Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein (original) (raw)
Abstract
Somatic gene therapy has been proposed as a means to achieve systemic delivery of therapeutic proteins. However, there is limited evidence that current methods of gene delivery can practically achieve this goal. In this study, we demonstrate that, following a single intramuscular administration of a recombinant adeno-associated virus (rAAV) vector containing the β-galactosidase (AAV-lacZ) gene into adult BALB/c mice, protein expression was detected in myofibers for at least 32 weeks. A single intramuscular administration of an AAV vector containing a gene for human erythropoietin (AAV-Epo) into mice resulted in dose-dependent secretion of erythropoietin and corresponding increases in red blood cell production that persisted for up to 40 weeks. Primary human myotubes transduced in vitro with the AAV-Epo vector also showed dose-dependent production of Epo. These results demonstrate that rAAV vectors are able to transduce skeletal muscle and are capable of achieving sustained expression and systemic delivery of a therapeutic protein following a single intramuscular administration. Gene therapy using AAV vectors may provide a practical strategy for the treatment of inherited and acquired protein deficiencies.
The use of therapeutic proteins in the treatment of disease is limited by the need for repeated protein administration and costly production methods. Lack of control over protein production within relevant tissues is especially important in diseases such as cystic fibrosis, sickle cell anemia, or β-thalassemia, because the therapeutic protein must be delivered directly to the target cell to correct the cellular defect. Somatic gene therapy provides a potential solution in that genes may be directly delivered to affected cells where the therapeutic protein will be produced. Alternatively, genes may be delivered to tissues which serve as sites for the synthesis and secretion of proteins that have effects elsewhere in the body. Gene therapy offers the potential for increased efficacy and more cost-effective production and administration of a variety of genes coding for therapeutic proteins.
Gene therapy has been proposed for several inherited and acquired protein deficiencies. The feasibility of this approach is supported by studies showing that gene delivery to skeletal muscle (1, 2), vascular smooth muscle (3), and liver (4) can result in systemic levels of therapeutic proteins. Skeletal muscle is a useful target to evaluate this approach because of its large mass, vascularity, and accessibility (5). Since muscle fibers are nondividing, effective gene delivery could potentially result in long-lived protein production. Posttranslational modification of proteins in muscle cells allows these proteins to be secreted with full potency and bioavailability (5). Indeed, genetically modified myoblasts are capable of long-term delivery of growth hormone (1, 2), coagulation factor IX (6), erythropoietin (Epo) (7), and β-glucuronidase (8). Although these studies are encouraging, there has been no convincing demonstration that this approach will be clinically useful or practical. More effective methods for gene delivery would be desirable to achieve sustained high-level expression of therapeutic gene products (9).
Currently available vectors for in vivo gene delivery to muscle include plasmid DNA, liposomes, protein–DNA conjugates, and vectors based on adenovirus or herpes virus (10). Plasmid DNA delivered directly to muscle generally results in persistent albeit low-level gene expression (11). Adenovirus vectors are efficient at delivering genes to many different tissues. However, the properties of current generation adenoviral vectors may result in only transient gene expression due to expression of viral genes leading to immune reactivity (12). Retroviral vectors require dividing cells for nuclear entry (13), thus cannot be used for efficient gene delivery to uninjured muscle (6, 14). Retroviral vectors have been used ex vivo, however, to genetically modify dividing myoblasts (2). Following surgical implantation, these modified cells fuse with established myofibers to produce hybrid muscle fibers. Gene therapy methods based upon surgical transplantation of myoblasts require substantial tissue-culture manipulation and surgical expertise (15). Therefore, the development of a simple and effective method of gene delivery to muscle resulting in long-term expression would be desirable.
Recombinant viral vectors derived from the nonpathogenic parvovirus, adeno-associated virus (AAV) (16, 17) are modified such that all viral genes are replaced with an eukaryotic expression cassette, including the appropriate regulatory elements and transgene. Recombinant AAV (rAAV) may elicit less of a host immune response due to the absence of viral genes. Viral particles are physically stable, thereby allowing them to be administered in vivo, and efficiently transduce a wide variety of dividing and nondividing cell types in vitro (18, 19, 20). Experience with AAV vectors in animal models has been limited, in part because of difficulties in producing large quantities of high-quality AAV vectors for in vivo studies. Previous_in vivo_ studies have demonstrated that direct administration of AAV vectors results in gene expression in nondividing or quiescent cells (21, 22, 23, 24, 25). In this study, we demonstrate that AAV vectors are an effective means for achieving gene expression in skeletal muscle both_in vitro_ and in vivo. Our findings indicate that following a single intramuscular administration of AAV vectors in mice, there is sustained expression (at least 32 weeks) and systemic delivery (up to 40 weeks) of biologically significant levels of a therapeutic protein.
MATERIALS AND METHODS
Vector Constructs, Viruses, and Cell Lines.
The AAV-lacZ vector was constructed by placing the Escherichia coli β-galactosidase (β-gal) gene under the transcriptional control of the cytomegalovirus (CMV) immediate early promoter (Fig.1A). A 2.7-kb_Kas_I–_Ear_I fragment from a partial digest of pUC119 (GenBank accession no. U07650U07650) was blunted and ligated to a multiple cloning sequence containing the following restriction enzyme sites (5′-_Not_I-_Mlu_I-_Sna_BI-_Age_I-_Bst_BI-_Bss_HII-_Nco_I-_Hpa_I-_Bsp_EI-_Pml_I-_Rsr_II-_Not_I-3′) to generate pUC-MCS. The following fragments were cloned into the multiple cloning site of pUC-MCS, successively; a 653-bp_Spe_I–_Sac_II fragment containing the CMV promoter was cloned into the _Sna_BI site, a 269-bp PCR-generated_Bst_BI–_Bst_BI fragment from the human growth hormone first intron was inserted into the _Bst_BI site, and the alcohol dehydrogenase–β-gal (lacZ) fusion gene (CLONTECH) was ligated into the _Bss_HII site. The 135-bp _Hpa_I–_Bam_HI blunt-end modified SV40 early polyadenylylation signal from pCMV-β plasmid (CLONTECH) was cloned into the _Hpa_I site. The resulting _Not_I–_Not_I expression cassette was inserted between the ITRs of a pUC-based plasmid containing both the AAV ITRs and the AAV rep and cap genes outside the ITRs (pAAV-lacZ). To construct pAAV-Epo, the pAAV-lacZ construct was modified as follows. The alcohol dehydrogenase–β-gal fusion gene was replaced with a 718-bp _Ppu_MI–_Nco_I fragment of the 1.34-kb human Epo cDNA (a kind gift of D. Kohn, Childrens Hospital, Los Angeles) along with the SV40 late polyadenylylation signal. A 2.1-kb _Cla_I–_Eco_RI noncoding segment from the lacZ gene was then cloned into the _Pml_I site as a spacer fragment.
Figure 1.
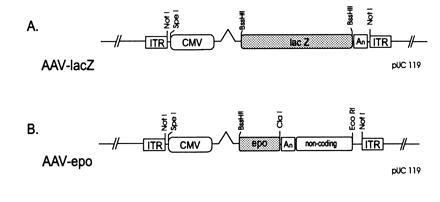
(A) pAAV-lacZ. ITR, inverted terminal repeat; CMV, CMV promoter; lacZ, bacterial β-gal; An, simian virus 40 (SV40) early polyadenylylation signal. (B) pAAV-Epo. Epo, human Epo coding sequence; An, SV40 polyadenylylation signal; noncoding, 2.2-kb noncoding fragment from the lacZ gene. See text for details.
Adenovirus type 2 (Ad2) from the American Type Culture Collection was used as helper virus to encapsidate AAV vectors. The 293 cell line (26), was cultured in complete DMEM (BioWhittaker) containing 4.5 g/liter glucose, 10% heat-inactivated fetal calf serum (FCS; HyClone), 2 mM glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin. The growth medium (GM) for C2C12 murine myoblast cell line (CRL 1772; American Type Culture Collection) was DMEM with 20% FCS, 1% chicken embryo extract, and 5 μg/ml gentamicin. Fetal human skeletal myoblasts (Clonetics, San Diego) were propagated in human GM, Ham’s F-12 medium containing 20% FCS and 5 μg/ml gentamicin. Murine differentiation medium (DM) containing DMEM with 2% horse serum was used to induce myoblast fusion and formation of differentiated myotubes; human DM was DMEM with 10% horse serum and 5 μg/ml gentamicin.
Vector Encapsidation.
Recombinant AAV virions were produced in human 293 cells as follows. Subconfluent 293 cells were transfected by calcium phosphate precipitation (27) with either the AAV-lacZ or AAV-Epo expression cassette flanked by ITRs. After 6 h, the cells were infected with adenovirus type 2 at a multiplicity of infection of 2 in fresh medium, and incubated at 37°C in 5% CO2 for 72 h prior to harvest. Pelleted cells were lysed in Tris buffer (10 mM Tris/150 mM NaCl, pH 8.0) by three cycles of freeze-thaw. The lysate was clarified of cell debris by centrifugation at 12,000 ×g followed by cesium chloride isopyknic gradient centrifugation. Recombinant AAV vectors were extracted from the resulting gradient by isolating the fractions with an average density of 1.38 g/ml, followed by resuspension in Hepes buffered saline containing 50 mM Hepes (pH 7.4) and 150 mM NaCl. The preparation was heated at 56°C for 1 h to inactivate adenovirus type 2.
Vector titer was determined by quantitative dot-blot hybridization of DNase-treated stocks and was routinely in the range of 1012–1013 particles per ml. In our experience, the particle to transduction unit (β-gal) ratio was between 102 and 103. Assessment of contaminating helper adenovirus in vector stocks was performed by scoring for cytopathic effect of viral stocks on 293 cells. Replication competent adenovirus was not detected by this bioassay, with a level of detection of 100 plaque forming units per ml.
In Vitro Transduction of Differentiated Myotubes and Transduction Assay.
Differentiation of mouse or human myoblasts was induced by seeding cells in 24-well plates at a density of 2 × 104 cells per cm2 and growing cells to confluence in GM, then substituting with DM. After 5 days (C2C12) or 14 days (primary human myoblasts) incubation in DM, respectively, in vitro transduction was performed by adding purified vector to murine or human skeletal myoblasts in OptiMEM (GIBCO/BRL). DM was added after viral adsorption.
Culture media of Epo transduced cells was changed 24 h prior to collection of supernatants at 3, 8, and 14 days following transduction. Human Epo was assayed by ELISA using the human Epo Quantikine IVD kit from R & D Systems according to manufacturer’s recommendations.
In Vivo Transduction of Murine Skeletal Muscle.
Animals were handled according to National Institutes of Health guidelines. For the administration of AAV-lacZ in vivo, 8-week-old male mice (Sprague–Dawley [Harlan]) received methoxyflurane anesthesia. The mid-portion of each tibialis anterior muscle was exposed via a 1-cm incision. Injections into the tibialis anterior were done by using a glass capillary pipette attached to a Hamilton syringe. A 10-μl volume of either PBS (pH 7.4), PBS containing 100 μg of plasmid DNA, or AAV-lacZ in PBS was injected. For AAV-Epo administration, i.m. injection into both hindlimbs of 6- to 8-week-old female BALB/c mice [The Jackson Laboratories or Simonsen Laboratories (Gilroy, CA)] was performed. After anesthesia with ketamine-xylazine, 100 μl of vector diluted in Hepes-buffered saline was administered percutaneously with a 28-gauge needle and syringe into each hindlimb, for a total of 200 μl per animal. Control animals received an equivalent dose of AAV-lacZ. Intravenous (i.v.) injections were performed with the single injection of 50 μl of vector in PBS in the tail vein.
Assays for in Vivo Transduction.
Tissue samples of tibialis anterior muscle, forelimb muscle, heart, brain, and liver were obtained for analysis of β-gal expression. One tibialis anterior muscle was processed for cross-sectional β-gal analysis, and total β-gal was determined from a crude homogenate of the other muscle using a chemiluminescent assay. For histochemical detection of β-gal, muscle was snap-frozen in dry ice-cooled isopentane, followed by serial transverse sectioning (10 μm), and processing as described by Sanes_et al._ (28). The cross-sectional area of the tibialis anterior expressing β-gal was determined as follows: after counter-staining with nuclear fast red (Vector Laboratories), the 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) stained tissue was digitally photographed and the cross-sectional area of stored images was determined using NIH image software. The Galacto-light (Tropix, Bedford, MA) chemiluminescent reporter assay kit was used for the detection of total β-gal activity in the entire tibialis anterior muscle. Standard curves were prepared from known amounts of purified β-gal (Sigma) resuspended in nontransduced muscle homogenate. β-Gal activity is expressed as nanograms of β-gal and was normalized for the entire muscle, minus background activity. Forelimb muscle, cardiac muscle, brain, and liver samples were assayed in the identical fashion.
The in vivo biological effect of gene delivery of AAV-Epo in BALB/c mice was analyzed at various times after i.m. or i.v. administration of vector. Blood was obtained from the orbital venous plexus under anesthesia. Hematocrit was determined by centrifugation of blood in a micro-capillary tube, and serum Epo levels were determined by ELISA as described above.
RESULTS
Expression of β-Gal After Intramuscular Injection of AAV-lacZ.
An AAV vector (AAV-lacZ) expressing the bacterial β-gal gene, controlled by the CMV promoter (Fig.1A), was delivered to adult male BALB/c mice by a single i.m. injection in the tibialis anterior muscle to investigate the time course of expression. The left and right tibialis anterior muscles of adult BALB/c mice were injected under direct vision with 8 × 109 vector particles of AAV-lacZ. Four animals were injected with vehicle alone. Animals were sacrificed at 2, 4, 8, 12, 24, and 32 weeks after injection, and the tibialis anterior muscle was excised and analyzed for the presence of bacterial β-gal (n = 5 for each group). Two measures of the efficiency of gene transfer were used: cross-sectional analysis of tissue sections stained for β-gal activity, and a chemiluminescent assay for β-gal activity. Gene expression persisted for at least 32 weeks (Fig.2 and Table 1): Two weeks after injection of 8 × 109 vector particles, 18% of the muscle cross-sectional area expressed β-gal, while at 32 weeks, 24% of the muscle cross-sectional area expressed β-gal. Sham-injected tibialis anterior muscle (n = 4) showed no background staining. Muscle β-gal activity was also determined in the contralateral injected muscle and revealed persistent expression for at least 32 weeks, in agreement with the cross-sectional fiber analysis (Table 1). In fact, β-gal expression was sustained with minimal inflammatory cell infiltrate. These data demonstrate that AAV-lacZ administration in muscle results in stable expression of the transgene for at least 8 months.
Figure 2.
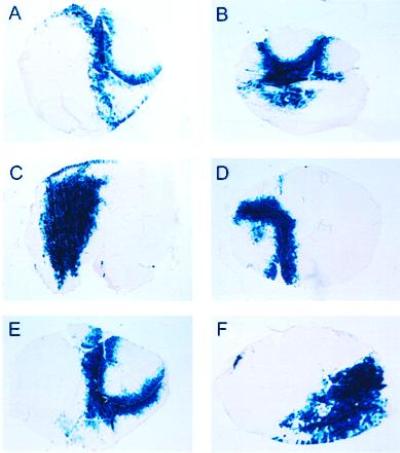
Histochemical detection of β-gal expression_in situ_ following injection of AAV-lacZ. The tibialis anterior of adult male BALB/c mice was injected with 8 × 109 vector particles. The animals were sacrificed at (A) 2, (B) 4, (C) 8, (D) 12, (E) 24, or (F) 32 weeks after the injection. The tibialis anterior was excised, and 10-mm sections were processed for β-gal histochemistry. (×25.)
Table 1.
Time course of β-gal expression following a single injection of AAV-lacZ
| Time, weeks | β-gal expression, ng/muscle | % cross-sectional area expressing β-gal |
|---|---|---|
| 2 | 1441 ± 458 | 18 ± 6 |
| 4 | 951 ± 176 | 20 ± 4 |
| 8 | 839 ± 436 | 24 ± 3 |
| 12 | 1878 ± 521 | 29 ± 5 |
| 24 | 2579 ± 1165 | 22 ± 3 |
| 32 | 1242 ± 484 | 24 ± 3 |
Positive staining filled the cytoplasm of the myofibers and was observed through large contiguous portions of muscle. Serial transverse sections revealed that blue staining extended throughout the length of the muscle fiber. This suggests that either the AAV vector transduced a number of muscle nuclei or the β-gal product had diffused throughout the fiber. Diffraction-interference contrast microscopy revealed a clear delineation between positively and negatively stained myofibers (Fig. 3), suggesting that vector delivery was limited by structural barriers such as epimyseal or perimyseal connective tissue. Homogenates prepared from brain, heart, liver, and forelimb muscle displayed no β-gal activity as compared with the background activity of sham-injected controls (data not shown).
Figure 3.
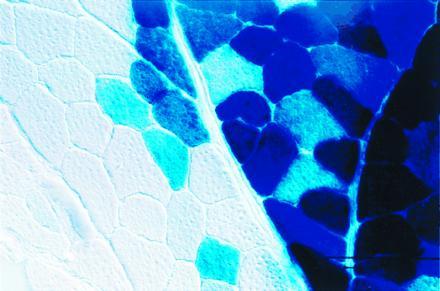
High-powered section of skeletal muscle 2 months after injection with AAV-lacZ. Tibialis anterior muscle was processed for in situ detection of β-gal and photographed with diffraction-interference contrast optics at ×400.
The efficiency of expression of the β-gal gene encapsidated in a viral particle was compared with the same construct delivered as plasmid DNA at 2 weeks following i.m. injection. Administration of 8 × 109 encapsidated vector particles resulted in 1441 ng β-gal/muscle (n = 5) (Table 1), while administration of 100 μg plasmid DNA, a typical plasmid DNA dose used_in vivo_ (29), produced 12 ng β-gal/muscle (n = 4) (data not shown). The naked DNA dose is equivalent to 2.2 × 1013 single-stranded genomes. Therefore, gene delivery by encapsidated vector was substantially more efficient than an equal molar quantity of vector DNA.
Transduced Murine Myotubes Secrete Human Epo in Vitro.
To determine if differentiated muscle cells in culture were targets for AAV vectors, the transduction efficiency of murine C2C12 myotubes was analyzed by using an AAV construct expressing the human Epo gene driven by the CMV immediate early promoter, AAV-Epo (Fig. 1B). These cells have been extensively studied as a model for mammalian myogenesis (30) and can be induced to differentiate by growth in reduced serum medium. Murine C2C12 myoblasts were maintained in growth media until confluent and then cultured for 5 days in DM. Differentiation was verified by the microscopic presence of multinucleate myotubes, representing fused myoblasts. There was a dose-dependent increase in the secretion of human Epo in cells transduced with AAV-Epo (Fig. 4). Eight days after addition of vector to mature myotubes, Epo levels peaked at >3400 milliunits/ml. This result indicates that in short-term myotube cultures, Epo is synthesized and secreted in a dose-dependent manner.
Figure 4.
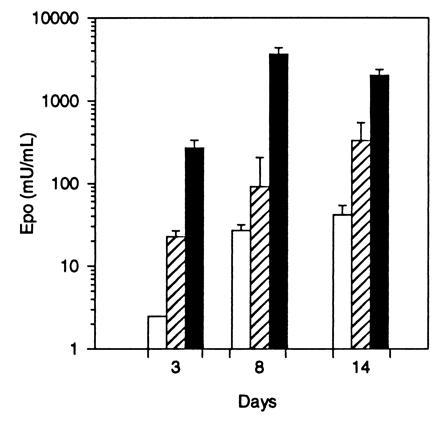
Secretion of human Epo by C2C12 myotubes transduced with AAV-Epo. Confluent C2C12 myoblasts were differentiated into myotubes and transduced with 3 × 108 (□), 3 × 109 (▨), or 3 × 1010 (▪) vector particles of AAV-Epo. Twenty-four hours following a complete change of media, the secretion of Epo was measured (3, 8, and 14 days after transduction). AAV-lacZ-transduced myotubes secreted <2.5 milliunits/ml Epo. The bar graph depicts mean concentration of Epo per well per 24 hr determined in triplicate cultures ± SEM.
Systemic Delivery of Human Epo in Vivo by Intramuscular Administration of AAV-Epo.
The AAV-Epo vector was administered to healthy adult mice to determine if systemic levels of human Epo and a biological response were obtained. Adult female BALB/c mice were injected i.m. in both hindlimbs with a single administration of AAV-Epo. A linear dose-response was obtained over the range from 3 × 109 to 3 × 1011 vector particles, evident at 20, 41, 62, and 83 days after injection (Table 2). The time course of Epo secretion is shown in Fig. 5. Serum levels of human Epo increased with time to a plateau at 6–8 weeks after injection. Biological activity of the recombinant Epo is shown by the elevation of hematocrit in the experimental animals. A comparison of circulating Epo levels versus hematocrit is shown in Table 2. Hematocrit increased with time and increasing vector dose. Control animals (AAV-lacZ injected) had undetectable levels of human Epo (<2.5 milliunits/ml, lower limit of detection for the assay). Stable elevation in hematocrit has been observed for up to 40 weeks in animals that received i.m. injection (data not shown). Late deaths occurred in this group, presumably related to complications of pronounced polycythemia. These results indicate that persistent and stable high-level secretion of Epo with a corresponding elevation in hematocrit is possible following a single i.m. administration of AAV-Epo.
Table 2.
Epo expression and hematocrit: AAV-Epo dose-response
| Dose | Days after administration | |||||||
|---|---|---|---|---|---|---|---|---|
| 20 days | 41 days | 62 days | 83 days | |||||
| EPO | HCT | EPO | HCT | EPO | HCT | EPO | HCT | |
| 3 × 1011 | 445 ± 98 | 74.2 ± 1.2 | 725 ± 112 | 82.3 ± 1.2 | 769 ± 61 | 86.5 ± 1.4 | 723 ± 253 | 88.5 ± 0.7 |
| 1 × 1011 | 85 ± 14 | 72.8 ± 1.5 | 212 ± 23 | 79.5 ± 1.7 | 234 ± 75 | 83.2 ± 0.2 | 220 ± 51 | 83.2 ± 2 |
| 3 × 1010 | 17 ± 5 | 60.0 ± 3.5 | 34 ± 17 | 74.7 ± 3.2 | 55 ± 28 | 78.7 ± 2.0 | 73 ± 45 | 80.0 ± 3 |
| 1 × 1010 | 3 ± 1 | 52.9 ± 1.8 | 11 ± 3 | 61.5 ± 1.9 | 12 ± 8 | 68.4 ± 4.6 | 15 ± 5 | 70.8 ± 8 |
| 3 × 109 | <2.5 | 49.9 ± 1.4 | <2.5 | 53.5 ± 2.5 | <2.5 | 57.0 ± 2.4 | 4 ± 4 | 57.5 ± 3 |
| i.v. | 7 ± 3 | 54.7 ± 3.2 | 13 ± 2.0 | 64.4 ± 5.3 | 10.1 ± 0.7 | 70.8 ± 8 | 21 ± 10 | 74.6 ± 7 |
| Control | <2.5 | 48.9 ± 1.0 | <2.5 | 49.1 ± 0.8 | <2.5 | 48.1 ± 0.7 | <2.5 | 48.2 ± 9 |
| Plasmid | 8 ± 10 | 50 ± 3.0 | <2.5 | 50.2 ± 1.0 | <2.5 | 47.8 ± 0.9 | ND | ND |
Figure 5.
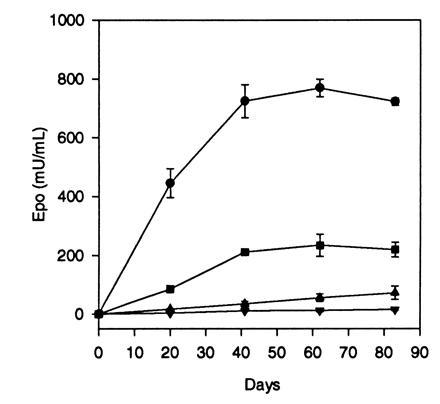
Dose-response and time course of Epo secretion in BALB/c mice after i.m. injection of AAV-Epo. Adult BALB/c mice were injected i.m. with 1 × 1010 (▾), 3 × 1010 (▴), 1 × 1011 (▪), or 3 × 1011 (•) vector particles at day 0, and serum Epo levels were measured at time points postinjection. Points represent means (n = 4) ± SEM.
An additional group of animals received i.v. injection of 3 × 1011 vector particles of AAV-Epo (n = 4). As shown in Table 2, the Epo levels resulting from the i.v. route were significantly lower than the group which received vector by the i.m. route. This result suggests that interstitial viral delivery in muscle, where there is a high local concentration of virus, results in improved transduction by AAV.
A comparison of the expression of AAV-Epo versus 100 μg of the AAV-Epo vector plasmid (equivalent to 2.8 × 1013 genomes) was performed following an i.m. injection (Table 2). At 20 days postinjection animals injected with plasmid had levels the same as those injected with 1–3 × 1010 vector particles. Calculated on the basis of number of genomes delivered, AAV-Epo results in at least 1000-fold greater Epo levels. At 41 and 62 days postinjection, the difference in Epo levels was even greater, since the serum Epo level in vector-injected animals increased slightly, while the Epo level in plasmid-injected animals declined to <2.5 milliunits/ml.
Human Myotubes Secrete Human Epo Following Transduction with AAV-Epo in Vitro.
To determine if differentiated primary human muscle cells are able to express Epo following transduction with AAV, primary human skeletal myotubes were induced to differentiate and transduced with AAV-Epo. The cells were confirmed to be differentiated by examination for multinucleate cells. Human myotube cultures transduced with AAV-Epo secreted Epo into the culture media in a dose-dependent manner (Fig. 6). No detectable Epo activity was measured in control cultures transduced with AAV-lacZ. The secretion of Epo increased over the 14-day interval posttransduction. These results show that primary human myotubes transduced by AAV vectors are fully capable of expressing and secreting Epo.
Figure 6.
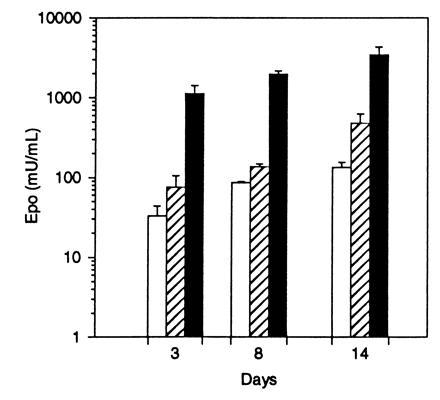
Epo secretion by primary human myotubes transduced with AAV-Epo. Confluent human myoblasts were differentiated into myotubes by culture for 14 days in differentiation media, then transduced with 3 × 108 (□:), 3 × 109 (▨), or 3 × 1010 (▪) vector particles of AAV-Epo. Twenty-four hour secretion of Epo was measured 3, 8, and 14 days after transduction. AAV-lacZ-transduced myotubes secreted <2.5 milliunits/ml Epo. Bar graph depicts mean concentration of Epo per well per 24 hr determined in triplicate cultures ± SEM.
DISCUSSION
Our results demonstrate that AAV vectors efficiently transduce skeletal muscle, resulting in long-term, stable protein expression. The dose-response of AAV-Epo was linear over a broad range of input vector. Lower doses resulted in systemic protein levels which would be within the therapeutic range for Epo delivered as recombinant protein. Higher doses, still on the linear portion of the dose-response curve, may be useful for the delivery of proteins requiring greater therapeutic concentration or with a short half-life. Both the rate of rise and the peak levels of Epo and hematocrit were proportional to input dose. Transduction of primary human myotubes also resulted in dose-dependent Epo secretion indicating that human muscle is also a potential target for AAV vectors.
The injected muscle was the predominant site of gene transfer and expression after i.m. delivery of vector. Histochemical staining for β-gal at the site of injection, and the absence of enzymatically detectable β-gal activity in other tissues, support this concept. Comparison of systemic levels of human Epo obtained from the i.m. and i.v. routes of administration of AAV-Epo also indicates that muscle was the predominant cellular target of the vector, since the biologic effect of i.m. injection was 50-fold greater than i.v. delivery. However, we cannot exclude the possibility that other cells present in muscle, such as endothelial cells, fibroblasts, or adipocytes (31) were transduced in vivo and expanded to fuse with existing myotubes.
Gene delivery and expression of encapsidated vector was more efficient than with plasmid DNA. This finding may be due to the ability of vector capsid proteins to enhance rapid nuclear translocation of viral particles (32). In contrast, plasmid DNA is sequestered outside the nucleus following microinjection (33). The observation that skeletal muscle is a suitable tissue for AAV-mediated gene transfer may reflect the presence of multiple target nuclei or improved cytoplasmic-nuclear transport of vector DNA in skeletal muscle.
High levels of transgene expression have been demonstrated following delivery of adenoviral vectors in vivo (14, 34), although most reports reveal relatively short-term duration of expression, due to immune-mediated loss of transduced cells (35, 36). Intramuscular AAV-lacZ administration resulted in sustained transgene expression, despite low-grade local immune reaction detected at later time points. It is unclear whether this reaction was due to AAV capsid proteins, other cellular contaminants, adenoviral proteins, or expression of bacterial β-gal. The late immunologic response suggests that antibody reactivity was elicited from increasing amounts of bacterial β-gal expressed.
Sustained expression of human Epo was also observed following administration of AAV-Epo. Since the human and murine Epo molecules share ≈80% identity in protein sequence (37), it was anticipated that human Epo would stimulate murine erythropoiesis (3, 7). Even the lowest vector dose resulted in slightly elevated hematocrit while Epo was undetectable by the human-specific ELISA, demonstrating high biological activity of the hormone. Despite the differences between the human and murine protein sequences, high levels of human Epo were maintained in the murine circulation, suggesting tolerance of BALB/c mice to human Epo. Recent results using adenoviral vectors suggest that viral expression results in host immune responses that may limit transgene stability (38, 39). Also, antibody response on first exposure to viral capsid protein eliminates the ability to readminister adenoviral vectors (40). Our preliminary data suggests that readministration of AAV vectors is possible. The expression of autologous proteins may be an important requirement for persistent expression (41). It is notable that injection of AAV-Epo into C57BL6, unlike BALB/c, resulted in a loss of circulating reticulocytes and fatal anemia (data not shown). However, the expression of autologous murine Epo from an AAV-vector resulted in equal increases in erythropoiesis in both BALB/c and C57BL6 mice (data not shown). This observation highlights the importance of autologous transgene protein expression in animal strains with differing immunological responsiveness (41).
The long-term expression after a single i.m. administration may be in part explained by vector delivery to nondividing cells. Transduction of differentiated nondividing cells by AAV in vivo is not without precedent. Kaplitt et al. (23) reported transduction of rat neurons in situ, and Ali et al. (25) have demonstrated successful gene transfer into the mouse retina. Importantly, our experiments demonstrate that primary cells are transduced in vitro and in vivo without efforts to promote DNA damage (20, 42) or to deliberately injure the muscle (14, 43). However, we cannot exclude the possibility that adult myoblasts, stimulated to divide by the injection protocol, were transduced in vivo.
The template for transcription in muscle was not defined in this study; however, the steady rise in circulating Epo over time may reflect the slow conversion of single-stranded vector DNA to transcriptionally active templates, either by second-strand synthesis (24, 44), or by reannealing of AAV strands of opposite polarity. The form of the vector in muscle, whether extra-chromosomal or integrated, is currently unknown.
Skeletal muscle gene therapy can result in sustained and systemic delivery of therapeutic proteins which may be used for the treatment of anemia, inherited coagulopathies, endocrinologic disorders, dyslipidemia, or metabolic storage diseases. Alternatively, AAV vectors may be utilized for the local delivery of therapeutic proteins directly to muscle. The novel finding that AAV vectors are capable of efficient gene transfer to murine and human muscle resulting in dose-dependent and persistent gene expression provides the basis for diverse gene therapy applications. It is also conceivable that high concentrations of locally secreted proteins, for example, in the coronary vasculature via smooth muscle cells, or directly to diseased cardiac muscle may prove to be beneficial. AAV-based vectors may be the basis for a new, practical and effective method of gene delivery to striated muscle_in vivo_, resulting in sustained delivery of a variety of therapeutic proteins.
Acknowledgments
We thank Susan Elliger, Carl Elliger for excellent technical assistance, Dr. Edward Aboujaoude, and Pat Logan for help with morphometric analysis. We thank Drs. Terry R. Flotte, William B. Guggino, Susan Craig, and David Sidransky for helpful discussions and review of this manuscript. This work was partially supported by the W. W. Smith Foundation (B.J.B. and P.D.K.), Clinical Research Grant 94–0431 from the National Foundation March of Dimes (P.D.K.), National Institutes of Health Grants HL-27867 (P.D.K.), HL-07227 (X.C.), and HL51811 (W. B. Guggino). B.J.B. is the recipient of a Richard Starr Ross Clinician Scientist Award from the Johns Hopkins University School of Medicine.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “_advertisement_” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: AAV, adeno-associated virus; rAAV, recombinant AAV; AAV-lacZ, rAAV encoding bacterial β-galactosidase; Epo, erythropoietin; AAV-Epo, rAAV encoding human Epo; DM, differentiation medium; CMV cytomegalovirus; β-gal, β-galactosidase; SV40, simian virus 40; ITR, inverted terminal repeat.
References
- 1.Barr E, Leiden J M. Science. 1991;254:1507–1519. doi: 10.1126/science.1962212. [DOI] [PubMed] [Google Scholar]
- 2.Dhawan J, Pan L C, Pavlath G K, Travis M A, Lanctot A M, Blau H M. Science. 1991;254:1509–1512. doi: 10.1126/science.1962213. [DOI] [PubMed] [Google Scholar]
- 3.Osborne W R, Ramesh N, Lau S, Clowes M M, Dale D C, Clowes A W. Proc Natl Acad Sci USA. 1995;92:8055–8058. doi: 10.1073/pnas.92.17.8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chowdhury J R, Grossman M, Gupta S, Chowdhury N R, Baker J R, Jr, Wilson J M. Science. 1991;254:1802–1805. doi: 10.1126/science.1722351. [DOI] [PubMed] [Google Scholar]
- 5.Blau H M, Springer M L. N Engl J Med. 1995;333:1554–1556. doi: 10.1056/NEJM199512073332308. [DOI] [PubMed] [Google Scholar]
- 6.Yao S N, Kurachi K. Proc Natl Acad Sci USA. 1992;89:3357–3361. doi: 10.1073/pnas.89.8.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tripathy S K, Goldwasser E, Lu M M, Barr E, Leiden J M. Proc Natl Acad Sci USA. 1994;91:11557–11561. doi: 10.1073/pnas.91.24.11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naffakh N, Pinset C, Montarras D, Li Z, Danos O, Heard J M. Gene Ther. 1996;7:11–21. doi: 10.1089/hum.1996.7.1-11. [DOI] [PubMed] [Google Scholar]
- 9.Leiden J M. N Engl J Med. 1995;333:871–873. doi: 10.1056/NEJM199509283331310. [DOI] [PubMed] [Google Scholar]
- 10.Ali N M, Lemoine N R, Ring C J A. Gene Ther. 1994;1:367–384. [PubMed] [Google Scholar]
- 11.Wolff J A, Malone R W, Williams P, Chong W, Acsadi G, Jani A, Felgner P. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 12.Acsadi G, Jani A, Massie B, Simoneau M, Holland P, Blaschuk K, Karpati G. Hum Mol Genet. 1994;3:579–584. doi: 10.1093/hmg/3.4.579. [DOI] [PubMed] [Google Scholar]
- 13.Roe T, Renolds T C, Yu G, Brown P O. EMBO J. 1993;12:2099–2108. doi: 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis H L, Demeneix B A, Quantin B, Coulombe J, Whalen R G. Hum Gene Ther. 1993;4:733–740. doi: 10.1089/hum.1993.4.6-733. [DOI] [PubMed] [Google Scholar]
- 15.Gussoni E, Pavlath G K, Lanctot A M, Sharma K R, Miller R G, Steinman L, Blau H M. Nature (London) 1992;356:435–438. doi: 10.1038/356435a0. [DOI] [PubMed] [Google Scholar]
- 16.Muzyczka N. Curr Top Microbiol Immunol. 1992;158:97–129. doi: 10.1007/978-3-642-75608-5_5. [DOI] [PubMed] [Google Scholar]
- 17.Berns K I, Linden R M. BioEssays. 1995;17:237–245. doi: 10.1002/bies.950170310. [DOI] [PubMed] [Google Scholar]
- 18.Flotte T R, Solow R, Owens R A, Afione S, Zeitlin P L, Carter B J. Am J Respir Cell Mol Biol. 1992;7:349–356. doi: 10.1165/ajrcmb/7.3.349. [DOI] [PubMed] [Google Scholar]
- 19.Podsakoff G, Wong K K, Jr, Chatterjee S. J Virol. 1994;68:5656–5666. doi: 10.1128/jvi.68.9.5656-5666.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexander I E, Russell D W, Miller A D. J Virol. 1994;68:8282–8287. doi: 10.1128/jvi.68.12.8282-8287.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flotte T R, Afione S A, Conrad C, McGrath S A, Solow R, Oka H, Zeitlin P L, Guggino W B, Carter B J. Proc Natl Acad Sci USA. 1993;90:10613–10617. doi: 10.1073/pnas.90.22.10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flotte T R, Afione S A, Zeitlin P L. Am J Respir Cell Mol Biol. 1994;11:517–521. doi: 10.1165/ajrcmb.11.5.7946381. [DOI] [PubMed] [Google Scholar]
- 23.Kaplitt M G, Leone P, Samulski R J, Xiao X, Pfaff D W, O’Malley K L, During M J. Nat Genet. 1994;8:148–154. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- 24.Fisher K J, Gao G P, Weitzman M D, Dematteo R, Burda J F, Wilson J M. J Virol. 1996;70:520–532. doi: 10.1128/jvi.70.1.520-532.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ali R R, Reichel M B, Thrasher A J, Levinsky R J, Kinnon C, Kanuga N, Hunt D M, Bhattacharya S S. Hum Mol Genet. 1996;5:591–594. doi: 10.1093/hmg/5.5.591. [DOI] [PubMed] [Google Scholar]
- 26.Graham F L, Smiley J, Russell W C, Naiva R. J Gen Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 27.Wigler M, Pellicer A, Silverstein S, Axel R, Urlaub G, Chasin L. Proc Natl Acad Sci USA. 1979;76:1373–1376. doi: 10.1073/pnas.76.3.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanes J R, Rubenstein J L R, Nicolas J. EMBO J. 1986;5:3133–3142. doi: 10.1002/j.1460-2075.1986.tb04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whalen R G, Davis H L. Hum Gene Ther. 1995;4:151–159. doi: 10.1089/hum.1993.4.2-151. [DOI] [PubMed] [Google Scholar]
- 30.Blau H M, Dhawan J, Pavlath G K. Trends Genet. 1993;9:269–274. doi: 10.1016/0168-9525(93)90012-7. [DOI] [PubMed] [Google Scholar]
- 31.Blau H M, Webster C. Proc Natl Acad Sci USA. 1981;78:5623–5627. doi: 10.1073/pnas.78.9.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hunter L A, Samulski R J. J Virol. 1992;66:317–324. doi: 10.1128/jvi.66.1.317-324.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dowty M E, Williams P, Zhang G, Hagstrom J E, Wolff J A. Proc Natl Acad Sci USA. 1995;92:4572–4576. doi: 10.1073/pnas.92.10.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stratford-Perricaudet L D, Makeh I, Perricaudet M, Briand P. J Clin Invest. 1992;90:626–630. doi: 10.1172/JCI115902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Y, Janich S, Cohn J A, Wilson J M. Proc Natl Acad Sci USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilgenkrantz H, Duboc D, Juillard V, Douton D, Pavirani A, Guillet J G, Briand P, Kahn A. Hum Gene Ther. 1995;6:1265–1274. doi: 10.1089/hum.1995.6.10-1265. [DOI] [PubMed] [Google Scholar]
- 37.Galson D L, Tan C C, Ratcliffe P J, Bunn H F. Blood. 1993;82:3321–3326. [PubMed] [Google Scholar]
- 38.Yang Y, Ertl H C, Wilson J M. Immunity. 1994;1:433–442. doi: 10.1016/1074-7613(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Nunes F A, Berencsi K, Gonczol E, Engelhardt J F, Wilson J M. Nat Genet. 1994;7:362–369. doi: 10.1038/ng0794-362. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, Jooss K U, Su Q, Ertl H C J, Wilson J M. Gene Ther. 1996;3:137–144. [PubMed] [Google Scholar]
- 41.Tripathy S K, Black H B, Goldwasser E, Leiden J M. Nat Med. 1996;2:545–550. doi: 10.1038/nm0596-545. [DOI] [PubMed] [Google Scholar]
- 42.Russell D W, Miller A D, Alexander I E. Proc Natl Acad Sci USA. 1994;91:8915–8919. doi: 10.1073/pnas.91.19.8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis H L, Whalen R G, Demeneix B A. Hum Gene Ther. 1993;4:151–159. doi: 10.1089/hum.1993.4.2-151. [DOI] [PubMed] [Google Scholar]
- 44.Ferrari F K, Samulski T, Shenk T, Samulski R J. J Virol. 1996;70:3227–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]