Therapeutic Antitumor Response After Immunization with a Recombinant Adenovirus Encodinga Model Tumor-Associated Antigen (original) (raw)
. Author manuscript; available in PMC: 2007 Aug 21.
Published in final edited form as: J Immunol. 1996 Jan 1;156(1):224–231.
Abstract
Recombinant adenovirus (rAd), deleted of critical genes that enable viral replication and replaced with genes encoding heterologous proteins, has been shown to be a safe and effective vector in gene therapy studies. To evaluate a potential role for rAd as an immunogen, we used two different replication-defective type 2 rAds encoding the model Ag, _β_-galactosidase (_β_-gal). To determine whether rAd elicited the kind of immune responses therapeutic in an anti-tumor setting, the _β_-gal-expressing adenocarcinoma, CT26.CL25, was used. Splenocytes from BALB/c mice immunized with 1 × 107 infectious units (iu) of rAd demonstrated anti-_β_-gal activity after in vitro culture with the relevant Ld _β_-gal peptide. Adoptive transfer of these same splenocytes produced dramatic regression of established pulmonary metastases. However, when tumor-bearing mice were treated with 1 × 107 iu of rAd, no reduction in established disease was observed even when rAd was given with exogenous IL-2. To increase the viral dose delivered to each animal, we used an E1/E4-deleted rAd that could be grown to much higher titers. Significant reduction occurred when 10-fold more rAd (1 × 108 iu) was administered. Exogenous IL-2 administration with 1 × 108 iu of rAd resulted in augmentation of this anti-tumor effect. These findings demonstrate that when using a nonreplicating virus, the viral dose is directly related to the immune response generated. These data constitute the first reported use of rAd in the treatment of an established experimental cancer and may have implications for the treatment of human cancer.
CTL recognizing tumor-associated Ags (TAA)3 can mediate tumor regression in human and murine systems (1-3). The genes encoding these TAA have been cloned recently (4-12), and recombinant viruses may be effective vectors capable of eliciting specific anti-neoplastic responses. Most recently, using a model tumor Ag system, immunizations with recombinant vaccinia virus and recombinant fowlpox virus have been shown to stimulate a CTL response sufficient to produce reductions of tumor burden in vivo. Moreover, these antitumor effects were significantly enhanced when viral vectors were administered with exogenous cytokines or when the recombinant virus simultaneously encoded cytokine genes (13, 14). After cell infection, these viral vectors presumably target the heterologous protein for the class I pathway of Ag processing and presentation and potential recognition by CD8+ T-lymphocytes (TCD8+). While this mechanism appears to be highly efficient based on the previous work with recombinant vaccinia and fowlpox viruses, it remains unclear how a particular vector with its unique kinetics of protein expression and ability to replicate influences the antitumor response.
Type 2 adenovirus is replication competent and ubiquitous among human beings. Adenovirus has been shown to be an effective immunizing vector with relatively low morbidity in humans. For over 30 yr, military recruits in the United States and Canada ingested adenovirus to vaccinate against adult respiratory disease. Little, if any, morbidity has been associated with adenovirus in this setting (15, 16).
More recently, recombinant adenovirus (rAd) has been used as a vector for the delivery of gene therapy in patients with cystic fibrosis, atherosclerotic disease, and _α_1-antitrypsin deficiency, among others. With genetic deletions that attenuate the virus and subsequent insertions of new genetic material, rAd effectively expresses these heterologous proteins in vitro and in vivo with relatively few apparent side effects (17-25). Although cellular immune responses against some rAd vectors have been reported, further genetic manipulation of the vector has been shown not only to improve transduction efficiency but also to decrease antiadenoviral responses (26, 27).
Multiple modes of administration of adenovirus have been used. Work in humans and mice has shown that adenovirus can express heterologous proteins when administered intranasally and i.m. (22-24, 28-30). The efficacy of the oral dosing among military recruits also offers a potential route of administration. Moreover, rAd can exhibit tissue tropism, which is dependent on the route of administration. Transduction efficiency of rAdin rats has been shown to vary with different routes of administration (31).
The purpose of this study was to examine how rAd’s heterologous protein expression and replication modulate an antitumor immune response. We used _β_-galactosidase (_β_-gal) as a model TAA in an _N_-nitroso-_N_-methylurethane-induced murine BALB/c (H-2d) murine colon adenocarcinoma and a replication-defective rAd that expressed _β_-gal. After in vitro stimulation with _β_-gal peptide, splenocytes from mice immunized with rAd generated an antitumor response in vitro, and their adoptive transfer was sufficient to reduce tumor burden in vivo. Active immunization to treat established tumor was enhanced by exogenous IL-2 and increasing the viral dose administered to the mouse. Independent of the effect of exogenous IL-2, our data suggested that changes in viral dose may modulate the immune response against _β_-gal-expressing tumors.
Materials and Methods
Cell lines
CT26.WT is a clone of the _N_-nitroso-_N_-methylurethane-induced BALB/c (H-2d) undifferentiated colonic adenocarcinoma, CT26 (32). After being transduced with LacZ, the tumor was subcloned to produce the _β_-gal-expressing CT26.CL25, as previously described (13). As a negative control in 51Cr release assays, a clone of the mouse thy moma EL4 (H-2b) that had been transfected with _β_-gal, called E22, was used. The human embryonic kidney cell line, 293 (American Type Culture Collection, Rockville, MD), was used to expand and titer all adenovirus.
All cell lines, except for 293, were grown and maintained in RPMI 1640 containing 10% heat-inactivated fetal bovine serum (Biofluids, Rockville, MD), 0.03% L-glutamine, 100 _μ_g/ml streptomycin, 100 U/ml penicillin, and 50 _μ_g/ml gentamicin sulfate (National Institutes of Health Media Center). The cell line 293 was grown in improved minimum essential medium that contained 10% heat-inactivated fetal bovine serum (Biofluids), 0.03% L-glutamine, 100 _μ_g/ml streptomycin, 100 U/ml penicillin, and 50 _μ_g/ml gentamicin sulfate (National Institutes of Health Media Center). CT26.CL25 and E22 were grown in the presence of 400 _μ_g/ml G418 (Life Technologies, Inc., Grand Island, NY).
rAd
Two types of recombinant adenoviridae, rAd-CMV1 and rAd-CMV2, expressing _β_-gal were used in these studies (Fig. 1). The backbone vectors for both recombinant adenoviridae were derived from the type 2 adenovirus. In both vectors, there was a complete E1A and a partial E1B deletion spanning 356 to 3327 bp. The E1 transcription unit normally expresses the major protein. 289R, which has the primary, if not exclusive, _trans_-activating function that induces the transcription of other early regions, i.e., delayed early. The deletion of E1 thus results in a nonreplicating viral construct.
FIGURE 1.
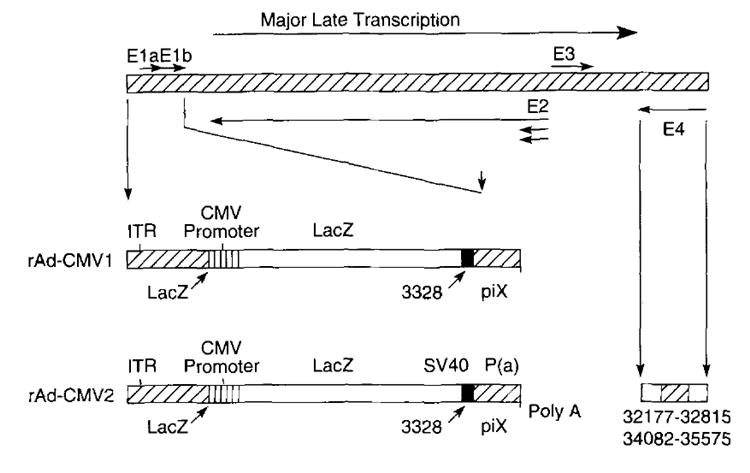
Two types of rAd, rAd-CMV1 and rAd-CMV2, were used in these studies. The back-bone vector is derived from the type 2 adenovirus. Both vectors have a complete E1A and partial E1B deletion spanning 356 to 3,327 bp. The LacZ gene is inserted into the E1 region under the control of the CMV immediate early promoter. rAd-CMV1 has no polyadenylation, while rAd-CMV2 has SV40 polyadenylation. In addition to the deletions in the E1 region, rAd-CMV2 has deletions in the E4 region spanning 32,177 to 32,815 bp and 34,082 to 35,575 bp.
In both rAd-CMV1 and rAd-CMV2, the LacZ gene is inserted into the E1 region and is under the control of the CMV immediate early promoter, one of the strongest promotional elements (33). rAd-CMV1 has no polyadenylation: rAd-CMV2 has SV40 polyadenylation. rAd-CMV2 vector also has deletions in the E4 region, covering 32,177 to 32,815 and 34,082 to 35,575 bp, while still retaining the portion in E4 needed for efficient viral assembly. These further deletions also provide more genetic space for potential dual and triple constructs.
rAd-mOV_γ_ was used as a negative control vector in experiments using rAd-CMV1. rAd-mOV_γ_ uses the same adenoviral backbone as rAd-CMV1 but expresses the mOV_γ_ chimeric T cell Ab receptor instead of the _β_-gal gene (34). rAd-Empty was used as a _β_-gal-negative control vector in those experiments using rAd-CMV2. rAd-Empty is identical with rAd-CMV2, except that both the E1-and the E4-deleted regions were not replaced with genetic material for heterologous proteins.
The viruses were propagated in line 293 cells, which constitutively express the E1 protein of Ad2. After amplification, the rAd-CMV1 and the rAd-mOV_γ_ viruses were purified using two rounds of cesium chloride gradient centrifugation. 293 cells were then infected, and plaques were counted by visual inspection approximately 2 wk after infection (35). After amplification and purification, available titers of rAd-CMV1 rose to 1 × 108 infectious units (iu)/ml.
To study the effects of higher viral titers, rAd-CMV2 was used. After amplification and purification, rAd-CMV2 was available in titers of up to 1 × 1011 iu/ml. rAd-CMV2 was titrated using serial dilutions of virus in a 96-well plate containing 293 cells. After approximately 4 days, an immunofluorescent anti-adenovirus Ab was added to the plates, and titer was calculated based on fluorescence (36). In titering calculations performed on identical samples of recombinant adenovirus, few, if any, differences were found between plaque-forming units and infectious units (data not shown); therefore, plaque-forming units and infectious units are considered equal, and for consistency, all viral doses will be reported as infectious units. All constructs used in these studies were generously supplied by Genzyme Corp. (Framingham, MA).
Peptide
The nonamer TPHPARIGL is the _β_-gal peptide naturally presented by the MHC class 1 Ld molecule (37) and spans amino acids 876–884 of _β_-galactosidase. The peptide used in these studies was synthesized by Peptide Technologies (Washington DC) to a purity >99% as determined by HPLC and amino acid analysis.
Effector cells
Effector cells were generated from female BALB/c mice, 8 to 12 wk old (Animal Production Colonies, Frederick Research Facility, National Institutes of Health, Frederick, MD). Mice were immunized with recombinant virus, and spleens were harvested 14 to 21 days after immunization. Single cell suspensions of splenocytes were then cultured in T-75 flasks at a concentration of 6 × 106 cells/ml. Cells were cultured in 30 ml of RPMI 1640 medium with 10% inactivated fetal bovine serum (Biofluids), 5 × 10−5 M 2-ME (Life Technologies), 0.03% L-glutamine, 100 _μ_g/ml streptomycin, 100 _μ_g/ml penicillin, and 50 _μ_g/ml gentamicin sulfate (National Institutes of Health Media Center). In addition, 1 _μ_g/ml of the _β_-gal peptide TPHPARIGL was added to the medium. Splenocytes were harvested from flasks after 6 or 7 days of in vitro culture. Before use, the cells were washed with HBSS.
51Cr release assay
51Cr release assays were performed over 6 h, as described previously (38). In brief, 2 × 106 target cells were incubated over 90 min with 200 _μ_Ci of Na51CrO4, (51Cr). During labeling, targets could also he pulsed with 1 _μ_g/ml (~1 _μ_M) of the relevant peptide or infected with viruses at an multiplicity of infection of 10:1.
After labeling, target cells were mixed with effector cells at the indicated E:T ratios. 51Cr release was determined by gamma counter and was calculated from samples in triplicate using the equation: [(experimental cpm − spontaneous cpm)/(maximal cpm − spontaneous cpm)] × 100. Data included in this report represent assays in which spontaneous release was <10% of maximum release.
In vivo adoptive immunotherapy studies
BALB/c mice were immunized with virus suspended in 0.5 ml of HBSS for i.v. tail vein injections, 0.1 ml of HBSS for left flank i.m. injections, and 0.02 ml of HBSS for left nares intranasal administration. All mice were randomized before immunization and received 1 × 107 iu rAd-CMV1 or rAd-mOV_γ_ via either i.v. or i.m. routes. Mice were immunized intranasally with 1 × 106 iu rAd-CMV1 or rAd-mOV_γ_. Twenty-one days after immunization, spleens were harvested, and effector cells were generated in vitro.
Pulmonary metastases were established by i.v. tail vein injections, as described previously (39). Five × 105 cells per mouse of either CT26.WT or CT26.CL25 were administered i.v.; 2 × 107 or 2 × 106 effector cells were then adoptively transferred into BALB/c mice bearing 3-day pulmonary metastases. In groups randomized to receive IL-2, i.p. administration of 90,000 IU of rIL-2 twice a day was started 12 h after adoptive transfer and was continued for 3 days.
On day 11 after tumor injection (8 days after the administration of effector cells), mice were killed. Lungs were excised and examined for the presence of pulmonary metastases in a randomized blinded fashion (39).
In vivo active treatment studies
BALB/c mice were challenged with 105 cells of either CT26.WT or CT26.CL25 by tail vein injection. Mice were then randomized to groups composed of five or six mice per group. On day 3 after tumor challenge, they were immunized i.v. with one of four doses (109, 108, 107, or 106 iu) of either rAd-CMV2 or rAd-Empty. Mice were then randomized to receive 1L-2. Administration of rIL-2 (90,000 IU) twice a day i.p. began 24 h after virus administration and was continued for 3 days. On day 14 after tumor challenge (day 11 after immunization), mice were killed, and their lungs were examined for metastases in a blinded manner (39).
Statistical analysis
Because some of the lungs of the mice had pulmonary metastases “too numerous to count,” an arbitrary cutoff wits set. When the evaluator reached a number >250, or in some experiments >500, he or she stopped counting and designated the lungs as having >250 (or >500) pulmonary metastases (shorter duration model allows the examiner to count an increased number of much smaller pulmonary metastases). Because of this arbitrary cutoff, the data did not follow a normal distribution. Thus, statistical evaluation of the data was performed using nonparametric two-tailed Wilcoxon’s test.
Results
rAd elicits specific CTL response after single immunization
To determine whether a specific lytic CTL response could be elicited against _β_-gal expressed by recombinant adenovirus, BALB/c mice were immunized i.v. with two doses of rAd-CMV1 or rAd-CMV2 (Fig. 2). Effector cells generated in vitro from splenocytes obtained 21 days after immunization were tested in a 51Cr release assay.
FIGURE 2.
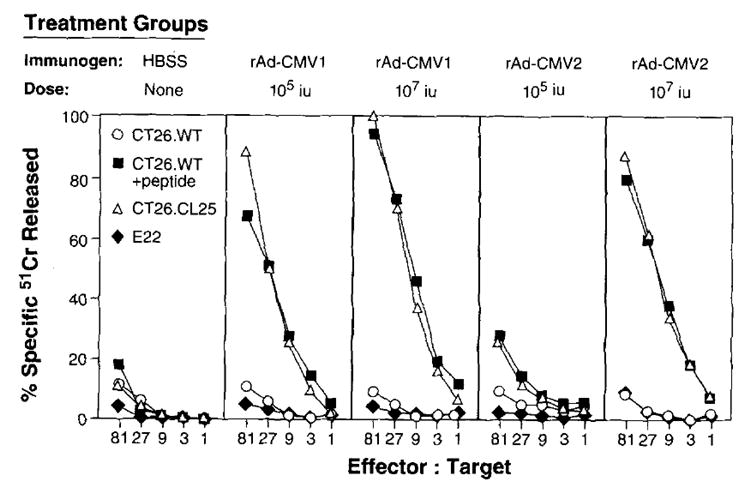
rAd can elicit a specific CTL response after a single immunization. BALB/c mice (three per group) were immunized i.v. with 1 × 105 or 1 × 107 iu of rAd-CMV1 and 1 × 105 or 1 × 107 iu of rAd-CMV2. Twenty-one days after immunization, splenocytes were harvested and cultured for 6 days in the presence of 1 _μ_g/ml of _β_-gal peptide. A 51Cr release assay was then performed. This experiment was repeated independently, and similar results were obtained.
Mice immunized with rAd-CMV1 and rAd-CMV2 generated highly specific CTL responses (Fig. 2). These responses are comparable to those seen in mice immunized with recombinant vaccinia virus and recombinant fowlpox virus (data not shown). The _β_-gal-producing tumor, CT26.CL25, and the peptide-pulsed parental CT26.WT tumor were lysed at 80 to 100% at the highest E:T ratio compared with less than 11% lysis of CT26.WT. The lysis of tumor in vitro appeared to be MHC class I restricted, since culturing the splenocytes from immunized mice with the Ld-restricted peptide stimulated CTL to lyse peptide-pulsed, MHC class I-matched, target cell lines expressing _β_-gal. Moreover, E22, an H-2b murine tumor that expresses _β_-gal, was not significantly lysed. Splenocytes from naive unimmunized mice were treated in vitro under identical conditions. These cells failed to exhibit any specific lytic activity, demonstrating that the in vitro sensitization alone could not account for the generation of lytic cells (Fig. 2, left panel).
A variety of routes of administration of rAd can be used to generate specific cytotoxic T lymphocytes effective in the adoptive immunotherapy of established pulmonary metastases
CD8+ T lymphocytes exhibiting specificity for tumor determinants have been shown to exhibit in vitro antitumor effects in adoptive immunotherapy (1, 2, 40). To examine this point in the setting of recombinant viral vectors, BALB/c mice were immunized with 1 × 107 iu, given either i.m. or i.v., or with 1 × 106 iu of rAd-CMV1 or rAd-mOV_γ_ intranasally. Three weeks after immunization, splenocytes were harvested from immunized and naive mice, and effector cells were generated by in vitro culture with _β_-gal peptide.
As shown in Figure 3, splenocytes from mice immunized with rAd-CMV1, regardless of the route of administration, exhibited specific lysis of the syngeneic targets CT26.CL25 and CT26.WT pulsed with peptide. No lysis was seen of the CT26.WT target or the allogeneic E22 cells. Furthermore, no specificity of lysis was seen with splenocytes from naive mice or those from mice immunized with rAd-mOV_γ_, indicating the need for Ag specificity during in vivo priming. Despite the high background lytic activity with splenocytes from intranasally immunized mice (Fig. 3_C_), adoptive transfer of these cells produced highly specific results in vivo (Fig. 4_C_).
FIGURE 3.
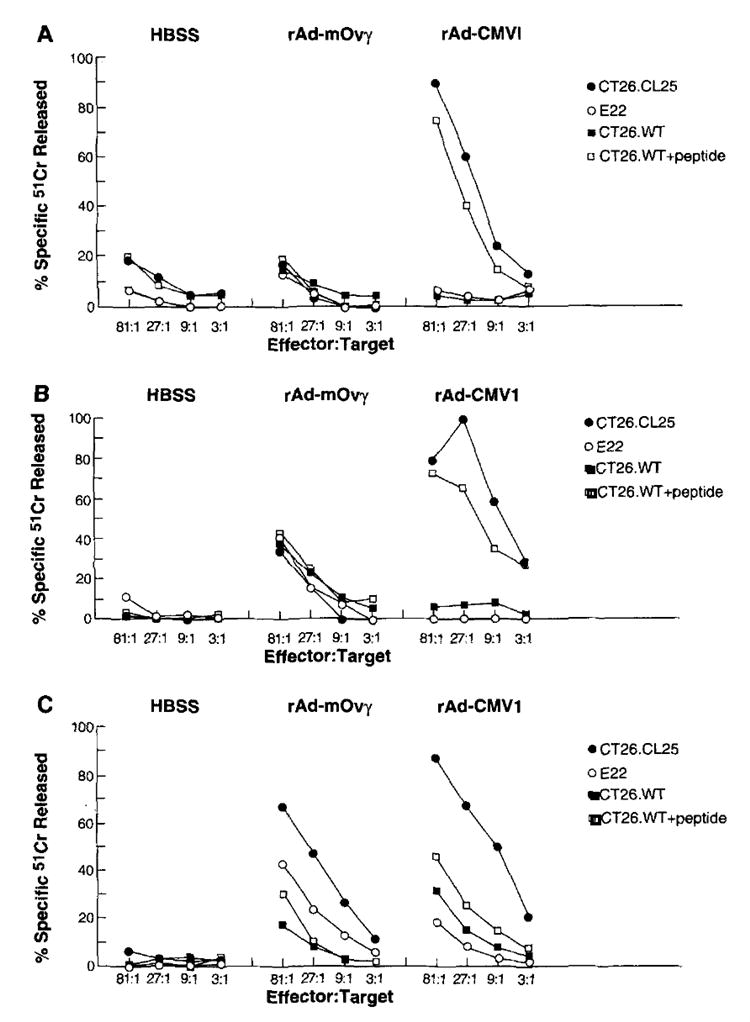
A–C, rAd administered i.v. (A), i.m. (B), and intranasally (C) can elicit specific CTL responses. BALB/c mice were immunized with 107 iu of rAd-CMV1 or rAd-mOV_γ_ i.v. (A) or i.m. (B), or with 106 iu of rAd-CMV1 or rAd-mOV_γ_ intranasally (C). Twenty-one days later, spleens were harvested and cultured in the presence of 1 _γ_g/ml of _β_-gal peptide. After 6 days in culture, a 51Cr release assay was performed on a portion of the splenocytes, while another portion was adoptively transferred by the i.v. route to mice bearing 3-day pulmonary metastases (see Fig. 4).
FIGURE 4.
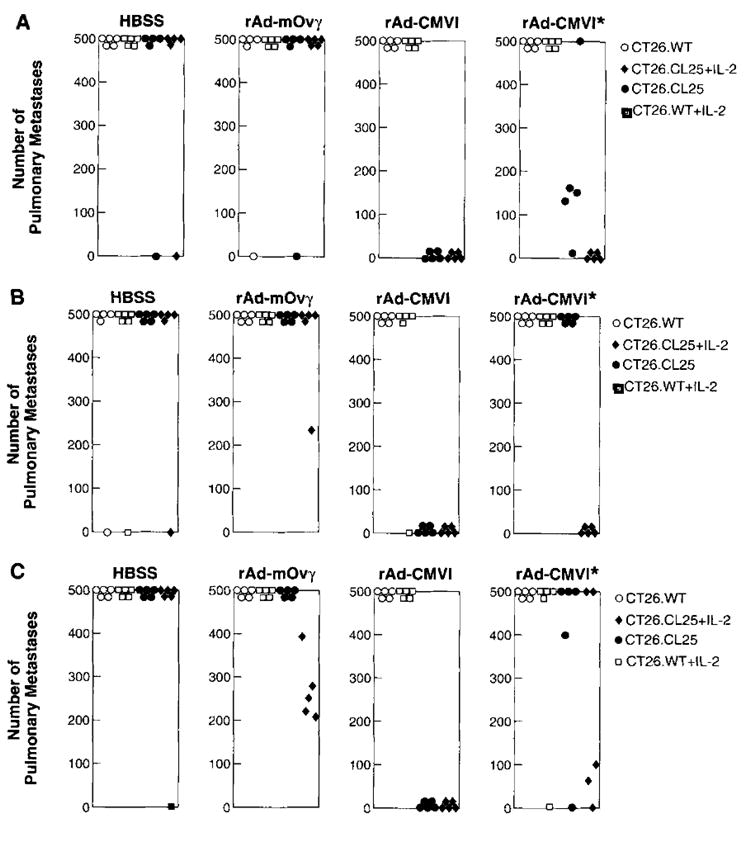
After in vitro stimulation with the relevant peptide, splenocytes from mice immunized 21 days previously were adoptively transferred into tumor-bearing mice. Immediately after adoptive transfer, those groups randomized to receive rIL-2 were given 90,000 IU of rIL-2 i.p. twice a day for 3 days. Eight days after the adoptive transfer of effector cells, mice were killed, lungs were excised, and metastases were counted in a blinded fashion. The results of the counts for each mouse are depicted in A, B, and C. The specific in vivo tumor response can be seen with splenocytes from mice immunized i.v. (A), i.m. (B), and intranasally (C). Moreover, in those groups that received only 2 × 106 effector cells that had been primed with rAd-CMV1 (all panels marked with *), the addition of exogenous rIL-2 gave antitumor responses similar to those seen in mice receiving the higher dose of effector cells. Nonparametric statistical analysis was performed using two-tailed Wilcoxon’s test. An independent repetition of these experiments yielded identical results.
In vivo, the efficacy of the effector cells for adoptive transfer was studied (Fig. 4). Lungs from the mice injected with effector cells from naive or rAd-mOV_γ_-immunized mice almost uniformly had >500 metastases/mouse. This tumor burden remained undiminished regardless of adjuvant rIL-2 administration. Mice bearing only the _β_-gal-expressing tumor that had received Ag-specific primed effector cells exhibited a significant reduction in metastases independent of the route of rAd in the immunization. Both i.v. (_p_2 = 0.0154) and i.m. priming (_p_2 = 0.0026) produced effector cells capable of significant reduction in tumor burden (Fig. 4, A and B) .This reduction was also seen in those animals that received effector cells from mice primed intranasally (Fig. 4_C_) with only 1 × 106 iu of a nonaerosolized virus. Despite the full log lower viral dose used to immunize this group, tumor-bearing mice that received these effector cells had a significant reduction of their tumor burden (_p_2 = 0.0026).
In all three groups, adoptively transferring 1 log lower effector cells eliminated the significant therapeutic effect (_p_2 > 0.05 for all groups) unless the mice received concomitant administration of 90,000 IU of rIL-2 i.p. twice daily over 3 days beginning on the day of adoptive transfer (i.v. _p_2 = 0.00154; i.m. _p_2 = 0.0026; intranasal _p_2 = 0.054). It appears that immunization with rAd-CMV1 is an effective immunogen, and that after in vitro restimulation, a sufficient number of effector cells are generated that enable these lymphocytes to reduce the in vivo tumor burden. These data also suggest the underlying importance of IL-2 in possible in vivo expansion of CTL, thus reducing the dose of effector cells needed.
In vivo immune response is a function of the rAd dose administered
We began active immunotherapy experiments using rAd-CMV1. Given the efficacy of the dose (1 × 107 iu/mouse) used in adoptive transfer experiments, all initial experiments were performed at this dosage, which was the highest available titer we could obtain and administer.
Active immunization with 1 × 107 iu/mouse did not affect tumor burden in mice inoculated with 5 × 105 tumor cells. In experiments examining hepatic _β_-gal expression after i.v. immunization, persistent expression could be seen up to 35 days (P. W. Chen, M. Wang, V. Bronte, Y. Zhai, S. A. Rosenberg, and N. P. Restifo, unpublished observations), corroborating data from other groups (28). When tumor inoculation was lowered to 1 × 105 cells, thus extending the time to death from disease onset, immunization with rAd-CMV1 was also not therapeutic regardless of whether IL-2 was administered contemporaneously.
We hypothesized that the viral dose was limiting the effectiveness of our active immunization protocol. To overcome this limitation, a newer rAd vector, rAd-CMV2, with both E1 and E4 deletions was used. Because of the increased genomic space created by the additional E4 deletion, rAd-CMV2 could be easily expanded and titrated up to 1 × 1011 iu/ml.
To test whether viral titer was critical to enhance the immune response, we inoculated tumor-bearing mice with varying titers of rAd-CMV2 and its control vector, rAd-Empty. The reduction in tumor burden correlated with the amount of virus administered in a dose-dependent fashion (Fig. 5). Immunization with 1 × 108 iu of rAd-CMV2 was able to reduce the mean number of metastases from 212 in the no treatment group to 74 in the treatment group (_p_2 = 0.003). One log lower of virus inoculum (1 × 107 iu) resulted in 157 mean metastases. Thus, a greater viral load and perhaps greater Ag presentation were able to significantly reduce the amount of tumor burden (_p_2 = 0.029). The addition of IL-2 dramatically enhanced the antitumor effect of rAd-CMV2 at the higher dose and was able to reduce the treatment group mean number of metastases to 8 (_p_2 = 0.0004 compared with the group that received 1 × 108 iu of rAd-CMV2 and no IL-2), consistent with the observation that IL-2 can enhance the function of other recombinant viral vaccines (14). IL-2 administration to both rAd-Empty control groups and mice bearing CT26.WT that were vaccinated with rAd-CMV2 did not generate a significant reduction in the number of CT26.CL25 pulmonary metastases compared with those in the groups that received IL-2 alone.
FIGURE 5.
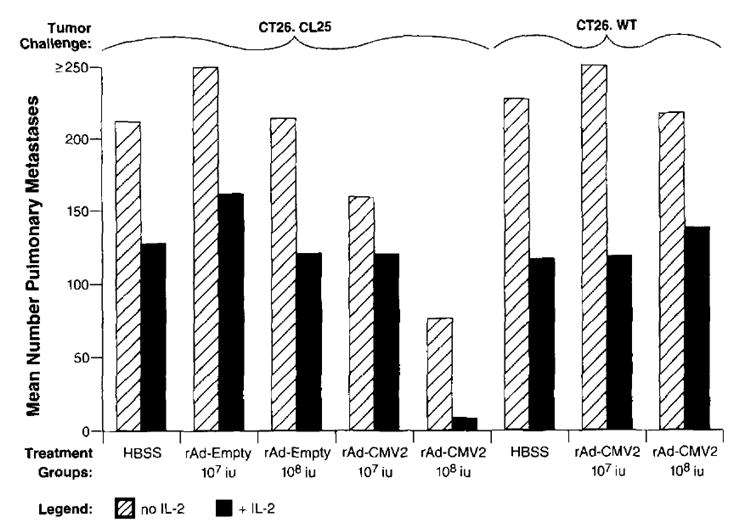
Active treatment of established pulmonary metastases correlated with viral titer administered and was improved with the addition of exogenous rIL-2. Groups of five or six mice were inoculated i.v. with 1 × 105 cells/mouse of CT26.CL25 or CT26.WT. Three days later, mice were immunized with rAd-CMV2 or rAd-Empty in doses of 1 × 107 and 1 × 108 iu/mouse. Additionally, some mice were randomized to receive 90,000 IU of exogenous rIL-2 twice a day for a total of 3 days. Fourteen days after tumor administration, mice were killed, and pulmonary metastases were counted blindly. The magnitude of the anti-tumor response, as represented by the mean number of pulmonary metastases, correlated in a dose-dependent fashion with the viral titer administered. Exogenous rIL-2 shifted the dose-response curve such that lower viral titers were needed to achieve equivalent results. Nonparametric statistical analysis was performed with a two-tailed Wilcoxon’s test. An independent repetition of this experiment confirmed these results.
Of therapeutic interest, mice tolerated rAd immunizations as high as 1 × 1010 iu. In mice immunized with 1 × 109 or 1 × 1010 iu of rAd-CMV2, complete tumor regression was noted, but the high nonspecific background killing by the anti-rAd immune response made the exact determination of the mechanism of regression unclear, although specific antitumor CTL presumably mediated this response. Evidence of nonspecific lysis can also be seen in mice receiving 1 × 108 iu of virus (Fig. 5). Nevertheless, high doses of rAd with or without IL-2 appeared to be therapeutically effective.
Preimmunization with rAd does not inhibit the immune response against a heterologous protein expressed during reimmunization with a rAd
Secondary immune responses elicited against some recombinant viruses may limit the immune response against the heterologous protein expressed by that virus upon repeated vaccination. This is particularly the case with recombinant vaccinia virus, since its nonrecombinant parent virus was used so extensively in the World Health Organization’s smallpox eradication program. To investigate the possibility that previous immunity to rAd may decrease the ability to elicit CTL after reimmunization with rAd, BALB/c mice were immunized with a control rAd-mOV_γ_. Two weeks later, preimmunized mice were reimmunized with rAd-CMV1. Fourteen days after the second injection, the splenocytes of all mice were harvested and placed in secondary culture in the presence of _β_-gal peptide. On day 7, specific _β_-gal target lysis was evaluated with a chromium release assay. As shown in Figure 6, preimmunization did not inhibit the immune response against a heterologous protein (_β_-gal) expressed by a rAd upon reimmunization. In separate experiments, rAd also did not cross-react or inhibit the anti-_β_-gal response of recombinant vaccinia virus and recombinant fowlpox virus in preimmunized or secondarily immunized animals (data not shown).
FIGURE 6.
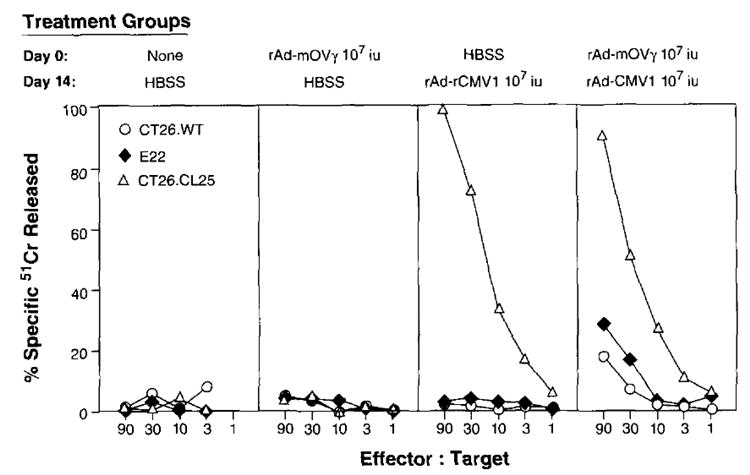
BALB/c mice were immunized with HBSS, 1 × 107 iu of rAd-CMV1 (_β_-gal-expressing rAd), or rAd-mOVγ (control). Fourteen days later, mice were reimmunized with HBSS or rAd-CMV1. On day 28, splenocytes from all mice were restimulated in culture with a 1 _μ_M concentration of the synthetic peptide TPHPARIGL for 7 days and then assayed for specific anti-_β_-gal lysis in a 51Cr release assay. The experiment was repeated with similar results.
Discussion
Previous work has shown that recombinant vaccinia and fowlpox viruses expressing TAA can mediate specific antitumor CTL responses. Recombinant viral vectors express heterologous TAA that have been inserted into the vector’s genome and introduce them into intracellular pathways of Ag procession and presentation. However, the influence of a particular vector’s Ag expression and replication in modulating an antitumor response remained unclear.
To examine these underlying mechanisms, we chose a nonreplicating recombinant adenovirus vector. rAd has been previously shown to be an immunogen in the context of anti-HBs Abs in immunized dogs, EBV envelope glycoprotein Abs in immunized cottontop tamarins, and specific CTL responses in mice immunized against the human CMV glycoprotein (41-43). These multiple in vivo studies have shown the immunogenicity of Ags expressed by recombinant adenovirus as well as their persistent expression. Given these characteristics, rAd could serve as a potentially powerful vaccination vector.
Because of the E1 deletion, the recombinant adenoviridae used in these studies cannot replicate. Yet this crippled virus is still able to infect the cell and present heterologous proteins using intracellular pathways. In contrast to vaccinia virus, rAd is unable to destroy the host cell (44) and has a prolonged period of protein expression. Recombinant vaccinia virus retains its ability to replicate, posing several potential issues. Its ability to replicate may magnify the response seen, presenting some level of unpredictability contingent on the immune status of the host organism. Additionally, its replication-competent nature may potentially pose hazards of disseminated viremia in hosts, particularly in those that are immunocompromised (45).
From the perspective of possible therapeutic implications, rAd has advantages over other vectors. Most notable is the safety of adenovirus in its clinical applications. Adenovirus is ubiquitous among humans, and its relatively benign symbiosis renders it an ideal vector. Adenovirus, in its unattenuated form, was used for over 30 yr as a vaccine among military recruits to prevent adult respiratory diseases. Little, if any, morbidity has been associated with adenovirus in this capacity (15, 16). Trials using rAd for gene therapy have also found little evidence of associated morbidity (17-25). Antiadenoviral cellular immune responses have been noted when using some adenoviral vectors (26, 46), but some of these responses were ameliorated with additional genetic manipulations of the viral vector (27). In experiments performed in this laboratory (data not shown), mice given repeated immunizations mounted responses against adenoviral proteins. However, previous exposure to a rAd did not attenuate in vitro CTL responses against the model TAA. While some types of adenovirus do have oncogenic potential in rodents, type 2 adenovirus has not been noted to be oncogenic.
The availability of multiple routes of administration using adenovirus also offers an advantage. In military recruits, adenovirus was administered orally. Although the oral route was not examined in these studies, it obviously has public health implications given the potential for facile dissemination. In the recent gene therapy trials addressing cystic fibrosis, recombinant adenovirus was delivered intranasally. The apparent differences in the transduction efficiency of tissues dependent on route of administration (31) suggests that the antitumor immune response generated could be rendered more tissue specific. In this paper, the murine work established effective priming using i.v., i.m., and intranasal routes of administration.
In the studies presented in this report rAd functioned as an immunogen. As assessed by in vitro 51Cr release assays and in vivo adoptive transfer studies, immunization with rAd specifically generated lymphocytes that lyse tumor targets which expressed the same TAA as the recombinant vector. Moreover, it appears that the priming of CTL occurs regardless of the route of rAd administration, implying that rAd may also be a highly efficient vector for intracellular Ag expression. Despite the relative paucity of APC in muscle, for example, those animals immunized i.m. are still able to generate CTL that causea significant reduction in tumor burden after adoptive transfer.
Intracellular pathways are important in the expression of heterologous proteins in rAd. We found that characteristics of the vector, such as replication competence and kinetics of expression, are crucial. With the rAd used in these studies, it became evident that increasing the viral load was a key factor in modulating the immune response. The nonreplicating property of these rAds seemed to render the viral titer a fine-tuned predictor of response. Presumbly, for every viral particle that would infect a host cell, TAA would be presented. Thus, viral load would be equivalent to Ag loading. Given the use of rAd in gene therapy studies, the influence of viral load on protein expression could be used to modulate the expression of the desired gene.
Several additional issues arise. While the doses we used were effectively immunogenic, could higher or lower doses lead to a tolerant state? Would the inundation of expressed Ag lead to high dose tolerance or would there continue to be further reductions in tumor burden? How do repeated boostings modulate the response? Moreover, while the persistence of Ag expression in adenovirus-infected cells may initially appear to render rAd a more potent vaccine, will the host eventually become tolerant to the Ag presented? Such questions are presently being addressed in this laboratory.
With the caveat in mind that the _β_-gal TAA model is an artificial one, this communication is the first, to our knowledge, to describe the use of rAd to elicit a specific CTL response that was therapeutic in both adoptive and active immunotherapy of established pulmonary metastases. While previously shown to bean effective vector for gene therapy and the prevention of infectious disease, rAd appears to have a role as an antitumor treatment modality in this murine setting. These findings suggest that recombinant adenovirus vectors expressing human melanoma TAA (35) may also enhance the generation of specific CTL responses against human tumors.
Acknowledgments
The authors thank the workers at Genzyme Corp. for providing us with recombinant adenoviruses. We also thank Paul Spiess for technical assistance, Dave Jones for help with animal experiments, and Martha Blalock for help with graphics.
Abbreviatlons used in this paper
TAA
tumor-associated Ag
TCD8+
CD8+ T-lymphocytes
rAd
recombinant adenovirus
_β_-gal
_β_-galactosidase
iu
infectious units
References
- 1.Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, Simpson C, Carter C, Bock S, Schwartzentruber D, Wei JP, White DE. Use of tumor infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. Preliminary report. N Engl J Med. 1988;319:1676. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 3.Alexander RB, Rosenberg SA. Long term survival of adoptively-transferred tumor infiltrating lymphocytes in mice. J Immunol. 1990;145:1615. [PubMed] [Google Scholar]
- 4.Boon T, Cerottini J, Van den Eynde B, van der Bruggen P, Van Pel A. Tumor antigens recognired by T lymphocytes. Annu Rev Immunol. 1994;12:337. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 5.Brichard V, Van Pel A, Wolfel T, Wolfel C, De Plaen E, Lethe B, Coulie P, Boon T. The tyrosinase genecodesfor an antigen recognired by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1993;178:489. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox AL, Skipper J, Chen Y, Henderson RA, Darrow TL, Shabanowitz J, Engelhard VH, Hunt DF, Slingluff CL., Jr Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science. 1994;264:716. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- 7.Houghton AN. Cancer antigens: immune recognition of self and altered self. J Exp Med. 1994;180:1. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardoll DM. Tumour antigens. A new look for the 1990s. Nature. 1994;369:357. doi: 10.1038/369357a0. [DOI] [PubMed] [Google Scholar]
- 9.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, Miki T, Rosenberg SA. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA. 1994;91:3515. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Roaenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robbins PF, El-Gamil M, Kawakami Y, Stevens E, Yannelli JR, Rosenberg SA. Recognition of tyrosinase by tumor-infiltrating lymphocytes from a patlent responding to immunotherapy. Cancer Res. 1994;54:3124. published erratum appears in Cancer Res 1994 Jul 15; 54(14):3952. [PubMed] [Google Scholar]
- 12.Wang RF, Robbins PF, Kawakami Y, Kang XQ, Rosenberg SA. Identitication of a gene encodinga melanoma tumor antigen recognized by HLA-A31-restricted tumor-intiltrating lymphocytes . J Exp Med. 1995;181:799. doi: 10.1084/jem.181.2.799. published erratum appears in J Exp Med 1995 Mar 1:181(3):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, Restifo NP. Active immunotherapy of cancer with a nonreplicatlng recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685. [PMC free article] [PubMed] [Google Scholar]
- 14.Bronte V, Tsung K, Rao JB, Chen PW, Wang M, Rosenberg SA, Restifo NP. IL-2 enhances the function of recombinant poxvirus-baced vaccines in the treatment of established pulmonary metastases. J Immunol. 1995;154:5282. [PMC free article] [PubMed] [Google Scholar]
- 15.Top FH., Jr Control of adenovirus acute respiratory disease in U.S. Army trainee. Yale J Biol Med. 1975;48:185. [PMC free article] [PubMed] [Google Scholar]
- 16.Chaloner-Larsson G, Contreras G, Furesz J, Boucher DW, Krepps D, Humphreys GR, Mohanna SM. Immunization of Canadian Armed Forces personnel with live types 4 and 7 adenovirus vaccines. Can J Public Health. 1986;77:367. [PubMed] [Google Scholar]
- 17.Rich DP, Couture LA, Cardoza LM, Guiggio VM, Armentano D, Espino PC, Hehir K, Welsh MJ, Smith AE, Gregory RJ. Development and fibrosis of recombinant adenoviruses for gene therapy of cystic fibrosis. Hum Gene Ther. 1993;4:461. doi: 10.1089/hum.1993.4.4-461. [DOI] [PubMed] [Google Scholar]
- 18.Kozarsky KF, Wilson JM. Gene therapy: adenovirus vectors. Curr Opin Genet Dev. 1993;3:499. doi: 10.1016/0959-437x(93)90126-a. [DOI] [PubMed] [Google Scholar]
- 19.Schneider MD, French BA. The advent of adenovirus. Gene therapy for cardiovascular disease. Circulation. 1993;88:1937. doi: 10.1161/01.cir.88.4.1937. [DOI] [PubMed] [Google Scholar]
- 20.Cristiano RJ, Smith LC, Kay MA, Brinkley BR, Woo SL. Hepatic gene therapy: efficient gene delivery and expression in primary hepatocytes utilizing a conjugated adenovirus-DNA complex. Proc Natl Acad Sci USA. 1993;90:11548. doi: 10.1073/pnas.90.24.11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen SJ, Wilson JM, Muller DW. Adenovirus-mediated gene transfer of soluble vascular cell adhesion molecule to porcine interposition vein grafts. Circulation. 1994;89:1922. doi: 10.1161/01.cir.89.5.1922. [DOI] [PubMed] [Google Scholar]
- 22.Zabner J, Petersen DM, Puga AP, Graham SM, Couture LA, Keyes LD, Lukason MJ, St George JA, Gregory RJ, Smith AE. Safety and efficacy of repetitive adenovirus-mediated transfer of CFTR cDNA to airway epithelia of primates and cotton rats. Nat Genet. 1994;6:75. doi: 10.1038/ng0194-75. [DOI] [PubMed] [Google Scholar]
- 23.Zabner J, Couture LA, Gregory RJ, Graham SM, Smith AE, Welsh MJ. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal eplthelia of patients with cystic fibrosis. Cell. 1993;75:207. doi: 10.1016/0092-8674(93)80063-k. [DOI] [PubMed] [Google Scholar]
- 24.Mastrangeli A, Danel C, Rosenfeld MA, Stratford-Perricaudet L, Perricaudet M, Pavirani A, Lecocq JP, Crystal RG. Diversity of airway epithelial cell targets for in vivo recomblnant adenovirus-mediated gene transfer. J Clin Invest. 1993;91:225. doi: 10.1172/JCI116175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crystal RG, McElvaney NG, Rosenfeld MA, Chu CS, Mastrangeli A, Hay JG, Brody SL, Jaffe HA, Eissa NT, Danel C. Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat Genet. 1994;8:42. doi: 10.1038/ng0994-42. see comments. [DOI] [PubMed] [Google Scholar]
- 26.Yang Y, Nunes FA, Berencsi K, Furth EE, Gonczol E, Wilson JM. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc Natl Acad USA. 1994;91:4407. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engelhardt JF, Ye X, Doranz B, Wilson JM. Ablation of E2A in recombinant adenoviruses improves transgene persistence and decreases inflammatory response in mouse liver. Proc Natl Acad Sci USA. 1994;91:6196. doi: 10.1073/pnas.91.13.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quantin B, Perricaudet LD, Tajbakhsh S, Mandel JL. Adenovirus as an expression vector in muscle cells in vivo. Proc Natl Acad Sci USA. 1992;89:2581. doi: 10.1073/pnas.89.7.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bessereau JL, Stratford-Perricaudet LD, Piette J, Le Poupon C, Changeux JP. In vivo and In vitro analysis of electrical activity-dependent expression of muscle acetylcholine receptor genes using adenovirus. Proc Natl Acad Sci USA. 1994;91:1304. doi: 10.1073/pnas.91.4.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallichan WS, Johnson DC, Graham FL, Rosenthal KL. Mucosal immunity and protection after intranasal immunization with recomhinant adenovirus expressing herpes simplex virus glycoprotein B. J Infect Dis. 1993;168:622. doi: 10.1093/infdis/168.3.622. [DOI] [PubMed] [Google Scholar]
- 31.Huard J, Lochmuller H, Acsadi G, Jani A, Massie B, Karpati G. The route of administration is a major determinant of the transduction efficiency of rat tissues by adenoviral recombinants. Gene Ther. 1995;2:107. [PubMed] [Google Scholar]
- 32.Brattain MG, Strobel-Stevens J, Fine D, Webb M, Sarrif AM. Establishment of mouse colonic carcinoma cell lines with different metastatic properties. Cancer Res. 1980;40:2142. [PubMed] [Google Scholar]
- 33.Boshart M, Weber F, Jahn G, Dorsch-Hasler K, Fleckenstein B, Schaffner W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovinls. Cell. 1985;41:521. doi: 10.1016/s0092-8674(85)80025-8. [DOI] [PubMed] [Google Scholar]
- 34.Hwu P, Shafer GE, Treisman J, Schindler DG, Gross G, Cowherd R, Rosenberg SA, Eshhar Z. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med. 1993;178:361. doi: 10.1084/jem.178.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhai Y, Yang JC, Kawakami Y, Spies P, Wadsworth C, Cardoza LM, Couture LA, Smith AE, Rosenberg SA. Antigen-specific tumor vaccines: development and characterization of recombinant adenoviruses encoding MART or gp100 for cancer therapy. J Immunol. 1996 In press. [PubMed] [Google Scholar]
- 36.Lynn DE. A BASIC computer program for analyzing endpoint assays. Biotechniques. 1992;12:880. [PubMed] [Google Scholar]
- 37.Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J Immunol. 1993;151:3971. [PubMed] [Google Scholar]
- 38.Restifo NP, Esquivel F, Kawakami Y, Yewdell JW, Mule JJ, Rosenberg SA, Bennink JR. Identification of human cancers deficient in antigen processing. J Exp Med. 1993;177:265. doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mule JJ, Yang JC, Lafreniere R, Shu S, Rosenherg SA. Identification of cellular mechanisms operational in vivo during the regression of established pulmonary metastases by the systemic administration of high-dose recombinant interleukin-2. J Immunol. 1987;139:287. [PubMed] [Google Scholar]
- 40.Greenberg PD. Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells. Adv Immunol. 1991;49:281. doi: 10.1016/s0065-2776(08)60778-6. [DOI] [PubMed] [Google Scholar]
- 41.Chengalvala M, Lubeck MD, Davis AR, Mizutani S, Molnar-Kimber K, Morin J, Hung PP. Evaluation of adenovirus type 4 and type 7 recombinant hepatitis B vaccines in dogs. Vaccine. 1991;9:485. doi: 10.1016/0264-410x(91)90033-3. [DOI] [PubMed] [Google Scholar]
- 42.Ragot T, Finerty S, Watkins PE, Perricaudet M, Morgan AJ. Replication-defective recombinant adenovirus expressing the Epstein-Barr virus (EBV) envelope glycoprotein gp340/220 induces protective immunity against EBV-induced lymphomas in the cottontop tamarin. J Gen Virol. 1993;74:301. doi: 10.1099/0022-1317-74-3-501. [DOI] [PubMed] [Google Scholar]
- 43.Berencsi K, Rando RF, deTaisne C, Paoletti E, Plotkin SA, Gonczol E. Murine cytotoxic T cell responqe specific for human cytomegalovirus glycoprotein B (gB) induced by adenovirus and vaccinia virus recombinants expressing gB. J Gen Virol. 1993;74:2507. doi: 10.1099/0022-1317-74-11-2507. [DOI] [PubMed] [Google Scholar]
- 44.Baxby D, Paoletti E. Potential use of non-replicating vectors as recombinant vaccines. Vaccine. 1992;10:8. doi: 10.1016/0264-410x(92)90411-c. [DOI] [PubMed] [Google Scholar]
- 45.Redfield RR, Wright DC, James WD, Jones TS, Brown C, Burke DS. Disseminated vaccinia in a military recruit wlth human immunodeficiency virus (HIV) disease. N Engl J Med. 1987;316:673. doi: 10.1056/NEJM198703123161106. [DOI] [PubMed] [Google Scholar]
- 46.Yang Y, Li Q, Ertl HCG, Wilson JM. Cellular and humoral immune responses to viral antigens create harriers to lung-directed gene therapy with recombinant adenoviruses. J Virol. 1995;69:2004. doi: 10.1128/jvi.69.4.2004-2015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]