa N-terminal fragment; therefore, the hVDRwt-c1 fragment does not migrate at its projected molecular weight (Fig. 3A). Nevertheless, the work of Vaisanen et al. [21] was substantiated by the R173Q-1,25D protease sensitivity assay (PSA) result, where the presence of hVDRwt-like ∼34 kDa (c1), ∼32 kDa (c2), or ∼30 kDa (c3) fragments were not observed. Alternatively, three new higher molecular weight bands were observed (a1-a3, Fig. 3B). As expected, the R173Q mutation did not effect 1,25D's ability to induce transcription (Fig. 2, Table 1), because R173 is an insertion loop residue.
Fig. 3.

Mapping the hVDR trypsin cleavage sites
(A) Protease sensitivity (PSA) footprint obtained from incubating [35S]-hVDRwt with the indicated vitamin D sterols (10−5 M). The three fragments generated are ∼34 (hVDRwt-c1), ∼32 (hVDRwt-c2), and ∼30 (hVDRwt-c3) kDa in size, based on their migration in a 14% SDS-PAGE gel relative to standards. (B) PSA results obtained in R173Q (lanes 1 and 2) and hVDRwt (lanes 3 and 4) incubated with 10−5M 1,25D. Two input bands are observed and labeled c' and c. The new 1,25D-induced bands produced in the R173Q mutant are labeled a1, a2 and a3. (C) PSA results obtained when hVDRwt and R402E were incubated with 1,25D, KH, and 23S,25R-lac (10−5 M). (D) PSA results obtained when R391Q was incubated with increasing concentrations of (10−5-10−9 M) KH and 1,25D. (E) PSA results obtained when hVDRwt, R158F and R158E were incubated with 10−5 M KH, 23S,25R-lac and 1,25D. (F) PSA results obtained for R402E/ K413Q and hVDRwt incubated with 10−5 M KH, 1,25D or BT. Collectively, the results indicate that both c' and c are capable of binding ligand and undergo similar cleavage [compare KH/23S,25R-lac with 1,25D upper bands (c1' and/or c3')].
It is noted that in all PSA gels, multiple input VDR bands are observed after in vitro expression in the rabbit reticulocyte lysate system; however, no contamination in the region of interest (∼34-30 kDa) is observed. The band labeled VDR-c' (Fig. 3B) is larger than VDR-c (hVDRwt). In addition the VDR-c' artifact binds vitamin D sterols, but the trypsin products migrate at much higher MW's (Figs. 3B-3E). This observation is supported by the evidence that the calculated MW ratios for c/c1, c/c3 and c'/c1', c'/c3' are not significantly different (UScanIt software package, data not shown).
The 1,25D-R173Q PSA result (Fig. 3B) confirms that all three hVDRwt conformers share the same N-terminus [21]. To identify the C-terminus of the hVDRwt-c2 and hVDRwt-c3 fragments, the cationic R-group of prospective trypsin sites in H11/H12 and/or the hinge domain (Figs 1B and 1C) were identified using in silico techniques (expasy.org) and mutated. Next the mutant and hVDRwt constructs were incubated with the ligands 1,25D, 1,23S,25R-lac and KH (Fig. 1A) and subjected to a partial trypsin digest. The KH and 1,23S,25R-lac ligands were chosen because they both preferentially stabilize the opened-like H12 conformer (hVDRwt-c3) instead of the closed H12 conformer (hVDR-c1) under steady state conditions (Fig. 3C) [27].
The 1,25D, KH and lactone-R402E/A PSA results show that no hVDR-c3 band is observed for all ligands, rather a band shift to hVDR-c2 occurs (Fig. 3C). In addition, when R391 was mutated to an Asn (R391Q), no change in the KH or 1,25D PSA footprint was observed (Fig. 3D). R391 was previously claimed to be the hVDR-c3 cleavage site [20,30]. Furthermore, a reduction in the stability (band density) of hVDR-c3 was observed in the R158E and F mutants (Fig. 3E, Table 2), but not a band shift; even though, MW analysis of the hVDRwt primary sequence would predict a ∼30 kDa hVDRwt fragment (aa159-427).
Table 2.
hVDRwt and R158F 1,25D, 1,23S,25R-lac, and KH PSA 34 kDa (c1)/ 30 kDa (c3) band density ratios
This table summarizes the 34 kDa (c1)/ 30 kDa (c3) in-lane band density ratios for 1,25D, KH, and 1,23S,25R-lac in hVDRwt and R158F (n = 4). The gel in figure 3E is a representative gel from one experiment. In each experiment four separate gels were run and the c1/c3 densitometry results averaged. The percent differences (%Δ) in the band density ratios for BS and KH are statistically significant when hVDRwt and R158F results are compared (p ≤ 0.05). NS indicates no significant change.
| Ligand | c1/ c3 ratio hVDRwt | c1/ c3 ratio R158F | %Δc1/c3 |
|---|---|---|---|
| 1,25D | 2.5 ± 0.41 | 1.9 ± 0.24 | NS |
| 1,23S,25R-lac | 0.63 ± 0.055 | 0.87 ± 0.04 | **25%**↑ |
| KH | 0.37 ± 0.03 | 0.52 ± 0.06 | **15%**↑ |
The only putative trypsin site remaining C-terminal to R402 is K413 (Fig. 1C); therefore, a R402E/K413Q double mutant was made. The comprehensive R402E/K413Q PSA results demonstrate that both KH and 1,23S,25R-lac have identical PSA footprints as 1,25D in the double mutant (Fig. 3F). In addition, the BT-hVDRwt-c2 and hVDRwt-c3 bands (Fig. 3A) were absent in the R402E/K413Q mutant (Fig. 3F). Of these three sites, only the R402 (H11) site is conserved through all known VDR primary sequences (expasy.org). Three species (Japanese quail, chicken and zebra-fish) lack K413 (Q in all three) and R173 trypsin sites; therefore, the pool of VDR tryptic isoforms in these three species could lead in-part to different VDR-regulated signaling specificities (e.g. genomic versus non-genomic pathways). It has been shown previously that exogenous truncation or removal of the AF2-domain (H12) does reduce hVDR's ability to transactivate in the presence of agonists [18]. It is noted that the K413Q mutation does not affect VDR transactivational properties (compare R402E with R402E/K413Q, Table 1 and Fig. 2).
The KH/1,23S,25R-lac PSA results coupled with their measured VDR affinities being ∼38% and 0.5% that of 1,25D's (Fig. 1A), indicate that these two sterols both effectively expose R402 to trypsin, but lack identical hVDRwt binding properties. In order to develop a potential mechanistic understanding for this conundrum we first assessed whether R402 becomes exposed when KH and 1,23S,25R-lac is bound to the VDR G-pocket (Fig. 1A). Then we tested the recently solved KH-zbVDR (zebra fish) crystal structure [12] by mutating residues that KH forms added or novel vdW interactions with when compared to 1,25D and/or 1,23S,25R-lac.
Comparison and functional evaluation of the 1,25D/KH/1,23S,25R-lac-hVDR complexes
When 1,25D and 1,23S,25R-lac were computationally docked to the hVDR molecule (pdb: 1DB1; see methods and [26]) both side-chains form H-bonds with H305 and H397 and thread through similar steric space within the ligand binding pocket (Fig. 4A). The same is true for one of the two side-chains of KH in the crystal structure and lowest energy computational model (Fig. 4B); however, the second KH side-chain forms additional vdW contacts with V300, H305 and L309 [12] not formed by 1,25D or 1,23S,25R-lac (compare Fig. 4B to 4A). In both the projected 1,23S,25R-lac and KH VDR G-pocket lowest energy complexes, the R402 R-group position and non-bonding is unchanged relative to 1,25D (Figs. 4A and 4B). In addition, the distance between K246 and E420 (salt-clamp forming hydrophobic cleft boundry; Fig. 1C) is not significantly altered over a 8.0 ns dynamics simulation by either ligand when compared to 1,25D (data presented elsewhere), providing evidence that KH and 1,23S,25R-lac do not alter H12 conformational dynamics relative to 1,25D, via alterations in their low energy, thermodynamically favored, VDR G-pocket complexes [15]. These structural results are consistent with KH, but not 1,23S,25R-lac VDR agonist potency in hVDRwt (Table 1). To continue to probe for the underlying variable influencing the unique hVDRwt KH and 1,23S,25R-lac PSA and RCI results, a series of hVDR point mutants were made to test the KH x-ray structure and the molecular models.
Fig. 4.

Molecular modeling of 1,25D side-chain analogs or metabolites in the VDR G- or A-pockets
(A) Backbone atom superimposition of the 23S,25R-lac (gray and red ball and stick) and 1,25D (green and red ball and stick)-hVDRwt lowest energy molecule models. Only the sterols' side-chains are shown in the figure. Both side-chains form H-bonds with H305 and H397, only those of 23S,25R-lac are highlighted. The potential interaction energy (Table 3) for each complex is indicated for each panel. The 1,25D complex is in strong agreement with the x-ray structure [11]. (B) Backbone atom superimposition of the lowest energy KH (gray) and 1,25D-hVDRwt (green) molecular models. Only H-bonds formed by KH are depicted as black dotted lines. This model is in strong agreement with the x-ray crystal structure [13], which was used as a guide in manual rotation of KH's second side-chain and L309 prior to assembly minimization (note the steric clash observed between KH's second side-chain and L309, black circle). (C) Backbone atom superimposition of 1,25D complexed to hVDRwt (orange) and hrVDR (C403S/C410N, green). There was no change in the 1,25D/ G-pocket potential interaction energy in these models. H-bonds formed only by the mutated residues are indicated by black dotted lines. The H-bond shared in both structures is indicated with a cyan dotted line. The hydrophobic contact formed between L404 and the C26 atom of 1,25D is highlighted by a black circle. (D) Backbone atom superimposition of 1,25D (green), 1,24R,25D (gray) and 1,24S,25D (cyan ball and stick structures). The 1,24S,25D side-chain does not thread through the same relative steric space as 1,24R,25D and 1,25D within the VDR G-pocket; however, all compounds form two H-bonds with H305 and H397 (only shown for 1,24R,25D, gray) and the C26-methyl of 1,24S,25D lies in similar steric space occupied by the 1,25D and 1,24R,25D C26-methyl groups. (E) Backbone atom superimposition of 1,25D (green), 1,23S,25D (gray) and 1,23R,25D (cyan ball and stick structures). All sterols form H-bonds with H305 and H397 and thread through the same relative steric space within the VDR G-pocket. Importantly, unlike their constitutional isomers (1,24R/S,25D) both the C25 and C23 hydroxyls coordinate to the H305 imidazole (black dotted lines). The calculated potential interaction energies are again provided. (F) Backbone atom superimposition of the 1,25D (green) and two 1,23S,25D (cyan and gray) VDR A-pocket (Fig. 1C) low energy molecular models. The green and cyan ball and stick models superimpose quite well with one another and both the 25-OH groups form an H-bond with R158 (green wireframe). It was also observed that the lowest energy side-chain conformation of 1,23S,25D from the dot map calculation (Fig. 7A) could form a favorable complex in the putative VDR A-pocket when R158 maintained its x-ray orientation and non-bonding contacts (gray wireframe, black dotted lines). H-bonds formed by 1,23S,25D, but not 1,25D, are highlighted with pink dotted line. (G) Backbone atom superimposition of the 1,25D (orange) and 2α-CH3-1,25D (green, β-chair) VDR G-pocket low energy models. The only difference between the two structures is the axial 2-methyl group forms additional vdW contacts with L233 and is proximal to F150 and Y236. (H) Backbone atom superimposition of the lowest energy 2α-CH3-1,25D (orange) and 2β-CH3-1,25D (green) VDR A-pocket complexes. Only the A-rings of the ligands are shown for simplicity. The two methyl groups are rendered in CPK and the atoms they are most proximal to highlighted with red (unfavorable) and violet (favorable) solid lines. Both the low energy structures are in the β-chair, 6-_s_-trans conformation (see Table 3) and both only form H-bonds through their 3β-OH group (Y143 and S278, black dotted lines).
CV1 cell transient transfection results obtained with the H305A, H305F and H305F/H397F VDR mutant constructs indicate that 1,25D's transactivational potential is blunted ∼10-fold, 2-fold (insignificant) and 0-fold respectively by the mutations (Table 1). Surprisingly, none of the mutations significantly altered KH's transcriptional potency/ efficacy (Table 1). Furthermore the V300Y mutation, designed off the KH-zbVDR crystal structure and hVDR molecular model to create a steric block for the second KH side-chain (Fig. 4B), potently hindered 1,25D induced reporter expression, but had only a minor effect on KH's (Table 1).
Based on our modeling results, loss of 1,25D agonism in V300Y may be explained by the 1,25D side-chain H-bonding with Y300 and H305 rather than H305/ H397 and pulling the C26 and C27 methyl groups away from H12 residues. Alternatively, KH is believed to bind in a different conformation than that observed in the x-ray structure (i.e. its lowest energy conformation, PC_Model v9.0) where vdW contacts with H305, H397 and H12 residues are maintained [27]. Nevertheless, removal of the H305 H-bonds and reducing (H305F or V300Y) or adding (H305A) steric volume to the hVDR G-pocket had a more detrimental functional effect on 1,25D than KH, an unexpected result based on the low energy x-ray and computational hVDR G-pocket complexes (Figs. 4A and 4B). However, closing the VDR G-pocket via a I268Y-V300Y mutation squelches all VDR agonist activity (Table 1). This mutation closes the pocket by I268Y and V300Y forming an intramolecular H-bond in the steric space occupied by the side-chain of 1,25D in the hVDRwt G-pocket complex (Fig. 4A). For 1,23S,25R-lac, its hVDRwt EC50 value is ∼1 μM, consistent with previous results [22]. In addition, unlike 1,25D and KH, the H305F/ H397F construct significantly enhanced (3-4 fold) 1,23S,25R-lac induced reporter activity (EC50 = 263 ± 102 nM, Table 1; p < 0.05).
From a structural perspective (PSA), the ability of both KH and 1,23S,25R-lac to stabilize the closed hVDR H12 conformation (VDR-c1) was enhanced significantly by H305F/H397F mutation (compare Figs. 3C and 5A), but remained unchanged in the H305A single mutant (compare Figs. 3C, 5C and 5D). The H305F and H397F single mutants had differential effects on the observed hVDR-c1 population stabilized by 1,23S,25R-lac and KH relative to hVDRwt; the H305F single mutant enhanced the KH-hVDR-c1 population (Fig. 5C), while the H397F mutation did so for the lactone (Fig. 5D). The observation that the H397F mutation increased the lactones hVDR-c1 density does not correlate to a change in the ligands ability to stimulate VDR-mediated reporter expression (Table 1). It is noted that conversion of H397 to Phe (F) increases the vdW attractive forces between the 397-locus and F422, an important AF2-domain residue [20,30].
Fig. 5.

PSA analysis of 1,25D, KH and 1,23S,25R-lac in various hVDR-H305/H397 point mutants
(A) Representative 14% SDS-PAGE gel of 1,25D, 23S,25R-lac and KH-H305F/H397F complexes after subjection to trypsin treatment (ligand concentration 10−5 M). In all gels the hVDR-c1 fragment is labeled (c1) and the hVDR-c3 fragment (c3). (B) PSA dose response experiment (10−5-10−9 M) for KH incubated with hVDR-H397F or H305F/H397F point mutants. (C) PSA results obtained when 10−5, 10−7 and 10−9 M KH was incubated with the hVDR-H305F or H305A constructs. (D) PSA results obtained when the hVDR-H305A or H397F mutants is dosed with 1,23S,25R-lac (10−5-10−9 M). (E) PSA results obtained when the 1,23S,25R-lac was incubated with the VDR H305F and H305F/H397F constructs. (F) Comparison of ±1,25D (10−5M) hVDRwt and H305F/H397F apo/holo-PSA results. (G) hVDRwt dose PSA result obtained with the 25-dehydro-26,23(S)-lactone (MK, Fig. 1A) as the ligand. At high concentrations MK covalently modifies the VDR molecule (Mizwicki et al., submitted). This is highlighted by the dashed black line inside the black box. (H) hVDR H305F/H397F- MK PSA dose experiment. (I/J) 1,25D:KH or 23S,25R-lac RCI-type PSA results. This experiment was carried out by making and incubating 1,25D:KH mixtures with the VDR prior to the partial trypsin digest. (K) A representative hVDRwt PSA result obtained with 1,25D and a series of its C23 and C24 side-chain metabolites at 10−6 and 10−8 M.
Based on the unexpected enhancement of 1,23S,25R-lac agonist potency and the unaltered ability of 1,25D and KH to transactivate in the H305F/H397F double mutant relative to hVDRwt, it is hypothesized that the H305F/H397F results may be best explained by a VDR intramolecular effect. Along these lines we have previously proposed that the VDR molecule samples the closed conformation in the absence of ligand (e.g. the VDR conformational ensemble model [25]).
The H305F/H397F point mutant enhances the intramolecular stability of H12 and the VDR molecule
In the absence of ligand, the H305F/H397F construct is substantially more stable than apo-hVDRwt (Fig. 5F). Thus it is proposed the H305F/H397F mutation shifts the thermodynamic equilibrium of hVDR H12 conformers towards the active H12 conformation (Fig. 1C), thereby energetically compensating for the loss of both the H305 and H397 H-bonds. This is supported by the evidence from comparison of apo-hVDRwt and apo-H305F/H397F molecular models that indicate the aromatic F305/397 loci make additional non-bond contacts with the π-systems of Y401 and F422. In fact, when 1,25D is removed from the VDR G-pocket, and the potential interaction energy (PIE, see methods) between hVDR aa402-aa427 and the remainder of the complex is computed, the apo-H305F/H397F/aa402-427 PIE is stronger than the apo-hVDRwt/ aa402-427 PIE (−405.2 and −398.6 kcal/mol respectively). However, it is noted that in our CV1 cell transient transfection assay system that the H305F/H397F mutation does not produce a constitutively active VDR construct (data not shown).
This same type of intramolecular effect on VDR H12 stability can be used to explain why the (23S)-25-dehydro-1α(OH)-26,23-lactone analog (MK, Fig. 1A) of 1,25D is a potent genomic antagonist of 1,25D-hVDRwt mediated transactivation, but is a pure agonist/ partial agonist in the rodent VDR (rVDR) [31,32]. In the rVDR the 403 and 410-loci are Ser (S403) and Gln (N410) rather than C403 and C410 in the human VDR (hVDR). When the low energy molecular models of the 1,25D-hVDR and hrVDR (C403S/C410N) complexes are compared, it is clear the rVDR S403 and N410 R-groups form additional H-bonds not formed by the hVDR C403 and C410 thiols (Fig. 4C). Thus it is clear aa402-427 (H12) of the rVDR form a more stable intramolecular complex with the body of the VDR protein when compared to aa402-427 (H12) of hVDR.
Quite surprisingly both the H305F and H305F/H397F mutations turn MK from being an antagonist into a VDR superagonist (∼50 to 80-fold more potent than 1,25D and KH, Fig. 6A, Table 1). The evidence that MK is more potent than KH in the H305F/H397F mutant is supported by the PSA dose experiments that show MK is better able to protect the protein against proteolysis than KH (compare Figs. 5B, 5G and 5H). This is also true for H305F (Mizwicki et al., submitted). Importantly, this switching of MK to a superagonist is not mediated solely by an intramolecular effect (Mizwicki, et al., submitted); however, it is clear that mutations that destabilize hVDR-c1 (R402E and R158F; Figs. 1C, 2 and Table 1), enhance MK's antagonistic potency relative to hVDRwt (Fig. 6B). Collectively, these results support the hypothesis that the apo-VDR molecule should not be considered a rigid body, rather a conformational ensemble of energetically similar conformational isomers [33,34,35]. The results also demonstrate that the MK analog may be a powerful pharmacological tool if these VDR mutants can be used therapeutically.
Fig. 6.
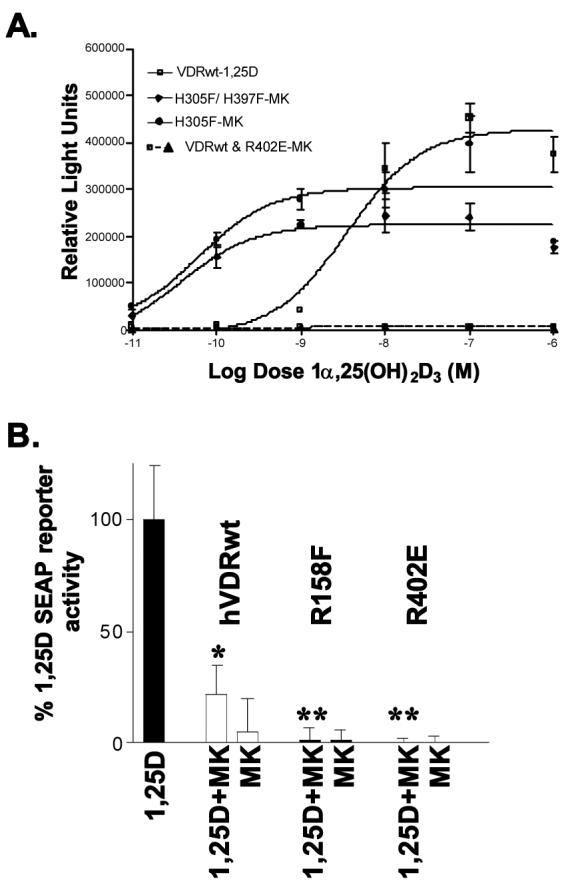
Conversion of MK into a superagonist or superantagonist
Representative MK dose-response curves for hVDRwt and single and double point mutants that turn MK into a significantly more potent agonist (H305F and H305F/H397F) or significantly more potent antagonist (R158F and R402E). All assays were performed in CV1 cells transiently transfected with the specified full length hVDR construct and an osteocalcin-VDRE driven SEAP-reporter (see methods). Effective concentrations (EC50) values for all mutants (assayed in triplicate; n ≥ 3) addressed in the text are summarized in table 1. (*) 10xMK significantly inhibits 1,25D induced reporter expression (p ≤ 0.05). (**) 10xMK antagonistic potency significantly greater relative to hVDRwt (p ≤ 0.05).
The presence of a VDR alternative pocket (A-pocket) can explain the PSA and functional profiles of KH, 1,23S,25R-lac and other 1,25D side-chain metabolites/ analogs
The KH and 1,23S,25R-lac-VDR structure-function results outlined above (Figs. 4, 5 and Table 1) indicate that both ligands do not induce a hVDRwt-c3 PSA fragment by sterically occluding H12 from sampling the closed conformation. Rather they both seem to have a lower, steady state, fractional occupancy [33] of the hVDR G-pocket (Fig. 1C), relative to 1,25D. This hypothesis would be consistent with the low VDR RCI values for both sterols. In addition, KH-VDR x-ray and in silico results suggest KH would be expected to have a much longer VDR half-life (t1/2) than 1,25D or the natural lactone, because it makes more attractive vdW interactions (non-bond contacts) with VDR G-pocket residues then the other two sterols (see Figs. 4A, 4B and Table 3), potentially explaining their different functional profiles (Table 1).
Table 3.
Calculated vitamin D sterol-VDR A- and G-pocket potential interaction energies
All values presented in this table were obtained by docking low energy side-chain conformations of the indicated vitamin D sterol generated using PC_Model v9.0 (see Fig. 7A and methods). All of the A- and G-pocket complexes tested showed total energy conservation over an 8 ns molecular dynamics simulation [26]. However, the potential interaction energy is an arbitrary value that only represents the non-bond potential energy between the ligand and the remainder of the assembly and is not an absolute value because the given sterols effective binding energy must be experimentally derived (see [33,53] for more information). However, the value does have meaning when models of constitutional isomers and/or conformational isomers are compared. The bold values represent the lowest energy complex obtained for sterols docked in the VDR G-pocket (see below). In assessing G-pocket potential stability all Pop. B side-chain conformers were docked
| Ligand | A-ring Chair/Orientation (a) | VDR Pocket (b) | PI E in kcal/ mol (c) |
|---|---|---|---|
| 1,25D | β/ trans | G | −107.9* |
| β/ trans | A | −92.7 | |
| α/ trans | A | −91.6 | |
| β/ cis | A | −93.7 | |
| KH (Gemini) | β/ trans | G | −130.3 |
| β/ trans | A | −106.7 | |
| α/ trans | A | −106.2 | |
| β/ between | A | −109.4 | |
| 1,23S,25R-lac | β/ trans | G | −109.7 |
| β/ trans | A | −100.7 | |
| α/ trans | A | −100.5 | |
| β/ cis | A | −104.9 | |
| 1,23R,25S-lac | β/ trans | G | −112.5 |
| β/ trans | A | −100.3 | |
| α/ trans | A | −100.0 | |
| β/ cis | A | −98.7 | |
| MK: (23S)-25- dehydro-26,23- lactone | β/ trans | G | −94.6 |
| β/ trans | A | ND | |
| α/ trans | A | −83.4 | |
| β/ cis | A | −88.6 | |
| 24-oxo-1,25D | β/ trans | G | −110.4 |
| β/ trans | A | −95.6 | |
| α/ trans | A | −95.1 | |
| β/ cis | A | −94.0 | |
| 1,24R,25D | β/ trans | G | −111.0 |
| β/ trans | A | −97.0 | |
| α/ trans | A | −98.1 | |
| β/ cis | A | −98.7 | |
| 1,24S,25D | β/ trans | G | −112.6 |
| β/ trans | A | −100.0 | |
| α/ trans | A | −98.9 | |
| β/ cis | A | −101.4 | |
| 1,23S,25D | β/ trans | G | −117.4 |
| β/ trans | A | −103.1** | |
| α/ trans | A | −102.8** | |
| β/ cis | A | −103.6** | |
| 1,23R,25D | β/ trans | G | −115.0 |
| β/ trans | A | −104.3** | |
| α/ trans | A | −103.3** | |
| β/ cis | A | −104.2** |
In order to begin testing the differential G-pocket fractional occupancy hypothesis, we designed a PSA experiment where RCI-like ratios of 1,25D:KH or 1,23S,25R-lac was incubated with hVDRwt and subjected to PSA. The results indicate that the switch between the predominant PSA fragment being hVDRwt-c1 and hVDRwt-c3 occurred at ratios between 1:4 and 1:8 for KH and 1:100 and 1:200 for 1,23S,25R-lac (Figs. 5I and 5J respectively), consistent with the measured KH and 1,23S,25R-lac RCI values (Fig. 1A). The result is also consistent with our previous exchange experiments demonstrating that KH had a slower exchange rate with [3H]-1,25D relative to 20-epi-1,25D, a well documented hVDRwt superagonist [18]. Based on these data it is posited that the reduced VDR G-pocket fractional occupancies of 1,23S,25R-lac and KH in the presence of equimolar 1,25D, is due to a mass action effect (e.g. reduction in free KH concentration available to the VDR G-pocket relative to equimolar 1,25D). This mass action effect can not be attributed to enhanced affinity of KH and 1,23S,25R-lac for the serum vitamin D binding protein (DBP), because both sterols have DBP RCI values below 5% (1,25D DBP RCI = 100%). Thus the only well characterized vitamin D sterol binding protein that remains a likely source of the mass action effect is the VDR molecule itself. To test this hypothesis all sterols were docked in the putative VDR alternative ligand binding pocket (A-pocket, Fig. 1C) [25,26,27,28].
Vitamin D sterols share the same A-ring domain when complexed to the VDR A- and G-pockets (Fig. 1C, black circle). We have previously demonstrated that the putative VDR A-pocket is capable of accepting/ binding more low energy side-chain, seco-B-ring and A-ring conformers of vitamin D sterols than the VDR G-pocket. In addition, ligand access to the R274 residue, the heart of both pockets, requires only minor movements in H2/ β-sheet R-groups to be accessed through the A-pocket portal, while large movements in H11/ H12 residues are required for ligand to access R274 through the G-pocket portal (Fig. 1C). These observations have led to the hypothesis that the putative VDR A-pocket represents a kinetically favored LBP and the G-pocket a thermodynamically favored LBP [25,26,27,28]. It is noted that the side-chain conformations of the sterols used in these modeling studies are generated and selected from side-chain dot maps [36,37] (Fig. 7A); therefore, no bias in the ligands starting side-chain orientation within either pocket is introduced if all conformers are tested.
When 1,25D, KH and 1,23S,25R-lac were docked in the VDR A-pocket it was observed that both sterols form additional vdW contacts not formed by 1,25D (Table 3 and [27]). All sterols form an H-bond with R158 when complexed to the putative A-pocket. When R158 is mutated to an E or F the A-pocket stability of both KH and 1,23S,25R-lac is reduced, but their G-pocket potential stabilities are not changed (data not shown). These in silico results are supported by the R158E and R158F PSA results (Fig. 3E) that indicate the R158 mutations reduce the hVDR-c3 density, but not the hVDR-c1 density relative to hVDRwt (Table 2). This parallel effect on PSA footprints was not observed with any other of the hVDRwt G-pocket mutants (see Fig. 5). In addition, the structural observation that the vitamin D sterol side-chain atoms do not directly contact H11/ H12 residues (e.g. AF2-domain residues) when bound to the putative A-pocket, suggests that ligands that show enhanced stability in the A-pocket relative to 1,25D would increase the exposure of R402 to trypsin, thereby enhancing the production of the hVDR-c3 PSA fragment. If this hypothesis is valid, then other vitamin D sterols that show increased VDR A-pocket potential stability, relative to 1,25D, should produce a higher density, hVDR-c3 PSA fragment.
Of the more than 30 sterols that have been docked in both the VDR G- and A-pockets, the 1,25D side-chain metabolites and KH all have enhanced A- and G-pocket potential interaction energies (PIE) when compared to 1,25D (Table 3). This was expected, because all the sterols are capable of forming more H-bonds than 1,25D in both VDR pockets, due to their chemistries. When PSA experiments were performed with 1,24S,25D3, 1,23R,25D3, 1,23S,25D3 and 1,24R,25D3 (Fig. 1A), all constitutional isomers showed enhancement in the hVDR-c3 band density with respect to 1,25D. The 23S and 24S-epimers show the greatest shift towards hVDR-c3 (Fig. 5K). It has been demonstrated by others, that of these constitutional isomers, only 1,24R,25D is as efficient a VDR transcriptional agonist as 1,25D [24,38,39]. These functional results are consistent with the PSA results which indicate that only 1,24R,25D is capable of effectively stabilizing a dense, 1,25D-like, hVDR-c1 band at 10−8 M (Fig. 5K). In addition, the enhanced hVDR G-pocket PIEs for the side-chain metabolites (Table 3) is generated purely from enhanced coulombic attractive forces, unlike the most potent genomic agonist KH (Table 1), whose enhanced G-pocket PIE is predominantly attributed to increased hydrophobic contacts. These fundamental differences in VDR G-pocket non-bond stabilizing forces provide a potential understanding for why KH is a 1,25D-like or better VDR transcriptional agonist in all assay systems, while the transactivational potency of 1,23S,25D, 1,24S,25D, and 1,23R,25D depends significantly on the cell-type and reporter system used [24,38,39].
The VDR A-pocket can be used to understand the lack of correlation between vitamin D sterol VDR RCI values and other structure-function results
The RCI values for 1,25D, 1,24S(OH)2D3 (1,24S-D), 1,24R(OH)2D3 (1,24R-D), 1,23S,25D, 1,23R,25D, 24-oxo-1,25D, 1,24S,25D, 1,24R,25D, 1,23R,25S-lac, 1,23S,25R-lac, KH, 19-nor-1,25D and 19-nor-KH are all less than 40%, with the exception of 1,24R-D and 24-oxo-1,25D (Fig. 1A). All these sterols have lower affinities for the serum vitamin D protein (DBP RCI < 100%) than 1,25D (data not shown). As stated previously, all sterols presented herein show similar to more stable VDR G-pocket complexes when compared to 1,25D (Table 3), with the exception of 1,25D's constitutional isomers (1,24R-D, not represented in Table 3). In addition, all the sterols have the same side-chain dot map profiles and/or population distributions (Fig. 7A). Yet three sets of epimers and many constitutional isomers have highly variable RCI values (Fig. 1A). Collectively the structure-function results suggest that these metabolites/ analogs would have higher VDR RCI values than 1,25D (e.g. out compete 1,25D). It is inferred from these results that no other known binding protein reduces the concentration of free sterol available to the VDR molecule with respect to [3H]-1,25D, and that the altered VDR RCI values are unlikely to be caused by altered side-chain conformational distribution or different solvation free energies of the sterols.
When 1,25D, 1,24R-D and 1,24S-D (constitutional isomers) were docked in the VDR G-pocket, 1,24S-D (RCI = 9%; PIE = −107.2 kcal/mol), but not 1,24R-D (RCI = 94%; PIE = −101.3 kcal/mol), formed an identical complex with the VDR G-pocket relative to 1,25D, a result seemingly inconsistent with a one pocket, induced-fit model. When these three sterols were docked in the putative VDR A-pocket, it was discovered that 1,24S-D formed an H-bond with R158, while 1,24R-D was unable to effectively contact R158, thereby reducing its vdW stability relative to 1,25D and 1,24S-D. Preliminary calculations indicate that the 1,24S-D H-bond with R158 is less susceptible to solvation than the 1,25D---R158 H-bond. These in silico results suggest that the 9% RCI value for 1,24S-D may be best explained by the kinetically favored A-pocket suppressing the concentration of free sterol available to the VDR G-pocket, relative to equimolar [3H]-1,25D. While the projected reduction in the VDR A-pocket fractional occupancy of 1,24R-D relative to [3H]-1,25D and 1,24S-D may explain how it can overcome its reduced VDR G-pocket PIE to yield a VDR RCI value of 94%. This same mass action argument has been used herein and elsewhere [26,27,28] to understand other vitamin D sterol-VDR structure-function conundrums.
Addition of a 25-OH group to both 1,24S-D (1,24S,25D) and 1,24R-D (1,24R,25D) further reduces the measured VDR RCI value (Fig. 1A), a result that is not consistent with the in silico VDR G-pocket complexes that indicate two side-chain hydroxyls enhance the attractive non-bond stabilizing forces formed between the side-chain and VDR G-pocket residues (Fig. 4D). When these two sterols were docked in the putative VDR A-pocket an increase in the electrostatic non-bond contacts made with A-pocket residues was observed relative to 1,24S-D, 1,24R-D and 1,25D (Table 3). Thus based on preliminary computations an inferred increased fractional occupancy of the VDR A-pocket by 1,24R/S,25D relative to 1,25D again may explain the reduction observed in their VDR affinities.
Interestingly, when the 24-OH group is oxidized to form 24-oxo-1,25D, the measured RCI value (98%) is nearly identical to 1,25D's (100%) and its PSA footprint is non-decipherable from 1,25D's (data not shown). When 24-oxo-1,25D was docked in the VDR A-pocket it was observed that the C24 carbonyl group did not alter the non-bond contacts when compared to 1,25D, as significantly as the other side-chain metabolites; therefore, of all the side-chain metabolites, 24-oxo-1,25D most closely mimics 1,25D's VDR binding and functional properties. Conversely, 1,24R,25D and 1,24S,25D both can have 1,25D-like transactivational potencies, but not a robust VDR affinity [24]. The in silico findings suggest the 1,24R,25D and 1,24S,25D functional properties are a product of their enhanced stabilities in the thermodynamically favored VDR G-pocket (e.g. reduced occupancy, but enhanced t1/2). Real-time kinetics experiments can be used to further test this hypothesis and are underway.
The C23-metabolite, 1,23S,25(OH)3D3, and the 1,23R,25(OH)3D3 synthetic analog are both constitutional isomers of the 1,24,25(OH)3D3 side-chain metabolites (Fig. 1A). A major difference between the C24-OH and C23-OH compounds is that the C23-OH sterols are 1,3-diols and therefore are capable of obtaining very similar side-chain conformations, where the steric space occupied by the terminal C26 and C27 methyls and the hydroxyl groups are nearly superimposible with 1,25D's (compare Figs. 4D and 4E). This may provide the principal reason C23-OH epimers have more similar VDR RCI (Fig. 1A) and PSA profiles (Fig. 5K) then the C24-OH epimers. When the C23-OH sterols were docked in the VDR G-pocket it was discovered that they both form more stable H-bonding networks with H305 and H397 than 1,25D or the C24-metabolites (Table 3, Fig. 4E). In addition, there is no significant change in the vdW contacts made with VDR AF2-domain residues. These in silico results (e.g. projected stability) are not consistent with the measured 10-fold reduction in VDR affinity (RCI value) for both 1,23S/R,25D (Fig. 1A). Again the reduced VDR RCI value may potentially be explained by the observation that both sterols form more non-bond contacts with VDR A-pocket residues than 1,25D or the C24-metabolites (Fig. 4F, Table 3). In addition, 1,23R/S,25D are able to form low energy, favorable, VDR A-pocket complexes when R158 maintains its orientation predicted by the 1,25D-VDR x-ray result [11] (Table 3) or when it moves into the position induced by 1,25D (Fig. 4F) and/or other vitamin D sterols when docked in the putative A-pocket [25,26,27,28].
Further oxidation of 1,23S,25D produces the (23S,25R)-1,25D-26,23-lactone (Fig. 1A). Its (23R,25S)-epimer has a measured RCI value that is over 20-fold better than the natural lactones (Fig. 1A). 1,23R,25S-lac is also a more potent VDR transcriptional agonist than the natural 1,23S,25R-lac, but not nearly as potent as 1,25D [22]. A distinct difference between the C23-lactones and other vitamin D sterol metabolites is the introduction of a C25 chiral center and lactonization essentially locks the orientation of all terminal side-chain atoms (Fig. 7A, table inset, total conformers); therefore, like the C24-OH metabolites, a distinct difference in the VDR RCI values is observed (Fig. 1A). When the lactones were docked in the VDR G- and A-pockets it was observed that the natural 1,23S,25R-lac formed a slightly more stable A-pocket complex than its synthetic epimer, while 1,23R,25S-lac formed a slightly more stable G-pocket complex than the natural epimer (Table 3). Thus the mass action effects projected from the low energy molecular models qualitatively support the VDR RCI and functional results.
Mechanistically understanding the 19-nor effect on vitamin D sterol VDR structure-function results
The removal of C19 (i.e. 19-nor modification) from 1,25D, KH and other vitamin D3 sterols reduces all their VDR RCI values approximately 5 to 10-fold (Fig. 1A and data not shown). Removal of C19 has no effect or increases (+)VDR-mediated cellular responses (e.g. differentiation/ anti-proliferation), while reducing the (−)VDR-mediated endocrine effect (e.g. induction of hypercalcemia) [40]. Removing C19 from 1,25D and KH does reduce the vdW attractive force between the ligand and VDR G-pocket residues by 0.61 ± 0.04 and 0.63 ± 0.07 kcal/mol respectively; therefore, a reduced vdW stability in the 19-norVDR complexes is observed, but it remains unclear if it and the hydrophobic effect together account for a 5 to 10-fold reduction in VDR affinity.
Dihedral driver experiments performed using 1,25D and 19-nor-1,25D demonstrate that the 19-nor modification reduces the rotational energy barrier about carbons 6 and 7 that must be overcome for the A-ring to transition between the α (below) and β-face (above) of the CD-ring [5]. We have previously demonstrated using in silico techniques and provided functional, but not structural validation, that the VDR A-pocket accepts at least three more A-ring/ seco-B-ring vitamin D sterol conformers than the VDR G-pocket [26,27,28]. Thus assuming the VDR A-pocket is kinetically favored, we propose a major variable associated with the 5 to 10-fold reduction in 19-nor VDR affinity can be attributed to the putative VDR A-pocket's ability to accept a higher population of 19-nor A-ring/ seco-B-ring rotomers relative to their 19-normal counterparts. On the other hand, the absence of alteration in exogenous and endogenous VDR-mediated gene activation by the 19-nor compounds, seems consistent with the <1% change in VDR G-pocket potential stability (data not shown, see recent x-ray results). Furthermore, any superagonist activity of 19-nor compounds and also the reduced hypercalcemic effect of some of these compounds, may in-part be a product of increased activation of non-genomic signaling imparted by vitamin D sterols through binding the VDR A-pocket/ molecule at or near the cell membrane. Nongenomic signals may ultimately cross-talk with or directly modulate the VDR and/or other gene regulators [25].
Addition of a 2α–CH3 to the vitamin D sterol significantly enhances VDR affinity and function
Addition of a methyl group at carbon-2 of 1,25D has been shown to have dramatic effects on the affinity and function of 1,25D and MK (Fig. 1A) [41,42]. For example the 2α-CH3 modification enhances 1,25D's VDR RCI value 4-fold (RCI = 400%), the potency of MK antagonism of 1,25D mediated gene transcription [42]; however, 2α-CH3-1,25D fails to stimulate apoptosis [43]. Conversely the 2β-CH3 modification reduces the VDR RCI value ∼10-fold (RCI = 13%) and MK's antagonistic potency [42], but it stimulates apoptosis [43]. Thus the effect the 2-methyl modification has on VDR affinity and function is not influenced by side-chain chemistry, or by metabolism of the side-chain through the C24-pathway [44]. According to VDR G-pocket docking experiments, the 2β-CH3 modification reduces the potential interaction energy by ∼0.3 kcal/mol, <1% relative to 1,25D, while the 2α-CH3 modification enhances the G-pocket PIE by approximately 1.8 kcal/mol (∼1% increase) [Fig. 4G]. The 2α-CH3-VDR G-pocket molecular model is in strong agreement with the recently published x-ray crystal structures and models that both demonstrate the axial 2α-CH3 forms vdW contacts or is proximal to F150, L233 and Y236 (Fig. 4G) [45]. Thus the differences in G-pocket stability qualitatively agree with the changes in the VDR RCI values and functional profiles; however, if presence of a VDR A-pocket is considered the effect of different VDR G-pocket stabilities would be amplified by a potential mass action effect (see below).
In silico results, suggest that a 2β-CH3 group increases the hydrophobic stability of the sterol in the VDR A-pocket relative to 1,25D, because the equatorial methyl group forms vdW contacts with S275 (3.85 Å) and R274 (3.55 Å) hydrophobic atoms (Fig. 4H). Conversely the axial 2α-CH3 does not allow the 6-_s_-cis or pre-vitamin conformations of 2α-CH3-1,25D to form a favorable complex with the VDR A-pocket (data not shown). In addition, in the 6-_s_-trans, β-chair, low energy conformation, the axial methyl is most proximal to the S237 hydroxyl (3.6 Å) and the R274 guanidinium (3.7 Å, Fig. 4H), both unfavorable non-bond interactions. Therefore it is proposed that the dramatically different structure-function profiles of the 2-CH3 epimers are best understood if the VDR A-pocket complexes are considered.
Potential importance of VDRA-pocket occupancy on G-pocket ligand accessibility
Recently we showed that in order for sterols to bind the putative A-pocket that an H-bond donor-acceptor exchange between H229 and Y295 is required and that this exchange is not possible under the hVDR x-ray pH of 6.5 [27]. When the H229 imidazole is charged, it forms an H-bond with the backbone carbonyl of R154 (Fig. 1C). The R154 residue was previously shown to be absolutely essential for optimal ligand-induction of VDR-mediated gene transcription (Fig. 2, Table 1). The importance of the H229-R154 intramolecular H-bond is verified by the H229Y transient transfection results that clearly demonstrate when the H229---R154 H-bond is fractured and the H229Y---Y295 H-bond is not, both 1,25D and KH are unable to induce VDRE dependent gene expression (Fig. 8A, Table 1). This functional result is consistent with the H229Y PSA results that indicate the mutation shifts the PSA footprint to hVDR-c3 whether KH or 1,25D is present (Fig. 8B).
Fig. 8.
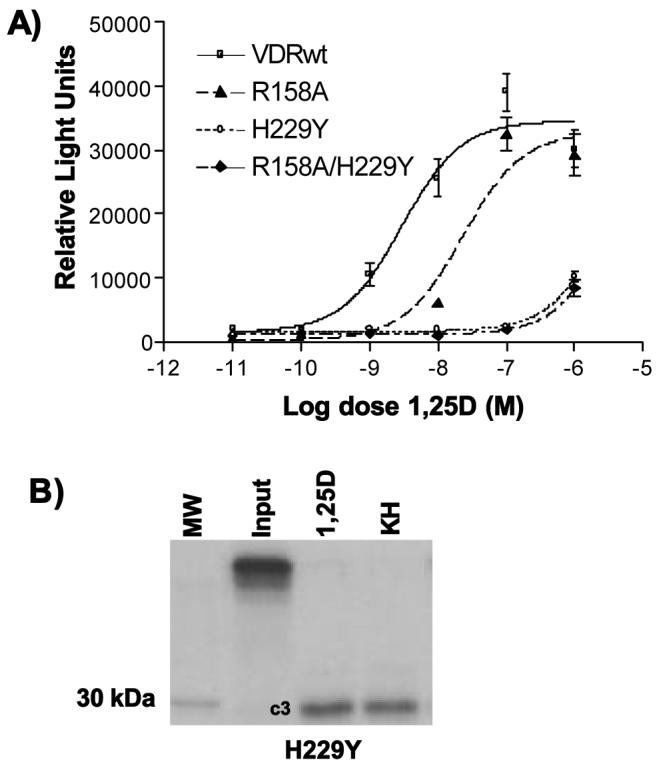
Importance of H229 locus in modulating A-pocket ligand accessibility and VDR potency
(A) CV1 cells were transiently transfected with the hVDRwt, R158A, H229Y or R158A/H229Y VDR construct. 1,25D dose response curves were generated using the osteocalcin-VDRE driven SEAP reporter system as previously described (see methods). (B) A partial trypsin digest experiment was performed by incubating H229Y with 10−5M 1,25D and KH.
Interestingly, upon A-pocket occupancy the H229-R154 H-bond and not the H-bond to Y295 is fractured [27], thus in theory mimicking the H229Y state (opened H12; hVDR-c3). This said, occupancy of the putative VDR A-pocket could alter R154's regiospecific localization and thereby serve in-part to modulate ligand accessibility to the VDR G-pocket via reducing the probability H12 will be clamped shut by the R154-D232/K413/L414 salt-bridge.
DISCUSSION
Over the past five years holo-VDR x-ray crystallographic results have not been able to provide a mechanistic understanding to inconsistencies between vitamin D sterol-VDR kinetics (RCI values), PSA footprints, functional assays (e.g. exogenous and endogenous VDR reporters) and/or non-genomic versus genomic cellular signaling; however they have definitively demonstrated that all VDR agonists stabilize the same thermodynamically favored VDR conformer (VDR-c1, Fig. 9). Like other nuclear receptors (NRs) with known ligands, the elementary controlling interaction governing VDR meditated genomic transactivation involves intermolecular interaction between the vitamin D sterol (e.g. 1,25D), and the VDR molecule. The current induced-fit mechanism used to describe NR-activation of gene transcription is based on comparison of apo/holo-NR x-ray crystallographic results [9]. It is inferred from such a model that the apo-NR molecule exists in a relatively rigid, opened conformation (Fig. 9, apo-VDR or c3) [10] where the C-terminal end of H11 must naturally move out of the way to expose the conserved H5 Arg residue (R274 of VDR, see Fig. 1C) and allow sterol to bind the G-pocket (Fig. 9, steps 1 and 2).
Fig. 9.

The putative vitamin D sterol (VDS)/ VDR conformational ensemble model
The traditionally accepted induced-fit mechanism (steps 1 & 2 of the thermodynamic cycle, blue arrows) occurs via a constructive collision between the apo-VDR molecule and 1,25D, producing a high energy intermediate (VDR-1,25D*) that rapidly transitions to the thermodynamically favored product. The black ribbon represents the position of helix-12 (H12) observed in various NR crystal structures and may represent the position of H12 that correlates with the representative PSA hVDRwt-c1, hVDRwt-c2 and hVDRwt-c3 apo/holo conformations. In the conformational ensemble model (steps 3 and 4, red arrows) the apo-VDR molecule is viewed as a Boltzman distribution of highly different, but energetically similar conformations. Therefore, the ligand serves to shift the Boltzman distribution of apo-VDR conformers, thus altering the affinity of the holo-VDR complex for other protein molecules with respect to the apostate. Collectively, these two mechanisms are identical from a thermodynamic perspective, but they differ in the following ways: 1) the conformational dynamics of the ligand are considered consistent with the mutual induced-fit model [48] (e.g. known importance of different 1,25D shapes); 2) the apo-VDR is envisioned to undergo rapid, reversible isomerization between a finite number of different, energetically similar conformations; 3) vitamin D sterols are proposed to initially prefer binding the kinetically favored VDR alternative ligand binding pocket (A-pocket, orange and white transparent Connolly surface) prior to accessing the G-pocket (black and white transparent Connolly surface), defined by x-ray crystallography; 4) what is perceived to be catabolism of the 1,25D sterol could be anabolic given all metabolites studied thus far show enhanced A-pocket non-bonding interactions relative to 1,25D; and 5) vitamin D sterol-VDR structure-function becomes more sensitive to mass action (i.e. a statistical model).
Recently, an alternative activation model has been proposed where the apo-VDR molecule is considered an ensemble of energetically similar conformations (Fig. 9, step 3). The fundamental difference between the induced-fit and conformational ensemble model is that the conformationally flexible vitamin D sterol serves to stabilize pre-existing helix-12 (H12) or apo-VDR conformers. Said differently, ligands do not induce a conformation change, rather they serve to shift the Boltzmann distribution of a pre-existing equilibrium of VDR conformers (Fig. 9) [25,26,27,28].
The first part of our study focused on variables other than the vitamin D sterol that influence H12 allostery. The majority of cellular signaling cascades modulated at least in-part by the VDR molecule require presence of the C-terminal activation helix (H12) and/or unique positioning of H12 (Fig. 9, c1-c3) [25]. Thus the findings that R402 (C-terminus of H11, is exposed when H12 is opened, apo-VDR or c3, Fig. 9) and K413 (H12, is posited to be exposed when H12 is partially closed, c2, Fig. 9) are apo- and holo-VDR trypsin sites not only supports the ensemble hypothesis, but also demonstrates that proteolysis could generate a dynamic pool of VDR isoforms in a cell type or tissue and species specific manner. Furthermore, unlike any other NR that requires hormone to be an active transcription factor, H12 (AF2-domain) of the VDR is stabilized in the closed conformation (Fig. 9; VDR-c1) by three salt-bridges that span the entire length of the activation helix. The most important of these salt-bridges is formed by R154, an H2-loop residue (Fig. 9) located in the hinge region of the VDR molecule (Fig. 1B). Ironically, the VDR trypsin site with highest affinity is the R173 locus. Cleavage here not only removes the DNA binding domain (Fig. 1B), but also removes the R154 locus; therefore, the observations that higher order species have a R173 site and all species have a R154 locus might indicate that VDR isoforms have physiological and/or species specific function. It is noted that all known full length and truncated VDR isoforms bind 1,25D. Collectively these results provide novel insight into VDR-H12 allostery (Fig. 9) and suggest western analyses must be carefully designed/ interpreted if VDR functional isomers lacking the DNA binding domain do exist [29].
In further support of the VDR-H12 conformational ensemble, we showed herein that the stability and population distribution of the apo-VDR molecule can be dramatically altered by mutation of the VDR (e.g. H305F/H397F). Furthermore, alterations in the intramolecular stability of H12 were shown to be consistent with the species specific antagonism of 1,25D genomic activities by its 25-dehydro-23S,26-lactone analog (MK, Fig. 1A). In addition, it was demonstrated that MK could be turned into a VDR superagonist or superantagonist by VDR mutations that enhance (e.g. H305F/H397F) or reduce (e.g. R402E) respectively the stability of hVDR-c1. Importantly, MK and its synthetic derivatives have been shown to have therapeutic promise in treating Paget's disease [31]. These findings would suggest that MK and its derivatives may have additional pharmacological usefulness if VDR mutants can be used therapeutically.
Finally, the concept that all apo-NR molecules exist as a Boltzmann distribution of energetically similar conformations is supported by NR-crystallographic results that demonstrate some apo-NR and apo-orphan NR crystal structures (pdb ID's: 3prg, 1ilg, 1pk5) harbor a closed, holo-like, H12 orientation. In addition, apo-PPARγ NMR results indicate no single unliganded conformation exists in solution [46]. Furthermore, orphan NRs as well as single point mutations made in the estrogen receptor (ER), a NR that ‘requires ligand’ for activation, show high levels of basal and/or constitutive activity [10,47]. Collectively the results therefore suggest that mechanistically a conformational ensemble model may be useful in understanding NR/ VDR signaling.
The second half of the manuscript applied two additional levels of complexity to the proposed VDR ensemble model a) the overlapping A-ring domain of the VDR G- and A-pockets (Fig. 9, large ribbon diagram) and b) the unique conformational flexibility of the vitamin D sterol [5]. The vitamin d sterol-VDR conformational ensemble model proposed herein is itself an extension of the mutual induced-fit model first proposed by Okamura et al. [48]. Both models are founded on the conformational mobility of the vitamin D sterol being able to selectively activate VDR dependent genomic and non-genomic signaling cascades [25,26,27,28]. Undoubtedly, the most important variable controlling the Boltzmann distribution of VDR ensemble members is the pool of vitamin D sterols available at any given time to the VDR molecule (Fig. 9). We demonstrated in this study that the presence of a putative VDR alternative ligand binding pocket (A-pocket) provides the first molecular model that is consistent with sterol specific and/or uniform alterations in the most elementary in vitro vitamin D sterol/ VDR structure-function studies [RCI (VDR affinity), PSA (VDR conformations) and CV1 cell transient transfection (VDR function)]. In specific, we were able to use a two pocket model to explain how KH and 1,23S,25R-lac stabilize the same VDR conformation (VDR-c3, see Fig. 3C), but have dramatically different functional profiles (see Figs. 1A, 3, 4A/B, 5C/D/E). In addition, the low affinity of KH and 1,23S,25R-lac for the VDR and their reduced G-pocket fractional occupancies in the presence of 1,25D was confirmed by RCI-like PSA experiments (see Fig. 5I/J).
Also, we were able to provide a proposed mechanism consistent with alterations in the VDR structure-function results obtained with vitamin D sterol epimers and/or constitutional isomers. In this study we demonstrated that all C24 and C23 side-chain metabolites of 1,25D, with the exception of 24-oxo-1,25D (Fig. 1A), showed significantly enhanced non-bond (vdW and Coulombic attractive forces) stability in the VDR A-pocket relative to 1,25D (see Table 3). While further calculations are still required to substantiate these preliminary in silico findings (see Table 3 legend), the projected increase in VDR A-pocket fractional occupancy of all side-chain metabolites correlates well with their reduced ability to compete with [3H]-1,25D (i.e. low RCI values) and increased presence of the VDR-c3 PSA fragment (i.e. enhanced exposure of R402). Thus the in silico results suggest the pool of vitamin D metabolites present in different assay systems should be considered when assessing differential VDR-mediated cellular activity. For example, it has already been definitively demonstrated that metabolism of many superagonists is either slowed dramatically and/or the superagonists selectively undergo C24-metabolism, stopping at the 24-OH compound. All these superagonist metabolites maintain strong VDR agonist profiles [23] and therefore the parent compound and its metabolites collectively are believed to generate the superagonist response/ profile.
According to our models and structure-function studies, the C24-selectivity would also contribute to the superagonist profile by reducing VDR A-pocket fractional occupancy of the collective pool of superagonist sterols relative to the pool of 1,25D metabolites. This is because C23-metabolites (1,25D only) were shown to have a higher A-pocket fractional occupancy than C24-metabolites (see Table 3). In fact, the enhanced A-pocket selectivity of the natural 1,23S,25R-lactone metabolite may provide a mechanistic understanding underlying its ability to block the 1,25D-induced increase in serum Ca+2 in vivo [49], while still promoting bone formation like 1,25D [50]. Thus there is a potential the model may provide an elementary mechanistic tool that is capable of separating the attractive therapeutic properties of vitamin D sterols (e.g. anti-proliferative and pro-differentiation stimulant) from their major pharmacological drawback (e.g. induction of hypercalcemia).
In closing, it is very well understood that reversible binding and differential recruitment of the cell specific spectrum of NCoA and NCoR molecules to the hydrophobic cleft of the VDR molecule (Fig. 1C) is a variable that dictates the efficacy of vitamin D sterol mediated expression of genes containing vitamin D response elements (VDREs) [51]. In these studies alterations in the conformational dynamics of the sterols, their often significantly reduced RCI values, and potential alterations in the elementary ligand-receptor interactions that trigger the genomic response are often overlooked. This is largely due to the assumption that VDR activation is governed by an induced-fit mechanism. It was demonstrated herein that the greater majority of vitamin D sterol-VDR structure-function conundrums can be potentially solved by presence of a kinetically favored alternative ligand binding pocket (A-pocket) and conformationally dynamic, rather than rigid apo-VDR molecule. In specific, the in silico results indicate the putative VDR A-pocket, vitamin D sterol fractional occupancy, is enhanced by reducing side-chain conformational dynamics (i.e. entropy), introduction of side-chain hydroxyl groups at C23 and C24-OH (S chirality preferred), reducing the rotational energy barrier about C6-C7 (e.g. 19-nor modification) and/or adding a 2β-CH3 group to 1,25D. Thus, it is posited the temporal order of recruitment of complexes and control of the VDR-mediated genomic and non-genomic signaling is precisely controlled by the pool of vitamin D sterol allosteric VDR regulators, which serve to alter the Boltzman distributions of VDR conformers/ isoforms.
The model presented herein could be relevant to other NR's, because they could also harness alternative binding sites. In addition, the intramolecular non-bonds that are formed between H12 and domain E (LBD) residues, are known to differ across the NR family (see NR pdb files); therefore, differences in the apo-H12 ensemble equilibrium could be a major factor in providing signaling specificity. For example, depending on the receptor, its environment (e.g. cellular localization or content and concentrations of co-regulatory molecules), and ligand-dependent or independent post-translational modifications, different concentrations of the ligand pools may be required to stabilize the ensemble threshold required to trigger or induce a given intracellular signaling cascade or global cellular event. Finally, while the model presented seems consistent with stereo and shape specific vitamin D sterol-VDR structure-function results and an overlapping two pocket allosteric model [52], it remains a hypothetical model until true kinetic and/or structural validation is obtained to further substantiate presence of a VDR A-pocket.
Acknowledgments
None of this work would have been possible without the work of Natasha Rochel, Fabrice Ciesielski and the rest of Dino Moras's laboratory. Supported in-part by USPHS grant DK-09012-39 to A.W. Norman.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Bouillon R, Okamura WH, Norman AW. Structure-function relationships in the vitamin D endocrine system. Endocr. Rev. 1995;16(2):200–257. doi: 10.1210/edrv-16-2-200. [DOI] [PubMed] [Google Scholar]
- 2.Sutton AL, MacDonald PN. Vitamin D: More Than a ‘Bone-a-Fide’ Hormone. Mol. Endocrinol. 2003;17:777–791. doi: 10.1210/me.2002-0363. [DOI] [PubMed] [Google Scholar]
- 3.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: The second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81(3):1269–1304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 5.Mizwicki MT, Norman AW. Two key proteins of the vitamin D endocrine system come into crystal clear focus: Comparison of the X-ray structures of the nuclear receptor for 1α,25(OH)2-vitmin D3, the plasma vitamin D binding protein, and their ligands. J Bone Miner Res. 2003;18(5):795–806. doi: 10.1359/jbmr.2003.18.5.795. [DOI] [PubMed] [Google Scholar]
- 6.Feng WJ, Ribeiro RC, Wagner RL, Nguyen H, Apriletti JW, Fletterick RJ, Baxter JD, Kushner PJ, West BL. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280(5370):1747–1749. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- 7.Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: Structure and function. Curr. Opin. Cell Biol. 1998;10(3):384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 8.Marimuthu A, Feng W, Tagami T, Nguyen H, Jameson JL, Fletterick RJ, Baxter JD, West BL. TR surfaces and conformations required to bind nuclear receptor corepressor. Mol. Endocrinol. 2002;16(2):271–286. doi: 10.1210/mend.16.2.0777. [DOI] [PubMed] [Google Scholar]
- 9.Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004;3(11):950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 10.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: Evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36(48):14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 11.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol. Cell. 2000;5(1):173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 12.Ciesielski F, Rochel N, Mitschler A, Kouzmenko A, Moras D. Structural investigation of the ligand binding domain of the zebrafish VDR in complexes with 1α,25(OH)2D3 and Gemini: purification, crystallization and preliminary X-ray diffraction analysis. J. Steroid Biochem Mol. Biol. 2004;89-90:55–59. doi: 10.1016/j.jsbmb.2004.03.109. [DOI] [PubMed] [Google Scholar]
- 13.Eelen G, Verlinden L, Rochel N, Claessens F, De CP, Vandewalle M, Tocchini-Valentini G, Moras D, Bouillon R, Verstuyf A. Superagonistic action of 14-epi-analogs of 1,25-dihydroxyvitamin D explained by vitamin D receptor-coactivator interaction. Mol. Pharmacol. 2005;67(5):1566–1573. doi: 10.1124/mol.104.008730. [DOI] [PubMed] [Google Scholar]
- 14.Tocchini-Valentini G, Rochel N, Wurtz JM, Mitschler A, Moras D. Crystal structures of the vitamin D receptor complexed to superagonist 20-epi ligands. Proc. Natl. Acad. Sci. USA. 2001;98(10):5491–5496. doi: 10.1073/pnas.091018698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tocchini-Valentini G, Rochel N, Wurtz JM, Moras D. Crystal structures of the vitamin D nuclear receptor liganded with the vitamin D side chain analogues calcipotriol and seocalcitol, receptor agonists of clinical importance. Insights into a structural basis for the switching of calcipotriol to a receptor antagonist by further side chain modification. J. Med. Chem. 2004;47(8):1956–1961. doi: 10.1021/jm0310582. [DOI] [PubMed] [Google Scholar]
- 16.Nakajima S, Yamagata M, Sakai N, Ozono K. Characterization of the activation function-2 domain of the human 1,25-dihydroxyvitamin D3 receptor. Molecular and Cellular Endocrinology. 1998;139(12):15–24. doi: 10.1016/s0303-7207(98)00077-x. [DOI] [PubMed] [Google Scholar]
- 17.Peleg S, Sastry M, Collins ED, Bishop JE, Norman AW. Distinct conformational changes induced by 20-epi analogues of 1α,25-dihydroxyvitamin D3 are associated with enhanced activation of the vitamin D receptor. J. Biol. Chem. 1995;270(18):10551–10558. doi: 10.1074/jbc.270.18.10551. [DOI] [PubMed] [Google Scholar]
- 18.Norman AW, Manchand PS, Uskokovic MR, Okamura WH, Takeuchi JA, Bishop JE, Hisatake JI, Koeffler HP, Peleg S. Characterization of a novel analogue of 1α,25(OH)2-vitamin D3 with two side chains: Interaction with its nuclear receptor and cellular actions. J Med. Chem. 2000;43(14):2719–2730. doi: 10.1021/jm0000160. [DOI] [PubMed] [Google Scholar]
- 19.Siu-Caldera M-L, Sekimoto H, Peleg S, Nguyen C, Kissmeyer A-M, Binderup L, Weiskopf A, Vouros P, Uskokovic M, Reddy GS. Enhanced biological activity of 1α,25-dihydroxy-20-epi-vitamin D3, the C-20 epimer of 1α,25-dihydroxyvitamin D3, is in part due to its metabolism into stable intermediary metabolites with significant biological activity. J. Steroid Biochem. Mol. Biol. 1999;71:111–121. doi: 10.1016/s0960-0760(99)00130-2. [DOI] [PubMed] [Google Scholar]
- 20.Carlberg C. Molecular basis of the selective activity of vitamin D analogues. J. Cell Biochem. 2003;88(2):274–281. doi: 10.1002/jcb.10337. [DOI] [PubMed] [Google Scholar]
- 21.Vaisanen S, Juntunen K, Itkonen A, Vihko P, Maenpaa PH. Conformational studies of human vitamin D receptor by antipeptide antibodies, partial proteolytic digestion and ligand binding. Eur. J. Biochem. 1997;248(1):156–162. doi: 10.1111/j.1432-1033.1997.t01-1-00156.x. [DOI] [PubMed] [Google Scholar]
- 22.Ishizuka S, Norman AW. The difference of biological activity among four diastereoisomers of 1α,25-dihydroxy-cholecalciferol-26,23-lactone. J. Steroid Biochem. 1986;25(4):505–510. doi: 10.1016/0022-4731(86)90395-x. [DOI] [PubMed] [Google Scholar]
- 23.Uskokovic MR, Norman AW, Manchand PS, Studzinski GP, Campbell MJ, Koeffler HP, Takeuchi A, Siu-Caldera M, Rao DS, Reddy GS. Highly active analogs of 1α,25-dihydroxyvitamin D3 that resist metabolism through C-24 oxidation and C-3 epimerization pathways. Steroids. 2001;66(35):463–471. doi: 10.1016/s0039-128x(00)00226-9. [DOI] [PubMed] [Google Scholar]
- 24.Harant H, Spinner D, Reddy GS, Lindley IJ. Natural metabolites of 1α,25-dihydroxyvitamin D3 retain biologic activity mediated through the vitamin D receptor. J Cell Biochem. 2000;78(1):112–120. [PubMed] [Google Scholar]
- 25.Norman AW, Mizwicki MT, Norman DPG. Steroid hormone rapid actions, membrane receptors and a conformational ensemble model. Nature Reviews Drug Discovery. 2004;3:27–41. doi: 10.1038/nrd1283. [DOI] [PubMed] [Google Scholar]
- 26.Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM, Moras D, Norman AW. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1α,25(OH)2-vitamin D3 signaling. Proc. Natl. Acad. Sci. USA. 2004;101(35):12876–12881. doi: 10.1073/pnas.0403606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizwicki MT, Bishop JE, Norman AW. Applications of the vitamin D sterol-Vitmain D receptor (VDR) conformational ensemble model. Steroids. 2005;70(57):464–471. doi: 10.1016/j.steroids.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Mizwicki MT, Bula CM, Bishop JE, Norman AW. A perspective on how the Vitamin D sterol/Vitamin D receptor (VDR) conformational ensemble model can potentially be used to understand the structure-function results of A-ring modified Vitamin D sterols. J. Steroid Biochem. Mol. Biol. 2005;97(12):69–82. doi: 10.1016/j.jsbmb.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 29.Bula CM, Huhtakangas JA, Bishop JE, Norman AW, Henry HL. Presence of a truncated form of the vitamin D receptor in VDR KO mice. Endocrinology. 2005;146(12):5581–5586. doi: 10.1210/en.2005-0806. [DOI] [PubMed] [Google Scholar]
- 30.Molnar F, Perakyla M, Carlberg C. Vitamin D receptor agonists specifically modulate the volume of the ligand-binding pocket. J. Biol. Chem. 2006;281(15):10516–10526. doi: 10.1074/jbc.M513609200. [DOI] [PubMed] [Google Scholar]
- 31.Ochiai E, Miura D, Eguchi H, Ohara S, Takenouchi K, Azuma Y, Kamimura T, Norman AW, Ishizuka S. Molecular Mechanism of the Vitamin D Antagonistic Actions of TEI-9647 Depends on the Primary Structure of the Carboxyl Terminal Region of the Vitamin D Receptor. Mol. Endocrinol. 2005;19(5):1147–1157. doi: 10.1210/me.2004-0234. [DOI] [PubMed] [Google Scholar]
- 32.Perakyla M, Molnar F, Carlberg C. A structural basis for the species-specific antagonism of 26,23-lactones on vitamin D signaling. Chem. Biol. 2004;11(8):1147–1156. doi: 10.1016/j.chembiol.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 33.Gutfreund H. Factors affecting rates of reactions. In: Gutfreund H, editor. Kinetics of the life sciences: receptors, transmitters and cataylsts. Cambridge Univ. Press; Cambridge: 1995. pp. 231–275. [Google Scholar]
- 34.Karplus M, McCammon JA. The dynamics of proteins. Sci. Am. 1986;254(4):42–51. doi: 10.1038/scientificamerican0486-42. [DOI] [PubMed] [Google Scholar]
- 35.Kenakin T, Onaran O. The ligand paradox between affinity and efficacy: can you be there and not make a difference? Trends Pharmacol. Sci. 2002;23(6):275–280. doi: 10.1016/s0165-6147(02)02036-9. [DOI] [PubMed] [Google Scholar]
- 36.Midland MM, Plumet J, Okamura WH. Effect of C20 stereochemistry on the conformational profile of the side chains of vitamin D analogs. Bioorg. Med. Chem. Lett. 1993;3:1799–1804. [Google Scholar]
- 37.Yamada S, Yamamoto K, Masuno H, Ohta M. Conformation-function relationship of vitamin D: Conformational analysis predicts potential side-chain structure. Journal of Medicinal Chemistry. 1998;41(9):1467–1475. doi: 10.1021/jm970761l. [DOI] [PubMed] [Google Scholar]
- 38.Sunita RD, Campbell MJ, Koeffler HP, Ishizuka S, Uskokovic MR, Spagnuolo P, Reddy GS. Metabolism of 1α,25-dihydroxyvitamin D3 in human promyelocytic leukemia (HL-60) cells: In vitro biological activities of the natural metabolites of 1α,25-dihydroxyvitamin D3 produced in HL-60 cells. Steroids. 2001;66(35):423–431. doi: 10.1016/s0039-128x(00)00230-0. [DOI] [PubMed] [Google Scholar]
- 39.Ishizuka S, Sumitani K, Hiura K, Kawata T, Okawa M, Hakeda Y, Kumegawa M. Biological activity assessment of 1α,25-dihydroxyvitamin D3-26,23-lactone and its intermediate metabolites in vivo and in vitro. Endocrinology. 1990;127:695–701. doi: 10.1210/endo-127-2-695. [DOI] [PubMed] [Google Scholar]
- 40.Olivera CJ, Bula CM, Bishop JE, Adorini L, Manchand P, Uskokovic MR, Norman AW. Characterization of five 19-nor-analogs of 1α,25(OH)2-vitamin D3 with 20-cyclopropyl-modified side-chains: implications for ligand binding and calcemic properties. J. Steroid Biochem Mol. Biol. 2004;89-90:99–106. doi: 10.1016/j.jsbmb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Konno K, Fujishima T, Maki S, Liu Z, Miura D, Chokki M, Ishizuka S, Yamaguchi K, Kan Y, Kurihara M, Miyata N, Smith C, DeLuca HF, Takayama H. Synthesis, biological evaluation, and conformational analysis of A-ring diastereomers of 2-methyl-1α,25-dihydroxyvitamin D3 and their 20- epimers: unique activity profiles depending on the stereochemistry of the A-ring and at C-20. J. Med. Chem. 2000;43(22):4247–4265. doi: 10.1021/jm000261j. [DOI] [PubMed] [Google Scholar]
- 42.Saito N, Matsunaga T, Fujishima T, Anzai M, Saito H, Takenouchi K, Miura D, Ishizuka S, Takayama H, Kittaka A. Remarkable effect of 2α-modification on the VDR antagonistic activity of 1α-hydroxyvitamin D-26,23-lactones. Org. Biomol. Chem. 2003;1(24):4396–4402. doi: 10.1039/b311107e. [DOI] [PubMed] [Google Scholar]
- 43.Konno K, Maki S, Fujishima T, Liu ZP, Miura D, Chokki M, Takayama H. A novel and practical route to A-ring enyne synthon for 1α,25-dihydroxyvitamin D3 analogs: Synthesis of A-ring diastereomers of 1α,25-dihydroxyvitamin D3 and 2-methyl-1,25-dihydroxyvitamin D3. Bioorg. Med. Chem. Lett. 1998;8(2):151–156. doi: 10.1016/s0960-894x(97)10204-9. [DOI] [PubMed] [Google Scholar]
- 44.Sunita RD, Siu-Caldera ML, Sekimoto H, Gennaro L, Vouros P, Takayama H, Konno K, Fujishima T, Reddy GS. Metabolism of 2-methyl analogs of 1α,25-dihydroxyvitamin D3 in rat osteosarcoma cells (UMR 106) Biol. Pharm. Bull. 2002;25(7):845–852. doi: 10.1248/bpb.25.845. [DOI] [PubMed] [Google Scholar]
- 45.Suhara Y, Nihei KI, Kurihara M, Kittaka A, Yamaguchi K, Fujishima T, Konno K, Miyata N, Takayama H. Efficient and versatile synthesis of novel 2α -substituted 1α,25- dihydroxyvitamin D3 analogues and their docking to vitamin D receptors. J Org. Chem. 2001;66(26):8760–8771. doi: 10.1021/jo010375i. [DOI] [PubMed] [Google Scholar]
- 46.Johnson BA, Wilson EM, Li Y, Moller DE, Smith RG, Zhou G. Ligand-induced stabilization of PPARγ monitored by NMR spectroscopy: Implications for nuclear receptor activation. J. Mol. Biol. 2000;298(2):187–194. doi: 10.1006/jmbi.2000.3636. [DOI] [PubMed] [Google Scholar]
- 47.Andersin T, Vaisanen S, Carlberg C. The critical role of carboxy-terminal amino acids in ligand-dependent and -independent transactivation of the constitutive androstane receptor. Mol. Endocrinol. 2003;17(2):234–246. doi: 10.1210/me.2002-0263. [DOI] [PubMed] [Google Scholar]
- 48.Okamura WH, Midland MM, Hill DK, Ringe K, Takeuchi JA, Vassar VC, Vu TH, Zhu G-D. Towards understanding the ‘mutually induced fit’ of vitamin D ligands and various proteins which bind metabolites and analogs. In: Norman AW, Bouillon R, Thomasset M, editors. Vitamin D, gene regulation, structure-function analysis and clinical application. 1997. pp. 11–18. [Google Scholar]
- 49.Ishizuka S, Ishimoto S, Norman AW. Biological activity assessment of 1α,25-dihydroxyvitamin D3-26, 23-lactone in the rat. J. Steroid Biochem. 1984;20:611–615. doi: 10.1016/0022-4731(84)90131-6. [DOI] [PubMed] [Google Scholar]
- 50.Seino Y, Ishizuka S. 23(S), 25(R)-1,25-dihydroxyvitamin D3-26,23-lactone in bone formation. In: Norman AW, Bouillon R, Thomasset M, editors. Vitamin D, gene regulation, structure-function analysis and clinical application. 1991. pp. 565–571. [Google Scholar]
- 51.Kahlen JP, Carlberg C. Allosteric interaction of the 1α,25-dihydroxyvitamin D3 receptor and the retinoid X receptor on DNA. Nucleic Acids Res. 1997;25(21):4307–4313. doi: 10.1093/nar/25.21.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308(5727):1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 53.Lazaridis T, Masunov A, Gandolfo F. Contributions to the binding free energy of ligands to avidin and streptavidin. Proteins. 2002;47(2):194–208. doi: 10.1002/prot.10086. [DOI] [PubMed] [Google Scholar]
- 54.Verboven C, Rabijns A, De Maeyer M, Van Baelen H, Bouillon R, De Ranter C. A structural basis for the unique binding features of the human vitamin D-binding protein. Nat. Struct. Biol. 2;2002;9:131–136. doi: 10.1038/nsb754. [DOI] [PubMed] [Google Scholar]