Transcriptional control of adipocyte formation (original) (raw)
. Author manuscript; available in PMC: 2007 Aug 29.
Abstract
A detailed understanding of the processes governing adipose tissue formation will be instrumental in combating the obesity epidemic. Much progress has been made in the last two decades in defining transcriptional events controlling the differentiation of mesenchymal stem cells into adipocytes. A complex network of transcription factors and cell-cycle regulators, in concert with specific transcriptional coactivators and corepressors, respond to extracellular stimuli to activate or repress adipocyte differentiation. This review summarizes advances in this field, which constitute a framework for potential antiobesity strategies.
Introduction
Obese individuals are more likely than their lean counterparts to develop cardiovascular disease and type 2 diabetes. The increase in adiposity in these individuals results from an upsurge in both adipocyte number and size of individual fat cells. Additionally, the disproportionate increase in the visceral adipose depots in some individuals is linked to development of certain metabolic disorders. Consequently, understanding the mechanisms regulating adipose formation should provide valuable information in the fight to combat the growing incidence of obesity in the modern world.
During the last several years, investigators have embarked on a detailed and systematic endeavor to define the transcriptional events regulating preadipocyte differentiation (adipogenesis) and adipocyte function. The differentiation of preadipocytes into adipocytes is regulated by an elaborate network of transcription factors that coordinate expression of hundreds of proteins responsible for establishing the mature fat-cell phenotype. At the center of this network are the two principal adipogenic factors, PPARγ and C/EBPα, which oversee the entire terminal differentiation process. PPARγ in particular is considered the master regulator of adipogenesis; without it, precursor cells are incapable of expressing any known aspect of the adipocyte phenotype (Rosen et al., 2002). On the other hand, cells deficient in C/EBPα are capable of adipocyte differentiation; however, these C/EBPα-deficient cells are insulin resistant (El-Jack et al., 1999; Wu et al., 1999). Much of our knowledge of this complex network and the importance of PPARγ and C/EBPα comes from studies performed in established preadipocyte cell lines as well as mesenchyme-derived precursor cells. More recently, data from a variety of knockout mice have confirmed these in vitro studies showing that many components of this network are required regulators of adipocyte development and function.
The 3T3-L1 and 3T3-F422A preadipocyte cell lines originally established by Green and associates have greatly facilitated our knowledge of the molecular mechanisms controlling adipogenesis (Green and Kehinde, 1975, 1976). Although committed to the adipocyte lineage, proliferating 3T3-L1 preadipocytes exert characteristics similar to those of other 3T3 fibroblasts. Confluent 3T3-L1 preadipocytes differentiate upon exposure to the adipogenic inducers fetal bovine serum (FBS), dexamethasone, isobutylmethylxanthine, and insulin. This cocktail activates an adipogenic program, which occurs in two well-defined phases. The stimulated cells immediately reenter the cell cycle and progress through at least two cell-cycle divisions, a phase often referred to as clonal expansion. During this time, the cells express specific adipogenic transcription factors as well as cell-cycle regulators that together facilitate expression of PPARγ and C/EBPα. Following this event, the committed cells undergo terminal differentiation manifested by production of lipid droplets as well as expression of multiple metabolic programs characteristic of mature fat cells. The validity of this 3T3-L1 system as an appropriate model of adipocyte formation in the animal has been supported by many studies performed in both mouse and human tissue.
The goal of this review is to discuss the transcriptional processes controlling the conversion of progenitor mesenchymal cells into fully functional adipocytes. Emphasis will be given to the transcription factors that have been shown to respond to various effectors and that induce a well-defined component of the adipogenic process. A number of factors attenuate adipogenesis and serve to function as molecular switches in controlling the fate of the progenitors; consequently, the mechanisms by which these negative regulators inhibit the activity of the proadipogenic factors will be discussed. Finally, the review will conclude with a discussion of the recent advances in our understanding of how various coactivators and corepressors control the activity of the adipogenic transcription factors and facilitate their communication with the transcriptional machinery.
Elucidation of the network of transcription factors regulating adipogenesis
PPARγ and C/EBPα: master regulators of adipogenesis
The role of PPARγ as the master regulator of adipogenesis is supported by overwhelming evidence from both in vivo and in vitro studies. Important early evidence of the critical role of PPARγ in regulating adipogenesis came from Spiegelman and collaborators, who had worked for several years to elucidate the transcription factors regulating expression of the adipose-specific fatty acid binding protein aP2/FABP4. This endeavor resulted in identification of a nuclear factor initially referred to as ARF6 that was later shown through cloning technology to correspond to PPARγ and its heterodimeric partner, RXR (Tontonoz et al., 1994a, 1994b). A series of gain-of-function studies in which _ppar_γ was ectopically expressed in nonadipogenic mouse fibroblasts showed that PPARγ alone can initiate the entire adipogenic program, giving rise to fat cells that are capable of many of the functions of mature adipocytes (Tontonoz et al., 1994c). In attempting to understand the importance of PPARγ in the development of adipocytes, investigators found that ablation of _ppar_γ in embryonic stem (ES) cells leads to embryonic lethality at E10 due to a defect in placentation as a result of PPARγ’s participation in formation of the trophoblast (Barak et al., 1999). To circumvent this problem, alternative strategies for obtaining knockout mice were developed that supported a role for PPARγ in the formation of all fat depots, including both brown and white (Barak et al., 1999; Rosen et al., 1999). These mouse models, however, provided only partial information concerning the function of PPARγ in adipocytes since both models were subject to significant limitations. In one case, the conclusions were based on chimeric mice derived from homozygously targeted ES cells (Rosen et al., 1999). In these animals, the knockout cells failed to develop into adipocytes; whereas the wild-type-derived cells gave rise to fully functioning adipose depots. Consequently, it was difficult to assess what impact the absence of PPARγ has on adipose tissue function. The tetraploid embryo strategy of Evans and coworkers (Barak et al., 1999) generated only one mouse, which died soon after birth, but allowed these investigators to observe that PPARγ deficiency in these animals resulted in failure to form adipose tissue. The establishment of white adipose tissue (WAT)-hypomorphic _ppar_γ knockdown mice resulted in animals that were severely lipodystrophic; these data authenticate PPARγ as the master regulator of adipogenesis (Koutnikova et al., 2003).
_ppar_γ is expressed as two isoforms, ppar_γ_1 and ppar_γ_2, generated by alternative promoter usage of the same gene, which gives rise to four distinct mRNAs. ppar_γ_1, ppar_γ_3, and ppar_γ_4 mRNAs all encode the PPARγ1 polypeptide, while ppar_γ_2 mRNA encodes the corresponding PPARγ2 polypeptide, which is identical to PPARγ1 with an additional 30 amino acids present at the N terminus (Fajas et al., 1997; Meirhaeghe et al., 2003; Tontonoz et al., 1994b). PPARγ1 is expressed in many tissues, whereas PPARγ2 expression is restricted almost exclusively to adipose. Studies performed in _ppar_γ−/− mouse embryonic fibroblasts (MEFs) demonstrate that ectopic PPARγ1 is as capable of inducing adipogenesis as PPARγ2 (Mueller et al., 2002). Furthermore, adipose-selective knockout of ppar_γ_2 in the mouse gives rise to insulin-insensitive animals with reduced fat; however, they still contain substantial amounts of adipose tissue, suggesting that PPARγ1 can compensate for many of the adipogenic functions of PPARγ2 (Zhang et al., 2004a). The fact that the PPARγ2-deficient mice are insulin resistant suggests that PPARγ2 may play a selective role in regulating insulin sensitivity.
Recognition that C/EBPα functions as a principal player in adipogenesis also resulted from gain-of-function studies in cultured cells as well as establishment of appropriate knockout mice. In the former case, Freytag and associates demonstrated that ectopic expression of C/EBPα in a variety of fibroblastic cells could induce adipogenesis (Freytag et al., 1994). Similar to the PPARγ studies, establishment of C/EBPα knockout mice was subject to significant setbacks since the animals die soon after birth due to the pups’ inability to produce glucose. This phenotype results from the requirement of C/EBPα for gluconeogenesis in the liver (Wang et al., 1995). Ablation of _c/ebp_α in all tissues except the liver revealed that C/EBPα is required for formation of WAT. Interestingly, C/EBPα is not required for the formation of brown adipose tissue (BAT), an observation that currently is not understood (Linhart et al., 2001).
PPARγ can induce adipogenesis in C/EBPα-deficient MEFs, whereas C/EBPα is incapable of driving the adipogenic program in the absence of PPARγ (Rosen et al., 2002). This observation suggests that C/EBPα and PPARγ participate in a single pathway of adipose development, in which PPARγ is the dominant factor. It must be mentioned that C/EBPα does provide a critical function during terminal adipogenesis since failure to express C/EBPα results in insulin resistance in cell culture models and an inability to develop WAT in vivo (El-Jack et al., 1999; Linhart et al., 2001; Wu et al., 1999). It has been suggested that, in addition to controlling insulin action, C/EBPα is required for maintaining expression of PPARγ in the mature fat cell (Wu et al., 1999). It is possible that establishment of the adipogenic phenotype in C/EBPα-deficient brown adipocytes is due to other mechanisms (possibly other C/EBPs) that function to maintain PPARγ production.
C/EBPβ and C/EBPδ
Well before the discovery of PPARγ as the master regulator of adipogenesis, several investigators attempted to identify the mechanisms responsible for determining the differentiation of precursor cells into adipocytes. It is now established that a cascade of transcription factors eventually leads to expression of PPARγ and C/EBPα. The first indication of such a network came from the work of McKnight and associates, which suggested that two other members of the C/EBP family, C/EBPβ and C/EBPδ, are expressed earlier than C/EBPα during adipogenesis in 3T3-L1 cells and that they are responsible for regulating C/EBPα expression (Cao et al., 1991; Yeh et al., 1995). Specifically, they demonstrated that ectopic expression of C/EBPβ and C/EBPδ in 3T3-L1 preadipocytes induces C/EBPα expression and the adipogenic program in the absence of extracellular hormones. They also showed that introduction of these C/EBPs into nonadipogenic NIH 3T3 fibroblasts can induce adipogenesis without stimulating C/EBPα expression.
These studies did not address, however, the mechanisms regulating PPARγ production. Other studies aimed at identifying the early events regulating adipogenesis demonstrated a direct link between the C/EBPs and PPARγ. Specifically, ectopic expression of C/EBPβ in NIH 3T3 fibroblasts, alone or in combination with C/EBPδ, induces expression of PPARγ2 and, following exposure to PPARγ ligands, in doing so, facilitates the conversion of the cells into adipocytes (Wu et al., 1995, 1996). In agreement with McKnight et al., these studies showed that NIH 3T3 cells do not express C/EBPα, even though they accumulate abundant amounts of triglyceride in response to activation of PPARγ. Additionally, both groups observed that C/EBPδ alone possesses minimal adipogenic activity. C/EBPβ and C/EBPδ play important roles in inducing expression of C/EBPα and PPARγ. This was shown by the identification of functional C/EBP regulatory elements in the promoters of _c/ebp_α and _ppar_γ (Christy et al., 1991; Clarke et al., 1997).
In an attempt to define the sequence of events leading to terminal adipogenesis, it was proposed that C/EBPβ and C/EBPδ simultaneously control expression of both PPARγ and C/EBPα. Alternatively, some investigators have suggested that C/EBPβ induces C/EBPα and that, together, these factors regulate PPARγ expression. More recently, studies have shown that ectopic expression of C/EBPβ in Swiss fibroblasts induces PPARγ as expected but is incapable of inducing C/EBPα to any significant extent in the absence of a potent PPARγ ligand. Moreover, retroviral expression of C/EBPβ in _ppar_γ−/− MEFs also shows that C/EBPβ, in the absence of active PPARγ, is incapable of stimulating expression of _c/ebp_α (Zuo et al., 2006). It appears, therefore, that the principal pathway of adipogenesis involves induction of C/EBPβ and C/EBPδ, which then facilitate expression of PPARγ. PPARγ along with these C/EBPs then activates C/EBPα expression.
The precise role of C/EBPβ and C/EBPδ in regulating this cascade of factors has been questioned, however, in knockout mice. Specifically, Tanaka et al. (1997) demonstrated that neonatal mice lacking both C/EBPβ and C/EBPδ have a defect in their ability to produce adipose tissue; however, this defect appears to be downstream of both PPARγ and C/EBPα since both factors are expressed in the poorly differentiated adipose tissue. In contrast, MEFs obtained from these knockout mice do not express C/EBPα or PPARγ and are incapable of undergoing adipogenesis in culture when compared to wild-type cells. These data suggest that there is some redundancy in the early steps of adipogenesis in vivo where alternative pathways operate to ensure expression of PPARγ and C/EBPα. Furthermore, it appears that C/EBPβ and C/EBPδ, in addition to inducing expression of PPARγ and C/EBPα, provide other functions during terminal adipogenesis since their absence prevents terminal adipogenesis at a step downstream of PPARγ or C/EBPα. One possible function might include induction of programs responsible for production of PPARγ ligands (Hamm et al., 2001).
Identifying the factors that regulate C/EBPβ and C/EBPδ expression as well as cooperate with these C/EBPs in an adipogenic-specific manner should provide additional insight into the mechanisms regulating the commitment of mesenchymal stem cells to the adipogenic lineage. Studies from Klemm and Lane provide convincing evidence that the cAMP regulatory element-binding protein, CREB, which is activated very early during adipogenesis in 3T3-L1 cells, participates in the induction of C/EBPβ expression (Zhang et al., 2004b). This observation is consistent with earlier studies showing a role for cAMP signaling in controlling C/EBPβ expression (Cao et al., 1991) and also explains the need for inducers of cAMP (isobutylmethylxanthine) in cocktails that initiate the adipogenic program. In contrast, induction of C/EBPδ is facilitated by glucocorticoids and C/EBPβ (Cao et al., 1991).
Other adipogenic factors
Recent quantitative expression profiling utilizing both microarray and qPCR analysis of mRNAs expressed during the early phase of adipogenesis in vitro and in adipose tissue in vivo suggests that many additional transcription factors are potential components of this complex network of factors responsible for inducing adipogenic gene expression (Fu et al., 2005b; Soukas et al., 2001). Investigators have identified Krox20 as a factor that acts early in the adipogenic program and appears to contribute to induction of C/EBPβ expression. Krox20 (also known as early growth response gene 2, or Egr2) is a transcription factor that is induced immediately following exposure of cells to mitogens. Krox20 is activated early in the adipogenic program of 3T3-L1 cells and not only promotes expression of C/EBPβ but also cooperates with C/EBPβ to facilitate terminal adipogenesis (Chen et al., 2005).
The fact that these early events, including activation of CREB, Krox20, and C/EBPβ, precede induction of PPARγ and C/EBPα transcription by 1 to 2 days suggests that additional processes are required in order to facilitate terminal adipogenesis. Lane and associates, in an attempt to explain this lag, suggested that C/EBPβ does not attain the capacity to bind to C/EBP response elements in the promoters of its target genes until several hours after its appearance in the nucleus because it is bound to satellite DNA (Tang and Lane, 1999). They proposed that its release from this compartment is facilitated by changes in chromatin structure that occur during clonal expansion and terminal adipogenesis. More recently, these investigators suggested that this lag in C/EBPβ activity also results from a delay in its phosphorylation by MAPKs and GSK3, which is required for its DNA-binding activity (Tang et al., 2005). Other studies have also identified an important site of phosphorylation within a regulatory domain of C/EBPβ, but, unlike the studies of Lane, these studies suggest that phosphorylation regulates C/EBPα expression (Park et al., 2004). More recent investigations suggest that the lag between the appearance of C/EBPβ and the expression of PPARγ2 results from the time required for synthesis of additional proteins that facilitate the activity of C/EBPβ. Specifically, transcription of the Kruppel-like factor KLF5 is activated by C/EBPβ and C/EBPδ and, in concert with these C/EBPs, contributes to induction of PPARγ2 (Oishi et al., 2005). Neonatal heterozygous KLF5 knockout mice have a significant deficiency in adipose tissue formation (Oishi et al., 2005). Additionally, MEFs obtained from these KLF5+/− mice are compromised in their ability to undergo adipogenesis in culture. Studies also suggest a role for other members of the KLF family including KLF6 and KLF15 in promoting adipogenesis (Li et al., 2005; Mori et al., 2005).
It is likely that additional factors of parallel pathways are induced early and converge on PPARγ at a stage downstream of C/EBPβ and C/EBPδ, such as the helix-loop-helix (HLH) transcription factor SREBP1c/ADD-1. A potential role for SREBP1c in regulating adipogenesis derives from studies showing that its expression is significantly enhanced in 3T3-L1 adipocytes in response to insulin (Kim et al., 1998a). Additionally, ectopic expression of a dominant-negative SREBP1c was shown to inhibit preadipocyte differentiation, while overexpression of this HLH protein significantly enhances the adipogenic activity of PPARγ (Kim and Spiegelman, 1996). Expression of SREBP1c alone, however, is only capable of inducing adipogenesis to a modest extent, and additional studies suggest that SREBP1c contributes to the production of PPARγ ligands, thereby facilitating the action of PPARγ (Kim et al., 1998b). There have been other investigations linking SREBP1c to the induction of PPARγ1 through SREBP binding sites within the ppar_γ_1 and γ_3_ promoters (Fajas et al., 1999). Support for an additional pathway regulating adipogenesis derives from recent investigations into the function of STAT5 proteins. STAT5A and STAT5B facilitate transmission of cytokine signaling to a host of target genes controlling many functions in several cell types. Ablation of these STATs in mice leads to a spectrum of pathological responses primarily associated with absence of growth hormone and prolactin signaling but also leads to a 5-fold reduction in adipose tissue mass compared to that of wild-type animals (Teglund et al., 1998). This phenotype could be due to the attenuation of proadipogenic prolactin signaling; however, recent studies suggest a direct role for STAT5 in adipogenesis. Specifically, ectopic expression of STAT5A in nonadipogenic fibroblasts induces preadipocyte differentiation, which includes activation of PPARγ activity as well as accumulation of multiple fat droplets (Floyd and Stephens, 2003). The mechanisms responsible for this activity of STAT5A, however, are not known since the direct target gene (or genes) has not been identified. There have been human genetic studies, however, supporting a role for STAT5 in regulating transcription from the ppar_γ_3 promoter (Meirhaeghe et al., 2003).
An interesting series of investigations show that components of the molecular clock might also have a role in regulating both adipocyte formation and function. Specifically, MEFs lacking BMAL1 (brain and muscle ARNT-like protein 1), a transcription factor known to regulate circadian rhythm, fail to differentiate into adipocytes, and ectopic expression of BMAL1 in these cells restores adipogenesis (Shimba et al., 2005). Similarly, another component of the molecular clock, Reverbα, is induced by BMAL1 and PPARγ during adipogenesis in 3T3-L1 preadipocytes and facilitates expression of several adipogenic genes (Fontaine et al., 2003; Shimba et al., 2005). A model for the transcriptional cascade regulating adipogenesis is illustrated in Figure 1, including those factors that induce expression or activity of other adipogenic transcription factors.
Figure 1. Induction of adipogenesis by a cascade of transcription factors.
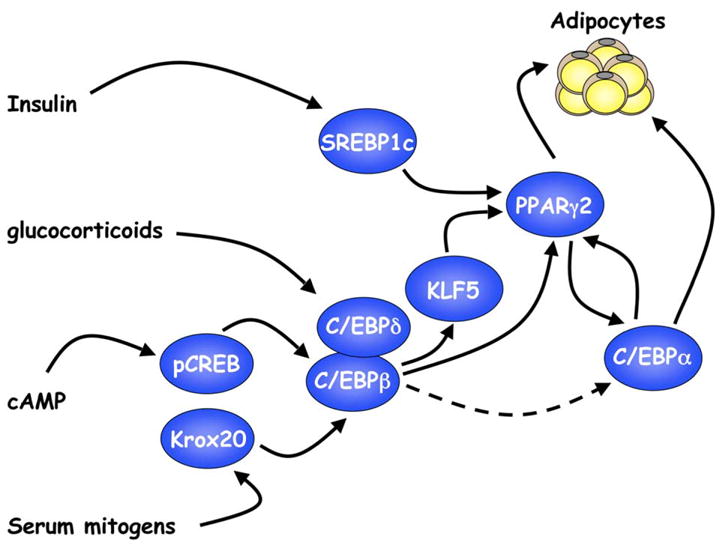
Exposure of preadipocytes to a cocktail of adipogenic inducers comprised of insulin, glucocorticoids, agents that elevate cAMP (isobutylmethylxanthine), and fetal bovine serum activates expression of several transcription factors that converge on PPARγ. PPARγ then induces C/EBPα expression, and together, these factors oversee terminal adipogenesis.
Role of clonal expansion and cell-cycle-related proteins in regulating adipogenesis
It is generally thought that clonal expansion of a population of preadipocytes is a prerequisite for their subsequent differentiation into adipocytes. Adipogenesis is induced in a confluent population of the cells by exposure to insulin, inducers of cAMP signaling, and glucocorticoids in 10% FBS. This medium, rich in mitogens, induces the entire population of cells to reenter the cell cycle (G0 to G1) and undergo at least two rounds of cell division before proceeding into terminal adipogenesis. Inhibition of cell proliferation with drugs that block S phase prevents adipogenesis, and it has therefore been suggested that adipogenesis requires mitosis to reorganize chromatin to facilitate induction of the adipogenic genes (Tang et al., 2003). Alternatively, the necessity for the clonal expansion phase may be due to a requirement for components of the cell-cycle machinery in promoting adipogenic gene expression. As mentioned above, Krox20 is an early growth response gene that is induced as confluent preadipocytes reenter the cell cycle and also plays a direct role in inducing C/EBPβ and PPARγ2 expression. The most notable cell-cycle proteins that regulate the adipogenic program are the E2F-family transcription factors and associated pocket proteins.
E2Fs, pocket proteins, and adipogenesis
Studies by Auwerx and associates have provided evidence suggesting that the E2F family of transcription factors regulate adipocyte differentiation (Fajas et al., 2002b). The data show that E2F1-3 and E2F4 have opposing effects on differentiation, which appears to be due to their differential regulation of ppar_γ_1 expression. In confluent preadipocytes, E2F4 represses PPARγ transcription through association with the pocket protein p130 and recruitment of the histone deacetylase HDAC3 to E2F response elements in the promoter of ppar_γ_1. As preadipocytes progress through clonal expansion, the abundance of E2F4/p130 complexes subsides, while E2F1/Rb complexes appear. Additionally, the cyclin-dependent kinase inhibitor p27KIP is downregulated (Morrison and Farmer, 1999; Patel and Lane, 2000), thereby facilitating activation of cyclin D/Cdk4/6, which corresponds with phosphorylation of Rb, resulting in the release of E2F1 to induce transcription of ppar_γ_1 (see Figure 2). These data demonstrating a function for E2Fs in adipogenesis correlate with a series of genetic studies performed in mice. _E2F1_−/− mice have a limited ability to accumulate adipose tissue in response to high-fat feeding, while _E2F4_−/− ES cells contribute more significantly to adipose tissue development than other tissues of chimeric mice. Consistent with the mouse models, _E2F1_−/− MEFs have a reduced capacity to differentiate into adipocytes, whereas E2F4-deficient MEFs and ES cells express an enhanced capacity for differentiation. Furthermore, the combined loss of the major E2F4-associated pocket proteins p107 and p130 leads to enhanced adipogenesis in corresponding MEFs (Classon et al., 2000), supporting the notion that E2F4/p107 or E2F4/p130 complexes and not E2F4 alone repress ppar_γ_1 transcription. One would predict that a deficiency in Rb would enhance adipogenesis by facilitating E2F1 activity; however, _Rb_−/− MEFs interestingly have a reduced capacity for differentiation into white adipocytes (Classon et al., 2000). This is likely due to the requirement of Rb in facilitating cell-cycle exit as well as cooperation with C/EBPs to induce adipogenic gene expression (Chen et al., 1996). It appears, therefore, that the E2Fs and pocket proteins regulate two separate but parallel pathways that result in the activation of PPARγ1 and PPARγ2 expression (Figure 2). Specifically, factors such as Rb, which channel through C/EBPβ, lead to PPARγ2 production, whereas factors that promote E2F1 activity lead to PPARγ1 expression. Since PPARγ2 and not PPARγ1 is considered to be the predominant regulator of adipogenesis, factors such as E2F that converge on PPARγ1 need to have a means of enhancing PPARγ2 expression. This process could be facilitated through C/EBPα whereby PPARγ1 induces C/EBPα, which in turn induces PPARγ2 expression (Wu et al., 1999; Zuo et al., 2006). Such a process could explain the redundancy in mechanisms regulating PPARγ2 expression in mice deficient in C/EBPβ and C/EBPδ (Tanaka et al., 1997). In these mice, it is conceivable that signals in the developing adipose depot act on E2F to stimulate PPARγ1, which then induces C/EBPα followed by PPARγ2 without the need for expression of C/EBPβ or C/EBPδ.
Figure 2. Role of cell-cycle proteins in regulating adipogenesis.
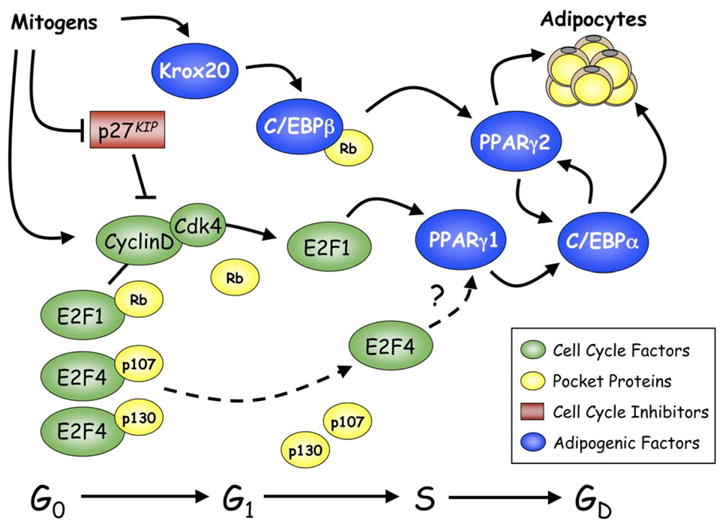
An alternative pathway to that presented in Figure 1 exists whereby E2Fs and associated pocket proteins regulate expression of PPARγ1. Activation of PPARγ2 likely occurs through the induction of C/EBPα by PPARγ1, and C/EBPα then induces PPARγ2 expression. This model is consistent with a role for clonal expansion in promoting adipogenesis. G0, G1, and S correspond to phases of the cell cycle, while GD is a term used to define the growth-arrested state of terminally differentiated cells.
Negative regulation of adipogenesis
The differentiation of mesenchymal stem cells along a particular lineage is regulated by both induction of various transcriptional activators and suppression of inhibitors. It is likely that the subtle balance in the activity of positive versus negative effectors determines whether adipogenesis proceeds within a particular population of progenitor cells. This concept is well illustrated by the studies of MacDougald and associates, who demonstrated that activation of the Wnt signaling pathway inhibits the differentiation of mesenchymal stem cells into adipocytes (Ross et al., 2000). Wnt signaling appears to favor differentiation of progenitor cells into bone or muscle, as opposed to adipocytes (Bennett et al., 2005). Wnts are a large family of extracellular effectors secreted by many different cell types and play a determining role during early development. The binding of various Wnts to corresponding Frizzled receptors and low-density lipoprotein receptor-related proteins (LRPs) activates signaling pathways that alter gene expression and cell function. The canonical Wnt pathway leads to mobilization of β-catenin into the nucleus, where it coactivates the TCF/LEF family of transcription factors. Exposure of preadipocytes to Wnts or ectopic expression of a constitutively active form of β-catenin inhibits adipogenesis by preventing induction of PPARγ and C/EBPα (Moldes et al., 2003; Ross et al., 2000). The precise mechanism involved is not known, but it likely involves expression of TCF/LEF target genes since expression of dominant-negative TCF (dnTCF) partially rescues the inhibitory effects of Wnt (Ross et al., 2000). Furthermore, expression of dnTCF causes spontaneous differentiation of preadipocytes, suggesting that the canonical Wnt signaling pathway acts in progenitor cells to suppress adipogenesis. An attractive candidate for a TCF-induced adipogenic inhibitor is cyclin D1 since its gene is a direct target of Wnt signaling, which has been shown to antagonize PPARγ activity (Fu et al., 2005a; Wang et al., 2003). It is also possible that β-catenin might contribute to the inhibition of PPARγ activity through mechanisms other than those involving TCF/LEF (Liu and Farmer, 2004; Liu et al., 2006), and it is worth noting that conditional deletion of β-catenin in the mesenchyme of the developing mouse results in a switch to adipogenesis in the myometrium (Arango et al., 2005).
Several studies have demonstrated that multiple effectors attenuate adipogenesis by compromising the activity of C/EBPβ. These observations not only identify the existence of negative regulators but also support a role for C/EBPβ in regulating preadipocyte differentiation. A series of these negative regulators, including GATA2/3, ETO/MTG8, CHOP10, GILZ, and Delta-interacting protein A (DIPA), are expressed in preadipocytes, and their expression is downregulated during differentiation. Ectopic expression of each of these proteins in preadipocytes inhibits adipogenesis through antagonism of C/EBPβ activity and thereby prevents the induction of PPARγ and C/EBPα (Batchnarova et al., 1995; Bezy et al., 2005; Rochford et al., 2004; Shi et al., 2003; Tong et al., 2000, 2005). It is worth noting that the vitamin D receptor, which is induced early in adipogenesis (Fu et al., 2005b), blocks preadipocyte differentiation by downregulating C/EBPβ through mechanisms that possibly involve induction of ETO/MTG8 (Blumberg et al., 2006). Similarly, Hedgehog signaling, which is known to regulate vertebrate development, plays a conserved role in inhibiting fat formation, possibly by inducing expression of GATA2 (Suh et al., 2006). Additionally, Notch signaling plays an important role in early development, and the Notch target Hes-1 blocks adipogenesis by mechanisms that possibly involve recruitment of members of the Groucho/TLE family of corepressors (Ross et al., 2006; Ross et al., 2004). Other investigators have hypothesized that oxygen tension might control adipose tissue function by regulating adipogenesis (Swiersz et al., 2004). Specifically, Yun et al. (2002) have demonstrated that hypoxia inhibits preadipocyte differentiation through a mechanism that involves repression of _ppar_γ expression by DEC1/Stra13. DEC1/Stra13 is a member of the Drosophila hairy/Enhancer of split transcription repressor family that is induced by hypoxia-inducible transcription factor 1α (HIF-1α). Stra13 is also induced by retinoic acid (RA) (Boudjelal et al., 1997) and, consequently, might also be the mediator by which RA inhibits adipogenesis (Schwarz et al., 1997).
As discussed above, insulin possesses significant proadipogenic activity in part by promoting expression of SREBP1c. Studies performed in animals as well as in cell culture demonstrate that insulin promotes adipogenesis by suppressing the inhibitory activity of the forkhead transcription factor FoxO1. Specifically, exposure of preadipocytes to insulin results in AKT-dependent phosphorylation of FoxO1, preventing its translocation into the nucleus and subsequent inhibition of adipogenic gene expression. To identify mechanisms responsible for this inhibitory activity, Accili and associates demonstrated that a constitutively active FoxO1, which is insensitive to AKT phosphorylation, inhibits the differentiation of 3T3-F422A preadipocytes by arresting the cells in clonal expansion. This block in the adipogenic progression is likely due to a FoxO1-associated induction of the cyclin-dependent kinase inhibitor p21CIP (Nakae et al., 2003). In support of an inhibitory function for FoxO1 in adipose tissue, additional studies showed that FoxO1 haploinsufficiency (foxo1+/−) protects against diet-induced insulin resistance and diabetes possibly by preventing adipocyte hypertrophy (Nakae et al., 2003). It is interesting that two additional members of the forkhead family, FoxA2 and FoxC2, also attenuate adipogenesis upstream of PPARγ (Davis et al., 2004; Wolfrum et al., 2003).
It is important to mention that, while three members of the KLF family are proadipogenic (KLF5, KLF6, and KLF15), at least one KLF acts as a suppressor of adipogenesis. Specifically, KLF2/lung Kruppel-like factor is abundantly expressed in adipose tissue in preadipocytes, and its expression is downregulated during adipogenesis (Banerjee et al., 2003; Wu et al., 2005). Ectopic expression of KLF2 in preadipocytes inhibits ppar_γ_2 transcription, possibly by binding to KLF regulatory elements in the same region of ppar_γ_2 that facilitates the proadipogenic activity of KLF5 (Banerjee et al., 2003; Oishi et al., 2005; Wu et al., 2005). The involvement of the different negative regulators in controlling adipogenesis is illustrated in Figure 3.
Figure 3. Negative control of adipogenesis.
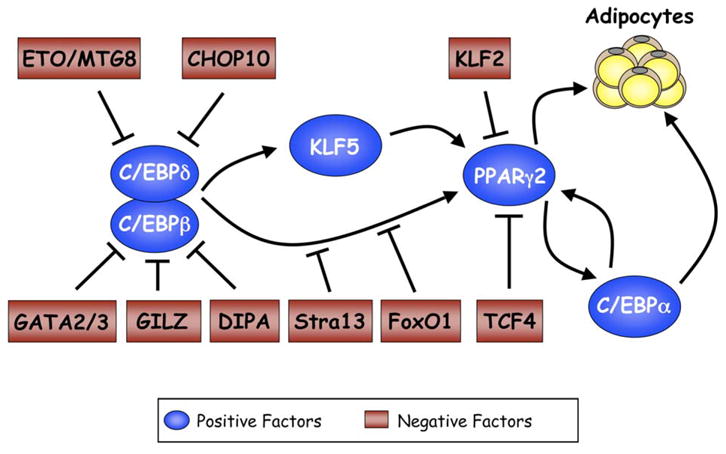
Negative regulators inhibit expression of PPARγ and C/EBPα by attenuating the activity of components of the cascade presented in Figure 1. Several of these negative factors appear to converge on C/EBPβ, supporting its role as a principal regulator of adipogenesis.
Role of coregulators in controlling the adipogenic transcription factors
All of the adipogenic transcription factors discussed above initiate their corresponding programs of gene expression by binding to response elements in target genes where they recruit appropriate coactivators following dissociation from corepressors. Most of these adipogenic coregulators are ubiquitously expressed and employed by other transcription factors in multiple cell types. Consequently, their selectivity in activating a specific gene is primarily defined by the interaction with the transcription factor that is docked on the response element within the promoter/enhancer of the target gene.
Coactivators
There is evidence suggesting that C/EBPβ can dock on the promoters of _c/ebp_α and _ppar_γ prior to their activation during the early phase of adipogenesis (Salma et al., 2006). Adipogenic effectors then facilitate association of the chromatin remodeling complex SWI/SNF with C/EBPβ on the ppar_γ_2 promoter (Salma et al., 2004). Glucocorticoid receptors (GRs) along with PPARγ are responsible for dislodging an mSin3a/HDAC-1 complex from C/EBPβ on the C/EBP response element in the _c/ebp_α promoter (Wiper-Bergeron et al., 2003; Zuo et al., 2006). Similarly, the adipogenic potential of C/EBPα depends on its interaction with SWI/SNF, which occurs through interaction with the transactivation element III (TEIII) domain in C/EBPα. This interaction mediates further association with TBP/TFIIB factors (Pedersen et al., 2001). C/EBPα can also associate with CBP/p300, but the precise role of this interaction during adipogenesis is not known (Erickson et al., 2001). PPARγ appears to be capable of interacting with several different coregulators, which explains how it functions to control expression of numerous gene programs in mature adipocytes. Notable among these coregulators is PPARγ coactivator 1α (PGC-1α), which coactivates a host of transcription factors in addition to PPARγ that collectively participate in energy balance (Lin et al., 2005). During development, PGC-1α and β regulate brown adipose formation by coactivating transcription factors including nuclear respiratory factor 1 (NRF-1) and PPARγ that regulate thermogenesis and mitochondrial biogenesis (Lin et al., 2005; Uldry et al., 2006). During adipogenesis, activation of most PPARγ target genes involves an elaborate process in which binding of PPARγ to corresponding ligands dislodges corepressor complexes (NCoR/SMRT with HDAC3) and recruitment of members of the p160 family of coactivators, usually TIF2 or SRC-1. Recent studies have shown that these p160 coregulators might also possess some nonredundant function since lack of TIF2 in mice decreases PPARγ activity in WAT and reduces fat accumulation. In BAT, the absence of TIF2 facilitates an interaction between SRC-1 and PGC-1α leading to an increase in thermogenic activity. Interestingly, _TIF2_−/− mice are protected against obesity and display enhanced adaptive thermogenesis, whereas _SRC-1_−/− mice are prone to obesity (Picard et al., 2002). The association of PPARγ with the p160 coregulators leads to further recruitment of histone acetyltransferases (HATs) that appropriately modify surrounding chromatin, allowing the transcriptional machinery access to the gene promoter. At present, most studies show that the p160 coactivators interact with the AF-2 domain of PPARγ in response to binding of appropriate ligands such as troglitazone. It is likely that other coactivators associate with the AF-1 domain at the N terminus of PPARγ. Investigations have shown that two homologous cofactors, p300 and CBP, bind to the N terminus of PPARγ2 in a ligand-independent manner, whereas binding to the C terminus is dependent on a ligand (Gelman et al., 1999). A possible role for CBP in regulating adipose tissue development has been supported by studies of CBP heterozygous mice, which show markedly reduced weight of adipose tissue but not of other tissues (Yamauchi et al., 2002).
PPARγ also communicates with the basal transcriptional machinery through its interaction with a large multicomponent Mediator complex that is required for adipogenesis. Specifically, PPARγ associates with the TRAP (thyroid hormone receptor-associated protein) coactivator-Mediator complex through binding to the TRAP220 subunit in a ligand-enhanced manner. MEFs lacking TRAP220 are resistant to PPARγ2-stimulated adipogenesis, but not to MyoD-stimulated myogenesis (Ge et al., 2002). These observations suggest that TRAP220 acts, via the Mediator complex, as a PPARγ2-selective coactivator; this interaction participates in commitment of mesenchymal cells along an adipogenic as opposed to myogenic lineage. This selectivity might also be facilitated by additional proteins that interact with the PPARγ-Mediator complex. Specifically, PPARγ-interacting protein (PRIP) can associate with CBP/p300 and TRAP130 of the Mediator complex. Supporting this notion, _PRIP_−/− MEFs are also resistant to PPARγ-induced adipogenesis (Qi et al., 2003). In addition, a recent study identified a novel human TAF (TBP [TATA-binding protein]-associated factor), hTAFII43 (TAF8), which is induced and sequestered within TFIID complexes during adipogenesis in 3T3-L1 cells. Furthermore, ectopic expression of a dominant-negative TAF8 blocks 3T3-L1 preadipocyte differentiation (Guermah et al., 2003).
Corepressors
Both NCoR and SMRT appear to function as negative modulators of PPARγ activity during adipogenesis since RNAi knockdown of these factors in 3T3-L1 cells leads to increased expression of PPARγ target genes and increased production of lipid droplets (Yu et al., 2005). Studies also suggest that Rb functions in a fashion similar to these corepressors by facilitating the docking of HDAC3 on PPARγ-driven promoters (Fajas et al., 2002a). Interestingly, other studies suggest that Rb acts as a molecular switch promoting brown versus white adipocyte formation (Hansen et al., 2004a, 2004b). In contrast, the corepressor protein RIP140 appears to function in regulating development of adipose tissue favoring a white phenotype. Specifically, mice devoid of RIP140 are lean, show resistance to high-fat-diet-induced obesity, and have increased oxygen consumption (Leonardsson et al., 2004). It appears that this phenotype stems from the capacity of RIP140 to suppress transcription factors regulating oxidative metabolism and mitochondrial biogenesis, characteristics of brown fat cells (Christian et al., 2005; Leonardsson et al., 2004; Powelka et al., 2006).
As mentioned above, deacetylases that modify histones as well as regulate activity of transcription factors are critical components of various corepressor complexes. The HDACs participate in adipogenic regulation, illustrated by the suppression of C/EBPβ and PPARγ by HDAC1 and HDAC3, respectively. Induction of adipogenesis includes dislodgement of these HDACs from their respective transcription factors by mechanisms that include their degradation in the 26S proteasome, and it is likely that the programmed turnover of these HDACs is an integral part of the adipogenic process (Yoo et al., 2006). Other deacetylases that target factors other than the histones might affect adipogenesis by altering the acetylation of coregulators of PPARγ. Specifically, Sirt1, the mammalian ortholog of the yeast longevity gene sir2, attenuates adipogenesis by repressing PPARγ activity leading to fat mobilization in white adipocytes, which triggers lipolysis (Picard et al., 2004). SIRT1 is a NAD-dependent protein deacetylase capable of monitoring cellular oxidative state and altering the activity of nuclear regulators in response to metabolites and nutrients (Imai et al., 2000; Rodgers et al., 2005). Repression of PPARγ activity appears to involve docking of SIRT1 with NcoR and SMRT on the promoters of PPARγ target genes in adipocytes (Picard et al., 2004). The interplay between various coactivators and corepressors in determining the differentiation of white versus brown adipocytes is shown in Figure 4.
Figure 4. Coregulators and adipogenesis.
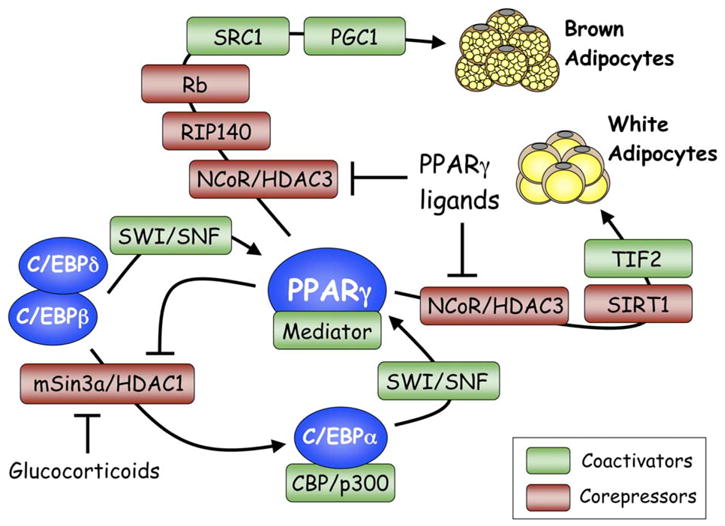
The activity of the adipogenic transcription factors is regulated by association with various corepressors (red boxes) or coactivators (green boxes) at different stages of differentiation of both brown as well as white preadipocytes. The Mediator shown to be associating with PPARγ corresponds to the TRAP (thyroid hormone receptor-associated protein) transcriptional coactivator complex, which interacts physically with PPARγ through the TRAP220 subunit. See text for detailed discussion of the participation of the various protein complexes in controlling each of the transcription factors.
Concluding remarks
In conclusion, it is quite apparent that significant progress has been made during the last few years in identifying the transcriptional processes controlling the differentiation of preadipocytes into mature fat cells. The challenge for the future is to understand the mechanisms governing the commitment of mesenchymal stem cells to the adipogenic lineage. There are certainly some indications of possible players in this process; however, the adipocyte field lags far behind that of other developmental systems since it has been difficult to locate adipogenic progenitors during early development. Additionally, there is a dearth of information concerning the mechanisms that give rise to the various white fat depots. Recent studies have suggested that there are significant differences between subcutaneous and visceral depots, particularly with regard to their role in cardiovascular disease and diabetes. The reasons for these differences are essentially unknown. It is possible that each of the depots arises from different progenitors controlled by a separate set of transcriptional processes, resulting in distinct depot-specific adipocytes. Future research will no doubt address some of these questions by investigating the role of developmental cues shown to participate in the formation of other tissues. Finally, most of what we know has come from studies of rodents either in vivo or in cell culture. There are examples of adipose function that do not translate from the mouse to the human; consequently, attention needs to be given to understanding the transcriptional control of adipocyte formation and function in human adipose tissue.
Acknowledgments
I am grateful to past and present members of my group who have contributed to the studies referenced in this review. I am particularly grateful to Dr. Kathryn Davis for valuable suggestions and constructive criticism. I am supported by National Institutes of Health grants DK51586 and DK58825.
References
- Arango NA, Szotek PP, Manganaro TF, Oliva E, Donahoe PK, Teixeira J. Conditional deletion of beta-catenin in the mesenchyme of the developing mouse uterus results in a switch to adipogenesis in the myometrium. Dev Biol. 2005;288:276–283. doi: 10.1016/j.ydbio.2005.09.045. [DOI] [PubMed] [Google Scholar]
- Banerjee SS, Feinberg MW, Watanabe M, Gray S, Haspel RL, Denkinger DJ, Kawahara R, Hauner H, Jain MK. The Kruppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-gamma expression and adipogenesis. J Biol Chem. 2003;278:2581–2584. doi: 10.1074/jbc.M210859200. [DOI] [PubMed] [Google Scholar]
- Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- Batchnarova N, Wang XZ, Ron D. Inhibition of adipogenesis by the stress-induced protein CHOP (Gadd153) EMBO J. 1995;14:4654–4661. doi: 10.1002/j.1460-2075.1995.tb00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, MacDougald OA. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA. 2005;102:3324–3329. doi: 10.1073/pnas.0408742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezy O, Elabd C, Cochet O, Petersen RK, Kristiansen K, Dani C, Ailhaud G, Amri EZ. Delta-interacting protein A, a new inhibitory partner of CCAAT/enhancer-binding protein beta, implicated in adipocyte differentiation. J Biol Chem. 2005;280:11432–11438. doi: 10.1074/jbc.M411741200. [DOI] [PubMed] [Google Scholar]
- Blumberg JM, Tzameli I, Astapova I, Lam FS, Flier JS, Hollenberg AN. Complex role of the vitamin D receptor and its ligand in adipogenesis in 3T3-L1 cells. J Biol Chem. 2006;281:11205–11213. doi: 10.1074/jbc.M510343200. [DOI] [PubMed] [Google Scholar]
- Boudjelal M, Taneja R, Matsubara S, Bouillet P, Dolle P, Chambon P. Overexpression of Stra13, a novel retinoic acid-inducible gene of the basic helix-loop-helix family, inhibits mesodermal and promotes neuronal differentiation of P19 cells. Genes Dev. 1997;11:2052–2065. doi: 10.1101/gad.11.16.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- Chen PL, Riley DJ, Chen Y, Lee WH. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 1996;10:2794–2804. doi: 10.1101/gad.10.21.2794. [DOI] [PubMed] [Google Scholar]
- Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab. 2005;1:93–106. doi: 10.1016/j.cmet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Christian M, Kiskinis E, Debevec D, Leonardsson G, White R, Parker MG. RIP140-targeted repression of gene expression in adipocytes. Mol Cell Biol. 2005;25:9383–9391. doi: 10.1128/MCB.25.21.9383-9391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christy RJ, Kaestner KH, Geiman DE, Lane MD. CCAAT/enhancer binding protein gene promoter: binding of nuclear factors during differentiation of 3T3-L1 preadipocytes. Proc Natl Acad Sci USA. 1991;88:2593–2597. doi: 10.1073/pnas.88.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SL, Robinson CE, Gimble JM. CAAT/enhancer binding proteins directly modulate transcription from the peroxisome proliferator-activated receptor gamma2 promoter. Biochem Biophys Res Commun. 1997;240:99–103. doi: 10.1006/bbrc.1997.7627. [DOI] [PubMed] [Google Scholar]
- Classon M, Kennedy BK, Mulloy R, Harlow E. Opposing roles of pRB and p107 in adipocyte differentiation. Proc Natl Acad Sci USA. 2000;97:10826–10831. doi: 10.1073/pnas.190343597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KE, Moldes M, Farmer SR. The forkhead transcription factor FoxC2 inhibits white adipocyte differentiation. J Biol Chem. 2004;279:42453–42461. doi: 10.1074/jbc.M402197200. [DOI] [PubMed] [Google Scholar]
- El-Jack AK, Hamm JK, Pilch PF, Farmer SR. Reconstitution of insulin-sensitive glucose transport in fibroblasts requires expression of both PPARγ and C/EBPα. J Biol Chem. 1999;274:7946–7951. doi: 10.1074/jbc.274.12.7946. [DOI] [PubMed] [Google Scholar]
- Erickson RL, Hemati N, Ross SE, MacDougald OA. p300 coactivates the adipogenic transcription factor CCAAT/enhancer-binding protein alpha. J Biol Chem. 2001;276:16348–16355. doi: 10.1074/jbc.m100128200. [DOI] [PubMed] [Google Scholar]
- Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R, Najib J, Laville M, Fruchart JC, Deeb S, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem. 1997;272:18779–18789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- Fajas L, Schoonjans K, Gelman L, Kim JB, Najib J, Martin G, Fruchart JC, Briggs M, Spiegelman BM, Auwerx J. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism. Mol Cell Biol. 1999;19:5495–5503. doi: 10.1128/mcb.19.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB, Miard S, Auwerx J. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002a;3:903–910. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. E2Fs regulate adipocyte differentiation. Dev Cell. 2002b;3:39–49. doi: 10.1016/s1534-5807(02)00190-9. [DOI] [PubMed] [Google Scholar]
- Floyd ZE, Stephens JM. STAT5A promotes adipogenesis in nonprecursor cells and associates with the glucocorticoid receptor during adipocyte differentiation. Diabetes. 2003;52:308–314. doi: 10.2337/diabetes.52.2.308. [DOI] [PubMed] [Google Scholar]
- Fontaine C, Dubois G, Duguay Y, Helledie T, Vu-Dac N, Gervois P, Soncin F, Mandrup S, Fruchart JC, Fruchart-Najib J, Staels B. The orphan nuclear receptor Rev-Erbalpha is a peroxisome proliferator-activated receptor (PPAR) gamma target gene and promotes PPAR-gamma-induced adipocyte differentiation. J Biol Chem. 2003;278:37672–37680. doi: 10.1074/jbc.M304664200. [DOI] [PubMed] [Google Scholar]
- Freytag SO, Paielli DL, Gilbert JD. Ectopic expression of the CCAAT/enhancer-binding protein alpha promotes the adipogenic program in a variety of mouse fibroblastic cells. Genes Dev. 1994;8:1654–1663. doi: 10.1101/gad.8.14.1654. [DOI] [PubMed] [Google Scholar]
- Fu M, Rao M, Bouras T, Wang C, Wu K, Zhang X, Li Z, Yao TP, Pestell RG. Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J Biol Chem. 2005a;280:16934–16941. doi: 10.1074/jbc.M500403200. [DOI] [PubMed] [Google Scholar]
- Fu M, Sun T, Bookout AL, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. A Nuclear Receptor Atlas: 3T3-L1 adipogenesis. Mol Endocrinol. 2005b;19:2437–2450. doi: 10.1210/me.2004-0539. [DOI] [PubMed] [Google Scholar]
- Ge K, Guermah M, Yuan CX, Ito M, Wallberg AE, Spiegelman BM, Roeder RG. Transcription coactivator TRAP220 is required for PPAR gamma 2-stimulated adipogenesis. Nature. 2002;417:563–567. doi: 10.1038/417563a. [DOI] [PubMed] [Google Scholar]
- Gelman L, Zhou G, Fajas L, Raspe E, Fruchart JC, Auwerx J. p300 interacts with the N- and C-terminal part of PPARgamma2 in a ligand-independent and -dependent manner, respectively. J Biol Chem. 1999;274:7681–7688. doi: 10.1074/jbc.274.12.7681. [DOI] [PubMed] [Google Scholar]
- Green H, Kehinde O. An established preadipose cell line and its differentiation in culture. II Factors affecting the adipose conversion. Cell. 1975;5:19–27. doi: 10.1016/0092-8674(75)90087-2. [DOI] [PubMed] [Google Scholar]
- Green H, Kehinde O. Spontaneous heritable changes leading to increased adipose conversion in 3T3 cells. Cell. 1976;7:105–113. doi: 10.1016/0092-8674(76)90260-9. [DOI] [PubMed] [Google Scholar]
- Guermah M, Ge K, Chiang CM, Roeder RG. The TBN protein, which is essential for early embryonic mouse development, is an inducible TAFII implicated in adipogenesis. Mol Cell. 2003;12:991–1001. doi: 10.1016/s1097-2765(03)00396-4. [DOI] [PubMed] [Google Scholar]
- Hamm JK, Park BH, Farmer SR. A role for C/EBPbeta in regulating peroxisome proliferator-activated receptor gamma activity during adipogenesis in 3T3-L1 preadipocytes. J Biol Chem. 2001;276:18464–18471. doi: 10.1074/jbc.M100797200. [DOI] [PubMed] [Google Scholar]
- Hansen JB, Jorgensen C, Petersen RK, Hallenborg P, De Matteis R, Boye HA, Petrovic N, Enerback S, Nedergaard J, Cinti S, et al. Retinoblastoma protein functions as a molecular switch determining white versus brown adipocyte differentiation. Proc Natl Acad Sci USA. 2004a;101:4112–4117. doi: 10.1073/pnas.0301964101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JB, te Riele H, Kristiansen K. Novel function of the retinoblastoma protein in fat: regulation of white versus brown adipocyte differentiation. Cell Cycle. 2004b;3:774–778. [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996;10:1096–1107. doi: 10.1101/gad.10.9.1096. [DOI] [PubMed] [Google Scholar]
- Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, Lowell BB, Spiegelman BM. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest. 1998a;101:1–9. doi: 10.1172/JCI1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPAR gamma through the production of endogenous ligand. Proc Natl Acad Sci USA. 1998b;95:4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutnikova H, Cock TA, Watanabe M, Houten SM, Champy MF, Dierich A, Auwerx J. Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPAR gamma hypomorphic mice. Proc Natl Acad Sci USA. 2003;100:14457–14462. doi: 10.1073/pnas.2336090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardsson G, Steel JH, Christian M, Pocock V, Milligan S, Bell J, So PW, Medina-Gomez G, Vidal-Puig A, White R, Parker MG. Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc Natl Acad Sci USA. 2004;101:8437–8442. doi: 10.1073/pnas.0401013101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Yea S, Li S, Chen Z, Narla G, Banck M, Laborda J, Tan S, Friedman JM, Friedman SL, Walsh MJ. Kruppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J Biol Chem. 2005;280:26941–26952. doi: 10.1074/jbc.M500463200. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Linhart HG, Ishimura-Oka K, DeMayo F, Kibe T, Repka D, Poindexter B, Bick RJ, Darlington GJ. C/EBPalpha is required for differentiation of white, but not brown, adipose tissue. Proc Natl Acad Sci USA. 2001;98:12532–12537. doi: 10.1073/pnas.211416898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem. 2004;279:45020–45027. doi: 10.1074/jbc.M407050200. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang H, Zuo Y, Farmer SR. A functional interaction between PPARgamma and beta-catenin. Mol Cell Biol. 2006 doi: 10.1128/MCB.00441-06. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirhaeghe A, Fajas L, Gouilleux F, Cottel D, Helbecque N, Auwerx J, Amouyel P. A functional polymorphism in a STAT5B site of the human PPAR gamma 3 gene promoter affects height and lipid metabolism in a French population. Arterioscler Thromb Vasc Biol. 2003;23:289–294. doi: 10.1161/01.atv.0000051382.28752.fe. [DOI] [PubMed] [Google Scholar]
- Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, Farmer SR. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J. 2003;376:607–613. doi: 10.1042/BJ20030426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Sakaue H, Iguchi H, Gomi H, Okada Y, Takashima Y, Nakamura K, Nakamura T, Yamauchi T, Kubota N, et al. Role of Kruppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem. 2005;280:12867–12875. doi: 10.1074/jbc.M410515200. [DOI] [PubMed] [Google Scholar]
- Morrison RF, Farmer SR. Role of PPARgamma in regulating a cascade expression of cyclin-dependent kinase inhibitors, p18(INK4c) and p21(Waf1/Cip1), during adipogenesis. J Biol Chem. 1999;274:17088–17097. doi: 10.1074/jbc.274.24.17088. [DOI] [PubMed] [Google Scholar]
- Mueller E, Drori S, Aiyer A, Yie J, Sarraf P, Chen H, Hauser S, Rosen ED, Ge K, Roeder RG, Spiegelman BM. Genetic analysis of adipogenesis through peroxisome proliferator-activated receptor gamma isoforms. J Biol Chem. 2002;277:41925–41930. doi: 10.1074/jbc.M206950200. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Kitamura Y, Biggs WH, III, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell. 2003;4:119–129. doi: 10.1016/s1534-5807(02)00401-x. [DOI] [PubMed] [Google Scholar]
- Oishi Y, Manabe I, Tobe K, Tsushima K, Shindo T, Fujiu K, Nishimura G, Maemura K, Yamauchi T, Kubota N, et al. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab. 2005;1:27–39. doi: 10.1016/j.cmet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Park BH, Qiang L, Farmer SR. Phosphorylation of C/EBP-beta at a consensus ERK/GSK3 site is required for the induction of adiponectin gene expression during the differentiation of mouse fibroblasts into adipocytes. Mol Cell Biol. 2004;24:8671–8680. doi: 10.1128/MCB.24.19.8671-8680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel YM, Lane MD. Mitotic clonal expansion during preadipocyte differentiation: calpain-mediated turnover of p27. J Biol Chem. 2000;275:17653–17660. doi: 10.1074/jbc.M910445199. [DOI] [PubMed] [Google Scholar]
- Pedersen TA, Kowenz-Leutz E, Leutz A, Nerlov C. Cooperation between C/EBPalpha TBP/TFIIB and SWI/SNF recruiting domains is required for adipocyte differentiation. Genes Dev. 2001;15:3208–3216. doi: 10.1101/gad.209901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Gehin M, Annicotte J, Rocchi S, Champy MF, O’Malley BW, Chambon P, Auwerx J. SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell. 2002;111:931–941. doi: 10.1016/s0092-8674(02)01169-8. [DOI] [PubMed] [Google Scholar]
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powelka AM, Seth A, Virbasius JV, Kiskinis E, Nicoloro SM, Guilherme A, Tang X, Straubhaar J, Cherniack AD, Parker MG, Czech MP. Suppression of oxidative metabolism and mitochondrial biogenesis by the transcriptional corepressor RIP140 in mouse adipocytes. J Clin Invest. 2006;116:125–136. doi: 10.1172/JCI26040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C, Surapureddi S, Zhu YJ, Yu S, Kashireddy P, Rao MS, Reddy JK. Transcriptional coactivator PRIP, the peroxisome proliferator-activated receptor gamma (PPARgamma)-interacting protein, is required for PPARgamma-mediated adipogenesis. J Biol Chem. 2003;278:25281–25284. doi: 10.1074/jbc.C300175200. [DOI] [PubMed] [Google Scholar]
- Rochford JJ, Semple RK, Laudes M, Boyle KB, Christodoulides C, Mulligan C, Lelliott CJ, Schinner S, Hadaschik D, Mahadevan M, et al. ETO/MTG8 is an inhibitor of C/EBPbeta activity and a regulator of early adipogenesis. Mol Cell Biol. 2004;24:9863–9872. doi: 10.1128/MCB.24.22.9863-9872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, Spiegelman BM. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002;16:22–26. doi: 10.1101/gad.948702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross DA, Rao PK, Kadesch T. Dual roles for the Notch target gene Hes-1 in the differentiation of 3T3-L1 preadipocytes. Mol Cell Biol. 2004;24:3505–3513. doi: 10.1128/MCB.24.8.3505-3513.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross DA, Hannenhalli S, Tobias JW, Cooch N, Shiekhattar R, Kadesch T. Functional analysis of Hes-1 in preadipocytes. Mol Endocrinol. 2006;20:698–705. doi: 10.1210/me.2005-0325. [DOI] [PubMed] [Google Scholar]
- Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- Salma N, Xiao H, Mueller E, Imbalzano AN. Temporal recruitment of transcription factors and SWI/SNF chromatin-remodeling enzymes during adipogenic induction of the peroxisome proliferator-activated receptor gamma nuclear hormone receptor. Mol Cell Biol. 2004;24:4651–4663. doi: 10.1128/MCB.24.11.4651-4663.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salma N, Xiao H, Imbalzano AN. Temporal recruitment of CCAAT/enhancer-binding proteins to early and late adipogenic promoters in vivo. J Mol Endocrinol. 2006;36:139–151. doi: 10.1677/jme.1.01918. [DOI] [PubMed] [Google Scholar]
- Schwarz EJ, Reginato MJ, Shao D, Krakow SL, Lazar MA. Retinoic acid blocks adipogenesis by inhibiting C/EBPbeta-mediated transcription. Mol Cell Biol. 1997;17:1552–1561. doi: 10.1128/mcb.17.3.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Shi W, Li Q, Song B, Wan M, Bai S, Cao X. A glucocorticoid-induced leucine-zipper protein, GILZ, inhibits adipogenesis of mesenchymal cells. EMBO Rep. 2003;4:374–380. doi: 10.1038/sj.embor.embor805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimba S, Ishii N, Ohta Y, Ohno T, Watabe Y, Hayashi M, Wada T, Aoyagi T, Tezuka M. Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc Natl Acad Sci USA. 2005;102:12071–12076. doi: 10.1073/pnas.0502383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukas A, Socci ND, Saatkamp BD, Novelli S, Friedman JM. Distinct transcriptional profiles of adipogenesis in vivo and in vitro. J Biol Chem. 2001;276:34167–34174. doi: 10.1074/jbc.M104421200. [DOI] [PubMed] [Google Scholar]
- Suh JM, Gao X, McKay J, McKay R, Salo Z, Graff JM. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 2006;3:25–34. doi: 10.1016/j.cmet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Swiersz LM, Giaccia AJ, Yun Z. Oxygen-dependent regulation of adipogenesis. Methods Enzymol. 2004;381:387–395. doi: 10.1016/S0076-6879(04)81026-7. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yoshida N, Kishimoto T, Akira S. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 1997;16:7432–7443. doi: 10.1093/emboj/16.24.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Lane MD. Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Dev. 1999;13:2231–2241. doi: 10.1101/gad.13.17.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, Lane MD. Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci USA. 2003;100:44–49. doi: 10.1073/pnas.0137044100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Gronborg M, Huang H, Kim JW, Otto TC, Pandey A, Lane MD. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc Natl Acad Sci USA. 2005;102:9766–9771. doi: 10.1073/pnas.0503891102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science. 2000;290:134–138. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]
- Tong Q, Tsai J, Tan G, Dalgin G, Hotamisligil GS. Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol Cell Biol. 2005;25:706–715. doi: 10.1128/MCB.25.2.706-715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Graves RA, Budavari AI, Erdjument-Bromage H, Lui M, Hu E, Tempst P, Spiegelman BM. Adipocyte-specific transcription factor ARF6 is a heterodimeric complex of two nuclear hormone receptors, PPAR gamma and RXR alpha. Nucleic Acids Res. 1994a;22:5628–5634. doi: 10.1093/nar/22.25.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPARγ2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994b;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ, a lipid-activated transcription factor. Cell. 1994c;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman B. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Wang C, Pattabiraman N, Zhou JN, Fu M, Sakamaki T, Albanese C, Li Z, Wu K, Hulit J, Neumeister P, et al. Cyclin D1 repression of peroxisome proliferator-activated receptor gamma expression and transactivation. Mol Cell Biol. 2003;23:6159–6173. doi: 10.1128/MCB.23.17.6159-6173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ND, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD, Taylor LR, Wilson DR, Darlington GJ. Impaired energy homeostasis in C/EBP alpha knockout mice. Science. 1995;269:1108–1112. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- Wiper-Bergeron N, Wu D, Pope L, Schild-Poulter C, Hache RJ. Stimulation of preadipocyte differentiation by steroid through targeting of an HDAC1 complex. EMBO J. 2003;22:2135–2145. doi: 10.1093/emboj/cdg218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C, Shih DQ, Kuwajima S, Norris AW, Kahn CR, Stoffel M. Role of Foxa-2 in adipocyte metabolism and differentiation. J Clin Invest. 2003;112:345–356. doi: 10.1172/JCI18698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Srinivasan SV, Neumann JC, Lingrel JB. The KLF2 transcription factor does not affect the formation of preadipocytes but inhibits their differentiation into adipocytes. Biochemistry. 2005;44:11098–11105. doi: 10.1021/bi050166i. [DOI] [PubMed] [Google Scholar]
- Wu Z, Xie Y, Bucher NLR, Farmer SR. Conditional ectopic expression of C/EBPβ in NIH-3T3 cells induces PPARγ and stimulates adipogenesis. Genes Dev. 1995;9:2350–2363. doi: 10.1101/gad.9.19.2350. [DOI] [PubMed] [Google Scholar]
- Wu Z, Bucher NLR, Farmer SR. Induction of peroxisome proliferator-activated receptor gamma during conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPβ, C/EBPδ and glucocorticoids. Mol Cell Biol. 1996;16:4128–4136. doi: 10.1128/mcb.16.8.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Rosen ED, Brun R, Hauser S, Adelmont G, Troy AE, McKeon C, Darlington GJ, Spiegelman BM. Cross-regulation of C/EBPα and PPARγ controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell. 1999;3:151–158. doi: 10.1016/s1097-2765(00)80306-8. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Oike Y, Kamon J, Waki H, Komeda K, Tsuchida A, Date Y, Li MX, Miki H, Akanuma Y, et al. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat Genet. 2002;30:221–226. doi: 10.1038/ng829. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Cao Z, Classon M, McKnight SL. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995;9:168–181. doi: 10.1101/gad.9.2.168. [DOI] [PubMed] [Google Scholar]
- Yoo EJ, Chung JJ, Choe SS, Kim KH, Kim JB. Downregulation of histone deacetylases stimulates adipocyte differentiation. J Biol Chem. 2006;281:6608–6615. doi: 10.1074/jbc.M508982200. [DOI] [PubMed] [Google Scholar]
- Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem. 2005;280:13600–13605. doi: 10.1074/jbc.M409468200. [DOI] [PubMed] [Google Scholar]
- Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell. 2002;2:331–341. doi: 10.1016/s1534-5807(02)00131-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fu M, Cui T, Xiong C, Xu K, Zhong W, Xiao Y, Floyd D, Liang J, Li E, et al. Selective disruption of PPARgamma 2 impairs the development of adipose tissue and insulin sensitivity. Proc Natl Acad Sci USA. 2004a;101:10703–10708. doi: 10.1073/pnas.0403652101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JW, Klemm DJ, Vinson C, Lane MD. Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. J Biol Chem. 2004b;279:4471–4478. doi: 10.1074/jbc.M311327200. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Qiang L, Farmer SR. Activation of C/EBPalpha expression by C/EBPbeta during adipogenesis requires a PPARgamma-associated repression of HDAC1 at the C/EBPalpha gene promoter. J Biol Chem. 2006;281:7960–7967. doi: 10.1074/jbc.M510682200. [DOI] [PubMed] [Google Scholar]