VEGFR1 (Flt-1+/−) GENE KNOCKOUT LEADS TO THE DISRUPTION OF VEGF MEDIATED SIGNALING THROUGH NITRIC OXIDE/HEMEOXYGENASE PATHWAY IN ISCHEMIC PRECONDITIONED MYOCARDIUM (original) (raw)
. Author manuscript; available in PMC: 2008 May 15.
Abstract
This report demonstrated that mice deficient in Flt-1 failed to establish ischemic preconditioned (PC) mediated cardioprotection in isolated working buffer-perfused ischemic reperfused (IR) hearts compared to wild type (WT) subjected to same PC protocol. Wild type (WT) and Flt-1+/− mice were divided into four groups: (1) WT IR (2) WT+PC (3) Flt-1+/− IR (4) Flt-1+/−+PC. Groups 1 and 3 mice were subjected to 30 min of ischemia followed by 2 hrs of reperfusion and groups 2 and 4 mice were subjected to four episodes of 4-min global ischemia followed by 6 min of reperfusion before ischemia reperfusion. For both wild type and Flt-1+/− mice, the postischemic functional recovery for the hearts was lower than the baseline, but the recovery for the knockout was less as compared to the WT mice even in preconditioning. The myocardial infarction and apoptosis were higher in Flt-1+/− compared to wild type I/R. Pronounced inhibition was observed in Flt-1+/− for p-AKT, p-eNOS, iNOS and most importantly HO-1 mRNA expression. Results demonstrates that Flt-1+/− mouse hearts are more susceptible to ischemia reperfusion injury and also documents preconditioning is not as effective as found in WT and therefore suggests the importance of VEGF/Flt-1 signaling in ischemic reperfused myocardium.
Keywords: Angiogenesis, Flt-1, nitric oxide, HO-1, myocardium, ischemia, reperfusion
INTRODUCTION
The essential role of vascular endothelial growth factor (VEGF) in angiogenesis and health and disease makes it attractive both as a therapeutic target for anti-angiogenic drugs and as a pro-angiogenic growth factor for the treatment of ischemic heart disease. Recent evidence suggests that VEGF may, in addition to promote angiogenesis, modulate various aspects of endothelial function and repair, leading to cardioprotection (1–5). Two receptors specific for endothelial tyrosine kinases have been identified for VEGF, Flt-1 or VEGFR1 and Flk-1/KDR or VEGFR2 which share 44% amino acid homology (6, 7). These two prime endothelial specific receptors are found to be important for VEGF mediated signaling in various systems. Some reports demonstrated that targeted disruption of Flt-1 and Flk-1 in mice prevents normal vascularization and embryonic development, however the two knockouts (Flk & Flt) have distinctive phenotypes (8, 9). Flk-1-deficient mice produce neither differentiated endothelial cells nor organized blood vessels whereas Flt-1 knock-out mice possesses mature, differentiated endothelial cells, but have large, disorganized vessels. Studies of Flt-1 signaling have yielded contrasting results. A soluble form of the extracellular Flt-1 domain occurs naturally and overexpression of this form inhibits VEGF-induced migration and proliferation of human microvascular endothelial cells and HUVECs by forming an inactive complex with VEGF and the full-length Flk-1 (10–13). Our recent study indicated reduced beneficial effects of ischemic preconditioning (PC) in Flt-1 heterozygous knockout mice compared to wild type. This observation may be due to down regulation of several important genes such as oncogene 1 (Gro1), heat shock proteins, I Kappa B Kinase β (IKKβ), colony stimulating factor (CSF-1) and annexin 7, suggesting the importance of VEGF/Flt-1 receptor signaling during ischemic preconditioning (2).
The VEGF mediated NO stimulation, increased endothelial constitutive nitric oxide synthase (eNOS) expression (14), Ca 2+ mobilization (15) and proliferation and migration (16) are observed in porcine aortic endothelial cells expressing Flk-1, but not in Flt-1 cells. Yeast two-hybrid system (17) analysis revealed interaction of Flt-1 along with p85 subunit of phosphatidylinositol 3′-kinase (PI3K), but this was never proven in Flt-1 expressing cells. Overall, most VEGF cell signaling described to date is mediated via Flk-1 or strongly suspected to involve Flk-1 instead of Flt-1 on the basis of ligand specificity (18).
Thus, it remains enigmatic whether Flt-1 is able to transmit a biologically meaningful signal in endothelial cells because almost all of the ensuing reports of biologically relevant VEGF signaling relates to Flk-1-mediated signaling. Downstream signaling of VEGF through Flt-1/VEGFR1 is not well studied both in vitro and in vivo. Therefore, in the present study, we explore PC mediated disruption of NO/HO-1(heme oxygenase) signaling and myocardial ischemia reperfusion injury using heterozygous Flt-1 knockout mouse ex-vivo. A striking instance in this report is that we documented PC mediated VEGF signaling through Flt-1 is important for cardioprotection by demonstrating significant reduction of DNA protein binding of CREB and STAT 3 transcription factors, inhibition of the extent of AKT and e-NOS phosphorylation. Most interestingly, we also found inhibition of iNOS as well as HO-1 and Bcl-2 followed by significant apoptotic cell death in heterozygous Flt-1 (Flt-1+/−) ischemic reperfused myocardium that lead to cardiac dysfunction.
EXPERIMENTAL PROCEDURES
Experimental animals
All animals received care in compliance with the principles of laboratory animal care formulated by the National Society for Medical Research and Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH). The heterozygous Flt-1 KO mice were generated as previously described (9) and maintained on a CD-1 background. Briefly, the first exon and splice donor site were replaced by a cassette containing neomycin phosphotransferase and the Escherichia coli Lac Z gene, resulting in disruption of Flt-1 coding and expression of beta-galactosidase (19). Flt-1+/− mice were identified by beta-galactosidase staining and age- and sex-matched CD-1 littermates were used as WT control. We have used heterozygous Flt-1 KO mice since the life span of Flt-1 homozygous KO mice is only 8 embryonic days.
Experimental protocol
Wild type (CD-1; WT) and Flt-1+/− (KO) mice were both randomized into two groups. For Group I (WT), after a 10-min stabilization, hearts were perfused for 40 min, followed by exposure to zero-flow normothermic global ischemia for 30 min followed by 120 min of reperfusion (I/R) and, for Group II (WT + PC), after stabilization, hearts were subjected to four episodes of 4-min global ischemia followed by 6-min reperfusion before I/R. For Group III (KO), hearts were perfused for 40 min before I/R and for Group IV (KO + PC), hearts were subjected to the same protocol as WT + PC.
Working Heart preparation
The mice were first anesthetized with sodium pentobarbital (200 mg/kg bw IP injection, Abbott Laboratories) and anticoagulated with Heparin (500 U/kg bw IP injection, Elkins-Sinn Inc). The heart was excised and immediately immersed in ice cold perfusion buffer. The aorta was cannulated and retrograde perfusion in the Langendorff mode through the aortic cannula was initiated at a perfusion pressure of 60 mm Hg. The perfusion buffer used in this study consisted of a modified Krebs-Henseleit Bicarbonate buffer [KHB: composed of (in mmol/L) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 25 NaHCO3, 10 glucose, and 1.7 CaCl2, gassed with 95% O2:5% CO2, filtered through a 5-μm filter to remove any particulate contaminants, pH 7.4] that was maintained at a constant temperature of 37°C and was gassed continuously for the entire duration of the experiment (20). After 10 minutes of retrograde perfusion, the heart was switched to antegrade perfusion mode where KHB buffer entered the cannulated left atrium at a pressure equivalent to 17 cm of water, and passed to the left ventricle from which it was spontaneously ejected through the aortic cannula. The mouse working heart system is a recirculating system at a constant pressure. At the top of the atrial reservoir, there is an overflow glass tube back to the ‘lung’ where the reoxygenation is happened. Thus, the isolated ‘working mouse heart system is operated at a constant pressure (1.5 kPa) instead of the constant flow system. Control measurements of heart rate, coronary flow, aortic flow, left ventricular developed pressure, and its first derivative dp/dtmax were monitored, analyzed, and recorded in real time using the Digitized data acquisition and analysis system (Micromed, Louisville, KY).
Infarct size
The infarct size was measured as previously described (2). After reperfusion, hearts were immediately perfused with 1% triphenyltetrazolium chloride. The hearts were excised and stored at −70°C. Sections of frozen hearts were fixed in 10% formalin, placed between two coverslips, and digitally imaged with the use of Epson scanner. To quantitate the areas of interest in pixels, Scion Image (Beta 4.03 for windows) analyzing software was used.
Determination of cardiomyocyte and endothelial cell apoptosis
The formaldehyde-fixed left ventricle was embedded in paraffin, cut into transverse sections (4 mm thick) and deparaffinized with a graded series of xylene and ethanol solutions. Immunohistochemical detection of apoptotic cells was carried out using TUNEL in which residues of digoxigenin-labelled dUTP are catalytically incorporated into the DNA by terminal deoxynucleotidyl transferase, an enzyme which catalyzes a template-independent addition of nucleotide triphosphate to the 3′-OH ends of double- or single-stranded DNA [21]. The incorporated nucleotide was incubated with a sheep polyclonal antidigoxigenin antibody followed by a FITC-conjugated rabbit anti-sheep IgG as a secondary antibody as described by the manufacturer (Apop Tag Plus, Intergen, Purchase, NY). The sections (n = 5) were washed in PBS three times, blocked with normal rabbit serum and incubated with mouse monoclonal antibody recognizing-a-sarcomeric actin (Sigma Japan, Tokyo, Japan) followed by staining with TRIRC-conjugated rabbit anti-mouse IgG (200:1 dilution, Dako Japan, Tokyo, Japan) [22]. For detection of apoptosis in endothelial cells the sections were first stained with TUNEL (FITC staining). The sections were then incubated with rabbit polyclonal anti-vonWillebrand factor (Dako) as a primary antibody followed by incubation with tetrarhodamine isothiocyanate-conjugated goat anti-rabbit IgG as a secondary antibody. The fluorescence staining was viewed with confocal laser microscopy (Fluoview, Olympas, Tokyo, Japan). For the quantitative purpose, the number of TUNEL-positive cardiomyocytes and endothelial cells were counted on 100 high power fields (HPF, magnification × 600) from the endocardium through the epicardium of the mid portion of the left ventricular free wall in five sections from each heart [17,23]. Representative confocal images show von Willebrand factor-positive endothelial cells (strong red staining in their cytosol) which are negative for TUNEL staining (absence of green staining in the nucleus) as well as those positive for TUNEL staining (magnification × 1200).
Quantitative real-time RT-PCR
Reverse transcription reaction (RT) was performed with 1 μg total RNA isolated from left ventricular tissue of WT and Flt-1+/− heterozygous KO mice subjected to I/R with or without PC. Real-time RT-PCR analysis was carried out with 10ng of RT product by iCycler iQ detection system (Biorad, Hercules, CA) using Syber Green I fluorescence and β-actin as reference control. The primer sequences used for real-time RT-PCR for HO-1 (NM-010442) is, forward, 5′-AGAGAAGGCTTTAAGCTG GTGAT-3′; reverse, 5′ GGAAGTAGAGTG GGGCATAGACT-3′ (1).
Western blot analysis
Cytosolic or nuclear protein was prepared from the left ventricular tissue 120 min after reperfusion using commercially available kit (Sigma, Saint Louis, MO). Standard Western Blot analysis was performed using antibodies to Ser473-phospho-Akt, Akt, ser1177 p- eNOS, e-NOS (Cell Signaling, Danvers, MA) & i-NOS (BD PharMingen, San Diego, CA). Primary antibody binding was visualized by horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence (1).
Protein DNA Array followed by Electrophoretic Mobility Shift Assay (EMSA) for validation
Nuclear extracts from the left ventricular tissue were prepared with the commercially available kit (Panomics Nuclear Extraction Kit). Each nuclear extract sample (3–5 μg/μl, 5 μl) were mixed with TranSignal Probe Mix (10 μl, from Panomics) and dH2O, 5 μl, this mixture was incubated at 15°C for 30 minutes. 2% agarose gel was prepared in 0.5 x TBE, sample was loaded and were run for 15 min at 120 V. The gel area was excised that contains protein/DNA complex and were transferred to 1.5 ml tube (4). One ml of Extraction buffer (supplied by the company, Panomics, Redwood city, CA) A was added and incubated at 55–60°C. 6 μl of gel extraction beads were added and incubated at room temperature for 10 min. The mixture was centrifuged at 10,000 rpm for 30 sec to pellet out the beads. The beads were washed and resuspended the pellet in 150 μl of extraction buffer B (supplied) and centrifuged at 10,000 rpm for 30 sec. Pellets were dried (air) for 10 min. Bound probe was eluted by resuspending the pellet in 50 μl of dH2O with a pipet and incubate at room temperature for 10 min. After centrifugation at 10,000 rpm for 1 min, supernatant was transferred to a fresh 0.2 tube. Eluted probe was stored on ice until proceeding to hybridization. This labeled probe was hybridized to the array membrane (Panomics, Catalog No. MA1010, Redwood City, CA). After hybridization and washing the membrane were treated with Enhanced ChemiLuminescence (ECL from Amersham) reagent and the transcription factors were detected by autoradiography for variable lengths of time with Kodak X-Omat film (4).
EMSA Analysis
Nuclear extracts were prepared as described above. Electrophoretic mobility shift assays (EMSA) were performed by using 5 μg of the nuclear extracts for 20 min at room temperature with 32P end-labeled oligonucleotides containing the putative CREB (5′AGAGAT TGC CTG ACG TCA GAG AGC TAG-3′), STAT 3 (5′GAT CCT TCT GGG AAT TCC TAG ATC-3′) and NFκB (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) binding site. Reaction products were resolved on 5% non-denaturing polyacrylamide gel. The specificity of the DNA–protein interaction was established by competition experiments using 10x cold CREB, STAT 3 and NFκB oligonucleotide as the competitor. After electrophoresis, gels were dried and visualized by autoradiography [24].
2.8. Statistical analysis
Results are expressed as mean ± standard error of the mean (± S.E.M.). Significance was determined using ANOVA followed by post hoc analysis with Bonferroni procedure. The level of statistical significance was taken as P < 0.05.
RESULTS
Effects of Flt-1 inhibition on the recovery of ventricular function
There were no difference in baseline function among the four groups. In general, there were no significant difference between Flt-1+/− and wild type mice in heart rate (Fig. 1B) and coronary flow (Fig. 1E) The functional parameters were decreased in all groups (after 30 minutes of ischemia) as expected compared with their respective baseline values. The post ischemic myocardial function was disrupted in the Flt-1+/− mice significantly which is evidenced by the significant decrease in the left ventricular developed pressure (LVDP), dp/dtmax and aortic flow (AF) as compared with the wild type mice.
Figure 1. A–E Effect of ischemia reperfusion and preconditioning on left ventricular function of wild type and Flt-1+/− mice.
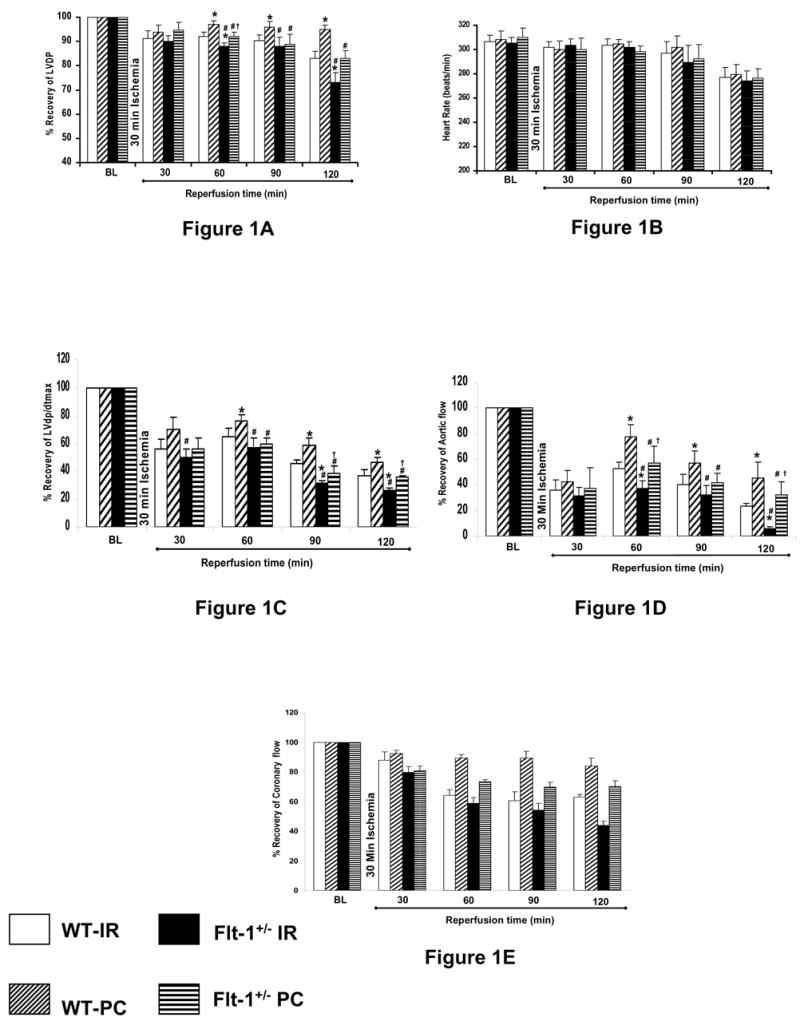
Post ischemic ventricular recovery of Flt KO and Wild type mouse hearts. The results (LVDP-A, heart rate-B, dP/dtmax C, aortic flow-D & coronary flow-E) are shown in % recovery of six animals per group. *P<0.05 compared with WT ischemia reperfusion, #P<0.05 compared with WT Preconditioning, †P<0.05 compared with KO ischemia reperfusion.
A significant decrease in the LVDP (Fig. 1A) after 120 minutes of reperfusion was found in both Flt-1+/− IR (73% ± 4.2) and Flt-1 +/− PC (83% ± 3.2) as compared with wild type IR (83%v± 2.9) and wild type PC (95% ± 1.9) respectively. Also a significant decrease in the dp/dtmax (Fig. 1C) after 120 minutes of reperfusion was found in both Flt-1+/− IR (25.87% ± 2.5) and Flt -1+/− PC (35.51% ± 1.3) as compared with wild type IR (36.88% ± 3.9) and wild type PC (46.76% ± 2.9) respectively. Similarly, aortic flow (Fig. 1D) also significantly decreased after 120 minutes of reperfusion in both Flt-1+/− IR (6.21% ± 0.8) and Flt-1+/− PC (32.49% ± 9.7) as compared with wild type IR (23.74% ± 1.8) and PC (45.34% ± 12.56) respectively. As expected a significant increase in the LVDP, dp/dtmax was also found in Flt-1+/− PC and wild type PC as compared with Flt-1+/− IR and wild type IR respectively after 120 minutes of reperfusion.
Effect of Flt-1 inhibition on Myocardial Infarct Size
Infarct size expressed as percent infarction to total area at risk was noticeably increased in Flt+/− knock out mouse hearts compared to the wild-type control. The results are shown in Fig. 2. Transversal cross sections from Flt+/− hearts which underwent ischemia reperfusion (38%) and ischemic preconditioning (27%) indicated more infarct size as compared to wild-type IR (29%) and PC (18.5%) heart sections. This difference was significant (P < 0.05) between these experimental groups.
Figure 2. Effect of ischemia reperfusion and preconditioning on infarct size of wild type and Flt-1+/− mice.
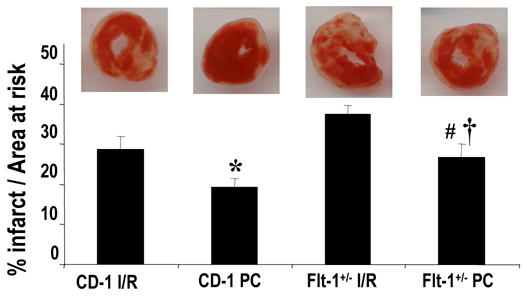
Infarct size of the hearts, expressed as a percentage of the area at risk in rat myocardium subjected to 30 min of ischemia followed by 2 hours of reperfusion. Results are expressed as mean ± standard error of six hearts/group. * P<0.01 compared with wild type ischemia reperfusion group, #P<0.01 compared with Flt-1+/− ischemia reperfusion group,†P<0.01 compared with wild type preconditioning group
Effect of Flt-1 inhibition on cardiomyocyte and endothelial cell apoptosis by Tunnel Assay
Apoptotic cardiomyocytes (Fig. 3A) and endothelial cells (Fig. 3B) were detected using TUNEL staining in conjunction with staining with antibody against α-sarcomeric actin and von Willebrand factor respectively. Apoptotic cardiomyocytes were significantly increased in Flt-1+/−IR and PC group when compared with the control IR and PC. Similar to cardiomyocyte apoptosis, the increased apoptotic endothelial cells were found in the Flt-1+/− IR and PC groups as compared to the corresponding controls. Hence disruption of Flt-1+/− increased cardiomyocyte and endothelial cell death due to apoptosis as compared to wild type.
Figure 3. TUNEL assay for apoptotic cardiomyocytes (A) and endothelial cells (B) after ischemia reperfusion and preconditioning of wild type and Flt-1+/− mice after ischemia.
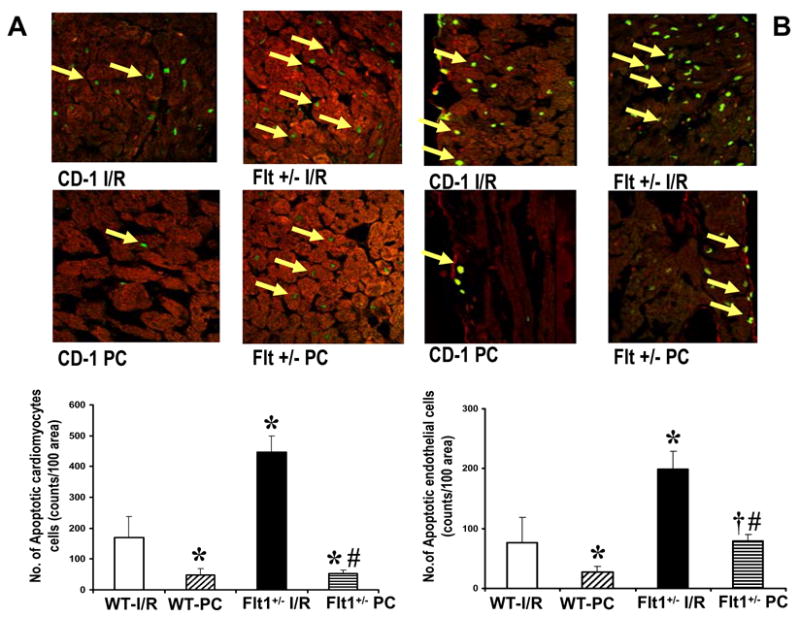
Tunnel assay for apoptotic cardiomyocyte (A) and endothelial cells (B) was performed as described in the methods section. Representative photographics show immunohistochemical staining of extended DNA. Results are expressed as means ± S.E.M of six hearts/group *P<0.01 compared with wild type ischemia reperfusion group, #P<0. 01 compared with Flt+/− ischemia reperfusion group, †P<0.01 compared with wild type preconditioning group.
Effect of Flt-1 inhibition on the mRNA expression of HO-1 by real-time RT-PCR
A significant increase in HO-1 mRNA (Fig. 4) levels was found in the Flt-1+/− (IR and PC) and Wild type (IR and PC) as compared with their respective baselines. The HO-1 level was also found to be increased in PC groups as compared with the IR groups in both Flt-1+/− and wild type mice. However HO-1 level was found to be significantly decreased in the Flt-1+/−.
Figure 4. Effect of Ischemia reperfusion and ischemic preconditioning on HO-1 mRNA expression.
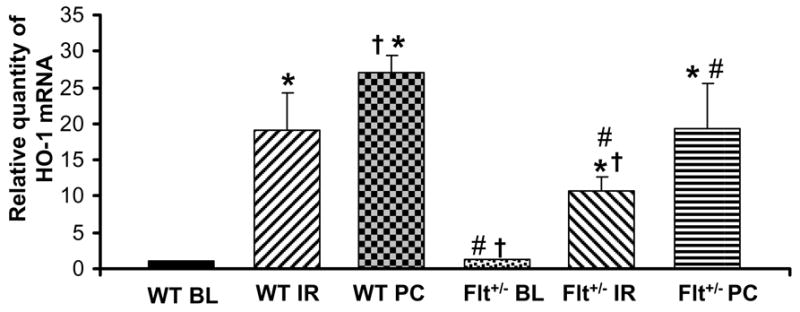
Bar graph showing relative abundance (arbitrary units) of HO-1mRNA levels after 30min of global ischemia and 2 hrs of reperfusion by quantitative real-time RT-PCR analysis (n=5). *p <0.05, compared with wild type baseline, †p <0.05, compared wild type IR, #p <0.05, compared wild type PC. BL: Baseline, I/R: Ischemia reperfusion, PC I/R: Ischemic preconditioning followed by ischemia reperfusion, WT: Wild type mice
Effect of Flt-1 inhibition on Transcription Factors – CREB, STAT 3 & NFκB
Protein/DNA array analysis revealed significant decrease in the DNA binding activity of CREB and STAT 3 transcription factors in Flt-1+/− after I/R and PC compared to the corresponding wild type. However there was significant DNA binding activity of NFκB observed in both I/R and PC groups compared to corresponding baseline samples. This is quite anticipated because I/R and PC might modulate the other VEGF receptor, Flk-1/KDR which might be responsible for increased NFκB DNA binding activity.
The protein/DNA results were validated by gel-shift analysis as shown in Figure 5 A–C. Ischemia and reperfusion decreases the CREB DNA activity in both Flt-1+/− and in wild type compared to corresponding baseline. However PC demonstrated increased CREB DNA binding activity in wild type as compared to wild type I/R but failed to do so in Flt-1+/− as shown in Figure 5A. Whereas STAT 3 DNA binding activity was found to be increased in both WT and Flt-1+/− PC compared to I/R but the extent of binding activity of STAT 3 is less in Flt-1+/− KO (Figure 5B). The extent of NFκB DNA binding activity was not affected because of the disruption of Flt-1. In other words both I/R and PC I/R demonstrated significant NFκB DNA binding activity in WT and Flt-1+/− compared to corresponding baseline (figure 5C).
Figure 5. Representative gel-shift analysis demonstrates and validates Protein/DNAarray results.
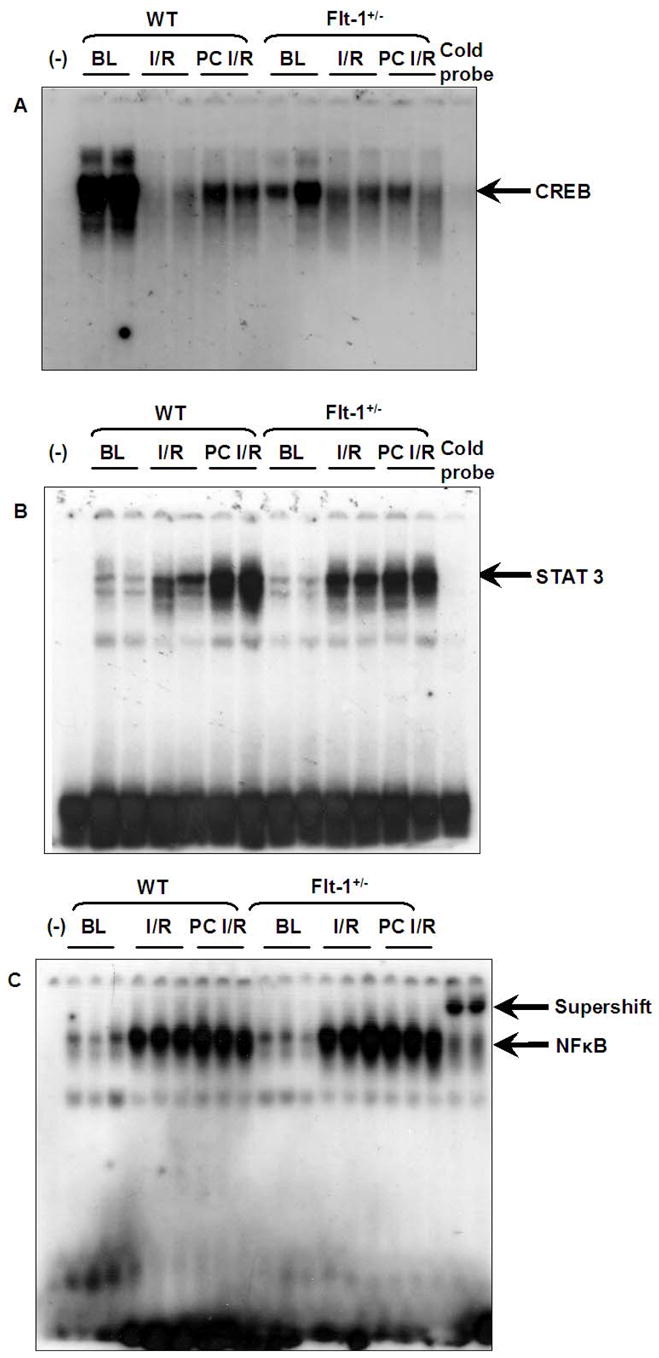
Representative Gel shift analysis for CREB (A), STAT 3 (B) & NFκB (C) (n=5) BL: Baseline, I/R: Ischemia reperfusion, PC I/R: Ischemic preconditioning followed by ischemia reperfusion, WT: Wild type mice, (−): Negative control.
Effect of Flt-1 inhibition on the phosphorylation of Akt, eNOS and iNOS expression
The phosphorylation of AKT (Fig. 6A) and eNOS (Fig. 6B) was decreased in all the three Flt-1+/− KO groups (Flt-1+/− BL, IR and PC) when compared to the corresponding wild type (BL, IR and PC group). Even though PC upregulated the phosphorylation of AKT and eNOS in both Flt-1+/− KO groups and wild type groups when compared to their respective baseline and ischemia/reperfusion groups, on the whole the phosphorylation of AKT and eNOS was found to be decreased in Flt-1 +/−KO groups when compared to the wild type. Similarly iNOS expression (Fig. 6C) was found to be decreased in the Flt-1+/− KO mice when compared to the wild type mice. But the iNOS expression is increased in the PC groups in both Flt-1+/− and wild type mice as compared with their respective baseline and ischemia/reperfusion groups.
Figure 6. Western blot analysis for iNOS, p-eNOS, e-NOS, p-AKT and AKT in wild type and Flt-1+/− mice.
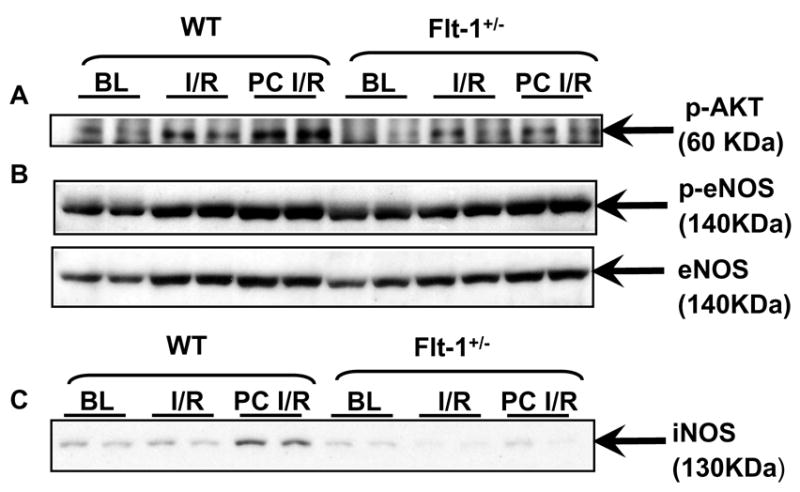
Representative Western blot showing the effect of I/R and PC on expression of (A) phosphorylated AKT (Immunoprecipitated with AKT and Immunoblotted with phosphorylated AKT), (B) phosphorylated and Non-phosphorylated eNOS and (C) iNOS (n=5). BL: Baseline, I/R: Ischemia reperfusion, PC I/R: Ischemic preconditioning followed by ischemia reperfusion, WT: Wild type mice.
DISCUSSION
Our previous study (2) demonstrated that PC significantly increased the expression of VEGF and Flt-1 during I/R associated with improvement of LV function and inhibition of infarct size however these effects were abolished in Flt-1+/− mice. Our present study extended these observations and provided for the first time a comprehensive evaluation of PC mediated signaling through VEGF-Flt-1/VEGFR1which triggered PI-3kinase survival pathway along with upregulation of NO/HO-1 molecules. We found this chain of signaling pathway along with cardioprotection was inhibited in heterozygous Flt-1+/− mice when subjected to PC protocol. These data suggests significant involvement of VEGF/Flt-1 receptor signaling in ischemia reperfusion injury. Little is known regarding the signaling and the importance of this particular pro-angiogenic ligand and its receptor, Flt-1 in ischemic myocardium. Therefore our present data in Flt-1 heterozygous KO mice carries significant message and information regarding VEGF/Flt-1 signaling in PC mediated cardioprotection.
The significant decrease of the postischemic myocardial function and the increase in infarct size in Flt-1+/− mice demonstrates the disruption of the VEGF induced Flt signaling in the ischemic reperfused heart. These results, however, differ somewhat from those obtained by the previous study (2) in that postischemic recovery of LV function and infarct size were comparable between Flt-1+/− and the wild mice without the PC challenge. Our previous study was done in the isolated Langendorff heart, a non working model using retrograde perfusion with a balloon inserted in the left ventricle as compared with the isolated working heart model which has been used in this study to examine the heart functions and infarct size. The working heart is more susceptible to I/R injury compared to the isolated Langendorff heart, because in the former heart coronary flow is markedly reduced during reperfusion in association with a decrease of aortic flow that further deteriorates LV function and aggravates cell injury while in the latter heart coronary perfusion pressure is kept constant throughout the experiment and coronary flow is less affected even though coronary artery resistance is increased during reperfusion. Therefore, a subtle difference in cardioprotective signaling through VEGF/Flt-1 receptors was masked in the isolated Langendorff heart and exaggerated in the working heart.
VEGF exerts its effect via two protein kinase receptors, Flt-1 and Flk-1. While there is abundant evidence that Flk-1 mediates the effect of VEGF in endothelial cell proliferation and permeability, the role of Flt-1 is somewhat more unknown and complicated. We think both the receptors are involved in PC mediated VEGF signaling in ischemic myocardium and there exists a significant cross-talk between the ligand, VEGF and tyrosine kinase receptors Flk-1 and Flt-1. The reason for this statement is from our PC results in Flt-1 KO mice which demonstrated less cardioprotection from wild type PC however the protection was found to be much significant than Flt-1 I/R as demonstrated by the expression and phosphorylation of HO-1, AKT and eNOS respectively. Significant inhibition of two survival transcription factors such as CREB and STAT3 were observed in Flt-1 KO. These two transcription factors transmit a survival signal through PI-3kinase-Akt-Bcl2 and CREB-dependent Bcl2 pathway. Partial inhibition of PC’s ability to precondition the heart, suggests involvement of PI-3kinase-AKT-CREB signaling pathway is at least partially responsible for the cardioprotection achieved by VEGF-Flt-1 signaling.
We documented and characterized already Flt-1 heterozygous mice in our previous study demonstrating 50% reduction in Flt-1 mRNA in rat myocardium as expected at the baseline level (2). Moreover, Flt-1 mRNA expression in the KO+PC compared with the WT+PC myocardium is significantly inhibited. Our previous study also demonstrated that expression of Flk-1 and VEGF mRNA is not affected in Flt-1+/− mice at the baseline level and after I/R, however, it is increased in KO+PC and WT+PC compared with KO and WT I/R mice as expected (2).
Again it is well established that VEGF increases the expression of endothelial NO synthase (eNOS) and inducible NO synthase (iNOS) (25). As previously described in ischemic hind limb, the angiogenic response to VEGF might involve the production of NO (26). There is little doubt that NO plays a significant role in the intracellular signaling in the cardiovascular system. Recent studies have demonstrated that eNOS/NO plays an important role in many VEGF-induced actions. VEGF has been shown to induce the production of nitric oxide (NO) from rabbit, pig, bovine, and human vascular endothelial cells (27–29). Inhibition of NO production by eNOS inhibitors significantly inhibited VEGF-induced mitogenic and angiogenic effects (30). Endothelial NOS/NO has been implicated as one of the important mediators for VEGF-induced hemodynamic changes and microvascular permeability. Although eNOS was originally described as a constitutive enzyme, recent studies indicated that a variety of stimuli including hypoxia, shear stress, inflammatory cytokines, high glucose, and injury could modulate eNOS expression and activity (31–32). In vitro experiments have shown that activation of KDR induces proliferation and migration of endothelial cells as well as expression of eNOS and iNOS (25, 33). So far, the role of VEGF receptors in the release of NO remains controversial. In human placental trophoblast it has been suggested that Flt-1 can induce the release of NO (34). The precise signaling pathways that mediate those responses have not yet been elucidated; however, phospholipase D and nitric oxide synthase have been identified as key enzymes activated by VEGF. In our present study we found significant inhibition of iNOS and partial inhibition of p-eNOS protein expression in Flt-1+/− mice subjected to I/R and also PC I/R when compared to wild type. Again real time RT-PCR for HO-1 mRNA level demonstrates significant inhibition of the expression of the same in Flt-1+/− after I/R and also PC I/R compared to corresponding wild type.
These data obviously denotes involvement of VEGF-Flt-1 signaling through NO and HO-1 in PC mediated cardioprotection. Two forms of HO have been identified including an inducible HO-1, that resembles stress-inducible protein HSP-32, and a constitutively expressed HO-2. Basal expression of HO-1 is either very low or even absent, but can be elevated during oxidative stress and hypoxia. This was exactly the case in our experimental conditions also. Both wild type and Flt-1+/− demonstrated very low level of HO-1 mRNA expression. HO-1 plays an important role in myocardial homoeostasis by protecting cardiomyocytes from ischemia/reperfusion-induced injury and secondary oxidative damage. Several reports demonstrate HO-1 mediated angiogenesis in various physiological conditions. A recent study documented improvement of blood flow by HO-1 gene transfer in ischemic hind limb, at least in part, via angiogenesis (35). Therefore, the complexity of VEGF biology is paralleled by the emerging complexity of interactions between VEGF ligands and their receptors, and the downstream signaling pathways they mediate in ischemic reperfused myocardium.
Therefore in conclusion, targeted disruption of tyrosine kinase receptor Flt-1 mouse hearts are more susceptible to ischemia reperfusion injury and fails ischemic preconditioning mediated cardioprotection which may be due to the downregulation of NO/HO-1 and therefore further supports the significance of VEGF-Flt-1/VEGFR1 receptor signaling in myocardial protection.
Acknowledgments
This study was supported by National Institutes of Health Grants HL 56803 to and HL 0169910.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaga S, Zhan L, Altaf E, Maulik N. Glycogen synthase kinase-3β/β-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and Survivin expression in rat ischemic preconditioned myocardium. J Mol Cell Cardiol. 2005;40:138–147. doi: 10.1016/j.yjmcc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Addya S, Shiroto K, Turoczi T, Zhan L, Kaga S, Fukuda S, Surrey S, Duan LJ, Fong GH, Yamamota F, Maulik N. Ischemic Preconditioning-mediated cardioprotection is disrupted in Flt-1 (VEGFR-1) heterozygous, targeted knockout mouse. J Mol Cell Cardiol. 2005;38:345–351. doi: 10.1016/j.yjmcc.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 3.Maulik N. Ischemic Preconditioning Mediated Angiogenic response in the heart. Antioxid Redox Signal. 2004;6(2):413–421. doi: 10.1089/152308604322899486. [DOI] [PubMed] [Google Scholar]
- 4.Fukuda S, Kaga S, Sasaki H, Zhan L, Zhu L, Otani H, Kalfin R, Das DK, Maulik N. Angiogenic signal triggered by ischemic stress induces myocardial repair in rat during chronic infarction. J Mol Cell Cardiol. 2004;36:547–559. doi: 10.1016/j.yjmcc.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Kaga S, Zhan L, Matsumoto M, Maulik N. Resveratrol Enhances Neovascularization in the infracted rat myocardium through the induction of Thioredoxin-1, Hemeoxygenase-1 and vascular endothelial growth factor. J Mol Cell Cardiol. 2005;39:813–822. doi: 10.1016/j.yjmcc.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Petrova TV, Makinen T, Alitalo K. Signaling via vascular endothelial growth factor receptors. Exp Cell Res. 1999;253:117–130. doi: 10.1006/excr.1999.4707. [DOI] [PubMed] [Google Scholar]
- 7.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor and its receptors. FESEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 8.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1 deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 9.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 10.Kendall R, Thomas K. Inhibition of vascular endothelial growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci USA. 1993;90:10705–10709. doi: 10.1073/pnas.90.22.10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roeckl W, Hecht D, Sztajer H, Wlaternberger J, Yayon A, Weich H. Differential binding characteristics and cellular inhibition by soluble VEGF receptors 1 and 2. Exp Cell Res. 1998;241:161–170. doi: 10.1006/excr.1998.4039. [DOI] [PubMed] [Google Scholar]
- 12.Jones CW, Ghazaleh RA, Cospedal R, Houliston RA, Martin J, Zachary I. Vascular endothelial growth factor stimulates prostacyclin production and activation of cytosolic phospholipase A2 in endothelial cells via p42/p44 mitogen activated protein kinases. FEBS Lett. 1997;420:28–32. doi: 10.1016/s0014-5793(97)01481-6. [DOI] [PubMed] [Google Scholar]
- 13.Kroll J, Walterberger J. A novel function of the vascular endothelial growth factor receptor-2 (KDR): rapid release of nitric oxide in response to VEGF-A stimulates in endothelial cells. Biochem Biophys Res Commun. 1999;265:636–639. doi: 10.1006/bbrc.1999.1729. [DOI] [PubMed] [Google Scholar]
- 14.Shen BQ, Lee DY, Zioncheck TF. Vascular endothelial growth factor governs endothelial nitric-oxide synthase expression via a KDR? Flk-1 receptor and protein kinase C signaling pathway. J Biol Chem. 1999;274:33057–33063. doi: 10.1074/jbc.274.46.33057. [DOI] [PubMed] [Google Scholar]
- 15.Cunningham SA, Waxham MN, Arrate PM, Brock TA. Interaction of the Flt-1 tyrosine kinase receptor with the p85 subunit of phosphatidylinositol 3-kinase. Mapping of a novel site involved in binding. J Biol Chem. 1995;270:20254–20257. doi: 10.1074/jbc.270.35.20254. [DOI] [PubMed] [Google Scholar]
- 16.Waltenberger J, Claesson-welsh L, Siegbahn A, Shibuya M, Heldin C. Different signal transduction properties of KDR and Flt-1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 17.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferra N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requiresment for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 18.He H, Venema VJ, Gu X, Venema RC, Marrero MB, Caldwell RB. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through Flk-1/KDR activation of c-Src. J Biol Chem. 1999;274:25130–25135. doi: 10.1074/jbc.274.35.25130. [DOI] [PubMed] [Google Scholar]
- 19.Ray PS, Estrada-Hernandez T, Sasaki H, Zhu L, Maulik N. Early effects of hypoxia/reoxygenation on VEGF, ang-1, ang-2 and their receptors in the rat myocardium: implications for myocardial angiogenesis. Mol Cell Biochem. 2000;213:145–153. doi: 10.1023/a:1007180518474. [DOI] [PubMed] [Google Scholar]
- 20.Turoczi T, Zhan L, Cordis G, Morris JE, Maulik N, Stevens RG, Das DK. HFE Mutation and Dietary Iron Content Interact to Increase Ischemia/Reperfusion Injury of the Heart in Mice. Circ Res. 2003;92:1240–1246. doi: 10.1161/01.RES.0000076890.59807.23. [DOI] [PubMed] [Google Scholar]
- 21.Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G, Anversa P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- 22.Maulik N, Yoshida T, Zu YL, Sato M, Banerjee A, Das DK. Ischemic preconditioning triggers tyrosine kinase signaling: a potential role for MAPKAP kinase 2. Am J Physiol. 1998;275:H1857–H1864. doi: 10.1152/ajpheart.1998.275.5.H1857. [DOI] [PubMed] [Google Scholar]
- 23.Sasaki H, Ray PS, Zhu L, Otani H, Asahara T, Maulik N. Hypoxia/Reoxygenation promotes myocardial angiogenesis via an NFκB–dependent mechanism in a rat model of chronic myocardial infarction. J Mol Cell Cardiol. 2001;33:283–294. doi: 10.1006/jmcc.2000.1299. [DOI] [PubMed] [Google Scholar]
- 24.Maulik N, Sato M, Price BD, Das DK. An essential role of NFκB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett. 1999;429:365–369. doi: 10.1016/s0014-5793(98)00632-2. [DOI] [PubMed] [Google Scholar]
- 25.Kroll J, Waltenberger J. VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR) Biochem Biophys Res Commun. 1998;252:743–746. doi: 10.1006/bbrc.1998.9719. [DOI] [PubMed] [Google Scholar]
- 26.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van der Zee R, Murohara T, Luo Z, Zollmann F, Passeri J, Lekutat C, Isner JM. Vascular endothelial growth factor/vascular permeability factor augments nitric oxide release from quiescent rabbit and human vascular endothelium. Circulation. 1997;95:1030–1037. doi: 10.1161/01.cir.95.4.1030. [DOI] [PubMed] [Google Scholar]
- 28.Parenti A, Morbidelli L, Cui XL, Douglas JG. Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal regulated kinase 1/2 activation in postcapillary endothelium. J Biol Chem. 1998;273:4220–4226. doi: 10.1074/jbc.273.7.4220. [DOI] [PubMed] [Google Scholar]
- 29.Morbedelli L, Chang CH, Douglas JG. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am J Physiol Heart & Circ. 1996;270:H411–H415. doi: 10.1152/ajpheart.1996.270.1.H411. [DOI] [PubMed] [Google Scholar]
- 30.Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sase K, Michel T. Expression and regulation of endothelial nitric oxide synthase. Trends Cardiovasc Med. 1997;7:28–37. doi: 10.1016/S1050-1738(96)00121-1. [DOI] [PubMed] [Google Scholar]
- 32.Le Cras TD, Xue C, Rengasamy A, Johns RA. Chronic hypoxia upregulates endothelial and inducible Nosynthase gene and protein expression in rat lung. Am J Physiol Hear & Circ. 1996;270:L164–L170. doi: 10.1152/ajplung.1996.270.1.L164. [DOI] [PubMed] [Google Scholar]
- 33.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDRand Flt-1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 34.Bussolati B, Dunk C, Mason M, Kontos CD, Ahmed A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor mediated angiogenesis via nitric oxide. Am J Pathol. 2001;159:993–1008. doi: 10.1016/S0002-9440(10)61775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki M, Takeshita S, Tsukamoto K, Mori I, Sato T, Ohno M, Nagai R, Ishizaka N. Facilitated angiogenesis induced by heme oxygenase –1 gene transfer in a rat model of hind limb ischemia. Biochem Biophys Res Commun. 2003;30:138–143. doi: 10.1016/s0006-291x(03)00114-1. [DOI] [PubMed] [Google Scholar]