Quantitative Characterization of Intrinsic Disorder in Polyglutamine: Insights from Analysis Based on Polymer Theories (original) (raw)
Abstract
Intrinsically disordered proteins (IDPs) are unfolded under physiological conditions. Here we ask if archetypal IDPs in aqueous milieus are best described as swollen disordered coils in a good solvent or collapsed disordered globules in a poor solvent. To answer this question, we analyzed data from molecular simulations for a 20-residue polyglutamine peptide and concluded, in accord with experimental results, that water is a poor solvent for this system. The relevance of monomeric polyglutamine is twofold: It is an archetypal IDP sequence and its aggregation is associated with nine neurodegenerative diseases. The main advance in this work lies in our ability to make accurate assessments of solvent quality from analysis of simulations for a single, rather than multiple chain lengths. We achieved this through the proper design of simulations and analysis of order parameters that are used to describe conformational equilibria in polymer physics theories. Despite the preference for collapsed structures, we find that polyglutamine is disordered because a heterogeneous ensemble of conformations of equivalent compactness is populated at equilibrium. It is surprising that water is a poor solvent for polar polyglutamine and the question is: why? Our preliminary analysis suggests that intrabackbone interactions provide at least part of the driving force for the collapse of polyglutamine in water. We also show that dynamics for conversion between distinct conformations resemble structural relaxation in disordered, glassy systems, i.e., the energy landscape for monomeric polyglutamine is rugged. We end by discussing generalizations of our methods to quantitative studies of conformational equilibria of other low-complexity IDP sequences.
INTRODUCTION
Intrinsically disordered proteins (IDPs) are functional proteins that do not fold into well-defined, ordered tertiary structures under physiological conditions (1–4). These proteins are termed intrinsically disordered because disorder prevails under nondenaturing conditions and amino acid sequence encodes the propensity to be disordered. Generic IDP sequences have a combination of low overall hydrophobicity (5) and low sequence complexity (6). The question of how disorder is used in function will remain unanswered pending the availability of accurate physical models for conformational equilibria of IDPs (4). Conformational equilibria refer to ensemble averages and spontaneous fluctuations of structural properties of IDPs in their native milieus.
In polymer physics, global descriptors provide a way to classify disorder
Theories based on the physics of polymer solutions are relevant for describing conformational equilibria of IDPs (7). The focus in these theories is on global measures such as the ensemble-averaged radius of gyration, 〈_R_g〉 (8). The balance between chain-chain and chain-solvent interactions is determined by the nature of solvent milieus, which are classified as being good or poor solvents (9,10). The scaling of 〈_R_g〉 with chain length N is written as 〈_R_g〉 = R_o_N ν. In a good solvent, the main repeating unit is chemically equivalent to the surrounding solvent, the effective chain-chain interactions are strictly repulsive, and 〈_R_g〉 ∼ _N_0.59. In a poor solvent, attractive interactions dominate and the result is a preference for an ensemble of compact conformations such that 〈_R_g〉 ∼ _N_0.33 (11). In the simplest of polymer frameworks, conformational ensembles for IDPs in aqueous milieus can be classified either as disordered swollen coils in a good solvent or compact, albeit disordered globules in a poor solvent. Which of these classifications best suits the description of conformational ensembles for archetypal IDP sequences in water? This question forms the focus of this work.
Rationale for studying monomeric polyglutamine
The relevance of monomeric polyglutamine is twofold: Homopolymers such as polyglutamine are archetypal IDPs because they are low complexity sequences and they are deficient in hydrophobic residues (5,6,12). Second, conformational fluctuations in monomeric polyglutamine are involved in seeding the aggregation of polyglutamine—a process that is relevant to the onset and progression of a class of hereditary neurodegenerative diseases (13–19). Ages-of-onset of disease in polyglutamine disorders show nonlinear, inverse correlation with the length of polyglutamine expansions (13). Different hypotheses have been put forth to explain both the toxicity associated with polyglutamine expansions and its chain length dependence (20). There is evidence for increased proteolytic processing of proteins with expanded polyglutamine tracts (20). Products of proteolysis are rich in polyglutamine (21) and their aggregation appears to be essential for toxicity (22). Inhibiting polyglutamine aggregation reduces neurodegeneration (23–25). Furthermore, the early species along aggregation pathways are viewed as being the most toxic (26,27). Obviously, monomeric, soluble polyglutamine is the starting point for the process of aggregation. An assessment of fluctuations that seed the formation of aggregates requires quantitative knowledge of conformational equilibria within the monomeric form and this topic is the focus of this work.
Monomeric polyglutamine is intrinsically disordered
Structural studies of monomeric polyglutamine suggest that these peptides are intrinsically disordered in aqueous milieus (28–30), although claims of short stretches of consensus polyproline II helix structure have been made (31). The absence of sequence specificity in a homopolymer explains the lack of preferred secondary and tertiary structures in polyglutamine (32–34). Analysis of data from our previous molecular dynamics (MD) simulations showed that monomeric polyglutamine is intrinsically disordered and favors collapsed conformations in water (32). However, we did not arrive at definitive conclusions regarding the solvent quality (good or poor) of water for polyglutamine because we conjectured that this would require simulations of conformational equilibria for multiple chain lengths. Instead, we sought quantitative adjudication using experimental methods.
Monomeric polyglutamine forms collapsed, spherical globules in water
Crick et al. (35) used fluorescence correlation spectroscopy (FCS) measurements to quantify the hydrodynamic sizes of monomeric polyglutamine as a function of chain length. They measured the scaling of translational diffusion times (〈_τ_D〉) for the peptide series (Gly)-(Gln)N-Cys-(Lys)2 in aqueous solution at room temperature (∼25°C). It was found that 〈_τ_D〉 scales with chain length N as τ_o_N ν where ν = 0.32 ± 0.02 and ln(_τ_o) = 3.04 ± 0.08. The measured value for ν supports the conclusion that water is a poor solvent for monomeric polyglutamine. The scope of these experiments is limited to quantifying scaling exponents, which is a necessary but not sufficient condition to assess the quality of a solvent (36). Conformational equilibria for polymers in poor solvents are distinguishable from those in good solvents based on the behavior of specific order parameters (36,37). Here, we complement the recent FCS studies by analysis of data from molecular simulations from which the relevant order parameters are directly accessible.
Questions of interest
This work focuses on answering three specific questions:
- Is it possible to make quantitative assessments regarding the quality of a solvent milieu for a single IDP sequence using data obtained from molecular simulations? To answer this question, we use the sequence Ac-(Gln)20-Nme (Q20) as our archetypal IDP sequence. Specifically, we compared results from analysis of multiple replica molecular dynamics (MRMD) for Q20 in water (T = 298K, P = 1 bar) to data from two sets of Metropolis Monte Carlo simulations for reference ensembles in good and poor solvents. The Monte Carlo simulations employed here are routinely used in the polymer physics literature and are based on the use of generic Hamiltonians that lack the specificities of chain-chain and chain-solvent interactions (38,39). The comparative analysis is guided by the use of polymer theories (36,37), which make specific predictions regarding variations of order parameters such as the scaling of internal distances, angular correlation functions, and radial density profiles as a function of solvent quality. We show that the comparative analysis leads unequivocally to the identification that water is a poor solvent for Q20. The main highlight of this analysis is that it can be adapted to classify the nature of disorder for any low-complexity IDP sequence (6).
- Why is water a poor solvent for polyglutamine? The observation that water is a poor solvent for polyglutamine can be inferred from its strong aggregation propensity (30,40,41). However, it seems counterintuitive that a system composed entirely of polar moieties readily forms aggregates given that the building blocks of polyglutamine, i.e., primary and secondary amides, are freely miscible with water (42,43). If anything, the high miscibility of model compounds suggests that water should be a good solvent for polyglutamine. Obviously, the concatenation into a polymer alters the solvation properties of amide groups. Here, we present a preliminary analysis based on comparisons of data from simulations of aqueous solutions of amide mixtures to that of Q20 in water. Based on this analysis, we propose that favorable intrabackbone interactions in the polymer provide at least part of the driving force for the collapse of polyglutamine in water.
- What is the nature of conformational relaxation dynamics for an IDP such as polyglutamine? Polyglutamine forms aggregates, albeit very slowly (44). Chuang et al. (45) have proposed that the rate limiting step for aggregation of polymers in poor solvents is conformational relaxation within polymer globules. Consistent with this prediction, we find that although the collapse transition for Q20 in water is rapid (∼5 ns), the timescales for conversion between distinct compact conformations are very slow, and the dynamics are akin to structural relaxation in glassy systems (46). We also show that the glassy behavior of Q20 in water is uncovered using the MRMD methodology employed in our work.
We organize the remainder of our presentation as follows: First, we describe details of the methods used in our work. Next, we describe the details of our results. Finally, we end with a summary and discussion of the main results.
MATERIALS AND METHODS
Potential functions for simulating conformational equilibria of polymeric reference states
Reference conformational equilibria of disordered polymers in good and poor solvents can be simulated using generic, implicit solvent models (38,39). In this approach (47), conformational equilibria for chains in good solvents are simulated using interatomic interactions based on a purely repulsive, inverse power potential as shown in Eq. 1.
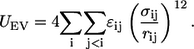 |
(1) |
|---|
Equation 1 corresponds to the so-called excluded volume (EV) limit, wherein only steric interactions are included. Simulations of conformational equilibria in the EV limit provide a good mimic for equilibria in good solvents. Conversely, the nonspecific drive of a chain to sequester itself from making contacts with a poor solvent can be captured by adding attractive van der Waals interactions to the repulsive potentials from the EV limit (38,39). This model, based on the Lennard-Jones functional form, is shown in Eq. 2, and is termed the LJ model.
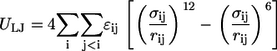 |
(2) |
|---|
In Eqs. 1 and 2, _r_ij denote distances between any two nonbonded atoms, _σ_ij are contact distances, and _ɛ_ij are the Lennard-Jones dispersion parameters. For the EV limit, Eq. 1, the parameters for _σ_ij and _ɛ_ij are those used in previous work (48). Conversely, for the LJ model, Eq. 2, we used the parameters from the OPLS-AA/L force field (49). These choices are justified on the following grounds: The _σ_ij values used in previous work were derived from Pauling's parameterization, which in turn reproduce heats-of-fusion data for model compounds. These _σ_ij values can be used in purely repulsive potentials and it has been shown that these parameters allow us to reproduce accurate Ramachandran maps (48). Conversely, the values of _σ_ij in the OPLS-AA/L force field are coparameterized with _ɛ_ij to reproduce the heats-of-vaporization and densities of neat liquids. Therefore, the _σ_ij values in OPLS-AA/L are too large for use in purely repulsive potentials. However, use of the OPLS-AA/L parameters for the LJ model guarantees that the densities of the maximally compact reference globules are similar to those expected for globules populated by chains in explicit water.
Simulations of reference conformational equilibria
We carried out Metropolis Monte Carlo simulations, as described in previous work (47,48), to simulate reference conformational equilibria for polyglutamine peptides using the EV and LJ models. In these simulations, the degrees of freedom were the backbone and side-chain dihedral angles of an isolated chain. We carried out two sets of simulations using each of the models shown in Eqs. 1 and 2. In the first set, we carried out simulations for a series of chain lengths to demonstrate that ensemble averaged radii of gyration scale (〈_R_g〉) with chain length as ∼_N_0.6 for the EV model and as ∼_N_0.33 for the LJ model. The scaling of 〈_R_g〉 with chain length N was obtained by gathering statistics for peptides of the form: Ac-(Gln)N-Nme, where Ac denotes the acetyl group and Nme stands for _N_-methylamide. For the EV limit, N = 50, 75, 100, 150, and 250 and for the LJ model, we simulated equilibria for N = 24, 27, 33, 36, 40, 47. The simulation temperatures were T = 298 K and T = 425 K for the EV and LJ models, respectively. We used a higher temperature in simulations based on the LJ model to improve the efficiency with which conformational space is sampled and to reduce the error bars in our estimates for polymeric properties. Given the high melting temperature for the LJ model, at T = 298 K we would have needed simulations that were orders of magnitude longer to obtain converged estimates, and hence the choice of T = 425 K as the simulation temperature.
As noted above, the purpose of the Monte Carlo simulations was to demonstrate that the two models, viz., EV and LJ, reproduce the scaling behaviors for polymers in good and poor solvents, respectively. The EV limit calculations were carried out for longer chains to overcome the finite size artifacts because the thickness of the polymer “tube” has to be negligible when compared to its contour length. For polyglutamine, this requirement does not hold true for chains with N < 50. In contrast, finite size effects play a minor role for quantifying the scaling law for chains in a poor solvent. This is true so long as N is larger than the length of locally stiff segments, approximately seven residues (47). The chain lengths used for calculations in the globular limit were therefore chosen in correspondence with recent FCS studies (35). In addition to the simulations used to quantify scaling laws, we also simulated conformational equilibria for Ac-(Gln)20-Nme, i.e., Q20 using both the EV and LJ models. As we will show in the Results section, the comparative analysis between ensembles obtained for Q20 using the EV, LJ, and molecular mechanics potentials in explicit solvent allows us to assess if the conformational equilibria for Q20 in water are congruent with those of chains in poor versus good solvents.
Setup of molecular dynamics simulations for Q20
To characterize conformational equilibria in water we used an approach that we refer to as multiple replica molecular dynamics (MRMD). This approach relies on the use of data from a large number of independent simulations and the advantage is that data are gathered using multiple independent simulations as opposed to a single, long, and potentially uninformative simulation. Conformational space is explored more efficiently by relying on the underlying stochasticity of phase space trajectories, given different initial positions and velocities.
We used the GROMACS simulation package (50) for all MD simulations. In this work, we report data from MRMD simulations applied to the peptide Q20 in water at T = 298 K. We simulated 60 independent replicas. For the peptide we used the OPLS-AA/L force field (49). The peptide was soaked in a bath of 8952 TIP3P water molecules (51). Boxes for individual simulations were prepared by soaking a random peptide conformation obtained in the EV ensemble, followed by adding or deleting water molecules such that we ended up with the same number of water molecules for all replicas. In each case, a steepest-descent minimization to remove steric clashes was followed by an equilibration run of 11 ns in the isothermal-isobaric ensemble (T = 298 K, P = 1 bar). The final configuration of the latter was used as the starting point for the production run of 50 ns length. Therefore, the total simulation time for each of the 60 independent simulations was 61 ns for a cumulative simulation time of ∼3.7 _μ_s.
The leap-frog integrator was used with a time step of 2 fs. The temperature was maintained through the Berendsen thermostat (52) with a coupling time of 0.2 ps. Similarly, constant pressure was maintained by the Berendsen manostat (52) with a coupling time of 1 ps and a compressibility of 4.5 × 10−5 bar−1. The average size of the cubic box throughout the simulations was roughly 65.4 Å with negligible volume fluctuations. Peptide bond lengths were constrained using the LINCS algorithm (53) and the rigidity of water molecules was achieved using the SETTLE algorithm (54). For nonbonded interactions, we employed 10 Å spherical cutoffs for van der Waals as well as for short-range Coulomb interactions. Long-range Coulomb interactions (10–14 Å) were recalculated every 10 steps, as were neighbor lists. The reaction field (RF) method (55) was used as a correction term for polar interactions beyond 14 Å. For each of the 60 independent simulations, structures of the peptide alone were saved once every 4 ps for subsequent analysis.
Setup of simulations for aqueous solutions of model compounds
To assess the differences between polyamides (such as polyglutamine) versus amides in water we carried out simulations of aqueous mixtures of amides. The systems studied were aqueous mixtures of _trans_-_N_-methylacetamide (NMA) and propionamide (PPA) in water; NMA is a model compound mimic of the peptide backbone (a secondary amide) whereas PPA is a mimic of the side chain (a primary amide). We followed the simulation protocol described for Q20. The amides were modeled using the OPLS-AA force field (56) and we used the TIP3P model for water molecules. To achieve concentrations of 1 m, 2 m, and 4 m, respectively, 15, 30, and 60 molecules of each amide were soaked in a box of 833 water molecules, and equilibrated for mixing purposes for 1 ns in the canonical (NVT) ensemble at T = 298 K. The production run was carried out in the isothermal-isobaric (T = 298 K, P = 1 bar) ensemble for 50 ns after an extra equilibration period of 200 ps. Ten such trajectories were run for each concentration and the snapshots of the amide configurations, which were saved every 10 ps, were analyzed to calculate site-site pair correlation functions.
Reliability analysis
Given _n_s independent trajectories, the standard error (SE) was estimated by computing the average of an observable for each trajectory. The SE is defined as the standard deviation in _n_s independent estimates for the mean. Our procedure for computing the SE is an adaptation of conventional block averaging methods. The difference is that the size of the block being averaged over is the length of an individual trajectory. The standard deviation of the trajectory-averaged structural quantities yields the SE indicated by error bars in the plots. This approach for calculating error bars is reasonable because data from different trajectories are in fact truly uncorrelated.
RESULTS
Demonstration of the validity of reference models
Fig. 1 shows the scaling of 〈_R_g〉 versus chain length N for polyglutamine in the EV and LJ limits, respectively. In the log-log plots shown in Fig. 1, the slopes provide an estimate of the scaling exponent. We find that slopes for polyglutamine in the EV and LJ limits are similar to the theoretical values of 0.59 and 0.33 in good and poor solvents, respectively. Deviations from theoretical values are primarily due to finite-size effects, i.e., the fact that we did not gather data for very long chains. In properly converged simulations, the scaling exponent in the EV limit will be overestimated when there are finite size artifacts. This is because short chains in the EV limit have a smaller, apparent 〈_R_g〉 when compared to the theoretical prediction. Conversely, finite size artifacts lead to an underestimation of the poor solvent exponent. This is because short chains have a larger apparent 〈_R_g〉, which is precisely what we find.
FIGURE 1.

Scaling laws for the two reference models (see Eqs. 1 and 2). The fit for the EV limit is done only over the last five points. As can be seen, finite-size effects cause the data for shorter chain lengths to fall off this line. Including these points would significantly overestimate the scaling exponent. In the globular reference state, finite-size effects are restricted to much shorter chain lengths. The theoretical exponent of ∼0.33 is slightly underestimated.
The preceding analysis demonstrates that conformational equilibria simulated using the EV and LJ models provide limiting distributions for disordered polypeptides in good versus poor solvents. Due to the extensive computational cost of the simulations in explicit water (see below) we cannot determine the scaling exponent, which requires very expensive simulations for multiple chain lengths. Instead, analyses of specific polymeric measures for Q20 in water were compared to those of Q20 in the EV and LJ limits, respectively. This allowed us to make definitive conclusions regarding the solvent quality of water for polyglutamine.
Comparative analysis of the distribution of shapes and sizes
For a specific conformation of a polymer, the shape and size are quantified using the gyration tensor defined as:
 |
(3) |
|---|
Here, _Z_m is the number of atoms in the molecule, ri are the position vectors of individual atoms, 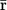 is the position vector of the centroid, and the symbol ⊗ refers to the dyadic product. If we use _λ_1,2,3 to denote the eigenvalues of T, the radius of gyration (_R_g), the measure of size, and asphericity (δ), which measures chain shape are given as:
is the position vector of the centroid, and the symbol ⊗ refers to the dyadic product. If we use _λ_1,2,3 to denote the eigenvalues of T, the radius of gyration (_R_g), the measure of size, and asphericity (δ), which measures chain shape are given as:
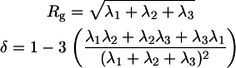 |
(4) |
|---|
For a perfect sphere, δ = 0, and for a perfect rod, δ = 1; for intermediate values, the chain assumes ellipsoidal shapes. Therefore, δ quantifies the degree to which chain shape deviates from that of a perfect sphere. This measure of shape has been very useful for analyzing asymmetry in protein structures (57) and for the analysis of average shapes of denatured proteins (47).
Two-dimensional histograms, i.e., ρ(_R_g, δ) in the space spanned by the two parameters _R_g and δ provide insights regarding the preferred shapes and sizes of a molecule (32). Fig. 2 shows these distributions for Q20 in water and for the two reference models. Conformations with low asphericity and low _R_g are favored for Q20 in water. This is suggestive of water being a poor solvent for Q20. This point is reinforced by favorable comparison of histograms in water to those obtained for the globular reference ensemble using the LJ model. The only difference is that the latter are characterized by smaller-scale fluctuations. In stark contrast, the peptides in the EV limit prefer conformations with larger _R_g and asphericity values. Even more importantly, there is no overlap between histograms obtained in the EV limit versus those for either Q20 in water or Q20 in the reference globule. Polymers, of the requisite length have access to three distinct phases, viz., the globule, coil, and rod phases (37). The data shown in Fig. 2 support the conclusion that conformational equilibria for Q20 in water and calculated using the LJ model are consistent with the globule phase whereas the equilibria in the EV limit are those of the coil phase.
FIGURE 2.

Two-dimensional histograms of the normalized radius of gyration and asphericity (see Eq. 4) for Q20 in water and the two reference models. The data are binned with a spacing of 0.05 Å on the _R_g axis and 0.02 on the δ axis, respectively. For the purpose of clarity, the colors are slightly offset from the white background.
Collapse does not mean order
One might be tempted to speculate that Q20 prefers a specific globular structure in water. If true, then such an observation would be incongruent with experimental observations, according to which soluble and monomeric polyglutamine peptides are described as being disordered by measures such as circular dichroism (30) or NMR (28). Fig. 3 shows that our results are consistent with interpretations of experimental data. The interresidue contact maps show no preference for specific contacts. We can, however, distinguish two classes of disorder: i), disorder under the constraint of dense packing results in relatively large contact probabilities (see panels B and C), and ii), disorder in the swollen-coil state with very low contact probabilities (see panel A). The preferred contacts in the EV limit are exclusively local. Conversely, in both the LJ globule as well as in water, long-range contacts (sequence spacing >10) are actually more likely than midrange contacts (sequence spacing 5–9). Local contacts are enhanced in the aqueous case vis-à-vis the LJ globule. We attribute these differences between the LJ globule and the aqueous globule to specific local interactions present in the latter (32), a feature that is missing in the case of the LJ globule. One might argue that our analysis of disorder observed for Q20 in water masks the identification of secondary structure, since _α_-helices or _β_-sheets with highly variable registry might be possible. However, previous analysis of backbone segments confirmed that there is little to no stable canonical secondary structure (32). Similar conclusions were drawn from the current dataset (data not shown).
FIGURE 3.

Contact maps for Q20 in water (B), in the EV limit (A), and in the globular limit (C). Grayscale indicates the frequency of observing a given residue-residue contact throughout the simulation. Short-range contacts are excluded to enhance the signal/noise ratio. A contact is defined by any two atoms k and l from residues i and j having a distance ≤3 Å. The maps are by definition symmetric.
Scaling of internal distances with sequence separation
The first polymeric measure we quantify is the scaling of internal distances with sequence separation:
 |
(5) |
|---|
In Eq. 5, the 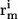 and
and 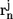 denote the position vectors of atoms m and n, which are part of residues i and j, respectively, and _Z_ij is the number of unique pairwise distances between the two residues. As in all equations, the angular brackets indicate the average over all trajectories and all saved snapshots. Plotted as a function of sequence separation, it is expected that for chains in a good solvent 〈_R_ij〉 ∼ |j − i|0.59 (58), which is also true in the EV limit (47). In a good solvent, polymers behave like fractal objects, i.e., internal distances scale with sequence separation the way end-to-end distances scale with chain length. Fig. 4 shows that the scaling of internal distances in the EV limit ensemble agrees with the theoretical scaling law. Significant deviations occur at small sequence separations, for which the local rigidity and detailed structure of the polymer modulate the limiting behavior. Similar observations were made by Ding et al. (59) in their analysis of the scaling behavior of proteins near and above the folding transition.
denote the position vectors of atoms m and n, which are part of residues i and j, respectively, and _Z_ij is the number of unique pairwise distances between the two residues. As in all equations, the angular brackets indicate the average over all trajectories and all saved snapshots. Plotted as a function of sequence separation, it is expected that for chains in a good solvent 〈_R_ij〉 ∼ |j − i|0.59 (58), which is also true in the EV limit (47). In a good solvent, polymers behave like fractal objects, i.e., internal distances scale with sequence separation the way end-to-end distances scale with chain length. Fig. 4 shows that the scaling of internal distances in the EV limit ensemble agrees with the theoretical scaling law. Significant deviations occur at small sequence separations, for which the local rigidity and detailed structure of the polymer modulate the limiting behavior. Similar observations were made by Ding et al. (59) in their analysis of the scaling behavior of proteins near and above the folding transition.
FIGURE 4.

The scaling of average internal distances as a function of sequence separation (see Eq. 5). A theoretical good solvent scaling law is indicated by the dotted line. SE are indicated by error bars for the data in water and the globular reference state. Errors are negligible for the EV ensemble and hence not shown. The polypeptide caps are included in this analysis, which is why there are effectively 22 residues in the chain.
Conversely, for chains in a poor solvent, theory tells us that ensemble-averaged internal distances should plateau to a constant value corresponding to the density of the collapsed species (37). The scaling of internal distances for Q20 in water and in the globular reference state is found to be consistent with this expectation. The plateau values achieved are in agreement with each other within error. Local length scales, also known as “blob” lengths are a characteristic of linear flexible polymers (11). Over this length scale, the scaling of internal distances as a function of sequence spacing is determined primarily by steric interactions, and it is not possible to distinguish good from poor solvents based on conformational equilibria over the “blob” length. Blob lengths can be deduced from the rising part of the curves shown in Fig. 4 and are found to be approximately seven to eight residues, consistent with previous findings (47,48).
Up-and-down topologies in water
Ensemble-averaged angular correlation functions, _c_ij, provide a way to quantify average topologies adopted by chains in different milieus. This function, analogous to a function proposed by Socci et al. (60), and computed as a function of sequence spacing, is defined as:
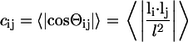 |
(6) |
|---|
Here, li(j) denotes the vector from the backbone nitrogen of residue i(j) to the carbonyl carbon on the same residue, and l is its length. Therefore, Θij is the effective angle between the direction of the chain at residues i and j. For chains in a good solvent _c_ij will decay exponentially as a function of sequence separation |i − j|. Conversely, chains in a poor solvent are under a packing constraint, and on average, the chain will reverse direction. This results in negative values for _c_ij. Fig. 5 shows precisely this behavior. In the EV limit, correlations slowly decay to zero as expected. In contrast, the data for the peptide in water and for the globular reference state are characterized by significant anticorrelation at approximately five to 10 residues of sequence separation. This is the aforementioned midrange length scale, over which the chain on average turns on itself. Beyond this length scale, correlations decay to zero. The large error bars for the data in water seen in Fig. 5 are due to two effects: i), every trajectory results in a distinct topology for the globule, and ii), on the timescale of the simulations, there is no interconversion between these distinct topologies indicating quenched disorder (see below).
FIGURE 5.

The angular correlation function (see Eq. 6) as a function of sequence separation. The polypeptide caps are excluded from this analysis. For details on errors see caption to Fig. 4.
Radial density profiles
Density profiles are another way to characterize the average shape of macromolecules, and form the basis for Lifshitz-type theories for the coil-to-globule transition (8,36):
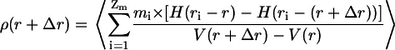 |
(7) |
|---|
Here, _r_i is the distance of atom i from the molecule's center of mass, _m_i is the mass of atom i, _Z_m is the number of atoms in the molecule, V(r) is the volume of a sphere with radius r, and H is the Heaviside step function. Fig. 6 shows that ρ(r) reaches a plateau value for short distances in both the globular reference state and for the peptide in water. The limiting density is ∼1.2 g/cm3. The most significant difference is in the long distance regime of the density profile. This implies that the peptides in water undergo larger-scale conformational fluctuations than in the globular reference state. The observed plateau value for the density of globules in water and in the LJ reference state is less than that of small, folded proteins (61). We attribute this difference to the presence of pronounced conformational fluctuations for an IDP such as Q20 when compared to stable, folded polypeptides. In the EV limit, the density profile is shallow, and reaches a plateau value of ∼0.4–0.5 g/cm3. Such a low value is possible, since chains in the EV limit are characterized by interior cavities of all sizes (47), and the density is averaged over both void spaces and the chain itself.
FIGURE 6.

The average density as a function of distance to the center of mass (see Eq. 7). For details on errors see caption to Fig. 4.
Kratky profiles
Finally, Kratky or scattering profiles, K(q), (62) provide a direct connection to experimental data, as they are available from small angle x-ray scattering (SAXS) measurements. If we assume homogeneous scattering cross sections across the molecule, the Kratky profile becomes an effective measure of the peptide's density as a function of a specific length scale:
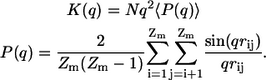 |
(8) |
|---|
Here, the _r_ij are pairwise atomic distances, _Z_m is the number of atoms in the molecule, N is chain length, and q are wavenumbers in units of Å−1. Large peaks in the low and intermediate _q_-regime (0.1 ≤ q ≤ 0.4) are indicative of compact geometries, as they result from a dense collection of scatterers. Conversely, if the Kratky profile is essentially flat with generally low amplitudes, we infer that the scatterers form a loosely packed object with low average density. This is the expected signature for chains in the EV limit. Fig. 7 shows that our expectation is again met by the actual data. The profile for the chain in water is very similar to that in the globular reference state, and is undoubtedly distinct from the profile for the EV chain. It is interesting to note that the Kratky profile shows significant quantitative differences between the globular reference and the water data in the high _q_-regime. This probes differences in local structural propensities between the two ensembles.
FIGURE 7.

Ensemble averaged Kratky profiles (see Eq. 8) calculated for the three different models. For details on errors see caption to Fig. 4.
Based on the preceding discussion, we conclude that polymer theory provides us with at least four distinct measures, which allow us to establish that water is a poor solvent for Q20. The four quantities we have used to make conclusive analyses are the scaling of internal distances, angular correlation functions to measure average topologies, radial density profiles, and Kratky profiles (closely related to radial density profiles). When these quantities are computed for data obtained from simulations in explicit water and compared to analysis of simulation data from reference states, we are able to make an unequivocal adjudication regarding the balance between chain-chain and chain-solvent interactions, i.e., solvent quality.
What are the driving forces for the collapse of polar polyglutamine in water?
Polyglutamine is a polyamide built of a repeat of secondary amides in the backbone and primary amides in the side chain. Fig. 8 shows a comparison of site-site pair correlation functions, g(r), for Q20 in water and for aqueous mixtures of dissociated primary and secondary amides. We normalized the intrachain and intermolecular pair correlation functions using different default models because the former is a polymer and the latter is a mixture of freely diffusing molecules. For the polymer, we used an ideal chain model, and for the model compounds, we used an ideal gas prior. Details are discussed in the Appendix. The model compounds chosen to represent the “dissociated” peptide are _trans_-_N_-methylformamide (NMF) and propionamide (PPA) mixing freely in solution. NMF, a secondary amide, is an analog of the backbone peptide unit, whereas PPA, a primary amide, is an analog of the polar side chain of glutamine.
FIGURE 8.

The left column shows site-site correlation functions for different atom pairs for Q20 in water. The data are normalized by an ideal chain prior (see Appendix). Dotted lines indicate SE intervals. The right column shows analogous site-site correlation functions for the solutions of NMF and PPA in water normalized by an ideal gas prior. Data for three different concentrations are shown (1 m, solid curves; 2 m, dashed curves; and 4 m dash-dotted curves). The sensitivity of the results to amide concentration is small. SE are negligible for these simulations.
The first row in Fig. 8 compares correlation functions between intrachain backbone donor and acceptor atoms to the site-site correlations between NMF donors (NNMF) and NMF acceptors (ONMF). The first peak around 3 Å is pronounced for the polymer and only weakly present for the model compound mixtures in solution. A different scenario holds for the comparison of pair correlations between backbone-donor and side-chain-acceptor atoms to those between NNMF and OPPA, which are shown in the second row of Fig. 8. There is a distinct, yet broad peak at 3 Å separation in the polymer, but general depletion otherwise. For the amide mixtures in solution, the situation is inverted in that there is relatively strong association at 4–5 Å, but no short-range peak at ∼3 Å. On the polymer side, the situation is very similar for the inverse pair correlation, viz., backbone acceptor and side-chain donor. Again, there is a weak, yet distinct peak around 3 Å, and a general depletion of density for short distances (third row of Fig. 8). For the model compounds, however, we observe a dominant peak at 3 Å followed by a broad second peak in the site-site correlation function for NPPA-ONMF. Finally, there is minimal deviation between pair correlations for the side chain–side chain donor-acceptor pair in the polymer and NPPA-OPPA (fourth row of Fig. 8). For the polypeptide, the correlation function is much smoother than that for other pairs. This is because the side chains have the most flexibility to rearrange with respect to one another. In both the polymer and for free amides we observe a distinct peak at 3 Å.
In summary, we can establish the following changes in the self-association behavior for amides in solution when compared to amides that are part of polyglutamine:
- For the model compounds in solution, we observe a marked preference for short-range correlations (∼3 Å) between donor atoms of primary amides (NPPA) and acceptor atoms of secondary amides (ONMF). Interrogation of the inverse pair correlations between sites NNMF and OPPA suggests favorable, solvent-separated intermolecular associations. These differences in donor-acceptor pair correlations are not preserved in the polymer. Instead, both types of pair correlations, viz., side-chain donor to backbone acceptor and backbone donor to side-chain acceptor, are equivalent.
- For the polymer, we observed a general trend that correlation function values are larger than unity for short (∼3 Å) and long distances (>6 Å) but are diminished over medium ranges (3.3–6 Å). This is due to excluded volume effects, which are absent in the ideal chain model used to normalize the pair correlations (see Appendix).
- The two pronounced terms in the polymer are the backbone-backbone and side chain–side chain correlation functions, which measure effective interactions between donor and acceptor atoms. Of these two correlation functions, only the pair correlations between backbone units are enhanced vis-à-vis the model compound counterparts. It appears that concatenated backbone units can solvate each other more favorably when compared to free secondary amides. Therefore, our preliminary conclusion is that the main driving force for collapse of polyglutamine in water derives from favorable intrabackbone correlations. This finding appears to be consistent with recent experimental data (63). There could be multiple sources for enhanced pair correlations. These include hydrogen bonding, the entropic benefits of releasing water molecules into the bulk, and the associated increase in chain packing density.
In the interest of clarity, we reiterate that the intrapolymer and model compound site-site pair correlations were normalized using different default models. Details of the normalization procedure are presented in the Appendix. For the polymer, we used an ideal chain model. This is different from the ideal gas model used as the default model for analyzing distance histograms for model compounds. Therefore, an intrapolymer site-site correlation is meaningful only if the peak or trough in the pair correlation function is greater than or less than unity, i.e., all enhancements and depletions in intrapolymer pair correlation functions arise due to specific multibody attractive/repulsive interactions. They should not be misinterpreted as being a consequence of elimination of entropic barriers via chain connectivity.
An alternative approach for making assessments regarding driving forces for collapse is to quantify the contributions of enthalpy and entropy to the free energy change associated with coil-to-globule transitions for polyglutamine. If this transition were to resemble hydrophobic collapse, the driving force would be primarily entropic in nature (64–68). The information necessary to make judgments regarding entropy and enthalpy is not available from simulations carried out for a single set of solution conditions. Free energy calculations on the solvation of collapsed versus extended states of Q20 would be able to address the above issue, but are intractable at this point.
Conformational relaxation dynamics: evidence for glassy kinetics and ruggedness of the energy landscape
Fig. 9 shows a checkerboard map of the average root mean square deviation (〈RMSD〉ij) calculated by superposition of all the structures in trajectory j onto the final structure in trajectory i. We find that the diagonal has a significantly lower average RMSD when compared to the off-diagonal elements, i.e., 〈RMSD〉ii < 〈RMSD〉ij. This is indicative of two features: First, there is strong residual correlation within each trajectory. Second, no pair of trajectories yields similar final structures, an observation that establishes the disordered nature of the ensemble. One might argue that inaccurate molecular mechanics force fields as well as the sluggishness of conformational sampling are the primary sources for our observation that the ensemble for polyglutamine is disordered. In other words, the MRMD simulation methodology applied to any polypeptide sequence with initial conformations drawn from the EV limit ensemble will yield a similar result. Although this skepticism is reasonable, it is also noteworthy that the ensemble dynamics methods of Pande and co-workers, which is similar in spirit to MRMD, have been used to successfully fold several small two-state proteins and obtain accurate estimates of their folding rates (69). Therefore, we propose that the congruence between our results and those based on spectroscopic experiments are robust because the homopolymeric nature of polyglutamine provides a reasonable physical basis for its intrinsic disorder. Of course, the concern expressed above can be addressed fully only through application of the MRMD approach to a wide range of sequences that have stable folds as well as to sequences that are predicted to be intrinsically disordered. These sorts of simulations are computationally challenging and may become feasible with appropriate methodological advances.
FIGURE 9.

Checkerboard map of the average all-atom RMSD in angstroms of the structures observed in trajectory j (y axis) from the final structure of trajectory i (x axis). This map is by construction not symmetric.
In Fig. 10, we show comparative analysis of the differences between the timescales for collapse versus the timescales associated with conformational relaxation. In panel A we plot 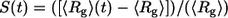 as a function of time. A single exponential fit for the decay of S(t) versus t is also shown. This function,
as a function of time. A single exponential fit for the decay of S(t) versus t is also shown. This function, 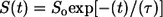 has the parameters _S_o = 0.40 and τ = 5 ns. In each of the trajectories, collapse from the relatively extended starting conformations, which are extracted from the EV ensembles is found to be a rapid process and occurs within the timescale of ∼5 ns, which is shorter than the equilibration times (11 ns) used in our analysis of MRMD data. This observation is robust across all trajectories. In the interest of clarity, we have added data from the equilibration periods. This was done for the analysis reported in Fig. 10 alone. For all other figures, only data from the production runs were used.
has the parameters _S_o = 0.40 and τ = 5 ns. In each of the trajectories, collapse from the relatively extended starting conformations, which are extracted from the EV ensembles is found to be a rapid process and occurs within the timescale of ∼5 ns, which is shorter than the equilibration times (11 ns) used in our analysis of MRMD data. This observation is robust across all trajectories. In the interest of clarity, we have added data from the equilibration periods. This was done for the analysis reported in Fig. 10 alone. For all other figures, only data from the production runs were used.
FIGURE 10.

(A) The time evolution of S(t), a normalized measure of 〈_R_g〉 as a function of time, t. The plot also shows the fit to a single exponential function 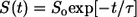 with _S_o = 0.40 and τ = 5 ns. The norm of the residuals between the raw data and the exponential function is 0.01. (B) RMSD of the structures within a trajectory from their final structure (gray diamonds) is compared to that of the structures within a trajectory to the final structure of other trajectories (gray circles). SE for the former could not be obtained because there is only one value per trajectory and per time point. For the cross-term, the 59 values per trajectory and per time point were preaveraged and SE could be obtained as usual. Data for the average conformational relaxation within a trajectory (gray diamonds) are fit to a stretched exponential function of the form described in the text. This is shown as the solid curve in the plot. Deviations from the stretched exponential function are largest for the earliest time points, t < 5 ns, and for the last 10-ns interval. The former is explained by the rapid collapse over short timescales, whereas the latter is entirely due to our choice of the final snapshot of the trajectory as the reference snapshot for analyzing conformational relaxation.
with _S_o = 0.40 and τ = 5 ns. The norm of the residuals between the raw data and the exponential function is 0.01. (B) RMSD of the structures within a trajectory from their final structure (gray diamonds) is compared to that of the structures within a trajectory to the final structure of other trajectories (gray circles). SE for the former could not be obtained because there is only one value per trajectory and per time point. For the cross-term, the 59 values per trajectory and per time point were preaveraged and SE could be obtained as usual. Data for the average conformational relaxation within a trajectory (gray diamonds) are fit to a stretched exponential function of the form described in the text. This is shown as the solid curve in the plot. Deviations from the stretched exponential function are largest for the earliest time points, t < 5 ns, and for the last 10-ns interval. The former is explained by the rapid collapse over short timescales, whereas the latter is entirely due to our choice of the final snapshot of the trajectory as the reference snapshot for analyzing conformational relaxation.
Although collapse is rapid, conformational relaxation is considerably slower. Panel B of Fig. 10 shows the time evolution of the average RMSD for superposition of structures within a trajectory i to the final structure of trajectory i, i.e., 〈RMSD(t)〉self. The temporal evolution of this parameter is described using a stretched exponential function: 〈RMSD(t)〉self = _R_oexp[−(t/τ)_β_], with _R_o = 22 Å and β = 0.15. Here, τ is set to be 5 ns, the timescale for collapse. The stretched exponential function, also known as the Kohlrausch-Williams-Watts (KWW) function (with 0 ≤ β ≤ 1), is used to describe structural relaxation in glassy systems (below the glass transition temperature) (46,70–72). If β assumes small values, then the system has access to a broad and heterogeneous distribution of relaxation times (70). Our discovery that conformational relaxation of Q20 follows nonexponential kinetics with a fairly small value of β is consistent with the postulate that distinct collapsed structures are likely to be of equivalent stability on account of the homopolymeric nature of polyglutamine, i.e., the energy landscape is rugged for Q20 in water at T = 298 K and P = 1 bar.
There are two predicted features for rugged energy landscapes: The first is slow, nonexponential relaxation within distinct basins, which is best described using a KWW function (46,70,71). Secondly, there should be evidence of even slower interconversion between distinct basins (70). Evidence for the latter is also shown in panel B of Fig. 10. Here, we track the temporal evolution of 〈RMSD(t)〉cross, which is the average RMSD for superposition of a snapshot from trajectory i upon the final structure of trajectory j, where j ≠ i. The desired average is calculated over all unique pairs of trajectories (i) and final structures (j). We find that, once the chain is collapsed, 〈RMSD(t)〉cross shows no significant time dependence over the remaining time scale of 50 ns. The time dependence of both 〈RMSD(t)〉cross and 〈RMSD(t)〉self taken together are interpreted as follows: Although collapse is rapid and the 〈_R_g〉 values across trajectories are similar to each other, each trajectory samples a distinct family of globular conformations, and there is no obvious interconversion between the distinct globules over the 50-ns timescale.
Our MRMD approach provides reliable information regarding global, polymeric order parameters, because this information is converged and roughly equivalent across all trajectories. Conversely, any analysis of specific structural propensities would yield entirely unreliable information because this requires interconversion between distinct conformational basins. To achieve this, each independent trajectory in the MRMD approach will need to be extended into the microsecond range or longer, without pruning, and perhaps increasing the number of independent trajectories. The impact of conformational heterogeneity and diminished conformational averaging is seen in the large error bars for the angular correlation function (see Fig. 5). This measure probes local conformational propensities as well as global properties and is therefore most sensitive to the quality of statistics we gather.
DISCUSSION, CONCLUSIONS, AND FUTURE WORK
We have analyzed MRMD simulations for a single polypeptide chain, Q20, in water. Our analysis, combined with polymer physics theories, and comparison to data from reference simulations, allows us to conclude that Q20 in water has all the characteristics of a chain in a poor solvent (Figs. 2–7). The physics of homopolymers allows us to generalize and conclude that water is a poor solvent for polyglutamine, i.e., at infinite dilution these systems form disordered globules and at finite concentrations, the stable thermodynamic state will be the phase separated aggregate (11,73). Implications of the poor solvent nature of aqueous solvents for the mechanism of aggregation have been discussed in detail (35) and will not be repeated here.
Polymer theory helps in making robust predictions
We borrowed the methods for analyzing conformational equilibria from the polymer physics literature (8,11,37,39,74). The motivation was to ask if the analysis of simulation data for a single chain length could lead to robust assertions about solvent quality. We showed that this is possible using comparative analysis of specific “order parameters” (36). Of particular relevance is the scaling of internal distances because it obeys a rigorous scaling law for fractal objects, i.e., chains in good and theta solvents. Departure from a scaling law must mean that the solvent is poor. Finite size effects limit the usefulness of such a measure only if the chain length drops below the “blob” length of seven to eight residues, since in this regime local structure overrides the mean polymeric behavior (11). The presence of two distinct length scales, viz., the blob length and a generic length, also means that the conclusions obtained from our analysis for N = 20 are robust and valid for all chain lengths N > 20. This point is emphasized in the development of modern theories for homopolymers (8,11,36) and in the observations of Crick et al. (35) who showed that the poor solvent scaling of chain size with length is obeyed for all lengths N ≥ 15. Our analysis was feasible due to low-sequence complexity, i.e., the homopolymeric nature of polyglutamine and the appropriate choice of chain length (longer than the blob length). The analysis methods are likely to be of general relevance for quantitative characterization of conformational equilibria for IDPs because many of these sequences are deficient in hydrophobic residues and are of sufficiently low sequence complexity (1,6).
In the preceding discussion, we proposed that our observations for Q20 are likely to be generic and valid for longer chains of monomeric polyglutamine. Although this statement is congruent with experimental data (28,30) and expectations based on polymer theories (37), recent results from coarse-grain simulations provide a different picture. Specifically, Khare et al. (75) used a coarse-grain model and showed that whereas polyglutamine peptides of length N < 37 are indeed disordered, chains of length _N_ > 37 are likely to form marginally stable _β_-helices. A similar proposal was put forth by Merlino et al. (76) who used an atomistic force field and explicit solvent to test the length-dependent stability of preformed _β_-helices for monomeric polyglutamine in short, 5-ns MD simulations. Although it might be argued that the simulations of Merlino et al. (76) were too short to be conclusive, the results of Khare et al. (75) are noteworthy. In light of their results, our predictions for N > 20 will need closer scrutiny. Toward this end, we are currently simulating conformational fluctuations and chain oligomerization as a function of chain length and concentration using molecular mechanics potentials and atomistic representations for chain molecules (X. Wang, A. Vitalis, and R. V. Pappu, unpublished data). A detailed comparison between our findings and those of Khare et al. (75) will be forthcoming in the near future.
Why is water a poor solvent for glutamine-rich peptides?
Combining experimental studies and our computational results, there remains little doubt that water is in fact a poor solvent for glutamine-rich peptides. These peptides are assumed to be in a “random-coil” state, the implication being that the ensemble is consistent with that of highly denatured proteins. Our results suggest that the absence of a consensus experimental signal is the result of a different type of disorder, i.e., of a heterogeneous ensemble of globular conformations. Given the polar nature of the side chain, and the infinite solubility of small amides in water, it is obvious that the solvation behavior changes upon transitioning from amides in water to a polyamide in water. To be able to compare the two cases, we remove effective concentration as an obvious factor by appropriate normalization. We conclude that the short-range steric and topological constraints in the polymer alter the solvation behavior primarily for the backbone unit, i.e., the secondary amides are more favorably solvated by themselves than by water. As a result, the chain collapses and minimizes its interface with water. This, however, does not imply that these peptides behave like classical hydrophobic solutes, such as polyethylene. At this point, we are unable to adjudicate the nature of the collapse transition, since we only have simulations of conformational equilibria for a single set of solution conditions.
Implications for the design of simulations aimed at quantitative characterization of conformational equilibria of IDPs
The SE for most of the data we presented are relatively large considering the investment of computational resources. This is a direct consequence of the very long interconversion times for different globular states of these peptides. Enhanced sampling techniques provide an obvious route to solve the sampling problem. Umbrella sampling (77–79) along _R_g as the reaction coordinate is a technique we are currently pursuing although the downside is that a large computational investment yields limited data, since it is nontrivial to recover quantities other than the potential of mean force along _R_g from these sets of simulations. This would render an analysis like the one presented here difficult. Conversely, the replica exchange method (80,81) uses high temperature replicas to enhance conformational rearrangement. Although this is useful in theory, we would suffer from the fact that we would need multiple replicas for each temperature. This is unavoidable for disordered systems such as polyglutamine in water, and therefore the required resources would actually increase.
Our MRMD methodology bears some resemblance to the ensemble dynamics methods of Pande and co-workers (82). To extract robust information regarding polymeric properties, we had to compare MRMD data to those obtained from simulations using two diametrically opposed reference states. As is the case in most molecular simulations of biomolecules, the choice of the force field will determine the details of simulation results (83). Since all force fields share similar features, our analysis methods applied to simulation data gathered using different force fields will in all likelihood lead to the conclusion that water is a poor solvent for polyglutamine. However, details such as the length scale for collapse transitions, and the stability of the collapsed states might vary from one force field to the next. Although comparative simulations with multiple force fields applied to the same problem have become more common in recent years (84–87), they are still prohibitively expensive for systems other than short peptides. For the data presented here, we used ∼1200 CPU days on a single 2.6-Ghz Intel Conroe Core with the fastest, freely available simulation engine, viz., GROMACS. Clearly, for expensive calculations such as these, simulations to compare different force fields are intractable without the use of distributed computing methods (69).
Besides our own work, few articles have been published, which study glutamine-based peptides in explicit solvent (76,88–90). In fact, coarse-graining and/or implicit solvent models have been a much more popular approach to answer questions about the structures of these peptides within intermolecular aggregates (91–98). In coarse-graining approaches, one obviously sacrifices details of the description for efficiency, which leads to reliable conclusions within the limits of the given model. However, the preference for collapsed states in polyglutamine is most convincingly established using explicit solvent models.
Acknowledgments
We are grateful to Professor Michael Rubinstein, Scott Crick, Alan Chen, Hoang Tran, and Matthew Wyczalkowski for helpful discussions.
This work was supported by grant MCB 0416766 from the National Science Foundation.
APPENDIX: DETAILS REGARDING CALCULATIONS OF INTRAPOLYMER SITE-SITE CORRELATION FUNCTIONS
Consider all unique pairs of backbone donor (N) and acceptor atoms (O), respectively. For generality, we shall use the labels A and B to refer to these atom pairs. Let _h_W(_r_AB) denote the histogram of interatomic distances obtained from analysis of MRMD simulation data for Q20 in water. Additionally, let _h_D(_r_AB) be the histogram obtained by gathering statistics from simulations based on an appropriate default model. Given the two histograms, _h_W(_r_AB) and _h_D(_r_AB), the desired site-site correlation function is defined as:
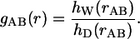 |
(9) |
|---|
It is important to emphasize that the choice for the default model determines the profile we obtain for _g_AB(r). The standard noninteracting model one uses in the theory of liquids is the so-called ideal gas prior. In this model, the sites are parts of rigid molecules that are free to translate and rotate around each other. The applicability of this default model for polymers is questionable because the resultant profiles one obtains for _g_AB(r) are dominated by the presence of chain connectivity in the real chain, which increases the effective concentration of repeating units with respect to each other. Therefore, we constructed intrachain site-site correlation functions using a so-called ideal chain model, which is analogous to the freely rotating chain model of Flory (9). In this model, bond lengths and bond angles are held fixed at equilibrium values (47) and the peptide unit is held fixed in the trans configuration. An ensemble of freely rotating chain conformations is generated by ignoring (turning off) all nonbonded interactions, including excluded volume effects. Histograms, _h_D(_r_AB), constructed using the resultant ensemble include the effects of chain connectivity, and exclude the effects of intrachain and chain-solvent interactions.
References
- 1.Dunker, A. K., C. J. Brown, and Z. Obradovic. 2002. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 62:25–49. [DOI] [PubMed] [Google Scholar]
- 2.Dunker, A. K., C. J. Brown, J. D. Lawson, L. M. Iakoucheva, and Z. Obradovic. 2002. Intrinsic disorder and protein function. Biochemistry. 41:6573–6582. [DOI] [PubMed] [Google Scholar]
- 3.Uversky, V. N. 2002. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dyson, H. J., and P. E. Wright. 2005. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6:197–208. [DOI] [PubMed] [Google Scholar]
- 5.Weathers, E. A., M. E. Paulaitis, T. B. Woolf, and J. H. Hoh. 2004. Reduced amino acid alphabet is sufficient to accurately recognize intrinsically disordered protein. FEBS Lett. 576:348–352. [DOI] [PubMed] [Google Scholar]
- 6.Weathers, E. A., M. E. Paulaitis, T. B. Woolf, and J. H. Hoh. 2007. Insights into protein structure and function from disorder-complexity space. Proteins: Struct. Funct. Bioinf. 66:16–28. [DOI] [PubMed] [Google Scholar]
- 7.Bright, J. N., T. B. Woolf, and J. H. Hoh. 2001. Predicting properties of intrinsically unstructured proteins. Prog. Biophys. Mol. Biol. 76:131–173. [DOI] [PubMed] [Google Scholar]
- 8.Grosberg, A. Y., and A. R. Khokhlov. 1994. Statistical Physics of Macromolecules. AIP Press, New York.
- 9.Flory, P. J. 1953. Principles of Polymer Chemistry. Cornell University Press, Ithaca, NY and London, UK.
- 10.Chan, H. S., and K. A. Dill. 1991. Polymer principles in protein structure and stability. Annu. Rev. Biophys. Biophys. Chem. 20:447–490. [DOI] [PubMed] [Google Scholar]
- 11.Rubinstein, M., and R. H. Colby. 2003. Polymer Physics. Oxford University Press, Oxford, UK and New York.
- 12.Romero, P. R., Z. Obradovic, X. Li, E. C. Garner, C. J. Brown, and A. K. Dunker. 2000. Sequence complexity of disordered protein. Proteins. 42:38–48. [DOI] [PubMed] [Google Scholar]
- 13.Bates, G. P. 2005. History of genetic disease: the molecular genetics of Huntington disease—a history. Nat. Rev. Genet. 6:766–773. [DOI] [PubMed] [Google Scholar]
- 14.Bates, G. 2003. Huntingtin aggregation and toxicity in Huntington's disease. Lancet. 361:1642–1644. [DOI] [PubMed] [Google Scholar]
- 15.Cummings, C. J., and H. Y. Zoghbi. 2000. Fourteen and counting: unraveling trinucleotide repeat diseases. Hum. Mol. Genet. 9:909–916. [DOI] [PubMed] [Google Scholar]
- 16.de Cristofaro, T., A. Affaitati, A. Feliciello, E. V. Avvedimento, and S. Varrone. 2000. Polyglutamine-mediated aggregation and cell death. Biochem. Biophys. Res. Commun. 272:816–821. [DOI] [PubMed] [Google Scholar]
- 17.Fischbeck, K. H. 2001. Polyglutamine expansion neurodegenerative disease. Brain Res. Bull. 56:161–163. [DOI] [PubMed] [Google Scholar]
- 18.Michalik, A., and C. Van Broeckhoven. 2003. Pathogenesis of polyglutamine disorders: aggregation revisited. Hum. Mol. Genet. 12:R173–R186. [DOI] [PubMed] [Google Scholar]
- 19.Wanker, E. E. 2000. Protein aggregation and pathogenesis of Huntington's disease: mechanisms and correlations. Biol. Chem. 381:937–942. [DOI] [PubMed] [Google Scholar]
- 20.Ross, C. A., and M. A. Poirier. 2004. Protein aggregation and neurodegenerative disease. Nat. Rev. Neurosci. 10:S10–S17. [DOI] [PubMed] [Google Scholar]
- 21.Venkataraman, P., R. Wetzel, M. Tanaka, N. Nukina, and A. L. Goldberg. 2004. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine containing proteins. Mol. Cell. 14:95–104. [DOI] [PubMed] [Google Scholar]
- 22.Haacke, A., S. A. Broadley, R. Boteva, N. Tzvetkov, F. U. Hartl, and P. Breuer. 2006. Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 15:555–568. [DOI] [PubMed] [Google Scholar]
- 23.Desai, U. A., J. Pallos, A. A. K. Ma, B. R. Stockwell, L. M. Thompson, J. L. Marsh, and M. I. Diamond. 2006. Biologically active molecules that reduce polyglutamine aggregation and toxicity. Hum. Mol. Genet. 15:2114–2124. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez, I., C. Mahlke, and J. Y. Yuan. 2003. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 421:373–379. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, X. Q., D. L. Smith, A. B. Merlin, S. Engemann, D. E. Russel, M. Roark, S. L. Washington, M. M. Maxwell, J. L. Marsh, L. M. Thompson, E. E. Wanker, A. B. Young, et al. 2005. A potent small molecule inhibits polyglutamine aggregation in Huntington's disease neurons and suppresses neurodegeneration in vivo. Proc. Natl. Acad. Sci. USA. 102:892–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoshino, M., K. Tagawa, T. Okuda, and H. Okazawa. 2004. General transcriptional repression by polyglutamine disease proteins is not directly linked to the presence of inclusion bodies. Biochem. Biophys. Res. Commun. 313:110–116. [DOI] [PubMed] [Google Scholar]
- 27.Arrasate, M., S. Mitra, E. S. Schweitzer, M. R. Segal, and S. Finkbeiner. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 431:805–810. [DOI] [PubMed] [Google Scholar]
- 28.Masino, L., G. Kelly, K. Leonard, Y. Trottier, and A. Pastore. 2002. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 513:267–272. [DOI] [PubMed] [Google Scholar]
- 29.Bennett, M. J., K. E. Huey-Tubman, A. B. Herr, A. P. West, S. A. Ross, and P. J. Bjorkman. 2002. A linear lattice model for polyglutamine in CAG-expansion diseases. Proc. Natl. Acad. Sci. USA. 99:11634–11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen, S., V. Berthelier, W. Yang, and R. Wetzel. 2001. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 311:173–182. [DOI] [PubMed] [Google Scholar]
- 31.Chellgren, B. W., A. F. Miller, and T. P. Creamer. 2006. Evidence for polyproline II helical structure in short polyglutamine tracts. J. Mol. Biol. 361:362–371. [DOI] [PubMed] [Google Scholar]
- 32.Wang, X. L., A. Vitalis, M. A. Wyczalkowski, and R. V. Pappu. 2006. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins. 63:297–311. [DOI] [PubMed] [Google Scholar]
- 33.Pande, V. S., A. Y. Grosberg, and T. Tanaka. 2000. Heteropolymer freezing and design: towards physical models of protein folding. Rev. Mod. Phys. 72:259–314. [Google Scholar]
- 34.Dill, K. A., and D. Stigter. 1995. Modeling protein stability as heteropolymer collapse. In Advances in Protein Chemistry, Vol 46. editors. 59–104. [DOI] [PubMed]
- 35.Crick, S. L., M. Jayaraman, C. Frieden, R. Wetzel, and R. V. Pappu. 2006. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. USA. 103:16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grosberg, A. Y., and D. V. Kuznetsov. 1992. Quantitative theory of the globule-to-coil transition. 1. Link density distribution in a globule and its radius of gyration. Macromolecules. 25:1970–1979. [Google Scholar]
- 37.Imbert, J. B., A. Lesne, and J. M. Victor. 1997. Distribution of the order parameter of the coil-globule transition. Phys. Rev. E. 56:5630–5647. [Google Scholar]
- 38.Reddy, G., and A. Yethiraj. 2006. Implicit and explicit solvent models for the simulation of dilute polymer solutions. Macromolecules. 39:8536–8542. [Google Scholar]
- 39.Steinhauser, M. O. 2005. A molecular dynamics study on universal properties of polymer chains in different solvent qualities. Part I. A review of linear chain properties. J. Chem. Phys. 122:094901. [DOI] [PubMed] [Google Scholar]
- 40.Krull, L. H., and J. S. Wall. 1966. Synthetic polypeptides containing side-chain amide groups. water-soluble polymers. Biochemistry. 5:1521–1527. [DOI] [PubMed] [Google Scholar]
- 41.Scherzinger, E., A. Sittler, K. Schweiger, V. Heiser, R. Lurz, R. Hasenbank, G. P. Bates, H. Lehrach, and E. E. Wanker. 1999. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proc. Natl. Acad. Sci. USA. 96:4604–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolfenden, R. 1978. Interaction of the peptide bond with solvent water: a vapor phase analysis. Biochemistry. 17:201–204. [DOI] [PubMed] [Google Scholar]
- 43.Wolfenden, R., L. Andersson, P. M. Cullis, and C. C. B. Southgate. 1981. Affinities of amino acid side chains for solvent water. Biochemistry. 20:849–855. [DOI] [PubMed] [Google Scholar]
- 44.Chen, S. M., F. A. Ferrone, and R. Wetzel. 2002. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. USA. 99:11884–11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chuang, J., A. Y. Grosberg, and T. Tanaka. 2000. Topological repulsion between polymer globules. J. Chem. Phys. 112:6434–6442. [Google Scholar]
- 46.Bryngelson, J. D., J. N. Onuchic, N. D. Socci, and P. G. Wolynes. 1995. Funnels, pathways, and the energy landscape of protein-folding: a synthesis. Proteins. 21:167–195. [DOI] [PubMed] [Google Scholar]
- 47.Tran, H. T., and R. V. Pappu. 2006. Toward an accurate theoretical framework for describing ensembles for proteins under strongly denaturing conditions. Biophys. J. 91:1868–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran, H. T., X. L. Wang, and R. V. Pappu. 2005. Reconciling observations of sequence-specific conformational propensities with the generic polymeric behavior of denatured proteins. Biochemistry. 44:11369–11380. [DOI] [PubMed] [Google Scholar]
- 49.Kaminski, G. A., R. A. Friesner, J. Tirado-Rives, and W. L. Jorgensen. 2001. Evaluation and reparameterization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 105:6474–6487. [Google Scholar]
- 50.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- 51.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 52.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 53.Hess, B., H. Bekker, H. J. C. Berendsen, and J. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 54.Miyamoto, S., and P. A. Kollman. 1992. Settle: an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 13:952–962. [Google Scholar]
- 55.Onsager, L. 1936. Electric moments of molecules in liquids. J. Am. Chem. Soc. 58:1486–1493. [Google Scholar]
- 56.Jorgensen, W. L., D. S. Maxwell, and J. TiradoRives. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118:11225–11236. [Google Scholar]
- 57.Dima, R. I., and D. Thirumalai. 2004. Asymmetry in the shapes of folded and denatured states of proteins. J. Phys. Chem. B. 108:6564–6570. [Google Scholar]
- 58.Schäfer, L. 1999. Excluded Volume Effects in Polymer Solutions as Explained by the Renormalization Group. Springer, Berlin, Germany.
- 59.Ding, F., R. K. Jha, and N. V. Dokholyan. 2005. Scaling behavior and structure of denatured proteins. Structure. 13:1047–1054. [DOI] [PubMed] [Google Scholar]
- 60.Socci, N. D., W. S. Bialek, and J. N. Onuchic. 1994. Properties and origins of protein secondary structure. Phys. Rev. E. 49:3440–3443. [DOI] [PubMed] [Google Scholar]
- 61.Fischer H., I. Polikarpov, and A. F. Craievich. 2004. Average protein density is a molecular-weight dependent function. Protein Sci. 13:2825–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glatter, O., and O. Kratky. 1982. Small Angle X-Ray Scattering. Academic Press, London, UK.
- 63.Moglich, A., K. Joder, and T. Kiefhaber. 2006. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc. Natl. Acad. Sci. USA. 103:12394–12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chandler, D. 2005. Interfaces and the driving force of hydrophobic assembly. Nature. 437:640–647. [DOI] [PubMed] [Google Scholar]
- 65.Southall, N. T., K. A. Dill, and A. D. J. Haymet. 2002. A view of the hydrophobic effect. J. Phys. Chem. B. 106:521–533. [Google Scholar]
- 66.Paulaitis, M. E., S. Garde, and H. S. Ashbaugh. 1996. The hydrophobic effect. Curr. Opin. Coll. Interf. Sci. 1:376–383. [Google Scholar]
- 67.Athawale, M. V., G. Goel, T. Ghosh, T. M. Truskett, and S. Garde. 2007. Effects of lengthscales and attractions on the collapse of hydrophobic polymers in water. Proc. Natl. Acad. Sci. USA. 104:733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmid, R. 2001. Recent advances in the description of the structure of water, the hydrophobic effect, and the like-dissolves-like rule. Monatsh. Chem. 132:1295–1326. [Google Scholar]
- 69.Pande, V. S., I. Baker, J. Chapman, S. P. Elmer, S. Khaliq, S. M. Larson, Y. M. Rhee, M. R. Shirts, C. D. Snow, E. J. Sorin, and B. Zagrovic. 2003. Atomistic protein folding simulations on the submillisecond time scale using worldwide distributed computing. Biopolymers. 68:91–109. [DOI] [PubMed] [Google Scholar]
- 70.Sastry, S., P. G. Debenedetti, and F. H. Stillinger. 1998. Signatures of distinct dynamical regimes in the energy landscape of a glass-forming liquid. Nature. 393:554–557. [Google Scholar]
- 71.Phillips, J. C. 1996. Stretched exponential relaxation in molecular and electronic glasses. Rep. Prog. Phys. 59:1133–1207. [Google Scholar]
- 72.Thirumalai, D., and R. D. Mountain. 1993. Activated dynamics, loss of ergodicity, and transport in supercooled liquids. Phys. Rev. E. 47:479–489. [DOI] [PubMed] [Google Scholar]
- 73.Raos, G., and G. Allegra. 1997. Macromolecular clusters in poor-solvent polymer solutions. J. Chem. Phys. 107:6479–6490. [Google Scholar]
- 74.Grosberg, A. Y., and D. V. Kuznetsov. 1992. Phase-separation of polymer-solutions and interactions of globules. Journal De Physique Ii. 2:1327–1339. [Google Scholar]
- 75.Khare, S. D., F. Ding, K. N. Gwanmesia, and N. V. Dokholyan. 2005. Molecular origin of polyglutamine aggregation in neurodegenerative diseases. Plos Computational Biology. 1:230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Merlino, A., L. Esposito, and L. Vitagliano. 2006. Polyglutamine repeats and beta-helix structure: molecular dynamics study. Proteins. 63:918–927. [DOI] [PubMed] [Google Scholar]
- 77.Kumar, S., J. M. Rosenberg, D. Bouzida, R. H. Swendsen, and P. A. Kollman. 1995. Multidimensional free-energy calculations using the weighted histogram analysis method. J. Comput. Chem. 16:1339–1350. [Google Scholar]
- 78.Sheinerman, F. B., and C. L. Brooks. 1998. Calculations on folding of segment B1 of streptococcal protein G. J. Mol. Biol. 278:439–456. [DOI] [PubMed] [Google Scholar]
- 79.Roux, B. 1995. The calculation of the potential of mean force using computer-simulations. Comput. Phys. Commun. 91:275–282. [Google Scholar]
- 80.Sugita, Y., and Y. Okamoto. 1999. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314:141–151. [Google Scholar]
- 81.Nymeyer, H., S. Gnanakaran, and A. E. Garcia. 2004. Atomic simulations of protein folding, using the replica exchange algorithm. Methods Enzymol. 383:119–149. [DOI] [PubMed] [Google Scholar]
- 82.Sorin, E. J., and V. S. Pande. 2005. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys. J. 88:2472–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sorin, E. J., and V. S. Pande. 2005. Empirical force-field assessment: the interplay between backbone torsions and noncovalent term scaling. J. Comput. Chem. 26:682–690. [DOI] [PubMed] [Google Scholar]
- 84.Hu, H., M. Elstner, and J. Hermans. 2003. Comparison of a QM/MM force field and molecular mechanics force fields in simulations of alanine and glycine “dipeptides” (Ace-Ala-Nme and Ace-Gly-Nme) in water in relation to the problem of modeling the unfolded peptide backbone in solution. Proteins. 50:451–463. [DOI] [PubMed] [Google Scholar]
- 85.Zagrovic, B., and V. S. Pande. 2006. Simulated unfolded-state ensemble and the experimental NMR structures of villin headpiece yield similar wide-angle solution X-ray scattering profiles. J. Am. Chem. Soc. 128:11742–11743. [DOI] [PubMed] [Google Scholar]
- 86.Gnanakaran, S., and A. E. Garcia. 2005. Helix-coil transition of alanine peptides in water: force field dependence on the folded and unfolded structures. Proteins. 59:773–782. [DOI] [PubMed] [Google Scholar]
- 87.Zaman, M. H., M. Y. Shen, R. S. Berry, K. F. Freed, and T. R. Sosnick. 2003. Investigations into sequence and conformational dependence of backbone entropy, inter-basin dynamics and the flory isolated-pair hypothesis for peptides. J. Mol. Biol. 331:693–711. [DOI] [PubMed] [Google Scholar]
- 88.Armen, R. S., B. M. Bernard, R. Day, D. O. V. Alonso, and V. Daggett. 2005. Characterization of a possible amyloidogenic precursor in glutamine-repeat neurodegenerative diseases. Proc. Natl. Acad. Sci. USA. 102:13433–13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zanuy, D., K. Gunasekaran, A. M. Lesk, and R. Nussinov. 2006. Computational study of the fibril organization of polyglutamine repeats reveals a common motif identified in beta-helices. J. Mol. Biol. 358:330–345. [DOI] [PubMed] [Google Scholar]
- 90.Starikov, E. B., H. Lehrach, and E. E. Wanker. 1999. Folding of oligoglutamines: a theoretical approach based upon thermodynamics and molecular mechanics. J. Biomol. Struct. Dyn. 17:409–427. [DOI] [PubMed] [Google Scholar]
- 91.Marchut, A. J., and C. K. Hall. 2007. Effects of chain length on the aggregation of model polyglutamine peptides: molecular dynamics simulations. Proteins. 66:96–109. [DOI] [PubMed] [Google Scholar]
- 92.Pellarin, R., and A. Caflisch. 2006. Interpreting the aggregation kinetics of amyloid peptides. J. Mol. Biol. 360:882–892. [DOI] [PubMed] [Google Scholar]
- 93.Marchut, A. J., and C. K. Hall. 2006. Spontaneous formation of annular structures observed in molecular dynamics simulations of polyglutamine peptides. Comput. Biol. Chem. 30:215–218. [DOI] [PubMed] [Google Scholar]
- 94.Cecchini, M., R. Curcio, M. Pappalardo, R. Melki, and A. Caflisch. 2006. A molecular dynamics approach to the structural characterization of amyloid aggregation. J. Mol. Biol. 357:1306–1321. [DOI] [PubMed] [Google Scholar]
- 95.Dokholyan, N. V. 2006. Studies of folding and misfolding using simplified models. Curr. Opin. Struct. Biol. 16:79–85. [DOI] [PubMed] [Google Scholar]
- 96.Nguyen, H. D., and C. K. Hall. 2004. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc. Natl. Acad. Sci. USA. 101:16180–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Peng, S., F. Ding, B. Urbanc, S. V. Buldyrev, L. Cruz, H. E. Stanley, and N. V. Dokholyan. 2004. Discrete molecular dynamics simulations of peptide aggregation. Phys. Rev. E. 69. [DOI] [PubMed]
- 98.Gsponer, J., U. Haberthur, and A. Caflisch. 2003. The role of side-chain interactions in the early steps of aggregation: molecular dynamics simulations of an amyloid-forming peptide from the yeast prion Sup35. Proc. Natl. Acad. Sci. USA. 100:5154–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]