Tyrosylprotein sulfotransferase: Purification and molecular cloning of an enzyme that catalyzes tyrosine O-sulfation, a common posttranslational modification of eukaryotic proteins (original) (raw)
Abstract
Tyrosine _O_-sulfation is a common posttranslational modification of proteins in all multicellular organisms. This reaction is mediated by a Golgi enzyme activity called tyrosylprotein sulfotransferase (TPST) that catalyzes the transfer of sulfate from 3′-phosphoadenosine 5′-phosphosulfate to tyrosine residues within acidic motifs of polypeptides. Tyrosine _O_-sulfation has been shown to be important in protein–protein interactions in several systems. For example, sulfation of tyrosine residues in the leukocyte adhesion molecule P-selectin glycoprotein ligand 1 (PSGL-1) is required for binding to P-selectin on activated endothelium. In this report we describe the purification of TPST from rat liver microsomes based on its affinity for the N-terminal 15 amino acids of PSGL-1. We have isolated human and mouse TPST cDNAs that predict type II transmembrane proteins of 370 amino acid residues with almost identical primary structure. The human cDNA encodes a fully functional N-glycosylated enzyme with an apparent molecular mass of ≈54 kDa when expressed in mammalian cells. This enzyme defines a new class of Golgi sulfotransferases that may catalyze tyrosine _O_-sulfation of PSGL-1 and other protein substrates involved in diverse physiologic functions including inflammation and hemostasis.
Tyrosine _O_-sulfation is a posttranslation modification of membrane and secretory proteins that occurs in all eukaryotes (1–3). The enzyme activity required for this reaction, called tyrosylprotein sulfotransferase (TPST), catalyzes the transfer of sulfate from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to tyrosines within highly acidic motifs of polypeptides (2, 4). Current evidence indicates that the enzyme is a membrane-associated protein with a lumenally oriented active site localized in the trans-Golgi network (5, 6).
Many proteins have been shown to contain tyrosine sulfate. Among these are several proteins involved in inflammation and hemostasis, including P-selectin glycoprotein ligand 1 (PSGL-1) (7), the α chain of complement factor C4 (8), coagulation factors V (9) and VIII (10, 11), platelet glycoprotein Ibα (12, 13), α2-antiplasmin (14), and heparin cofactor II (15). Although the role of tyrosine _O_-sulfation is incompletely understood, it is clear that tyrosine _O_-sulfation plays a role in protein–protein interactions in several systems. Tyrosine _O_-sulfation is required for the optimal interaction between factor VIII and Von Willebrand factor (10, 11), PSGL-1 and P-selectin (7), GPIbα with Von Willebrand factor and α-thrombin (12, 13), and complement factor C4 and C1s (8). However, in most cases a functional role for tyrosine _O_-sulfation has not been established.
Although tyrosine _O_-sulfation is required for optimal function for some proteins, the structural basis for this requirement is established only for hirudin, a potent anticoagulant protein secreted in the saliva of the medicinal leech. Hirudin is sulfated at Tyr63 and has a 10-fold higher affinity for thrombin than unsulfated hirudin (16). In cocrystals with α-thrombin, the sulfated C-terminal dodecapeptide peptide of hirudin makes a number of ionic and hydrophobic interactions with anion binding exosite 1 of α-thrombin. In this complex the sulfate group of Tyr63 is involved in an extensive intermolecular hydrogen-bonded network (17). Because hirudin is the only tyrosine-sulfated protein that has been crystallized with a ligand, it is not known whether tyrosine _O_-sulfation plays a direct role in protein–protein interactions in other systems.
The kinetics of the TPST reaction has been studied with crude and partially purified enzyme preparations from a variety of mammalian tissues (18, 19). However, it is not clear whether TPST activity is due to one enzyme or a family of enzymes. Two groups have previously reported the purification of TPST (20, 21). However, neither group has reported amino acid sequence, nor have cDNAs encoding the enzyme been isolated.
In this article, we report the purification of a TPST from rat liver and the molecular cloning of human and mouse cDNAs encoding the enzyme.
MATERIALS AND METHODS
Assay of Sulfotransferase Activity.
TPST activity was determined by measuring the transfer of [35S]sulfate from [35S]PAPS (Dupont/NEN) to an immobilized peptide. The peptide (QATEYEYLDYDFLPEC) represents the N-terminal 15 residues of the mature PSGL-1 polypeptide to which a C-terminal cysteine residue was added. It spans three potential tyrosine sulfation sites that have been shown to be sulfated in mammalian cells (7, 22, 23). The peptide was linked via the cysteine residue to iodoacetamide-activated resin (UltraLink Iodoacetyl, Pierce) at a density of 1.5–3.0 μmol/ml of resin. The enzyme assay was performed by combining 10 μl of peptide-derivitized beads with 2–20 μl of sample in 40 mM Pipes, pH 6.8/0.3 M NaCl/20 mM MnCl2/50 mM NaF/1% Triton X-100/1 mM 5′-AMP in a final volume of 130 μl. The assay was initiated by addition of 0.5 μCi of [35S]PAPS (≈1.7 μM; 1 Ci = 37 GBq). After 30 min at 37°C, the reaction beads were washed extensively with 6 M guanidine at 65°C and the radioactivity incorporated into the beads was determined by liquid scintillation counting. The reaction rate was optimal at pH 6.8–7.0, 0.3 M NaCl, and 2 μM PAPS and was linear with respect to time and sample input. Transfer of [35S]sulfate was inhibited by free peptide with an IC50 equimolar to the concentration of immobilized peptide in the assay. One unit of activity was defined as 1 pmol of product formed per min.
Purification of TPST.
Male 200- to 300-g Sprague–Dawley rats (Harlan–Sprague–Dawley) were anesthetized with CO2 and decapitated, and the livers were excised and immersed in ice-cold homogenization buffer [10 mM Tris⋅HCl, pH 7.5/1.5 mM MgCl2/250 mM sucrose/0.5 mM DTT/0.5 mM phenylmethylsulfonyl fluoride (PMSF)]. All further steps were performed at 4°C. Livers were minced, suspended in 30 ml of buffer per g of liver and passed twice through a Zeigler–Pettit continuous-flow homogenizer (24), and the homogenate centrifuged (10 min, 800 × g). The postnuclear supernatant was centrifuged (90 min, 28,000 × g), the microsomal pellet was suspended in 2% Triton X-100/20 mM _N_-tris[hydroxymethyl]- methyl-3-amino propane sulfonic acid (TAPS), pH 9.0/0.5 mM PMSF/leupeptin (10 μg/ml)/antipain (10 μg/ml) at 1.5 ml/g of liver and stirred for 1 h. PMSF was added to 0.5 mM and the microsomal extract was clarified by centrifugation (60 min, 40, 200 × g). The supernatant was adjusted to 10% glycerol (wt/vol), 50 mM Mops (pH 7.5), and 0.5 mM PMSF.
Extract from 120 livers was applied at 25 cm/h to a 5 × 20 cm Toyopearl SP-550C column (Tosohaas, Montgomeryville, PA) equilibrated with 50 mM Mops, pH 7.5/10% glycerol/0.05% Triton X-100 (buffer A). The column was washed with buffer A and then eluted with 0.25 M NaCl in buffer A, followed by 1 M NaCl in buffer A. After this and subsequent steps, fractions were frozen in liquid N2 and stored at −80°C.
Enzyme eluted from Toyopearl SP-550C was pooled and diluted with buffer A to a conductivity equivalent to 0.15 M NaCl. The material was applied at 110 cm/h to a PSGL-1 peptide column (1.5 × 6 cm, 1.5 μmol of peptide per ml) equilibrated with 0.1 M NaCl in 50 mM Mops, pH 7.5/10% glycerol/0.02% Triton X-100 (buffer B). The column was washed with 0.1 M NaCl in buffer B and then step-eluted with 0.35 M NaCl, followed by 1 M NaCl in buffer B at 55 cm/h.
Fractions from the PSGL-1 peptide column were pooled and dialyzed against buffer B until the conductivity was equivalent to 0.2 M NaCl. The material was applied at 150 cm/h to an ethanolamine UltraLink precolumn (1 × 10 cm) in series with a PSGL-1 peptide column (0.5 × 20 cm, 2.7 μmol of peptide per ml) equilibrated with 0.15 M NaCl in buffer B. After washing with 0.2 M NaCl in buffer B, the precolumn was removed from the circuit. The column was eluted with 0.3 M NaCl in buffer B and then developed with a 20-ml linear 0.3–1 M NaCl gradient at 30 cm/h.
In-Gel Tryptic Digestion, HPLC Separation, and Microsequencing.
Proteins were separated by SDS/PAGE and stained with Coomassie blue. Protein bands were excised and subjected to in-gel reduction, S-carboxyamidomethylation, and tryptic digestion (Promega). Ten percent of the resultant mixture was analyzed as follows. Sequence information was determined by capillary reverse-phase chromatography (180 μm × 15 cm, LC Packings, San Francisco) coupled to the electrospray ionization source of a Finnigan (San Jose, CA) LCQ ion trap mass spectrometer. The instrument was programmed to acquire successive sets of three scan modes consisting of full scale MS over the m/z range of 395–1,200 atomic mass units, followed by two data-dependent scans on the most abundant ion in the full scan. These data-dependent scans allowed the automatic acquisition of a high-resolution scan to determine charge state and exact mass and MS/MS spectra for peptide sequence information. Interpretation of the MS/MS spectra of the peptides was facilitated by searching the National Center for Biotechnology Information nonredundant and expressed sequence tag (EST) databases with the algorithm sequest (25). The remainder (90%) of the peptide mixture was separated by microbore HPLC using a 1 mm × 150 mm Zorbax C18 reverse-phase column on a Hewlett–Packard 1090 HPLC/1040 diode array detector. Optimum fractions were chosen based on differential UV absorbance at 205, 277, and 292 nm, peak symmetry, and resolution and then further screened for length and homogeneity by matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-MS) on a Thermo BioAnalysis Lasermat 2000 (Hemel, England). Strategies for peak selection, reverse-phase separation, and Edman microsequencing have been described (26). Tryptic peptides were submitted to automated Edman degradation on an Applied Biosystems model 477A protein sequencer.
Expression of Recombinant TPST in Mammalian Cells.
The pcDNA3.1(+) vector (Invitrogen) was modified for expression of fusion proteins containing an N-terminal epitope for HPC4, a Ca2+-dependent monoclonal antibody to protein C (27). The _Nhe_I and _Bam_HI fragment in the multiple cloning site of the vector was replaced with a 48-bp double-stranded oligonucleotide with a 5′ _Nhe_I half site and a 3′ _Bam_HI half site containing an ideal Kozak sequence immediately upstream to the sequence encoding the HPC4 epitope.
The human TPST coding sequence was amplified by Advantage KlenTaq polymerase using EST clone 116978 as template. The following primers were used: top strand, 5′-CGGGATCCGGTTGGGAAGCTGAAGCAGAAC-3′; bottom strand, 5′-GGACTAGTATTACTCCACTTGCTCCGTCTG-3′. The PCR introduced a _Bam_HI site at the initiation codon and an _Spe_I site after the termination codon (underlined). The cycling parameters were as follows: 25 cycles, denaturation, 94°C for 30 s; annealing, 55°C for 30 s; extension, 68°C for 2 min. The product was gel-purified, ligated into the pGEM-T (Promega), and sequenced on both strands. The insert was excised by using _Bam_HI and _Apa_I and directionally cloned into unique _Bam_HI and _Apa_I sites in the multiple cloning site of the modified pcDNA3.1(+) vector. In the fusion protein, the native initiating methionine is replaced with 15 residues, containing the HPC4 epitope (MEDQVDPRLIDGKDP, underlined).
Chinese hamster ovary cells (CHO-K1) were grown in high-glucose α-modified Eagle’s medium containing 10% fetal calf serum (FCS) and 2 mM glutamine at 37°C and 5% CO2/95% air. The human embryonic kidney cell line 293-T was grown in low-glucose DMEM containing 10% FCS and 2 mM glutamine at 37°C and 5% CO2/95% air. Cells were transfected with empty vector or vector containing cDNAs encoding human TPST-HPC4 fusion protein or mouse TPST by using Lipofectamine (GIBCO/BRL) according to the instructions of the supplier. The medium was changed at 24 h and after an additional 24 h the conditioned medium was collected. The cell monolayers were washed with Ca2+/Mg2+-free Hanks’ balanced salt solution and the cells were released from the plates. The cells were pelleted by centrifugation and extracted with 1% Triton X-100/0.1 M NaCl/20 mM TAPS, pH 9.0/leupeptin (10 μg/ml)/antipain (10 μg/ml)/5 mM benzamidine. Extracts and conditioned media were clarified by centrifugation (15 min, 10,000 × g) and stored at −80°C.
Purification of TPST Fusion Protein.
Human TPST-HPC4 was purified from extracts of ten 162-cm2 dishes of transiently transfected 293-T cells. The extract was adjusted to 10% glycerol, 50 mM MOPS (pH 7.5), and 5 mM CaCl2 and incubated with 0.5 ml of HPC4-UltraLink (5 mg of antibody per ml of resin) for 15 h at 4°C. The resin was packed into a column and washed with 2 M NaCl/20 mM Mops, pH 7.5/2 mM CaCl2/0.1% Triton X-100 followed by 0.15 M NaCl in the same buffer. Bound protein was eluted with 10 mM EDTA/0.15 M NaCl/20 mM Mops, pH 7.5/0.1% Triton X-100/10% glycerol. Fractions were assayed for protein content and TPST activity. Samples were electrophoresed on SDS/10% polyacrylamide gels and proteins were transferred to Hybond-P membranes (Amersham). The membranes were blocked and probed with HPC4, and bound antibody was detected with enhanced chemiluminescence using horseradish peroxidase-conjugated anti-mouse Ig (Amersham).
Purified human TPST fusion protein, prepared as described above, was treated with 0.3 M 2-mercaptoethanol/5 mM EDTA (2 min, 100°C) and incubated in the presence or absence of 2.5 units of peptide N-glycosidase F (Oxford Glycosciences, Bedford, MA) for 12 h at 37°C. Samples were analyzed by SDS/PAGE followed by Western blotting with HPC4.
Northern Blot Hybridization.
For human TPST a 1,138-bp partial cDNA, corresponding to nucleotides 1–1,138, was excised from EST 116978 by using _Eco_RI. For mouse TPST, a 1,560-bp _Eco_RI_–Xho_I fragment of EST 567635, which corresponds to nucleotides 321-1881 of the cDNA, was used as a probe. Probes were labeled with [α-32P]dCTP (Dupont/NEN) and random hexamer priming with the Klenow fragment of DNA polymerase I (Pharmacia). Multiple tissue Northern blots of poly(A)+ RNA (CLONTECH) were prehybridized with for 60 min at 68°C and hybridized with 32P-labeled probe overnight at 68°C. The blots were washed for two 20-min periods with 2× SSC/0.1% SDS at 22°C and two 20-min periods with 0.1× SSC/0.1% SDS at 50°C. The membrane was exposed to a phosphorimager screen (Molecular Dynamics) for 16 h at room temperature.
RESULTS
Validation of TPST Assay.
Crude rat liver microsomal extract or buffer were combined with peptide-derivitized beads and [35S]PAPS and incubated under standard assay conditions. After washing, the beads were treated with proteinase K (1 mg/ml, 37°C, 15 h) in 50 mM Tris⋅HCl, pH 8.0/1 mM CaCl2. Released material was hydrolyzed in 1 M NaOH at 110°C for 24 h under N2 and the hydrolysate was analyzed by HPLC as described (7). This analysis revealed two 35S-labeled peaks that comigrated with tyrosine sulfate and free sulfate standards, respectively (Fig. 1). In the absence of enzyme, 35S-labeled products were not detected. This proves that peptidyl [35S]tyrosine sulfate was formed and that nonenzymatic tyrosine _O_-sulfation of substrate does not occur under these assay conditions.
Figure 1.
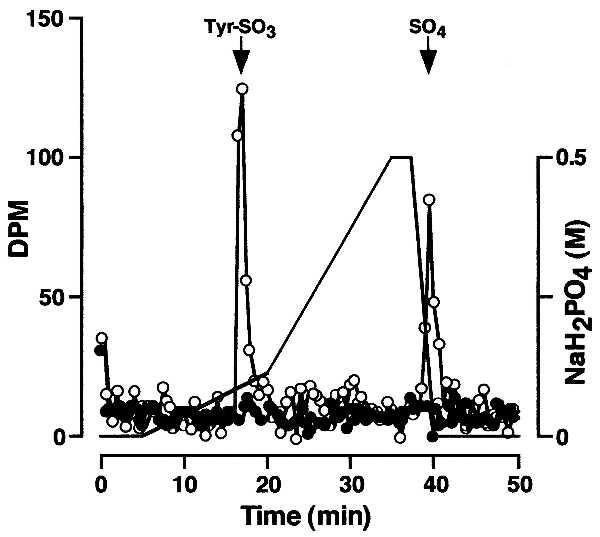
Sulfotyrosine analysis. Microsomal extract (○) or buffer (•) was combined with PSGL-1 peptide-derivitized beads in the presence of [35S]PAPS and incubated under standard assay conditions. The beads were washed extensively and then treated with proteinase K. Released material was hydrolyzed under alkaline conditions and analyzed by HPLC.
Rat Liver TPST Copurifies with an ≈50-kDa Polypeptide.
To determine whether rat liver TPST activity was membrane-associated, postnuclear supernatants were centrifuged (60 min, 100,000 × g) and the supernatant (cytosol) and pellet (microsomes) were collected and assayed. More than 98% of the TPST activity in crude homogenate was in the microsomal fraction. When microsomes were solubilized with 2% Triton X-100/20 mM TAPS, pH 9.0, and centrifuged (60 min, 100,000 × g), >95% of the enzyme activity was recovered in the 100,000 × g supernatant. TPST activity in postnuclear supernatant of rat liver homogenate was not detectable when detergent was excluded from the assay mixture, consistent with a luminal orientation of the enzyme active site.
The 18,400-fold enrichment of TPST from 2 kg of rat liver is summarized in Table 1. Microsomal extracts were prepared from 120 rat livers and applied to a Toyopearl SP-550C column. The column was washed and sequentially eluted with 0.35 M NaCl and 1 M NaCl. The bulk of the bound enzyme activity was eluted with 1 M NaCl. Enzyme activity eluted from Toyopearl SP-550C from two 120 rat preparations was diluted and applied to a PSGL-1 peptide column. The column was washed and sequentially eluted with 0.35 M NaCl and 1 M NaCl. Most of the bound enzyme activity was eluted with 1 M NaCl.
Table 1.
Purification of rat liver TPST
| Step | Protein | Sulfotransferase | |||||
|---|---|---|---|---|---|---|---|
| Volume, ml | Concentration, mg/ml | Total, mg | Total activity, units | Specific activity, units/mg | Yield, % | Purification, fold | |
| Postnuclear superanant | 4,060 | 34.6 | 140,400 | 23,080 | 0.16 | — | — |
| Microsomal extract | 3,720 | 11.6 | 43,000 | 36,510 | 0.85 | 100 | 5.3 |
| Toyopearl SP-550C | 300 | 12.6 | 3,780 | 8,880 | 2.35 | 24 | 14.7 |
| Peptide column 1 | 60 | 0.38 | 23 | 4,600 | 200 | 13 | 1,250 |
| Peptide column 2 | 30 | 0.026 | 0.8 | 2,360 | 2,950 | 8 | 18,440 |
Fractions from the first peptide column were pooled, dialyzed, and applied to a second PSGL-1 peptide column. The column was washed, eluted with 0.3 M NaCl, and then developed with a linear 0.3–1 M NaCl gradient (Fig. 2A). Enzyme activity eluted as a broad peak between 0.7 and 1.0 M NaCl, resolved from the bulk of the protein. After the second peptide column, the enzyme was enriched by ≈18,440-fold to a specific activity of ≈2,950 units/mg, with a yield of ≈8%. Enzyme-containing fractions eluted from the second peptide column were subjected to SDS/PAGE. This showed a major protein band at ≈50-kDa that coeluted with enzyme activity and thus was a candidate for further analysis (Fig. 2B, arrow). This polypeptide had a slightly faster electrophoretic mobility under nonreducing conditions (data not shown). In addition, its mobility is similar to that previously reported for bovine and rat TPSTs (20, 21).
Figure 2.
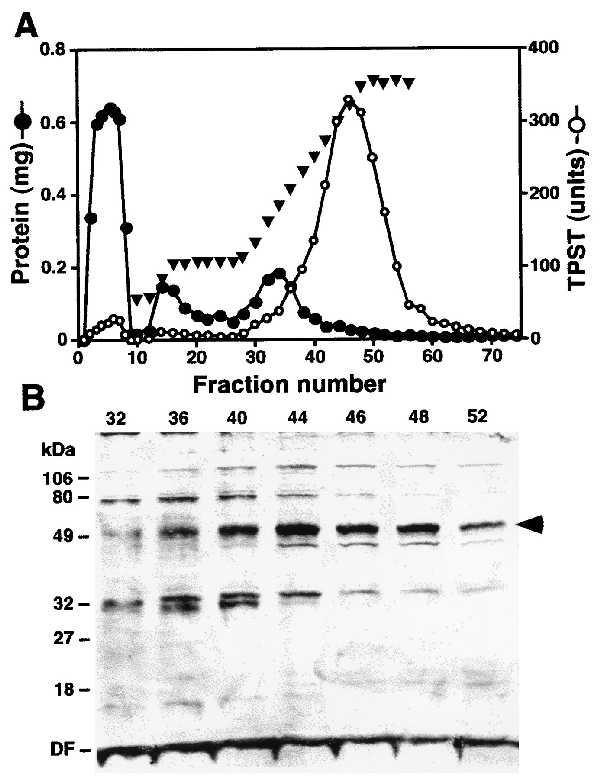
Purification of rat liver TPST. (A) PSGL-1 peptide column 2 chromatogram. TPST eluted from the first PSGL-1 peptide column was applied to the column and the column was eluted with 0.3 M NaCl followed by a linear 0.3–1 M NaCl gradient in buffer B. (B) SDS/PAGE of PSGL-1 peptide column fractions. Aliquots of the indicated fractions were electrophoresed on SDS/10% polyacrylamide gels under reducing conditions. Proteins were visualized by silver staining. The arrow indicates the protein band that was sequenced. DF, dye front.
Molecular Cloning of TPST.
Eluate from the second peptide column was subjected to preparative SDS/PAGE. The ≈50-kDa band was excised and subjected to in-gel tryptic digestion and HPLC separation. Four peaks were selected and sequenced by automated Edman degradation. Eight additional peptide sequences were obtained by on-line ion trap HPLC/MS/MS sequencing. These sequences did not match known protein sequence in the National Center for Biotechnology Information database. The peptide sequences were used to perform reiterative searches of the EST database using the tblastn and blastn algorithms. Confirmatory searches were performed with the MS/MS data by using the sequest algorithm. Searches identified 27 human and 15 mouse ESTs that formed contigs spanning 1,110-nucleotide ORFs. I.M.A.G.E. Consortium cDNA clones (28) were obtained (Research Genetics, Huntsville, AL), and the nucleotide sequences of both strands were determined.
The most 5′ human EST clone (clone 116978, GenBank accession no. T93946) had a 1,795-bp insert containing an 81-nucleotide 5′ untranslated region, a 1,110-nucleotide coding region, and a 604-nucleotide 3′ untranslated region. The most 5′ EST mouse TPST clone (clone 567635, GenBank accession no. AA183558) had a 1,560-bp insert that includes 999 nucleotides of coding sequence and a 561-nucleotide 3′ untranslated region. From the sequence of the mouse EST, a primer was designed to amplify the 5′ end of the cDNA from mouse liver Marathon-Ready cDNA (CLONTECH). The following primers were used: top strand, 5′-CCATCCTAATACGACTCACTATAGGGC-3′ (AP-1), and bottom strand, 5′-GCGCACAGACACTCCTTGTCGCAG-3′. The cycling parameters were 30 cycles of denaturation at 94°C for 30 s and annealing/extension at 68°C for 3 min. An ≈1.4-kb product was gel-purified, ligated into the pGEM-T, and sequenced on both strands. A full-length mouse TPST cDNA was constructed by splicing the 425-nucleotide 5′ end of the PCR product to the 1,456-nucleotide 3′ end of the EST clone by blunt-end ligation at a unique _Ssp_I restriction site.
The predicted amino acid sequence of human TPST is shown in Fig. 3. The nucleotide sequence and the predicted amino acid sequence of the human TPST ORF are 89% and 96% identical to the mouse sequences, respectively (data not shown). For both the human and mouse cDNAs, the sequences surrounding the proposed initiating ATG codons have an adenosine in position −3 and a guanosine in position +4, thereby conforming to Kozak consensus features. Both cDNAs have polyadenylylation signals upstream from the beginning of the poly(A) tail.
Figure 3.
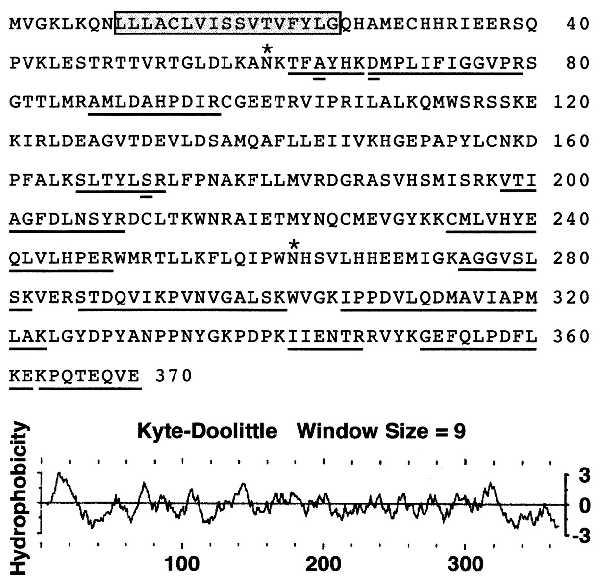
Predicted amino acid sequence and hydropathy plot of human TPST. Peptides sequenced from the rat protein are underlined. Difference between rat peptide sequences and the predicted human protein are indicated (double underlined). In the rat protein Ala64 is a Thr, Asp68 is an Asn, and Ser171 is an Ala. Two potential sites for N-linked glycosylation are indicated (asterisk) and the putative transmembrane membrane is boxed. The hydropathy plot was calculated by the method of Kyte and Doolittle with the pepplot program (Genetics Computer Group).
The cDNAs predict proteins of 370 amino acids with molecular masses of 42,185 Da for the human and 42,129 Da for the mouse protein. All 12 peptide sequences from the rat protein are represented in the predicted amino acid sequence of TPST. Of the 128 amino acids of rat peptide sequence, the predicted human amino acid sequence is different at only three positions (Fig. 3), whereas the mouse sequence differs at only one position.
All known glycosyltransferase and Golgi sulfotransferase cDNAs, with the exception of heparan sulfate d-glucosaminyl 3-_O_-sulfotransferase (29), predict proteins with type II transmembrane topology with short cytoplasmic domains and luminal catalytic domains. Kyte–Doolittle hydrophobicity plots of human (Fig. 3) and mouse (data not shown) TPST reveal a prominent hydrophobic segment of 17 residues near the N terminus. This segment is preceded by basic residues and is not followed by a suitable signal peptidase cleavage site. This indicates that TPST has type II transmembrane topology and, therefore, predicts that the catalytic domain resides in the lumen of the Golgi. This prediction is supported by the observation that TPST activity in rat liver microsomes is detectable only after detergent lysis of the microsomes. Both polypeptides are predicted to have six luminal cysteine residues and two potential sites for the addition of N-linked glycans. No other protein motifs were found by the motifs program (Genetics Computer Group).
Expression of Recombinant TPST in Mammalian Cells.
Human TPST was expressed in CHO-K1 and 293-T cells from a vector modified for expression of fusion proteins with an N-terminal HPC4 epitope. Cells transfected with empty plasmid or plasmid encoding human TPST-HPC4 were extracted and assayed for TPST activity and protein content. Compared with mock-transfected cells, TPST activity was overexpressed by a factor of 9 in CHO-K1 and 80 in 293-T cells transfected with human TPST cDNA. Mouse TPST expressed in 293-T cells from the unmodified pcDNA3.1(+) vector was overexpressed by 74-fold (Table 2). Note that the specific activity of TPST in mock-transfected cells is comparable to the specific activity observed in the postnuclear supernatant of rat liver homogenate. TPST activity was not detectable in cell-free conditioned medium from CHO-K1 and 293-T cells transfected with empty plasmid or plasmid encoding mouse or human TPST.
Table 2.
Expression of TPST in mammalian cells
| Cell | Transfection | Specific activity, units/mg | Induction, fold |
|---|---|---|---|
| CHO-K1 | Mock | 0.25 ± 0.11 (3) | — |
| hTPST-HPC4 | 2.31 ± 0.89 (3) | 9 | |
| 293-T | Mock | 0.11 ± 0.03 (9) | — |
| hTPST-HPC4 | 8.84 ± 1.53 (5) | 80 | |
| mTPST | 8.10 ± 1.37 (4) | 74 |
Purification and Characterization of Recombinant Human TPST.
To demonstrate that TPST was encoded by the transfected cDNA, TPST-HPC4 fusion protein was purified from extracts of 293-T cells. By using HPC4 affinity chromatography, the TPST fusion protein was enriched ≈750-fold to a specific activity of ≈6,300 units/mg. Silver staining of a reduced SDS/polyacrylamide gel revealed a major ≈54-kDa protein and a minor contaminant with slightly slower electrophoretic mobility (Fig. 4A). Western blot analysis of nonreduced and reduced TPST-HPC4 revealed a single polypeptide with calculated molecular masses of 48 kDa under nonreducing and 54 kDa under reducing conditions (Fig. 4B). The slower electrophoretic mobility of TPST under reducing conditions indicates that TPST contains disulfide bonds. A minor ≈100-kDa HPC4-reactive protein was observed under nonreducing that likely represents TPST dimer. In addition, purified TPST was treated with peptide N-glycosidase F and analyzed by SDS/PAGE followed by Western blotting using HPC4 (Fig. 4B). Peptide N-glycosidase F treatment resulted in a decrease of ≈7 kDa in the apparent molecular mass of recombinant TPST, consistent with the removal of one or two complex N-linked glycans.
Figure 4.
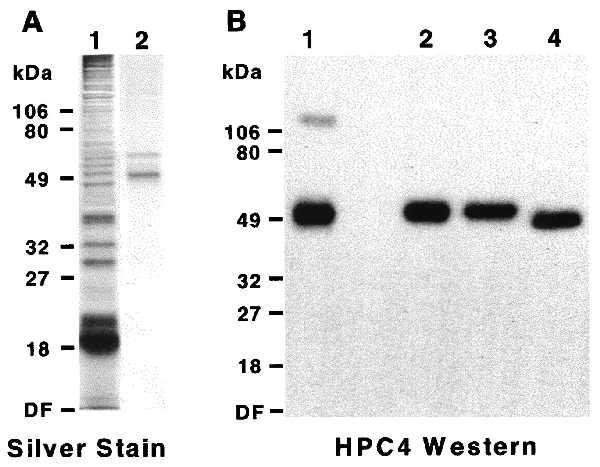
Characterization of recombinant human TPST. Human TPST-HPC4 fusion protein was transiently expressed in 293-T cells and purified. (A) Extracts of transfected cells (lane 1) and purified fusion protein (lane 2) were electrophoresed on SDS/10% polyacrylamide gels under reducing conditions and proteins were visualized by silver staining. (B) Purified TPST-HPC4 was electrophoresed on SDS/10% polyacrylamide gels under nonreducing (lane 1) and reducing (lane 2–4) conditions. Additional samples were either sham-treated (lane 3) or treated with peptide N-glycosidase F (lane 4). Fusion proteins were visualized by Western blotting using HPC4. DF, dye front.
Northern Blot Analysis.
Northern blots were probed with 32P-labeled cDNA probes to determine the pattern of mRNA expression. This analysis showed a single ≈1.8- to 2.0-kb transcript in all human and mouse tissues examined (Fig. 5). The tissue sources of the overlapping EST clones from the human (brain, liver/spleen, heart, placenta, uterus, and adipose tissue) and mouse (brain, thymus, mammary gland, spleen, and testis) are consistent with a widespread tissue distribution of TPST transcripts.
Figure 5.
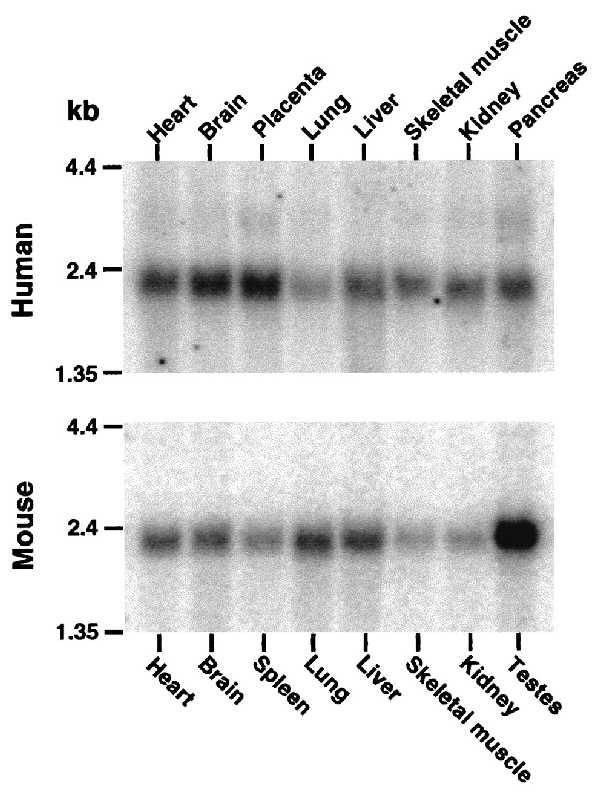
Northern blot analysis. Northern blots of poly(A)+ mRNA from multiple human and mouse tissues were probed with 32P-labeled partial cDNA probes.
DISCUSSION
TPST is an enzyme that catalyzes the posttranslational sulfation of tyrosine residues within acidic motifs of many polypeptides in all multicellular organisms. Tyrosine _O_-sulfation is a common posttranslational modification shown to be important in protein–protein interactions in several systems. We have purified TPST from rat liver microsomes based on its affinity for the N terminus of PSGL-1, a known TPST substrate. Tryptic peptides derived from the rat protein were used to isolate human and mouse cDNAs that predict type II transmembrane proteins of 370 amino acid residues. All 12 tryptic peptides derived from the purified rat protein, comprising ≈35% of the protein, are represented in the deduced amino acid sequence of the human and mouse cDNA. Furthermore, the predicted molecular weight of the TPST coding region, in conjunction with the two potential N-glycosylation sites, is consistent with the size of the purified enzyme as assessed by SDS/PAGE. Both human and mouse TPST cDNAs induce overexpression of TPST activity when transfected into mammalian cells. These data conclusively demonstrate that the cDNAs encodes a TPST. Further investigations will be required to determine the preferred macromolecular substrate(s) for this enzyme.
Previous work has demonstrated that TPST activity is localized to the trans-Golgi in mammalian cells (5, 19, 30). Several lines of evidence strongly support the conclusion that the TPST cDNAs we isolated encode membrane-bound Golgi enzymes. We have shown that the TPST activity in crude liver homogenates is sedimentable and is present exclusively in the microsomal fraction. We provide evidence that the active site of the enzyme is lumenally oriented in microsomes, consistent with previous observations (19, 20). Furthermore, the observation that TPST carries N-linked glycans demonstrates that it transits the Golgi compartment. In addition, we were unable to detect TPST activity in culture supernatants of mammalian cells transfected with TPST cDNA, indicating that the enzyme is not secreted in an active form even when overexpressed. Finally, the predicted domain structure of TPST is similar to known Golgi glycosyltransferases and sulfotransferases, which are almost invariably type II transmembrane proteins with short cytoplasmic domains and luminal catalytic domains.
TPST exhibits homology to a large family of cytosolic sulfotransferases, including phenol and hydroxysteroid sulfotransferases. The known members of this family contain two regions that are highly conserved throughout phlogeny, called region I and region IV (31). These regions are involved in cosubstrate binding (32–35). Kakuta et al. (35) have recently solved the crystal structure of mouse estrogen sulfotransferase in the presence of the cosubstrate analog PAP and the substrate 17β-estradiol (35). Alignment of mouse TPST and mouse estrogen sulfotransferase reveals a 20% identity and 52% similarity with 19 alignment gaps, 12 of which are ≤3 residues long (Fig. 6). Notably, TPST has a 35-residue N-terminal extension that includes the putative noncleavable signal peptide/membrane anchor. In estrogen sulfotransferase, the residues that contact the 5′ phosphate of the cosubstrate (PKSGTTW, underlined) form a loop between β-sheet 3 and α-helix 3 that corresponds to region I (35). This region is highly conserved in TPST and corresponds to residues 78–84 (PRSGTTL, underlined). The residues involved in binding the 3′ phosphate of PAP are located in two discontinuous regions of estrogen sulfotransferase. The first region includes two residues, Arg130 and Ser138, located just before and within α-helix 6. The second is composed of residues 257–259 (Arg-Lys-Gly). The corresponding residues in TPST are Arg184, Ser192, Ala322, Lys323, and Leu324. Thus, although the degree of identity is limited, most of the residues involved in cosubstrate binding in the estrogen sulfotransferase structure are predicted to be conserved in TPST (Fig. 6). TPST exhibits a similar degree of homology to Golgi sulfotransferases, including heparan sulfate 2-sulfotransferase (36), chondroitin 6-sulfotransferase (37), and the C-terminal domain of heparan sulfate _N_-deacetylase/_N_-sulfotransferase (38).
Figure 6.

Alignment of the amino acid sequences of mouse estrogen sulfotransferase and mouse TPST. The alignment was produced by using the bestfit program (Genetics Computer Group). Amino acid identities between mouse estrogen sulfotransferase and mouse TPST are indicated by a bar, whereas similarities are indicated by double and single dots. Residues involved in cosubstrate binding in the estrogen sulfotransferase crystal structure are highlighted. Differences between mouse and human TPST are indicated (asterisk).
In summary, we have purified and cloned a member of a class of novel sulfotransferases that catalyzes tyrosine _O_-sulfation. This enzyme may catalyze tyrosine _O_-sulfation of PSGL-1 and other protein substrates involved in diverse physiologic functions including inflammation and hemostasis.
Acknowledgments
We thank Jana Beihl and Vickie Gurfinkel for technical assistance, Caroline Thompson for automated DNA sequencing, Tom Zamborelli at Amgen, Inc., for peptide synthesis, and Reneé Robinson, John Neveu, and Derrick Arnelle for HPLC, mass spectrometry, and peptide sequencing. This work was supported by a grant from the Oklahoma Center for the Advancement of Science and Technology (H97–035).
ABBREVIATIONS
PAPS
3′-phosphoadenosine 5′-phosphosulfate
TPST
tyrosylprotein sulfotransferase
PSGL-1
P-selectin glycoprotein ligand-1
EST
expressed sequence tag
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AF038008 and AF038009).
References
- 1.Huttner W B. Nature (London) 1982;299:273–276. doi: 10.1038/299273a0. [DOI] [PubMed] [Google Scholar]
- 2.Huttner W B, Baeuerle P A. Mod Cell Biol. 1988;6:97–140. [Google Scholar]
- 3.Niehrs C, Beibwanger R, Huttner W B. Chem Biol Interact. 1994;92:257–271. doi: 10.1016/0009-2797(94)90068-x. [DOI] [PubMed] [Google Scholar]
- 4.Bundgaard J R, Vuust J, Rehfeld J F. J Biol Chem. 1997;272:21700–21705. doi: 10.1074/jbc.272.35.21700. [DOI] [PubMed] [Google Scholar]
- 5.Baeuerle P A, Huttner W B. J Cell Biol. 1987;105:2655–2664. doi: 10.1083/jcb.105.6.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosa P, Mantovani S, Rosboch R, Huttner W B. J Biol Chem. 1992;267:12227–12232. [PubMed] [Google Scholar]
- 7.Wilkins P P, Moore K L, McEver R P, Cummings R D. J Biol Chem. 1995;270:22677–22680. doi: 10.1074/jbc.270.39.22677. [DOI] [PubMed] [Google Scholar]
- 8.Hortin G L, Farries T C, Graham J P, Atkinson J P. Proc Natl Acad Sci USA. 1989;86:1338–1342. doi: 10.1073/pnas.86.4.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hortin G L. Blood. 1990;76:946–952. [PubMed] [Google Scholar]
- 10.Pittman D D, Wang J H, Kaufman R J. Biochemistry. 1992;31:3315–3325. doi: 10.1021/bi00128a003. [DOI] [PubMed] [Google Scholar]
- 11.Leyte A, van Schijndel H B, Niehrs C, Huttner W B, Ph. Verbeet M, Mertens K, van Mourik J A. J Biol Chem. 1991;266:740–746. [PubMed] [Google Scholar]
- 12.Dong J-F, Li C Q, Lopez J A. Biochemistry. 1994;33:13946–13953. doi: 10.1021/bi00250a050. [DOI] [PubMed] [Google Scholar]
- 13.Marchese P, Murata M, Mazzucato M, Pradella P, De Marco L, Ware J, Ruggeri Z M. J Biol Chem. 1995;270:9571–9578. doi: 10.1074/jbc.270.16.9571. [DOI] [PubMed] [Google Scholar]
- 14.Hortin G, Fok K F, Toren P C, Strauss A W. J Biol Chem. 1987;262:3082–3085. [PubMed] [Google Scholar]
- 15.Hortin G, Tollefsen D M, Strauss A W. J Biol Chem. 1986;261:15827–15830. [PubMed] [Google Scholar]
- 16.Stone S R, Hofsteenge J. Biochemistry. 1986;25:4622–4628. doi: 10.1021/bi00364a025. [DOI] [PubMed] [Google Scholar]
- 17.Skrzypczak-Jankun E, Carperos V E, Ravichandran K G, Tulinsky A. J Mol Biol. 1991;221:1379–1393. [PubMed] [Google Scholar]
- 18.Niehrs C, Kraft M, Lee R W H, Huttner W B. J Biol Chem. 1990;265:8525–8532. [PubMed] [Google Scholar]
- 19.Rens D S, Roth J A. J Biol Chem. 1989;264:899–905. [PubMed] [Google Scholar]
- 20.Niehrs C, Huttner W B. EMBO J. 1990;9:35–42. doi: 10.1002/j.1460-2075.1990.tb08077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.William S, Ramaprasad P, Kasinathan C. Arch Biochem Biophys. 1997;338:90–96. doi: 10.1006/abbi.1996.9800. [DOI] [PubMed] [Google Scholar]
- 22.Pouyani T, Seed B. Cell. 1995;83:333–343. doi: 10.1016/0092-8674(95)90174-4. [DOI] [PubMed] [Google Scholar]
- 23.Sako D, Comess K M, Barone K M, Camphausen R T, Cumming D A, Shaw G D. Cell. 1995;83:323–331. doi: 10.1016/0092-8674(95)90173-6. [DOI] [PubMed] [Google Scholar]
- 24.Zeigler D M, Pettit F H. Biochemistry. 1966;5:2932–2938. doi: 10.1021/bi00873a024. [DOI] [PubMed] [Google Scholar]
- 25.Eng J K, McCormick A L, Yates J R., III J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 26.Lane W S, Galat A, Harding M W, Schreiber S L. J Protein Chem. 1991;10:151–160. doi: 10.1007/BF01024778. [DOI] [PubMed] [Google Scholar]
- 27.Stearns D J, Kurosawa S, Sims P J, Esmon N L, Esmon C T. J Biol Chem. 1988;263:826–832. [PubMed] [Google Scholar]
- 28.Lennon G, Auffray C, Polymeropoulos M, Soares M B. Genomics. 1996;33:151–152. doi: 10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]
- 29.Shworak N W, Liu J, Fritze L M S, Schwartz J J, Zhang L, Logeart D, Rosenberg R D. J Biol Chem. 1997;272:28008–28019. doi: 10.1074/jbc.272.44.28008. [DOI] [PubMed] [Google Scholar]
- 30.Lee R W H, Huttner W B. Proc Natl Acad Sci USA. 1985;82:6143–6147. doi: 10.1073/pnas.82.18.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinshilboum R M, Otterness D M, Sksoy I A, Wood T C, Her C, Raftogianis R B. FASEB J. 1997;11:3–14. [PubMed] [Google Scholar]
- 32.Komatsu K, Driscoll W J, Koh Y, Strott C A. Biochem Biophys Res Commun. 1994;198:1119–1127. doi: 10.1006/bbrc.1994.2587. [DOI] [PubMed] [Google Scholar]
- 33.Marsolais F, Varin L. J Biol Chem. 1995;270:30458–30463. doi: 10.1074/jbc.270.51.30458. [DOI] [PubMed] [Google Scholar]
- 34.Driscoll W J, Komatsu K, Strott C A. Proc Natl Acad Sci USA. 1995;92:12328–12332. doi: 10.1073/pnas.92.26.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kakuta Y, Pedersen L G, Carter C W, Negishi M, Pedersen L C. Nat Struct Biol. 1997;4:904–908. doi: 10.1038/nsb1197-904. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi M, Habuchi H, Yoneda M, Habuchi O, Kimata K. J Biol Chem. 1997;272:13980–13985. doi: 10.1074/jbc.272.21.13980. [DOI] [PubMed] [Google Scholar]
- 37.Fukata M, Uchimura K, Nakashima K, Kato M, Kimata K, Shinomura T, Habuchi O. J Biol Chem. 1995;270:18575–18580. doi: 10.1074/jbc.270.31.18575. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto Y, Orellana A, Gil G, Hirschberg C B. J Biol Chem. 1992;267:15744–15750. [PubMed] [Google Scholar]