Mechanism of chromosome compaction and looping by the E. coli nucleoid protein Fis (original) (raw)
. Author manuscript; available in PMC: 2007 Sep 20.
Published in final edited form as: J Mol Biol. 2006 Sep 22;364(4):777–798. doi: 10.1016/j.jmb.2006.09.043
Abstract
Fis, the most abundant DNA-binding protein in E. coli during rapid growth, has been suspected to play an important role in defining nucleoid structure. Using bulk-phase and single-DNA molecule experiments we analyze the structural consequences of nonspecific binding by Fis to DNA. Fis binds DNA in a largely sequence-neutral fashion at nanomolar concentrations, resulting in mild compaction against applied force due to DNA bending. With increasing concentration, Fis first coats DNA to form an ordered array with one Fis dimer bound per 21 bp and then abruptly shifts to forming a higher-order Fis-DNA filament, referred to as a ‘low mobility complex’ (LMC). The LMC initially contains two Fis dimers per 21 bp, but additional Fis dimers assemble into the LMC as the concentration is further increased. These complexes, formed at or above 1 μM Fis, are able to collapse large DNA molecules via stabilization of DNA loops. The opening and closing of loops on single DNA molecules can be followed in real time as abrupt jumps in DNA extension. Formation of loop-stabilizing complexes is sensitive to high ionic strength, even under conditions where DNA bending-compaction is unaltered. Analyses of mutants indicate that Fis-mediated DNA looping does not involve tertiary or quaternary changes in the Fis dimer structure but that a number of surface-exposed residues located both within and outside the helix-turn-helix DNA binding region are critical. These results suggest that Fis may play a role in vivo as a ‘domain barrier element’ by organizing DNA loops within the E. coli chromosome.
Keywords: chromosome structure, nonspecific DNA binding, DNA looping, nucleoprotein filament, single-DNA molecule micromanipulation
INTRODUCTION
The prokaryotic nucleoid is a condensed but highly dynamic nucleoprotein structure. The E. coli nucleoid contains a single circular chromosome containing about 1.5 mm of DNA packaged into a volume of about 0.2 μm3. Even further condensation is required during rapid growth when several chromosome equivalents are present in the cell due to multiple ongoing replication cycles. Because bacteria do not contain histones or stably wrapped nucleoprotein complexes analogous to nucleosomes, packaging of the chromosome into the prokaryotic nucleoid must differ radically from DNA packaging in eukaryotic cells. The E. coli chromosome is thought to be organized as a dynamic ensemble of topologically-isolated supercoiled loops or ‘domains’ 1; 2. Several independent lines of evidence suggest the connections isolating these domains are spaced every 10 kb on average during rapid growth, but determination of the ‘domainin’ factors responsible for this organization has been elusive 3; 4.
Additional compaction is believed to be mediated by a set of abundant nucleoid-associated DNA bending proteins. The dominant nucleoid proteins in growing E. coli are HU, IHF, H-NS (and its paralog SptA) and Fis 5; 6; 7. Single-DNA studies have shown that HU compacts DNA when bound at subsaturating concentrations 8; 9; 10. Sequence-independent binding by IHF has also been shown to compact λ phage chromosomes in vitro 11. AFM experiments have shown that H-NS can form filaments containing two DNA duplexes, suggesting that H-NS may have a domainin activity 12. Cellular levels of Fis vary widely depending on growth conditions but, as noted below, Fis can be the dominant DNA binding and bending protein in the cell 13; 14. A nucleoid architecture role for Fis was suggested by Schneider et al. 15 based on observations of enhanced branching of Fis-bound supercoiled DNA. Here we show that Fis can function both to directly stabilize loops in linear DNA (i.e., has domainin activity via stabilizing DNA nodes) as well as to promote DNA compaction by means of its bending activity.
Fis has been most extensively studied in the context of its ability to regulate specific DNA transactions. The protein was initially identified as a factor required for the Hin and Gin-catalyzed site-specific DNA inversion reactions and as such was named factor for inversion stimulation 16; 17. Subsequently, Fis was found to regulate λ phage site-specific recombination reactions, oriC and plasmid replication, and specific transcription reactions 5; 18. Each of these diverse reactions are typically controlled by one or more Fis dimers binding specific sites on DNA. Selective Fis binding to specific DNA sites at the low nanomolar range can be demonstrated by nuclease and chemical footprinting assays in the presence of excess mixed-sequence or homopolymeric competitor DNA. These sites are related by a highly degenerate 15 bp sequence motif, which can specify a stable Fis binding site when embedded in a larger DNA segment 19; 20; 21.
Fis binds DNA by means of helix-turn-helix (HTH) structure motifs located at the C-termini of each subunit within the homodimer (Fig. 1A) 22; 23; 24. A distinguishing feature of the Fis dimer structure is that the two DNA recognition α-helices are spaced only about 25 Å apart from each other, thereby preventing Fis from binding to a straight DNA duplex. Since the separation between HTH motifs does not appear to change upon binding, DNA bending is required in order to form a complex 25. A variety of lines of evidence suggest that the DNA is bent at least 40–50° upon Fis binding 20; 22; 23; 26; 27; 28. Thus, Fis selects specific binding sites by means of a degenerate code of functional groups on bases within a 15 bp core sequence and by the ability of a particular DNA segment to conform to the Fis binding surface. Additional bending of DNA, estimated to extend up to 90° depending upon the DNA sequence flanking the 15 bp core binding site, occurs through contacts with residues on the “sides” of Fis 20.
Figure 1. Sequence-independent Fis binding to DNA.
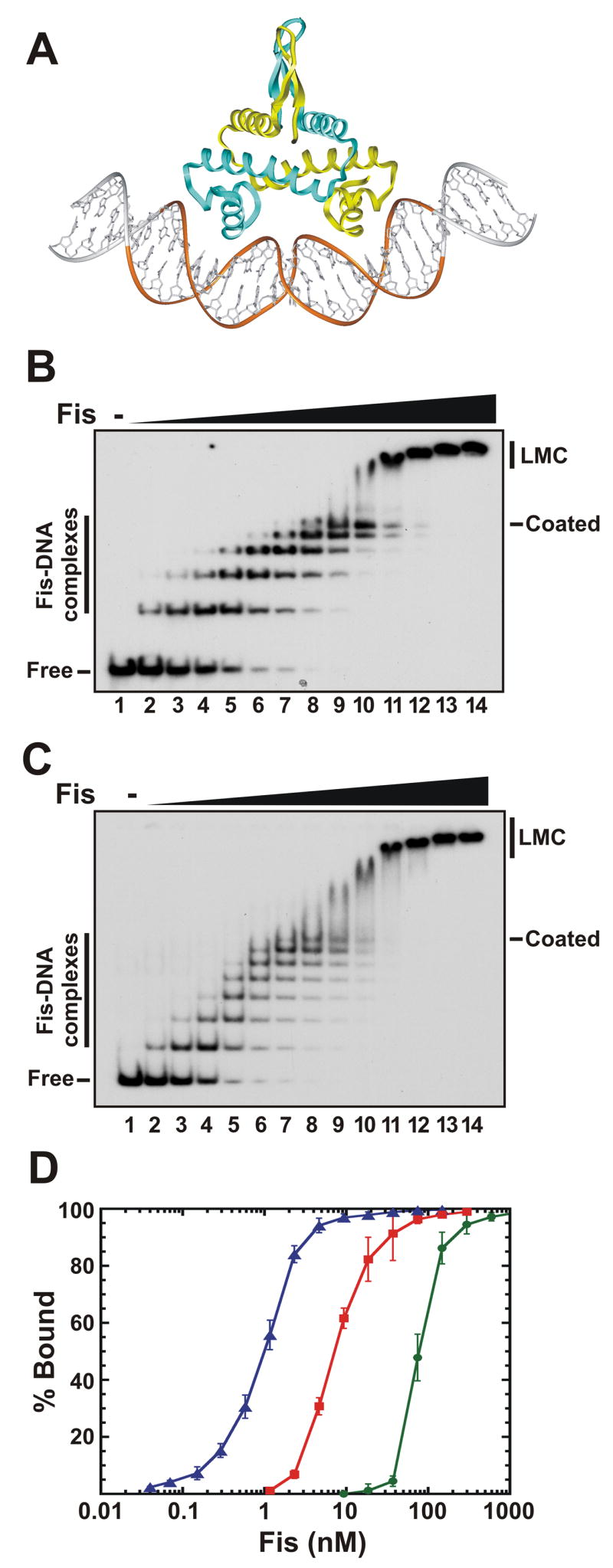
A. Model of a Fis-DNA complex. The two helix-turn-helix motifs, spaced by 25 Å, bind successive major grooves on one side of the DNA helix, forcing a bend into the double helix. The mobile N-terminal β-hairpin arms protrude from the side opposite to the DNA. The gold ribbon over the DNA backbone denotes the 21 bp minimal binding site. The structure of Fis in this and subsequent figures is from the K36E mutant, in which residues 10–98 that include the β-hairpin arms are resolved 24, and it is modeled onto DNA with an overall curvature of about 50°20.
B. Gel mobility shift assays of Fis binding to a 100 bp DNA fragment from phage λ Fis was added in 2-fold increments beginning at 0.15 nM (lane 2) and ending at 600 nM (lane 14). No Fis was added in lane 1. The locations of individual Fis-DNA complexes, the “coated” complex, the “low mobility complex” (LMC), and the unbound (Free) DNA are denoted.
C. Same as panel B, except that a 149 bp fragment from the yeast actin gene was the DNA substrate.
D. Fis binding isotherms of the complex bound by one or more Fis dimers (left curve, blue triangles), the coated complex containing 7 or more Fis dimers (middle curve, red squares), and the LMC complex (right curve, green circles) assembled on the actin DNA fragment. Points represent the mean and standard deviations obtained from ≥4 independent experiments. Hill coeficients for the first complex, the coated complex, and the LMC are 1.5, 1.5, and 3.5, respectively.
When E. coli is growing rapidly in the presence of ample nutrients, Fis is the most abundant DNA binding protein in the cell 13; 14; 29. Fis levels in cells growing in MOPS-buffered rich media or LB after nutrient upshift can exceed 40,000 dimers/cell or 50 μM. Fis levels roughly correspond to growth rate, and are lower in poorer media; little Fis is detectable in stationary phase. The abundance of Fis under optimal growth conditions and its robust sequence-independent binding [this report and 30] led us to investigate its role in organizing and compacting the chromosome. In a preliminary report, we used single-DNA micromanipulation to show that Fis is capable of stabilizing loops along 48.5 kb λ-DNA molecules, resulting in dramatic compaction of the DNA molecules against applied force 31. Here, we investigate the sequence-independent binding properties of wild-type and mutant Fis proteins using a combination of bulk-phase and real-time single-DNA molecule approaches. Our results reveal multiple modes of non-specific Fis-DNA binding, each with distinct micromechanical properties. These findings suggest that Fis may play a prominent role in organizing the bacterial nucleoid into its topologically-isolated domain structure as a function of cell physiology.
RESULTS
Binding of Fis to DNA and formation of Fis-DNA complexes
Two bulk-phase approaches were employed to evaluate nonspecific binding activity of Fis. Polyacrylamide gel mobility shift assays were performed on a collection of different DNA fragments revealing Fis-DNA complexes as a series of slower migrating bands. DNA scission patterns generated from Fis-chemical nuclease chimeras identified the positions of bound Fis dimers. Both methods demonstrated that Fis binds DNA in a sequence-independent manner such that DNA molecules can be completely coated by Fis along their entire length. Additional Fis molecules cooperatively bind to these Fis-DNA filaments to generate higher-order complexes that are likely to be important for the stabilization of DNA loops observed in single-DNA molecule experiments described below.
Individual Fis dimers bind DNA at subnanomolar concentrations
Increasing concentrations of Fis protein were incubated with 84 – 262 bp 32P-labeled DNA fragments from E. coli, S. cerevisiae, and phage λ and the products subjected to native polyacrylamide gel electrophoresis. Examples of Fis titrations are shown in Figs. 1B and C, and the binding characteristics of these and four other DNA fragments are summarized in Table 1. Among the fragments used, the yeast MET14c and E. coli lacZ fragments contain weak specific Fis sites as judged by the presence of 1–2 complexes when 50 μg/ml herring testes DNA fragments are included in reactions (not shown). In the absence of competitor DNA, Fis binds to the DNA segments beginning with an apparent Kd for the first complex around 0.1 nM for the fragments containing a weak specific Fis site to about 1 nM for the fragments that do not contain a demonstrable Fis site.
Table 1.
Sequence-independent binding by Fis to different DNA fragments
| Kd | |||||||
|---|---|---|---|---|---|---|---|
| DNA1 | size (bp) | stable site2 | cmpl 1 3 | coated4 | # complexes5 | bp/complex6 | [Fis]LMC 7 |
| λ attR | 100 | 0 | 0.8 | 12 | 5 | 20 | 75–150 |
| _y_ACT1 | 149 | 0 | 0.75 | 10 | 7 | 21 | 75–150 |
| _y_MET14a | 84 | 0 | 0.9 | 10 | 4 | 21 | 75–150 |
| yMET14b | 116 | 1 | 0.1 | 12 | 5 | 23 | 75–150 |
| _y_MET14c | 150 | 1–2 | 0.25 | 12 | 7 | 21 | 75–150 |
| _ec_LacZ | 262 | 1 | 0.5 | 12 | ~12 | 22 | 75–150 |
At higher concentrations, Fis first coats DNA, and then forms a low-mobility complex (LMC)
For each of the DNA fragments studied, a sequential series of discrete complexes form with increasing Fis concentrations up to a point, which we will refer to as a “Fis-coated complex”, where the maximum number of discrete complexes are observed. Although formation of the first complex varied with respect to Fis concentration, half of the DNA fragments become coated by Fis when incubated with 10 ± 2 nM Fis, and nearly 90% of the molecules are fully coated by 20 nM (Table 1 and Fig. 1D, red squares).
Further increase in Fis concentration resulted in an abrupt decrease in electrophoretic migration of the DNA complex; we call this the “low mobility complex” (LMC). Migration of the LMC gradually further decreases as the Fis concentration is further increased, implying increasingly higher-order Fis-bound complexes are generated. The transition from Fis-coated DNA to the LMC occurs over a narrow range of Fis concentrations, with 90% LMC formation at about 100 – 150 nM for all fragments tested (Fig. 1D, green circles and data not shown).
The number of Fis molecules in the Fis-DNA complexes resolved by native gel electrophoresis was quantified using 32P-labeled FisHMK and fluorescein-labeled DNA fragments. Molar ratios of Fis dimers per DNA molecule for the 5 discrete complexes formed on either a 100 bp fragment from phage λ or a 149 bp fragment from the yeast actin gene correspond to sequential binding of Fis dimers, beginning with 1 dimer per DNA fragment in the first complex (Table 2). Given the size of the different DNA fragments and the number of discrete complexes, the Fis-coated complexes therefore contain a Fis dimer every 20–23 bp (Table 1). This DNA length corresponds to the predicted minimal binding site of the Fis-DNA complex (see Discussion).
Table 2.
Stoichiometry of Fis-DNA complexes1
| Number of Fis dimers/DNA molecule | ||
|---|---|---|
| Complex2 | 100 bp (phage λ) | 149 bp (Actin) |
| 1 | 1.0 ± 0.1 | 1.1 ± 0.1 |
| 2 | 2.1 ± 0.1 | 2.0 ± 0.1 |
| 3 | 2.9 ± 0.1 | 2.9 ± 0.1 |
| 4 | 3.9 ± 0.1 | 4.1 ± 0.1 |
| 5 | 5.0 ± 0.2 | 5.1 ± 0.4 |
| LMC3 | ||
| 0.2 μM | 11 ± 2 | 15 ± 24 |
| 0.3 μM | 17 ± 2 | |
| 0.6 μM | 23 ± 2 | 25 ± 3 |
| 1.2 μM | 30 ± 3 |
The low mobility complex (LMC) when first formed on the 100 bp λ fragment contains 10–11 Fis dimers per DNA molecule (Table 2). Occasionally, about 10 discrete complexes leading up to the initial LMC can be discerned, confirming that the LMC contains approximately twice the number of Fis dimers as the Fis-coated complex. The LMC initially formed on the 149 bp actin fragment contains about 15 Fis dimers per DNA molecule. The numbers of dimers in the LMC for both DNAs continue to increase when higher concentrations of Fis are present in the binding reaction. These values suggest that the abrupt shift from the Fis-coated DNA molecules to the LMC corresponds to approximately a doubling of the numbers of bound Fis dimers with additional Fis dimers subsequently recruited onto the Fis-DNA filament.
Several experiments showed that the LMC does not contain two or more DNA fragments (data not shown). These included performing Fis titrations on the 149 bp 32P-labeled actin probe in the presence of a 10-fold molar excess of an unlabeled 276 bp fragment. No change in the pattern or migration of the labeled complexes as compared to the competitor-free reactions was observed, except that about four times more Fis protein was required to form individual complexes and two times more Fis was required to form the LMC. In addition, when increasing amounts of unlabeled 276 bp fragments were added to reaction in which sufficient Fis was present to shift >90% of 32P-labeled actin fragments into the LMC, no change in the migration of the complexes, including the formation of a supershifted LMC, were detectable. Instead, the amount of the labeled LMC gradually decreased as the concentration of the unlabeled fragment increased. We conclude that the LMC contains one DNA bound by 1 Fis dimer per 10 bp.
DNA scission patterns by Fis-1,10 phenanthroline chimeras
Sequence-independent binding by Fis was also investigated by analyzing DNA scission patterns generated by Fis site-specifically coupled to the chemical nuclease 1,10 phenanthroline-copper (Fis-OP). In earlier studies scission patterns by different Fis-OP copper chimeras at specific Fis binding sites were examined 20; 27. These studies found that 1,10 phenanthroline copper tethered to position 98 located at the C-terminal end of the helix-turn-helix motif cleaved DNA on either side of a variety of Fis binding sites with nearly equivalent efficiencies (Fig. 2A). Moreover, unlike other Fis-OP chimeras the efficiency of cleavage by Fis N98C-OP was insensitive to Fis-induced bending of DNA flanking the core binding site.
Figure 2. DNA cleavage by Fis-OP nuclease chimeras when bound nonspecifically.
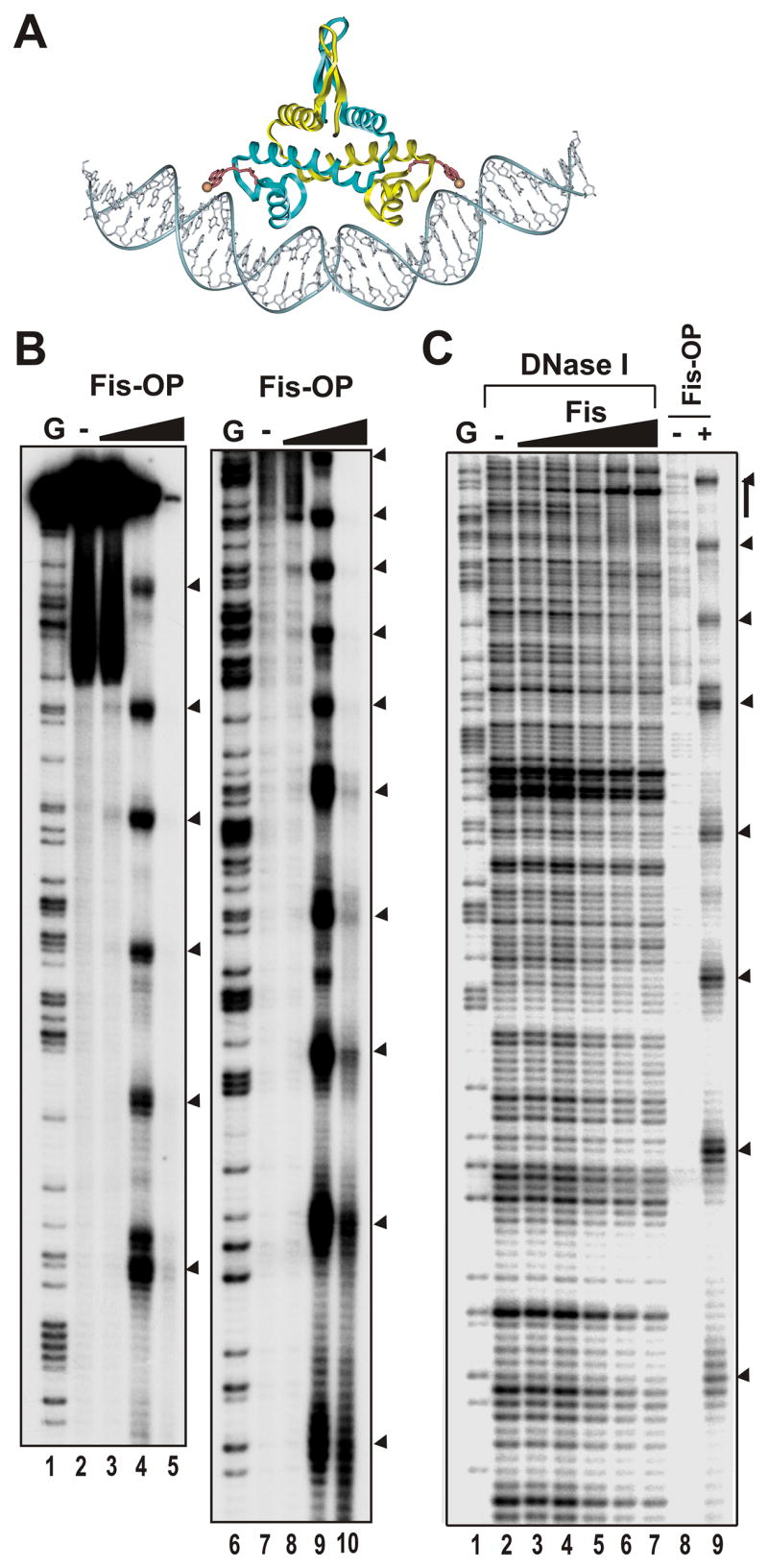
A. Model of Fis98-OP bound to DNA. The 1,10 phenanthroline copper moiety linked to the C-terminus (residue 98) of Fis through an acetamido linker is shown in red with the copper as an orange sphere.
B. A 262 bp fragment from the 5′ end of the lacZ gene and 32P-labeled at the 5′ end of the coding strand was incubated with 0 and approximately 5, 10, and 20 nM Fis98-OP (lanes 2–5, 7–10). Lanes 1 and 6 are G chemical sequencing ladders 56. The left and right panels are the same samples electrophoresed for longer or shorter time periods, respectively, on the same gel to resolve most of the DNA length.
C. DNase I and Fis98-OP cleavage. The fragment used in panel B was subjected to DNase I footprinting with 0, and approximately 1, 2.5, 5, 10, and 20 nM Fis (lanes 2–7). Lanes 8 and 9 are incubation with no protein or 10 nM Fis98-OP, respectively. Lane 1 is a G sequencing ladder. Arrowheads denote major Fis98-OP cleavage sites, and the vertical bar denotes a weak DNase I protected region by Fis about 150 bp into the lacZ coding region.
To probe nonspecific binding of Fis, increasing amounts of Fis N98C-OP were incubated with a 32P end-labeled 262 bp fragment from lacZ, and DNA scission was activated by addition of copper and reducing agent. As shown in Fig. 2B, electrophoresis of the products in a denaturing polyacrylamide gel revealed a regular pattern of scission throughout the fragment (lanes 4 and 9). Cleavages are present every 19.7 ± 1.3 bp, suggesting that Fis dimers are positioned adjacent to each other in a regular array, consistent with the Fis-coated complexes observed by gel mobility shift experiments. At higher Fis N98C-OP concentrations only the smaller end-labeled fragments remained (lanes 5 and 10), suggesting multiple cleavages occurred within each fragment.
Fis binding arrays are not readily detectable by standard DNase I footprinting experiments on the lacZ fragment (Fig. 2C). However, the DNase I digestion pattern does reveal a weak Fis binding site near the 3′ end of the DNA segment. The pattern of Fis N98C-OP cleavages (lane 9) is consistent with the phase of the Fis binding array being set by preferential binding at this site. A similar situation where high affinity specific Fis binding sites appear to phase arrays of nonspecific Fis binding has been observed in the xylAF region (data not shown).
Compaction of DNA against force for different Fis concentrations
We recently reported preliminary results of experiments studying sequence-independent DNA binding by Fis to single λ-DNA molecules 31. In those ‘magnetic tweezer’ experiments a 48.5 kb λ-DNA was tethered to a glass surface, while its other ‘free’ end was attached to a magnetic particle, allowing controlled forces to be applied to the molecule (Fig. 3A) 8. For low (sub-μM) Fis concentrations a mild compaction of the DNA against applied force occurred, similar to that observed for other DNA-bending proteins including IHF and HU 8; 9; 11; 32; 33. However, at higher (μM) concentrations, we observed that below a well-defined force threshold, dramatic compaction of the DNA molecule occurred: over roughly a minute, the entire molecule was folded to nearly zero length. When the force was increased to a large (10 pN) level after DNA collapse, we observed opening via a series of discontinuous jumps, suggesting that the DNA had been condensed by formation of loops stabilized by Fis. Here we report experiments examining the mechanism of the ”bending” and “looping” modes of DNA condensation.
Figure 3. Single-DNA study of Fis-DNA interactions.

A. Vertical magnetic tweezer setup. A single DNA molecule is tethered to the top surface inside the flow cell; a paramagnetic bead attached at the other end of the molecule provides a ‘handle’ to which controlled forces can be applied using permanent magnets on a translator. The bead is imaged using a 100x objective on a piezoelectric positioner (bottom panel), allowing a computer to track the bead in three dimensions.
B. Force-extension curves for bare DNA (black) in buffer with glycerol and BSA (see text), and with Fis at 10 nm (light green) 20 nM (orange), 50 nM (dark green), 100 nM (violet) and 200 nM (blue) concentrations. At 10 nM, little effect is observed; at 20 nM, maximum compaction occurs. As concentration is increased further, a gradual reduction of compaction occurs. Over this (low) concentration range, all force-extension curves are entirely reversible, indicating no tendency for Fis to stabilize DNA crossings.
C. Collapse of DNA against applied force driven by Fis. Naked DNA in buffer (open circles) and DNA incubated with 200 nM Fis (blue crosses) and 500 nM Fis (green circles) show reversible force-extension response over the full range of forces, but for 1 μM Fis concentration (orange diamonds) the Fis-DNA complex undergoes a collapse (horizontal dashed line) when force is reduced to 0.2 pN. For larger Fis concentrations of 6 μM (blue triangles), and 13 μM (red squares) similar collapse transitions occur at higher forces of 0.3 and 0.6 pN, respectively.
Fis-coated λ-DNA molecules at low concentrations: mild DNA compaction due to bending
Fig. 3B shows effects of Fis binding on the DNA force-extension response at relatively low Fis concentrations in 100 mM KGlu buffer, with 500 ng/ul BSA and 5% glycerol added to approximate conditions used in the gel experiments, alongside a curve for naked DNA in protein-free buffer. For low Fis concentrations (10 nM or below), little change in the force-extension curve is observed. At 20 nM there is a prominent shift of the force-extension curve to shorter extensions at each force measured. Between 20 nM and 100 nM there is a partial return of the force curve back towards that of naked DNA, indicating less compaction. The 200 nM Fis-DNA complex shows the same force-extension curve as that measured for 100 nM.
All these low-concentration (200 nM Fis and below) force-extension curves are mechanically reversible (subsequent scans down and back up in force yield coincident curves), indicating that Fis in this concentration range does not stabilize DNA crossings or otherwise tightly condense DNA. Instead, in this concentration range, Fis binds along DNA and the bends act to compact the DNA 34, with larger forces required to achieve a given extension. The 20 nM concentration where the bending-compaction is maximal corresponds to the concentration where formation of near fully-Fis-coated DNA occurs in the gel experiments. The 20 to 200 nM range, where subsequent reversal of the force-extension curve occurs, corresponds to the concentration range where the LMC forms in gel experiments (see Fig. 1B-D). In these low-concentration regimes the Fis-DNA complex does not reach the full DNA contour length under high forces (10 pN); the complex has a maximum length that is 0.8 ± 0.1 microns shorter than that of the naked λ-DNA.
Fis-DNA complexes at high concentrations: DNA collapse due to loop formation
Fig. 3C shows the effects on DNA when larger concentrations of Fis were exchanged into the flow cell. For forces larger than 1 pN, complexes assembled with up to 13 μM Fis showed reversible force-extension behavior with moderate compaction relative to bare DNA, similar to that observed in the lower-concentration experiments (Fig. 3B). Then, as the force was gradually decreased below 1 pN, a threshold was reached in which the λ-DNA complexes assembled with ≥ 1 μM Fis abruptly collapsed. With 1 μM Fis, the DNA molecule condensed from a length of 8 μm to <0.5 μm over a one minute period after force was lowered to 0.2 pN. At 6 μM Fis, the threshold force for DNA collapse was 0.3 pN and at 13 μM the threshold was at 0.6 pN. Collapse from about 12 to 0 μm at a constant force of 0.6 pN with 13 μM Fis occurred over about 3.5 min. Collapse was not observed to occur with 500 nM or lower Fis concentration.
Loop size distribution during force-controlled condensation and decondensation
This abrupt DNA condensation is characteristic of the formation of DNA loops, that are generated by Brownian motion only for low forces < 1 pN 31; 35. Previous preliminary results showed that following this collapse, re-opening by a force of 9 pN proceeded in a stepwise manner, indicative of a looping mechanism for condensation. We have now collected more extensive and precise data on loop sizes using shorter DNA molecules (plasmid pBS290-2, a 10.5 kb E. coli plasmid unrelated to λ-DNA), using the setup of Fig. 3A. Shorter DNAs give the advantage of a reduction in end fluctuation due to random bending fluctuations, yielding higher spatial resolution. Also, since this plasmid is unrelated to λ-DNA, experiments with it provide a check that the looping-condensation effect is sequence-independent. Finally, the use of short DNA molecules allows us to determine extension from the out-of-focus bead image, which enables us to collect slightly more than 10 measurements/sec.
Fig. 4 shows data for the 10.5 kb molecules, which when fully extended as naked DNA are slightly more than 3 μm long. As in the experiments of Fig. 3, Fis was allowed to bind at 5 pN tension (enough to keep the molecule essentially straight) before the force was reduced to permit looping-condensation. Fig. 4A shows condensation of the DNA by 10 μM Fis, against a 0.5 pN force. Because of the short DNA length, the extension measurement noise is greatly reduced to about 100 nm per single measurement (spread in data, Fig. 4A). Given our data rate of 10 measurements/sec, the noise level is 30 nm·sec1/2, so after smoothing over a 1 sec time window, we can detect 30 nm steps that are more than 1 sec apart. The raw data (Fig. 4A) indicate that condensation proceeds via a step-wise route, providing further evidence for a looping-mediated mechanism. Furthermore, occasional reopening events (steps to longer extension) can also be observed, as expected near the condensation force threshold 35.
Figure 4. Step observation and analysis during collapse and subsequent reopening of Fis-DNA complexes.

A. Extension versus time for a 10.5 kb DNA in 10 μM Fis solution and under a 0.5 pN constant force. The ~100 nm rapid fluctuation in the extension around its local average is due to thermal fluctuations. Steps down, and occasionally back up, are observed, with amplitudes in the 200 nm range.
B. Reopening of the Fis-DNA complex following the collapse shown in A, driven by a force of 7 pN. Opening proceeds via a series of jump events of roughly 200 nm amplitude, consistent with reversal of the step-wise condensation shown in A.
C. Collapse of a 10.5 kb DNA in 1 μM Fis solution, against a 0.2 pN force. Note the larger thermal noise amplitude relative to A, and also the relatively small initial extension. However, step-like features are still observable during condensation.
D. Reopening of the Fis-DNA complex following the collapse shown in C, driven by a force of 4 pN; as in B, opening proceeds via a series of step events of roughly 200 nm size.
E. Step size distribution obtained from closing traces from a series of 10 μM Fis experiments similar to that shown in A. The average step size is 225 nm.
F. Step size distribution obtained from opening traces from a series of 10 μM Fis experiments similar to B. The average step size is 275 nm.
G. Step size distribution obtained from closing traces from a series of 1 μM Fis experiments similar to C. The average step size is 250 nm; after correction for the low force involved, the DNA length involved in each step is roughly double the extension step observed.
H. Step size distribution obtained from opening traces from a series of 1 μM Fis experiments similar to D. The average step size is 210 nm.
Following closing, the 10.5 kb DNA was reopened using a 7 pN force (Fig. 4B), and a series of upward steps were observed with a typical size of 200 nm (600 bp). The large force suppresses thermal fluctuations, indicating that the spread in the data of Fig. 4A is thermal rather than instrumental in origin. We estimate our instrumental single-extension measurement error to be 20 nm (for our 10 measurements/sec data rate, this corresponds to 7 nm·sec1/2).
Fig. 4C shows a separate condensation experiment where 1 μM Fis was used, and therefore, a lower force of 0.2 pN was required for condensation. The thermal noise level is larger than in Fig. 4A, but steps down are still evident. Importantly, even before loops form, at 0.2 pN the Fis-DNA complex is only 50% of the total DNA length (see Fig. 3B), so a 100 nm jump in Fig. 4C corresponds to closure of a 200 nm (600 bp) loop. Following condensation, this molecule could be decondensed by a 4 pN force (Fig. 4D), with opening proceeding by a series of jumps.
We carried out a series of experiments to collect sufficient data for statistical analysis of the jump size distributions. Four separate experiments (different flow cells) were performed, each involving between four and seven opening-closing cycles. Steps in both closing and opening traces were measured, and are shown as histograms in Fig. 4E–H. Fig. 4E shows the step distribution during closing against a 0.5 pN force for 13 μM Fis; the distribution shows a clear peak at 150 nm, with an average step size in the distribution of 225 nm; the distribution width is 115 nm. After correcting for the reduced extension of the Fis-DNA complex at this force (see Fig. 3C), we conclude that the mean DNA length per step is 270 nm (810 bp); the distribution width is 140 nm (420 bp).
From the opening data following this closing at 0.5 pN, we obtain the opening step distribution shown in Fig. 4F. The average step is 275 nm and the distribution width is 170 nm. After correcting for the small difference between the extension of the Fis-DNA complex and the total DNA length at 7 pN (see Fig. 3C), we conclude that the opening steps involve DNA loops of mean length 290 nm (870 bp) and a distribution width of 180 nm (540 bp). Comparing the corrected means and widths of the closing and opening distributions, we find that they are nearly the same, indicating that little tight crosslinking occurs after the initial condensation.
Similar analysis of 1 μM closing data is shown in Fig. 4G. In this case, a mean step size of 250 nm and a distribution width of 160 nm was measured. After correction for the short extension of the Fis-DNA complex at 0.2 pN (Fig. 3C), we conclude that the mean step size has a DNA length of 500 nm (1500 bp) and a width of 320 nm (960 bp). During opening following this low-force condensation, the loop distribution of Fig. 4H was obtained, with a mean step size of 210 nm and a width of 150 nm; we estimate corresponding DNA loop sizes of 220 nm (660 bp) and distribution width of 160 nm (480 bp). The large difference between the loop distributions during closing and opening suggests that in this case, there may be appreciable crosslinking of the complex following its initial condensation.
While the loop-opening traces are unambiguous, there is appreciable thermal fluctuation in the lower-force loop-closing trace, eliminating our ability to detect steps (at the rate they occur in these experiments) smaller than 50 nm in size during closure at 0.5 pN, or smaller than 100 nm during closure at 0.2 pN. Furthermore, at any force, pairs of loops which close or open in rapid succession may be erroneously identified as single loops. Both these sources of error will result in us missing small steps, tending to shift the peaks of the histograms of Fig. 4 to larger step sizes. This source of systematic error will be larger for data taken at lower force where thermal fluctuations have larger amplitude (i.e., Fig. 4C and G). However, in these experiments, >80% of the loop events are 100 nm or larger in size and are separated by apparent mean pause times >5 sec. This indicates that our step-size analysis is not severely affected by small or rapid loop events.
Fis complexes on λ-DNA are stable in protein-free buffer
We evaluated the stability of Fis complexes formed on λ-DNA by exchanging the surrounding solution within the flow cell and observing effects on single tethered DNA molecules (Fig. 5A). Naked λ DNA (open circles) was incubated with 50 nM Fis, causing a decrease in DNA extension at stretching forces between 10 and 0.1 pN (open squares). Then, the protein solution was replaced with protein-free buffer, incubated for 20 min, and a final force-extension series was measured (filled squares). The absence of any change in the Fis-induced elastic properties of the λ-DNA indicates that at least the fraction of Fis responsible for the shift of the force curve remained bound.
Figure 5. Stability of Fis-DNA complexes.

A. Naked λ-DNA in buffer (open circles) was incubated with 50 nM Fis (filled squares). Then, the protein solution was replaced with protein-free buffer and incubated for 20 min, and the force-extension response was re-measured (open squares). The lack of any change indicates that Fis which is “coating” DNA does not spontaneously dissociate into buffer.
B. Naked λ-DNA in buffer (open circles) was incubated with 6 μM Fis while held extended by a 10 pN force. Then, the protein solution was replaced by protein-free buffer; finally force was gradually reduced and extensions were measured (filled circles); Fis-induced collapse was observed at 0.3 pN, indicating that looping-competent Fis bound to DNA does not spontaneously dissociate into buffer.
C. Naked λ-DNA in buffer (open circles) was incubated with 50 nM Fis, resulting in the expected high-force compaction effect (open triangles). Then, the protein solution was replaced by protein-free buffer containing 50 μg/ml nonspecific DNA fragments, which resulted in no change in the high-force compaction (filled triangles). Higher DNA fragment concentration of 250 μg/ml (filled circles) led to a shift of the force curve back towards that of naked DNA indicating that Fis may transfer to competitor DNA at sufficiently high concentration.
D. In an experiment similar to that shown in C, initially naked DNA (open circles) was incubated with 5 μM Fis solution, resulting in the expected shift of the force-extension curve above 1 pN (filled squares; data for the force shift generated by 200 nM Fis is shown for comparison, filled triangles). After washing with 50 μg/ml DNA fragments the complex retained the force-extension behavior shifted relative to naked DNA (in accord with C), but did not undergo looping-condensation at low forces (open squares). Thus, DNA-bending Fis is stable in the presence of nonspecific DNA, but DNA-looping Fis can be removed (or quenched) by nonspecific DNA.
The stability of looping-competent Fis complexes was similarly evaluated. Fig. 5B shows an experiment where initially naked λ DNA (open circles) was reacted with 6 μM Fis while held at a high force of 10 pN; this high force prevented looping-condensation. Then, the protein solution was replaced with protein-free buffer, and incubated for 20 min. Finally the force was gradually reduced, and extensions were measured (filled circles). For forces above 0.3 pN the Fis-coated λ-DNA exhibited a decrease in extension similar to that observed in experiments where Fis was in solution during the elasticity measurements (Fig. 3B), indicating that Fis is able to bind normally to a straight DNA molecule extended by 10 pN tension. Then, at 0.3 pN, an abrupt collapse at 0.3 pN occurred, exactly as in the experiment that employed no intermediate wash by protein-free buffer (compare to Fig. 3C). We conclude that DNA-bound Fis molecules that promote both DNA bending (coating) and looping are stable against spontaneous dissociation.
DNA fragments in solution eliminate DNA looping and can suppress DNA bending
We examined whether addition of DNA fragments in solution would be able to dislodge bound Fis or compromise its ability to loop DNA. A tethered and initially naked λ-DNA (Fig. 5C, open circles) was reacted with 50 nM Fis; binding was confirmed by the expected compaction of DNA at forces between 0.05 and 10 pN (Fig. 5C, open triangles). Protein-free buffer was introduced to remove free protein, and then the wash buffer was replaced with protein-free buffer containing 50 μg/ml herring sperm DNA fragments (Promega, Madison WI). After a further incubation for 20 minutes, we measured a force-extension curve (filled triangles). The presence of this level of competitor DNA had no effect on the high force behavior, indicating that the Fis which is bound so as to generate a bending-compaction shift does not easily transfer to competitor DNA. Finally, we introduced a higher concentration of DNA fragments (250 μg/ml) and incubated for 20 minutes. This higher concentration of DNA in the surrounding buffer resulted in a partial shift of the force-extension curve towards that of naked DNA (filled circles); longer incubation times with competitor DNA had no additional effect (data not shown). We conclude that at least part of the Fis responsible for DNA bending can be transferred to competitor DNA when present at high concentration.
A similar experiment was carried out for higher Fis concentrations to examine the stability of the molecules responsible for DNA looping (Fig. 5D). Naked DNA (open circles) was reacted with 5 μM Fis; the resulting force-extension curve (filled squares) showed a compaction shift over the range 1 to 10 pN similar to that seen in other experiments (200 nM Fis data are shown for comparison, filled triangles). Following this, protein-free buffer, and then buffer containing 50 μg/ml DNA fragments were introduced. Then, a final force-extension curve was measured (open squares). There was no change in the curve for the force range from 1 to 10 pN, in accord with the lower-concentration experiment of Fig. 5C. However, there was no looping-mediated collapse at low forces. Thus, the looping function of Fis is eliminated by the presence of 50 μg/ml of competitor DNA even though the bending-compaction activity is not affected.
Increased salt inhibits DNA looping but does not affect DNA bending
Experiments over a range of KGlu concentrations revealed different salt dependences of bending and looping activities of Fis. Fig. 6 shows results for different KGlu concentrations with the naked DNA response (open circles) shown for reference; the naked DNA force-extension response is independent of KGlu concentration (data not shown) in accord with other studies of other univalent salts 36. As discussed above, after incubation with 5 μM Fis in 100 mM KGlu buffer, a modest compaction of DNA at high forces, and an abrupt collapse of DNA at about 0.3 pN due to looping was observed (triangles). However, in a separate experiment where DNA was incubated with Fis at 500 mM KGlu (filled squares), no low-force looping-collapse was observed, even though the high-force compaction was nearly the same as in the 100 mM buffer. In that experiment the protein solution was subsequently replaced with protein-free 100 mM KGlu buffer, with the result that there was still no looping-condensation (filled circles). Therefore, at sufficiently high KGlu concentration, Fis is unable to bind to DNA so as to subsequently trap loops. In a further series of experiments, we determined that looping-condensation occurred when DNA was incubated with 5 μM Fis in 200 mM KGlu, but 300 mM KGlu eliminated looping (data not shown).
Figure 6. Effects of salt on Fis-DNA interactions.

Naked λ-DNA (filled circles) undergoes a modest compaction at high forces, and collapse at low forces after incubation with 5 μM Fis in our standard-salt buffer (100 mM KGlu, triangles). In a second experiment using 5 μM Fis in high-salt buffer (500 mM KGlu, filled squares) the high-force compaction occurs but no collapse occurs at low force. Washing the sample with the standard-salt buffer (100 mM KGlu, filled circles) did not lead to looping-condensation. In a third experiment, DNA was first incubated with 5 μM Fis in 100 mM KGlu buffer, and then the protein solution was replaced with 500 mM KGlu protein-free buffer and finally force was reduced; the result was collapse at 0.5 pN (filled diamonds). Thus, salt concentration at the time of binding controls the ability for Fis-DNA complexes to undergo looping-condensation.
Given the above results, we carried out experiments where Fis was bound initially in low salt and then the protein solution was replaced with high-salt buffer. A DNA molecule bound by 5 μM Fis at 100 mM KGlu was allowed to undergo looping-condensation at 0.3 pN (Fig. 6, open triangles) and then re-extended by a large 12 pN force. The protein solution in the flow cell was then replaced with protein-free 500 mM KGlu buffer, and the force was again gradually decreased (filled diamonds). Remarkably, looping-collapse was observed to occur at 0.5 pN. These experiments indicate that the salt-sensitive step in the looping reaction is at the initial Fis binding stage and not during the looping reaction itself.
We also examined whether bending or looping activities of Fis would be affected by detergent. Experiments were performed with 5 μM Fis in 100 mM KGlu plus 1% Triton X-100, a nonionic surfactant. The presence of the detergent had no effect on either bending or looping by Fis (data not shown), indicating that hydrophobic interactions are not playing a major role in these activities and that nonspecific protein aggregation is not a likely cause of the DNA collapse.
Effects of Fis mutations on sequence-independent DNA binding, compaction, and looping-condensation
To identify which residues on Fis play important roles in long range DNA bending and looping, a series of mutant derivatives of Fis were analyzed for their sequence-independent DNA binding activities and their effect on force responses on single λ-DNAs. We also considered whether altered tertiary or quaternary forms of Fis were functioning to mediate DNA looping. All single-DNA experiments reported in this section used 5 μM Fis mutant in the 100 mM KGlu buffer, conditions under which wild-type Fis generates a robust looping-condensation reaction at a threshold force of 0.3 pN (Fig. 3C). The locations of residues studied are illustrated in Fig. 7A. Table 3 summarizes results of single-DNA bending/looping experiments as well as both their specific (binding to the λ F site) and nonspecific binding properties by gel-based assays.
Figure 7. Behavior of single-site mutants of Fis.

A. Surface representation of the Fis dimer in two orientations showing the locations of mutated residues studied. The two subunits of the dimer are in light and dark grey unless otherwise colored. Individual amino acid residues analyzed in this study are colored with respect to their looping and compaction activities: similar to wild-type (blue), looping defective (red), and DNA stiffening (green). Proteins containing substitutions at residues with asterisks also exhibit reduced DNA compaction.
B. Deletion of N-terminal arm residues 2 to 26 does not affect looping-compaction. Naked λ-DNA (open circles) was incubated with 5 μM Fis mutant Δ(2-26) (filled circles). Although the DNA extensions in the high force range approach the naked DNA in this experiment, other experiments with this mutant exhibit a high force compaction shift similar to the wild-type.
C. Fis residue Arg 71 is plays an important role in looping-collapse. Naked λ-DNA (open circles) was incubated with Fis mutant R71A (filled squares) with the result that high-force compaction was observed, but no low-force collapse occurred. In a separate experiment with R71K (open squares), high-force compaction and low-force looping-collapse were nearly indistinguishable from wild-type. In a third separate experiment, R71Y (open diamonds) generated high-force compaction and promoted looping-collapse at a higher threshold force than observed with wild-type Fis.
D. Micromechanical properties of Fis mutants K36E, R76A, K90A, and G72D. Complexes on λ-DNA were formed with 5 μM of each mutant. The force-shift by Fis K36E (filled triangles) is nearly indistinguishable from the naked DNA (open circles), and no looping-condensation occurred. Mutants R76A (open triangles) and K90A (filled squares) exhibited no looping-condensation, but did generate a compaction force-shift. Fis G72D (filled diamonds) exhibits both bending-compaction and looping-condensation.
E. Fis residue Asn 73 plays a key role in DNA bending-compaction. Naked λ-DNA (open circles) was incubated with 5 μM Fis mutant N73S; the resulting N73S-DNA complex (filled circles) was “anti-compacted” at high forces (shift to right), indicating that N73S stiffens DNA; looping-collapse at low forces occurred. In a separate experiment with 5 μM N73A (filled squares) a similar anti-compaction effect was observed, along with an incomplete looping-condensation reaction at very low force (below 0.1 pN).
F. Crosslinking of Fis dimers does not affect looping-condensation activity. Naked DNA (open circles) was incubated in separate experiments with 5 μM V58C in the presence of diamide to maintain disulfide crosslinks or in the presence of DTT (unlinked). In both cases, looping-condensation occurred.
Table 3.
Sequence-independent DNA binding properties of Fis mutants
| Mutant | Kd (sp)1 | Kd (ns)2 | High force Z shift3 | Loop stabilization (force threshold)4 | Remarks |
|---|---|---|---|---|---|
| WT | 3.0 | 0.75 | compacts | + (0.3 pN) | wild-type |
| S18CS-S | 3.5 | 0.75 | compacts | ++ (0.6 pN) | disulfide-linked β-arms |
| Δ(2–26) | 5.0 | 5.0 | compacts | + (0.4 pN) | β-arm deletion; DNA inversion |
| S30CS-S | 3.5 | 2.5 | compacts | + (0.4 pN) | disulfide-linked dimmer |
| K36E | 3.0 | 0.75 | none | - | faster LMC migration |
| Q45C | 3.5 | 0.75 | compacts | - | (+DTT) |
| N48C | 3.0 | 1.0 | compacts | + (0.3 pN) | (+DTT) |
| V58CS-S | 4.0 | 12 | compacts | ++ (0.9 pN) | disulfide-linked dimmer |
| V58C | 3.0 | 11 | compacts | ++ (0.7 pN) | unlinked dimer (+DTT) |
| Q68A | 4.0 | 2.5 | none | - | transcription faster LMC migration |
| R71A | 4.5 | 2.0 | compacts | - | transcription reduced DNA bending |
| R71L | 4.5 | 5.0 | compacts | - | transcription reduced DNA bending |
| R71Y | 4.5 | 3.0 | compacts | ++ (0.8 pN) | reduced DNA bending |
| R71K | 5.0 | 0.75 | compacts | + (0.3 pN) | transcription |
| G72A | 5.5 | 7.0 | compacts | + (0.3 pN) | transcription |
| G72D | 5.5 | 16 us | compacts | + (0.5 pN) | transcription |
| N73A | 85 | >150 us | stiffens | + (weak, 0.1 pN) | reduced DNA bending |
| N73S | 7.0 | 14 | stiffens | + (0.2 pN) | |
| T75A | 7.0 | 8.0 | compacts | ++ (0.8 pN) | transcription, λ excision |
| R76A | 4.0 | 8.0 us | compacts | - | |
| R85C | >150 | 100 us | compacts(weak) | - | (+DTT) DNA binding helix faster LMC migration |
| R89C | 6.0 | 40 us | none | - | (+DTT) DNA binding helix |
| K90A | 4.0 | 100 us | compacts | - | DNA binding helix |
| N98C | 2.0 | 0.75 | compacts | - | (+DTT) |
In general, we find that a subset of changes in surface-exposed residues located both within and outside the DNA binding region abolish detectable looping-condensation and alter the elastic properties of the Fis-DNA filament. Changes within the DNA binding region tend to compromise nonspecific binding activities to a greater extent than specific binding. Each of the mutants tested, including those that were severely impaired for formation of discrete nonspecific DNA complexes, form LMCs on gels; in some cases, the electrophoretic migrations of the LMCs are qualitatively different, implying differences in the Fis-DNA stoichiometry or structure of the filaments. Moreover, there is no direct correspondence between the robustness of nonspecific DNA binding (at low Fis concentrations) and looping (at high Fis concentrations). Taken together, these findings point to the importance of protein-protein interactions involving residues distributed on the Fis surface for the cooperative formation of the higher-order Fis-DNA filaments and for looping-condensation activity.
Fis N-terminal ‘arms’ do not play a role in looping
A prominent feature of the Fis protein structure is the N-terminal mobile β-hairpin arms, which are reminiscent of the β-arms of HU and IHF that interact with DNA 24; 37. It seemed possible that the Fis arms could function to mediate DNA looping by either directly interacting with a second segment of DNA or through β-strand interactions with a second Fis dimer. Fis S18C contains the N-terminal arms crosslinked together by a disulfide bridge under mild oxidizing conditions. Fis Δ(2–26) has the entire β-hairpin arm region (residues 2 through 26) deleted. Both these mutants induce DNA bending-compaction and looping-collapse similar to wild-type (Table 3; Fig. 7B, filled circles). These results rule out models where the N-terminal β-arms play a role in DNA looping.
Substitutions of Arg71 eliminate or enhance DNA looping without affecting high-force DNA compaction
Arg 71 (R71) is located shortly before the HTH DNA binding motif on the ‘sides’ of the Fis dimer (Fig. 7A). Previous studies have shown that R71 is a critical determinant for wrapping flanking DNA within specific Fis-DNA complexes, presumably because of formation of a salt link with the DNA backbone 20. We therefore assayed several chemically different substitutions of R71 and found that these had profound and variable effects on looping.
Alanine and leucine substitutions at residue 71 have been previously shown to have only a small effect on equilibrium binding to specific sites but largely eliminate the wrapping of flanking DNA (Table 3) 20; 38. Formation of discrete DNA complexes by Fis mutants R71A and R71L is moderately impaired, but both form LMCs on native gels (e.g., Fig. 8B). R71A (Fig. 7C, filled squares) or R71L (not shown) induced high force DNA compaction in a manner that was nearly indistinguishable from wild-type. However, no DNA collapse was observed with R71A. Furthermore, no hysteresis (lack of reversible extension when force was cycled) was observed when the R71A complexes were re-extended after incubation at 0.05 pN. With R71L, there was a very slow collapse observed at forces below 0.05 pN in one experiment, but in other experiments no collapse was observed. Therefore, alanine or leucine substitutions at Arg 71 are able to assemble rigidly bent DNA filaments, but these mutant-DNA complexes are nearly or completely incapable of looping-condensation.
Figure 8.

Gel mobility shift assays evaluating nonspecific binding properties of selected Fis mutants. A. Fis mutant R71K behaves indistinguishably from wild-type Fis. B. R71A, which fails to loop, weakly forms discrete complexes at high protein concentration. C. K90A, which fails to loop, is very defective in nonspecific binding. D. N73S, which forms stiffer complexes that promote weak looping, poorly forms discrete complexes at high protein concentrations. E. K36E, which does not change the elastic properties of DNA at 5 μM, forms nearly a full complement of discrete complexes, but its LMC migrates faster than the wild-type LMC. F. G72D, which exhibits normal compaction and looping at 5 μM, is very defective for in nonspecific binding. For each mutant, Fis was added at 2-fold increasing concentrations beginning at 0.125 nM (lane 1) to 256 nM (lane 12). The DNA probe for each experiment was the 149 bp fragment from the yeast actin gene.
Fis R71Y also reduces bending of DNA at specific Fis sites 38. DNA filaments generated with this mutant also exhibit a compaction shift at high forces similar to wild-type (Fig. 7C, diamonds). At low forces we find the surprising result that Fis R71Y exhibits a more robust looping activity than wild-type Fis; the threshold force for DNA collapse (0.9 pN) is over twice that for wild-type.
We also tested Fis R71K, which maintains the long basic character of the side chain and has site-specific and sequence-independent DNA binding properties that are indistinguishable from wild-type (see Fig. 8A) (Table 3) 20; 38. Fis R71K forms complexes on λ DNA whose bending and looping properties are identical to wild-type (Fig. 7C, open squares).
Mutations in the DNA binding domain
A number of mutations within the HTH DNA binding region had an appreciable effect on nonspecific DNA binding and elastic properties even though all formed an LMC. In the ‘positioning’ helix C, alanine substitutions of Thr 75 and Arg 76 have little to no effect on specific binding but reduce nonspecific binding about 10-fold, with R76A impaired in formation of multiple discrete complexes on gels (Table 3). T75A exhibits robust DNA looping-collapse activity (not shown), whereas R76A has no detectable looping-collapsing activity (Fig. 7D, open triangles). Arg 85 and Arg 89 are two residues located within the recognition helix D, that are critical for binding specific Fis sites, although R89C remains capable of binding to the strong F site on phage λ 39. As expected, R85C and R89C display extremely poor nonspecific binding activity but are still capable of forming an LMC at Fis concentrations around 300 nM. Neither of these mutants are able to promote looping-condensation but both weakly compact DNA, which presumably reflects their ability to form an LMC. Lys 90 is also located in the recognition helix and displays very poor nonspecific binding (Fig. 8C), even though binding to specific sites is near wild-type (Table 3). DNA filaments containing K90A show normal compaction but no looping-condensation (Fig. 7D, filled squares). Asn 98 is the C-terminal residue on Fis that extends 4 residues after the recognition helix. Nonspecific binding and DNA compaction by N98C is indistinguishable from wild-type yet this mutant is also unable to promote looping-condensation.
Asn73 substitutions form stiff complexes with DNA
Asn 73 (N73) is located just prior to the HTH on Fis and is believed to form a hydrogen bond with the DNA backbone that is important for Fis binding and bending 23; 27. A Fis mutant with this residue changed to a serine (N73S) binds specific sites with an equilibrium Kd similar to wild-type, although these complexes are much less stable to challenge with competitor DNA 20. Nonspecific DNA binding by Fis N73S is reduced by about 10-fold (Fig. 8D) (Table 3). In single-DNA experiments, N73S led to larger-than-naked-DNA extensions above 0.2 pN (Fig. 7E, filled circles). This distinct “anti-compaction” effect is characteristic of an increase in persistence length, i.e., formation of an unbent complex which is more stiff than naked DNA 34; a similar effect has been observed to occur in experiments with HU at few-μM concentrations 8; 9; 33; 40. At high forces (10 pN) the N73S complex extends to only slightly more than the naked DNA length; allowing us to ensure that the anti-compaction effect is not a result of a large increase in DNA length, but instead is due to a change in persistence length. Fitting of the N73S data to the semiflexible polymer model 41 indicates a net persistence length of 170±40 nm, a little more than three times that of naked DNA.
N73S-DNA collapsed between 0.1 and 0.2 pN (Fig. 7E, closed circles below 0.2 pN), indicating that this mutant is able to promote DNA looping. The low force required for looping-condensation (nearly three-fold smaller than the 0.3 pN threshold for wild-type protein) is consistent with the inverse proportional dependence of looping-condensation on persistence length expected theoretically 35. Given this, the reduction of threshold force arises simply from the reduced probability of thermal bending fluctuations in the case of a stiffer polymer, and does not necessarily indicate weakening of the chemical interactions responsible for stabilizing loops.
An alanine substitution at this position (N73A) decreases specific binding 10-fold and nonspecific binding >100-fold (Table 3). As with other severe DNA binding mutants, an LMC still formed in the presence of high Fis concentration (300 nM), even though only 1 – 2 N73A dimer-DNA complexes were resolved on polyacrylamide gels. N73A also stiffens DNA at high forces, but to a slightly lower degree than N73S, and near 0.1 pN we observed the onset of hysteresis, indicative of a limited degree of looping-condensation. We conclude that Fis-DNA filaments assembled by Asn73 mutants generate extended and stiffer DNA molecules relative to naked DNA, but that these filaments are still able to support some looping.
Mutations outside of the DNA binding domain
We also investigated effects of mutations at several residues that are well outside the Fis DNA binding region. K36E, Q45C, N48C, and Q68A have little to no difference from wild-type on forming nonspecific DNA complexes (Table 3). However, K36E, Q45C, and Q68A fail to promote looping-condensation (Fig. 7D and data not shown). Complexes assembled with 5 μM K36E (Fig. 7D) and Q68A (not shown) exhibit force-extension curves that are nearly indistinguishable from naked DNA, implying that the DNA filaments are stiffer than those containing wild-type Fis. The electrophoretic migrations of the LMCs formed by K36E and Q68A are reproducibly faster than the wild-type LMC (Fig. 8E), consistent with a different structure or Fis content of the filaments. Fis N48C behaves like the wild-type protein in all respects tested.
Mutant Fis proteins affecting tertiary and quaternary structure
We considered the possibility that looping activity of Fis might be mediated by dimers bound to DNA in an alternate conformation. Previous studies have shown that Fis protomers readily exchange between dimers in solution and that the folding of the Fis dimer occurs in at least two temporally distinct events 25 42; 43. Conceivably, an occasional Fis dimer might be bound in a filament such that one of the DNA binding motifs was associated with DNA and the other HTH motif was exposed, enabling it to interact with a second DNA segment. Alternatively, perhaps Fis protomers could occasionally bind within a filament as monomers, which would expose a dimerization interface for interaction with a second monomer.
To test such models, we prepared dimers that contained disulfide links between the subunits within the core dimerization region. Oxidized Fis S30C contains a disulfide bond linking the two A helices, and oxidized V58C has the center of the B helices crosslinked to each other. V58C exhibited a high-force shift indicative of DNA bending similar to wild-type and a low-force collapse indicative of DNA looping-condensation at a 0.9 pN threshold force (Fig. 7F, open triangles), above the force threshold for wild-type protein. Separate experiments with non-crosslinked V58C led to a similar result with a slightly reduced collapse threshold force (Fig. 7F, open squares). Looping-condensation by 5 μM disulfide-linked S30C was nearly indistinguishable from wild-type. We conclude that crosslinking of the two subunits in the homodimer does not interfere with the looping function of Fis.
Lastly, we tested substitutions at Gly 72 which is located at the three amino acid residue β-turn between helix B and the C+D HTH DNA binding motif. The phi and psi peptide bond angles for Gly 72 correspond to values in native proteins where only glycine is normally represented. Other amino acids at this position may be expected to partially destabilize the positions of the HTH motifs in the dimer, and indeed, Fis mutants G72A and G72D decrease the Tm by 8.7° and 15.4°, respectively 44. However, the crystal structures of these two mutants are indistinguishable from wild-type within their DNA binding regions, and they bind normally to specific DNA sites at 23° (Table 3). Nonspecific binding of G72A is reduced about 14-fold, and G72D only weakly forms discrete complexes on polyacrylamide gels (Fig. 8F). Nevertheless, both proteins form the LMC, and single-DNA experiments generate force-extension curves that are similar to wild-type Fis (Fig. 7D, filled diamonds and not shown). We conclude that mutations that destabilize tertiary interactions that could alter the spacing of the two HTH motifs in the Fis dimer do not significantly alter the formation of bending-compaction or looping-collapsing Fis-DNA filaments.
DISCUSSION
We have shown that Fis is able to bind nonspecifically to DNA so as to carry out two distinct architectural functions on chromosome-length DNA molecules. First, single-DNA experiments verify that Fis introduces rigid bends into DNA, leading to chromosome compaction even at relatively high forces. More strikingly, we find that Fis can trap DNA loops, resulting in dramatic condensation of single chromosomes against an applied force. Bulk solution experiments show that Fis can coat DNA to form a nucleoprotein filament. Increasing concentrations of Fis result in higher-order filaments that correlate with the looping activity revealed by single-DNA assays. Looping, and high force DNA compaction by Fis are independent functions that can be further distinguished by their sensitivities to salt and competitor DNA and by the effects of single residue substitutions. As elaborated below, these multiple modes of Fis-DNA binding may have important consequences for gene regulation and chromosome packaging within the bacterial nucleoid.
Nonspecific DNA binding at low Fis concentrations: formation of coated filaments inducing maximal DNA compaction
Native gel electrophoresis assays demonstrate that Fis binds DNA in a largely sequence-neutral manner beginning at sub-nM concentrations (Fig. 9A). We imagine that individual Fis dimers dynamically distribute over the DNA, sampling the many potential binding sites. DNA segments that more optimally conform to the Fis binding surface will be occupied for a greater time than other sites. Binding to high affinity sites can occur with an apparent Kd as low as 10 pM (measured in the absence of competitor DNA). As the Fis concentration is increased above 10-20 nM, a “coated filament” assembles, containing Fis dimers bound at about 21 bp intervals (Fig. 9B). Formation of an organized Fis-DNA filament is supported by the discrete ladder obtained from the Fis-nuclease assays, where the 1,10-phenathroline-copper cleavage moiety is linked to the end of the HTH motif. On average, the cleavage sites are spaced 20 bp apart, consistent with linear arrays of Fis dimers bound along the DNA. The positions of the Fis dimers within a given DNA segment is presumably phased by the presence of preferential Fis binding site(s) within the region, as suggested by the pattern obtained on the lacZ fragment (Fig. 2).
Figure 9. Sketches of Fis binding DNA in different concentration regimes.

A. Disperse binding (≤10 nM); bends generated by bound Fis dimers (filled circles) are well separated from one another.
B. Coated filament (~20 nM): Fis dimers bound adjacent to one another, roughly 21 bp apart, such that the bends introduced into the DNA are largely in phase (see panel F).
C. The “LMC” (≥75 nM): Fis dimers cooperatively assemble onto the Fis-coated complex to form an LMC beginning with a density of about 2 dimers per 21 bp; at higher Fis concentrations increasing numbers of Fis dimers are associated with the LMC.
D. Fis-looped DNA (≥1 μM): Fis dimers bound in the LMC are able to stabilize DNA crossings.
E. Sketch of Fis dimer clusters organizing a supercoiled E. coli chromosome domain. Localized regions of high density Fis binding that are postulated to occur at clusters of high affinity Fis binding sites within the chromosome. Two remote clusters associate to stabilize a loop containing the intervening DNA. Fis and other DNA binding/bending proteins (grey circles) bound within the intervening DNA will further compact the DNA.
F. Structural model of 3 tandem Fis dimers within a coated complex. Each Fis dimer is rendered as yellow and blue subunits and are positioned to account for the DNA cleavages (green nucleotides) generated by Fis98-OP chimeras. Where visible, acetamido-1,10-phenanthroline are red sticks with copper ions as orange spheres.
The maximum shift of force-extension curves performed on single DNA molecules occurs around 20 nM Fis, which coincides with the concentration just prior to or at the point where coated filaments form in the bulk experiments. In this concentration regime, the elastic behavior of the Fis-DNA complexes is fully reversible, as revealed by identical force-extension curves as the force is cycled up and down. The lack of hysteresis indicates that there is no tendency for DNA molecules coated with Fis in this regime to stick to themselves. Binding of additional Fis proteins results in a modest reduction of chromosome compaction as evidenced by a shift in the force extension curves slightly towards naked DNA. The elastic properties of the DNA molecules bound by Fis over all concentration ranges tested remained constant after washing with protein-free buffer, indicating that Fis remains associated with the DNA complex. However, high amounts of DNA in trans can promote dissociation, presumably by a direct transfer mechanism.
The low-concentration behavior of Fis-DNA complexes is reminiscent of results obtained for other nonspecific DNA binding and bending proteins, e.g., HU, IHF, HMGB1/2 and NHP6A 8; 9; 11; 32; 33 and are consistent with theoretical expectations 34. The complexes formed with the HMGB family of proteins or moderate concentrations of HU/IHF can be stretched to their full contour length with forces above 5 pN, implying that the DNA is more flexibly associated with these proteins. In contrast, Fis-DNA complexes are appreciably shorter than the naked DNA, even at forces above 10 pN, indicating Fis form stiff bends along DNA.
The micromechanical properties and binding stoichiometries of the Fis-DNA filaments are fully consistent with our current understanding of how Fis binds DNA. The close spacing between DNA recognition helices within the Fis dimer, in relation to adjacent DNA major grooves on B DNA, demands that the DNA must be bent in order for a complex to form 22; 23. Hence, DNA within the Fis interface would be expected to behave as a rigidly bent segment. The magnitude of DNA bending has been estimated from gel electrophoresis experiments, Fis-OP cleavage data, and molecular modeling to be a minimum of about 50°, with additional dynamic bending generating up to 90° of DNA curvature. The length of DNA required for contacts between the two HTH motifs in the dimer is modeled to extend over 21 bp, as highlighted in Fig. 1, which fits with the 21 bp minimal binding site length experimentally determined from oligonucleotide binding studies.
The distribution of orthophenathroline-copper (OP-Cu) induced cleavages at each scission site within the Fis-DNA arrays is no broader than that observed for specific Fis-OP complexes 27. The tight cleavage pattern suggests that only one OP-Cu moiety from adjacent dimers can insert into the minor groove to cleave DNA; the OP-Cu linked to residue 98 of the proximal subunit of other dimer would be sterically excluded from the same minor groove. Molecular modeling of Fis-DNA arrays based on a 21 bp binding site and Fis-OP cleavage data imply that the Fis dimers must be immediately adjacent to each other on the same side of the DNA helix (Fig. 9F). The ~50° bends introduced by adjacent Fis dimers are therefore predicted to be in phase to give an overall solenoidal structure to the DNA molecule (sketched in Fig. 9B). In the 21 bp spacing model, contacts with DNA flanking the minimal binding site by residues such as Arg 71 and Arg 76, are sterically prevented. Although an arginine at these positions has been shown to enhance DNA bending within high affinity complexes, there is not a significant change on DNA compaction by filaments of Fis mutants R71A and R76A in single-DNA assays. On the other hand, Fis mutants containing substitutions at Asn 73 had the unique property of forming complexes that behaved as if they were stiffer than naked DNA. Models of Fis-DNA complexes show that the Asn 73 side chain from each subunit engages in the most distal contacts within the bent 21 bp minimal binding site (Fig. 7A), and as would therefore be expected, complexes of Fis mutant N73A bound to a specific Fis binding site migrate as if the DNA is less curved as compared to wild-type Fis-DNA complexes 20.
Higher-order Fis-DNA filaments promote DNA looping
As the Fis concentration is raised above 75 nM, the electrophoretic migration of the DNA complexes abruptly shift to a distinct low mobility form in a manner reflecting strong cooperativity (Fig. 9C). The low mobility complex (LMC) initially contains about one Fis dimer every 10 bp, or double the density of the coated filament, but as the Fis concentration is raised, increasingly higher-density complexes are generated. Formation of the higher-order LMCs coincides with the ability of Fis-DNA complexes to capture and stabilize DNA loops (Fig. 9D). Strong evidence for loops is provided by the stepwise condensation of single-DNA molecules bound by DNA when the applied force drops below a well-defined threshold (Fig. 4A). This threshold force increases with Fis concentration, while the size of loops captured during condensation goes down with Fis concentration. Both these effects can be explained by supposing that the density of looping-competent sites along the complex increases with Fis concentration 31; 45. DNA looping by Fis has yet to be detected in bulk phase experiments, although enhanced branching of plasmid DNA by Fis has been observed by electron microscopy 15.
Complete condensation of 50 kb λ chromosomes occurs within seconds to a few minutes, depending upon the Fis concentration and the level of the applied force in relation to the threshold. Further evidence for DNA looping driving the condensation is provided by the subsequent stepwise decondensation of the complexes driven by large force (Fig. 4B, D). The fact that entire DNA molecules can be condensed indicates that looping-competent sites are distributed along the entire length of the DNA molecules that we have used. The opening distributions (Fig. 4B,D) indicate that the final condensed DNA forms loops that average 250 nm (750 bp) in DNA length. The peak of the opening jump distribution (most probably loop size) is roughly 200 nm (600 bp), appreciably larger than the < 3 persistence length size expected theoretically for closure against applied forces of 0.2 to 0.5 pN 35. This difference is even more striking when the reduced apparent persistence length of the Fis-DNA complex of 20±6 nm 31 is taken into account. This suggests that not every location along the Fis-DNA complex is competent for loop capture.
The magnitude of the threshold force is always in the sub-piconewton range, indicative of the role of thermal motion of the DNA in creating the loops since high stretching forces on DNA create a large barrier for loop formation. The free energy associated with this barrier can be estimated to be the product of the applied force and the length of DNA involved in a loop 46. To illustrate, for a 150 nm (450 bp) loop under a 1 pN force, this product is roughly 150 pN nm, or in more familiar units, about 25 kcal/mol; this barrier is too large to be crossed via thermal motion on laboratory experiment time scales (detailed calculations including the energy and entropy of DNA loop formation agree with this simple estimate, see 35).
The structural nature of the LMC remains to be elucidated. The additional Fis molecules may be bound, at least in part, to the exposed DNA surface opposite the resident Fis dimers within the coated molecule, although the direction of DNA curvature would be predicted to discourage such binding. Alternatively, the additional Fis molecules may associate with the filament primarily by protein-protein interactions. Support for the latter model comes from the observation that Fis mutants which are strongly defective for DNA binding remain capable of assembling LMCs at Fis concentrations similar to or only slightly higher than wild-type. In these cases, strong cooperative interactions between Fis proteins must compensate for the weak direct DNA interactions. Recruitment of Fis molecules into the LMC appears to be enhanced in buffer containing KGlu instead of NaCl as evidenced by a more prominent decrease in the mobility of the LMC with increasing Fis concentrations (data not shown). The looping activity observed in the single-DNA experiments is also more robust in buffer containing KGlu (data not shown), providing further support for the higher-order LMC complexes being the active species for looping. Looping activity, but not compaction due to Fis-mediated DNA bending, appears relatively sensitive to salt concentrations since complexes assembled in buffer containing 0.3 M KGlu do not undergo collapse even at forces well below the threshold for complexes assembled at lower salt concentrations. By contrast, chromosomal compaction by Fis-mediated bending is unaltered at KGlu concentrations up to at least 0.5 M.
We presume that loop capture is mediated by interactions between Fis proteins bound within the LMC, but interactions directly involving DNA binding cannot be ruled out. Mutants such as K36E, Q45C, and Q68A that are defective for looping-collapse, but whose single residue changes are located well outside the DNA binding region, provide strong evidence for the importance of Fis-Fis interactions. Remarkably, substitutions of a single residue, Arg 71, can abolish (e.g., alanine), enhance (e.g., tyrosine) or cause no measurable change (e.g., lysine) on looping activity. As noted above, Arg 71 can contact DNA when bound in an isolated complex, but it is not expected to be capable of binding DNA in coated filaments or LMCs. Interestingly, this residue has also been found to be critical for interactions with the C-terminal domain of the E. coli RNA polymerase α subunit (αCTD). To emphasize the selective nature of these mutations, we point out that mutants containing changes at the adjacent residue Gly 72, which also is critical for αCTD interactions, exhibited looping-collapsing activity that was essentially identical to wild-type Fis.
Biological implications
Fis has been extensively studied as a specific regulator of transcription, recombination and replication reactions 5. In most of these cases, one or a small number of Fis dimers bind in a sequence-specific manner to a DNA segment(s) within a regulatory (e.g., promoter) region to control the reaction. In vitro experiments demonstrate that optimal Fis to DNA ratios for activation of Hin-catalyzed DNA inversion, phage λ excision, or transcription through αCTD binding, are all well below ratios required to form Fis-DNA filaments. Indeed, higher amounts of Fis often lead to inhibition of activity. A mechanism for Fis-mediated activation of transcription has also been proposed to involve the formation of DNA microloops within the promoter region 47. Our results provide a potential mechanism for the creation of these loops, but as discussed below, the formation of high density Fis-DNA complexes required for looping may compete with RNA polymerase binding and thus more likely lead to localized silencing of gene expression.
Although cellular Fis concentrations under moderate to rapid growth conditions are well above those required to coat DNA in vitro, the DNA content and hence the number of potential binding positions in the bacterial nucleoid greatly exceeds the number of Fis molecules, assuming a 21–27 bp Fis binding site and 2–3 genome equivalents per cell. Thus, coating of the entire chromosome by Fis in vivo cannot occur. However, a number examples of intergenic regions harboring multiple high affinity Fis binding sites dispersed over 100–200 bp have been reported 5; 47; 48. Clusters of specific Fis sites may function to nucleate high density Fis arrays in vivo. DNase I footprinting and Fis-OP nuclease cleavage assays of Fis complexes assembled in vitro within DNA segments containing multiple Fis sites often exhibit patterns that reflect binding not only over the specific Fis sites but throughout the region, indicative of higher order coated complexes. In most cases, these regions would be expected to become transcriptionally “silenced”, as experimentally observed for the dusB-fis, aldB, and xylAF operons, in a manner reminiscent of silencing by H-NS filaments 5; 6.
The robust condensation activity that we observe for Fis-coated single-λ chromosomes in vitro, leads us to further postulate that looping between localized regions of high density Fis binding may contribute to the looped-domain organization of the E. coli chromosome (Fig. 9E). Under rapid growth conditions, where Fis is among the most abundant nucleoid proteins in the cell, domain barrier elements are spaced roughly 10 kb apart 3; 4. Genetic assays indicate that this distance increases considerably under growth conditions where Fis levels are low. Fis may thus provide a mechanism for controlling chromosome structure as a function of cell physiology. Recent in vivo support for Fis functioning as a domain barrier element comes from the observation that a number of supercoiling sensitive genes that are distributed around the chromosome exhibit altered expression patterns in a fis mutant 50.
METHODS
Fis proteins
Wild-type and mutant Fis proteins were overexpressed using the T7 promoter and purified from RJ3387 (BL21 (DE3) fis::kan-767 endA8::tet) using SP-sepharose and FPLC monoS chromatography essentially as described in 20. FisHMK contained the sequence coding for GARRASVA, representing the heart muscle kinase substrate, added to the C-terminal end of Fis. All other mutants, including their binding properties to specific sites, have been described previously 20; 24; 25; 29; 38; 51. Long term storage at −20°C of Fis S18C and Fis S30C resulted in the preparations used in this report being >95% disulfide-linked dimers. In order to achieve a high percentage of disulfide-linked dimers for V58C, the protein was first incubated for 5 min at 0°C with 0.1 mM diamide (Sigma) and then add to the binding reactions, which also included 0.1 mM diamide. Under these conditions, >85% of the Fis V58C is in a disulfide-linked dimer form, as measured by SDS-PAGE.
Gel mobility shift assays
Standard Fis-DNA binding reactions contained 20 mM HEPES (pH 7.5), 100 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 5% glycerol, 500 μg/ml BSA, and 5 fmol 32P-labeled DNA. Fifty μg/ml sonicated herring sperm DNA (Promega) was included when binding to specific sites was measured. DNA probes were typically prepared by PCR using 5′-32P-labeled primers. Fis was diluted into the same buffer but with 20% glycerol. Fis was incubated with the DNA for 10 min at 23°C prior to loading on a native polyacrylamide gel. Gels were 5% polyacrylamide (19:1 acrylamide:bisacrylamide) in 0.5x TBE buffer and were electrophoresed at 5V/cm, dried under vacuum and subjected to phosphorimaging. Quantitations were performed using ImageQuant (Amersham).
For Fis-DNA stoichiometry determinations 10 μg FisHMK, containing a kinase tag at its C-terminus, was labeled with γ[32P]-ATP using heart muscle kinase as described 52. The specific activity of the labeled Fis was determined after SDS-PAGE by relating the 32P Cerenkov counts from Fis bands with the amount of Fis determined by co-electrophoresing lysozyme standards and coomassie blue staining. Fis-DNA binding reactions contained from 0.5 nM to 1.2 μM Fis and 25 fmol of DNA fragments, which were generated by PCR using 5′-fluorescein labeled primers (Sigma-Genosys). The reactions were electrophoresed in a 5% polyacrylamide gel (19:1 acrylamide:bisacrylamide) together with different amounts of unbound fluorescein-labeled DNA probe. After electrophoresis, the gel was fluoroimaged with a Typhoon (488 nm laser, Amersham), and the amount of fluorescence in each band was determined by comparison to a standard curve of the unbound DNA to obtain the number of moles of DNA in each complex band. The gel was also subjected to phosphorimaging (Typhoon), and the Fis-DNA complex bands were excised to determine the amount of Fis by Cerenkov counts. To account for unbound Fis that overlapped the low mobility complex near the top of the gel, samples containing 32P-labeled Fis without DNA were electrophoresed in parallel. The 32P counts from gel slices in the Fis-only lanes were subtracted from the counts obtained from the Fis-DNA complexes. The direct quantitation revealed that the individual Fis-DNA complexes formed from the lower amounts of Fis contained incremental numbers of Fis dimers to DNA, beginning with one Fis dimer in the first complex. Therefore, the ratio of phosphor to fluorescence counts was also used to determine the Fis-DNA stoichiometry within the complex bands.
The DNA fragments used for the binding studies are as follows: the phage λ attP fragment spans sequences from −220 to −121 with respect to the attP core and does not contain the F site; yeast actin is from the S. cerevisiae ACT1 gene, MET14a, b, c are 3 non-overlapping segments from the S. cerevisiae MET14 gene. The ec_lacZ_ 262 bp fragment containing the N-terminal coding region of E. coli lacZ was obtained from an _EcoR_1-_Nde_I digest of pUC18. The λ F-site probe was a 263 bp PCR fragment from −220 to +43 with respect to the λ attR core. The radiolabeled probes were obtained by PCR using primers labeled with γ-32P-ATP using polynucleotide kinase, or in the case of the eclacZ fragment, by end-filling with α-32P-ATP using Klenow.
DNA cleavage by Fis-phenanthroline copper chimeras
Fis-OP was prepared as follows. 150 μg Fis mutant Asn98Cys was reduced with 10 mM DTT for 15 min on ice followed by 1 min at 37°C and then subjected to batch chromatography with cellulose phosphate (Whatman P11) to remove the DTT. Fis was eluted with 100 μl buffer (10 mM Tris-HCl, pH 7.5, 1 M NaCl and 20% glycerol), followed by addition of 80 μl of the same buffer without NaCl plus 20 μl of 7 mM 5-iodoacetamido-1,10-phenanthroline (gift of the late David Sigman, UCLA) and incubation at 4°C overnight 27. The modified Fis was again subjected to cellulose phosphate chromatography to remove unreacted phenanthroline, and recovery was estimated by the Bradford reaction using BSA as a standard. Different concentrations of Fis were incubated with 32P-end labeled DNA fragments in a 20 μl reaction containing 20 mM Tris-HCl, pH 7.5, 80 mM NaCl, and 50 μM CuSO4. After 10 min at 23°C, mercaptopropionic acid was added to 3 mM, and the DNA cleavage reaction proceeded for an hour, followed by quenching with 1 mM neocuprine. Five μg sonicated salmon sperm carrier DNA were then added to the samples which were then precipitated with ethanol, and the resulting pellets resuspended in deionized formamide plus 1 mM NaOH. Electrophoresis was in an 8% polyacrylamide-urea sequencing gel.
Single-DNA micromanipulation
Single-DNA experiments were carried out with linearized DNAs of two sizes. In either case, the basic method used end-labeling of the DNAs. This allowed magnetic particles to be attached to one DNA end, and allowed the bead-DNA structures to then be tethered to a suitably prepared cover glass or other large object. The magnetic particles were then used as ‘handles’ to stretch the molecules. This general ‘magnetic tweezers’ approach has been reviewed in 36; 53; our approach is discussed in 8.
End-labeling of DNAs
The DNA used in most of the experiments of this paper was 48.5 kb λ-DNA (Promega, Madison WI). 12-base oligomers carrying single biotin or digoxygenin labels were ligated onto the ends as described previously 8; 54 to obtain linear DNA with a biotin at one end and a digoxygenin at the other. The biotin and digoxygenin labels allowed the DNAs to be bound at one end to 2.8-micron-diameter streptavidin-coated magnetic particles (M-280, Dynal), and at the other to a surface coated with antidigoxygenin (Roche). Since these molecules carry only one label at each end, they are effectively ‘nicked’ and cannot be supercoiled.
The other DNA used was a 10.7 kb plasmid pBS290-2 which was linearized using unique BamHI and HindIII restriction sites to obtain a 10.5 kb linear DNA fragment with defined 4-base 5′ overhang ends. These molecules were end-ligated to 1 kb PCR products made using a reaction including biotin- and digoxygenin-labeled dUTP (Roche) following the approach of 33; 53. PCR fragments were cut using restriction enzymes to be compatible with the linearized pBS290-2 ends. The resulting 12.5 kb molecules carry multiple biotins at one end and multiple digoxygenins at the other; either nicked (used in this study) or supercoilable tethered molecules can be obtained 53.
Vertical magnetic tweezer experiments
All experiments were carried out in flow cells assembled by hand, with an upper surface which was functionalized with anti-digoxygenin (Roche). Labeled DNAs were incubated with paramagnetic beads, and then the bead-DNA constructs were injected into the flow cell, and allowed to settle to the anti-dig-coated surface. After a 5-minute incubation, the flow cell was inverted and placed on the experiment. In experiments with the short 10.5 kb DNAs, the flow cell was pre-incubated with 0.5 mg/ml BSA solution to suppress nonspecific adsorption of the beads to the glass surface.
Experiments with the long λ-DNAs proceeded by first, a series of measurements of tether extensions and forces for naked DNA in buffer (generally the 100 mM KGlu buffer). Extensions were measured for λ-DNA using the piezoelectric objective positioner to focus on the bead 8. These measurements are essential since they provide a check that the tether has the mechanical properties established for single DNAs 36, and also provide the information necessary to convert the position of the magnets (Fig. 2) to force (see 8 for further discussion of this calibration procedure).
Following verification that we had an acceptable single-DNA tether, protein solution was introduced to the flow cell; for our 50 μl flow cell, 200 μl of solution was generally used. Then, either time series of extension measurements, or force-extension measurements were made. Further experiments (buffer wash, addition of competitor DNA, etc.) were then done as required.
Experiments with the shorter 10.5 kb linearized pBS290-2 plasmid were carried out slightly differently, using a variation on the methods introduced by 33; 53; 55. For that length of DNA (roughly 3 μM maximum extension), the tethered bead may be observed by positioning the microscope focal plane slightly below the maximum extension position. When the molecule is relaxed, the bead moves near the surface, and appears as an out-of-focus image. The image power spectrum at an empirically determined wave number changes monotonically with bead position, and provides a bead position measurement. By measuring the power spectrum component against known bead position (by moving the objective to known positions using the piezoelectric positioner) we obtain a calibration allowing dynamic measurement of the bead position. A similar measurement is done on a nearby surface-adsorbed bead, to correct for instrument drift. Software to carry out all this is implemented in Labview, and measures the bead positions at 10 to 12 measurements/sec. The single-measurement error is roughly 10 nm, estimated from the fluctuation in extension observed for beads adsorbed to the surface (data not shown). Measurements on thermally fluctuating beads attached via DNAs have a larger noise component (see Fig. 4A–D) of thermal origin. Experiments proceeded by first, the type of auto-focus calibration of 8 in protein-free buffer, followed by a calibration of the out-of-focus bead positions, and then finally roughly acquisition of extension time series.
Buffers
The basic buffer used in all single DNA molecule experiments was 20 mM HEPES (pH 7.6) containing potassium glutamate (KGlu) at 0.1 M unless otherwise specified. In experiments employing low Fis concentrations (< 1 μM), 5% glycerol plus 500 ng/μl BSA (Roche) was included to more closely match the gel-shift assay conditions. Fis binding in gel-shift experiments containing 0.1 M KGlu in the binding buffer was identical to the standard reactions containing 0.1 M NaCl, except that that the LMC tended to decrease in mobility with increasing Fis to a greater extent. We infer that formation of higher-order coated complexes may be enhanced in the presence of KGlu.
Acknowledgments
Work at UIC was supported by NSF grants DMR-0203963, MCB-0240998 and PHY-0445565. Work at UCLA was supported by NIH grant GM038509.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deng S, Stein RA, Higgins NP. Organization of supercoil domains and their reorganization by transcription. Mol Microbiol. 2005;57:1511–1521. doi: 10.1111/j.1365-2958.2005.04796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Higgins NP, Deng S, Pang Z, Stein RA, Champion K, Manna D. Domain behavior and supercoil dynamics in bacterial chromosomes. In: Higgins NP, editor. The Bacterial Chromosome. ASM Press; Washington, D.C.: 2005. pp. 133–153. [Google Scholar]
- 3.Postow L, Hardy CD, Arsuaga J, Cozzarelli NR. Topological domain structure of the Escherichia coli chromosome. Genes Dev. 2004;18:1766–1779. doi: 10.1101/gad.1207504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein RA, Deng S, Higgins NP. Measuring chromosome dynamics on different time scales using resolvases with varying half-lives. Mol Microbiol. 2005;56:1049–1061. doi: 10.1111/j.1365-2958.2005.04588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson RC, Johnson LM, Schmidt JW, Gardner JF. Major nucleoid proteins in the structure and function of the Escherichia coli chromosome. In: Higgins NP, editor. The Bacterial Chromosome. ASM Press; Washington, D.C.: 2005. pp. 65–132. [Google Scholar]
- 6.Dame RT. The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin. Mol Microbiol. 2005;56:858–870. doi: 10.1111/j.1365-2958.2005.04598.x. [DOI] [PubMed] [Google Scholar]
- 7.Travers A, Muskhelishvili G. Bacterial chromatin. Curr Opin Genet Dev. 2005;15:507–514. doi: 10.1016/j.gde.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Skoko D, Wong B, Johnson RC, Marko JF. Micromechanical analysis of the binding of DNA-bending proteins HMGB1, NHP6A, and HU reveals their ability to form highly stable DNA-protein complexes. Biochemistry. 2004;43:13867–13874. doi: 10.1021/bi048428o. [DOI] [PubMed] [Google Scholar]
- 9.van Noort J, Verbrugge S, Goosen N, Dekker C, Dame RT. Dual architectural roles of HU: Formation of flexible hinges and rigid filaments. Proc Natl Acad Sci USA. 2004;101:6969–6974. doi: 10.1073/pnas.0308230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sagi D, Friedman N, Vorgias C, Oppenheim AB, Stavans J. Modulation of DNA conformations through the formation of alternative high-order HU-DNA complexes. J Mol Biol. 2004;341:419–428. doi: 10.1016/j.jmb.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 11.Ali BM, Amit R, Braslavsky I, Oppenheim AB, Gileadi O, Stavans J. Compaction of single DNA molecules induced by binding of integration host factor (IHF) Proc Natl Acad Sci USA. 2001;98:10658–10663. doi: 10.1073/pnas.181029198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dame RT, Wyman C, Goosen N. H-NS mediated compaction of DNA visualised by atomic force microscopy. Nucleic Acids Res. 2000;28:3504–3510. doi: 10.1093/nar/28.18.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ball CA, Osuna R, Ferguson KC, Johnson RC. Dramatic changes in Fis levels upon nutrient upshift in Escherichia coli. J Bacteriol. 1992;174:8043–8056. doi: 10.1128/jb.174.24.8043-8056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talukder AA, Iwata A, Nishimura A, Ueda S, Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol. 1999;181:6361–6370. doi: 10.1128/jb.181.20.6361-6370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider R, Lurz R, Luder G, Tolksdorf C, Travers A, Muskhelishvili G. An architectural role of the Escherichia coli chromatin protein FIS in organising DNA. Nucleic Acids Res. 2001;29:5107–5114. doi: 10.1093/nar/29.24.5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koch C, Kahmann R. Purification and properties of the Escherichia coli host factor required for inversion of the G segment in bacteriophage Mu. J Biol Chem. 1986;261:15673–15678. [PubMed] [Google Scholar]
- 17.Johnson RC, Bruist MF, Simon MI. Host protein requirements for in vitro site-specific DNA inversion. Cell. 1986;46:531–539. doi: 10.1016/0092-8674(86)90878-0. [DOI] [PubMed] [Google Scholar]
- 18.Finkel SE, Johnson RC. The Fis protein: it’s not just for DNA inversion anymore. Mol Microbiol. 1992;6:3257–3265. doi: 10.1111/j.1365-2958.1992.tb02193.x. [DOI] [PubMed] [Google Scholar]
- 19.Bruist MF, Glasgow AC, Johnson RC, Simon MI. Fis binding to the recombinational enhancer of the Hin DNA inversion system. Genes Dev. 1987;1:762–772. doi: 10.1101/gad.1.8.762. [DOI] [PubMed] [Google Scholar]
- 20.Pan CQ, Finkel SE, Cramton SE, Feng JA, Sigman DS, Johnson RC. Variable structures of Fis-DNA complexes determined by flanking DNA-protein contacts. J Mol Biol. 1996;264:675–695. doi: 10.1006/jmbi.1996.0669. [DOI] [PubMed] [Google Scholar]
- 21.Hengen PN, Bartram SL, Stewart LE, Schneider TD. Information analysis of Fis binding sites. Nucleic Acids Res. 1997;25:4994–5002. doi: 10.1093/nar/25.24.4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kostrewa D, Granzin J, Koch C, Choe HW, Raghunathan S, Wolf W, Labahn J, Kahmann R, Saenger W. Three-dimensional structure of the E. coli DNA-binding protein FIS. Nature. 1991;349:178–180. doi: 10.1038/349178a0. [DOI] [PubMed] [Google Scholar]
- 23.Yuan HS, Finkel SE, Feng JA, Kaczor-Grzeskowiak M, Johnson RC, Dickerson RE. The molecular structure of wild-type and a mutant Fis protein: relationship between mutational changes and recombinational enhancer function or DNA binding. Proc Natl Acad Sci USA. 1991;88:9558–9562. doi: 10.1073/pnas.88.21.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safo MK, Yang WZ, Corselli L, Cramton SE, Yuan HS, Johnson RC. The transactivation region of the Fis protein that controls site-specific DNA inversion contains extended mobile beta-hairpin arms. EMBO J. 1997;16:6860–6873. doi: 10.1093/emboj/16.22.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merickel SK, Sanders ER, Vazquez-Ibar JL, Johnson RC. Subunit exchange and the role of dimer flexibility in DNA binding by the Fis protein. Biochemistry. 2002;41:5788–5798. doi: 10.1021/bi020019+. [DOI] [PubMed] [Google Scholar]
- 26.Thompson JF, Landy A. Empirical estimation of protein-induced DNA bending angles: applications to lambda site-specific recombination complexes. Nucleic Acids Res. 1988;16:9687–9705. doi: 10.1093/nar/16.20.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan CQ, Feng JA, Finkel SE, Landgraf R, Sigman D, Johnson RC. Structure of the Escherichia coli Fis-DNA complex probed by protein conjugated with 1,10-phenanthroline copper(I) complex. Proc Natl Acad Sci USA. 1994;91:1721–1725. doi: 10.1073/pnas.91.5.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perkins-Balding D, Dias DP, Glasgow AC. Location, degree, and direction of DNA bending associated with the Hin recombinational enhancer sequence and Fis-enhancer complex. J Bacteriol. 1997;179:4747–53. doi: 10.1128/jb.179.15.4747-4753.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osuna R, Lienau D, Hughes KT, Johnson RC. Sequence, regulation, and functions of Fis in Salmonella typhimurium. J Bacteriol. 1995;177:2021–2032. doi: 10.1128/jb.177.8.2021-2032.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Betermier M, Galas DJ, Chandler M. Interaction of Fis protein with DNA: bending and specificity of binding. Biochimie. 1994;76:958–967. doi: 10.1016/0300-9084(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 31.Skoko D, Yan J, Johnson RC, Marko JF. Low-Force DNA condensation and discontinuous high-force decondensation reveal a loop-stabilizing function of the protein Fis. Phys Rev Lett. 2005;95:208101. doi: 10.1103/PhysRevLett.95.208101. [DOI] [PubMed] [Google Scholar]
- 32.McCauley M, Hardwidge PR, Maher LJ, 3rd, Williams MC. Dual binding modes for an HMG domain from human HMGB2 on DNA. Biophys J. 2005;89:353–364. doi: 10.1529/biophysj.104.052068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnurr B, Vorgias C, Stavans J. Compaction and supercoiling of single, long DNA molecules by HU protein. Biophys Rev Lett. 2006;1:29–44. [Google Scholar]
- 34.Yan J, Marko JF. Effects of DNA-distorting proteins on DNA elastic response. Phys Rev E. 2003;68:011905. doi: 10.1103/PhysRevE.68.011905. [DOI] [PubMed] [Google Scholar]
- 35.Sankararaman S, Marko JF. Formation of loops in DNA under tension. Phys Rev E Stat Nonlin Soft Matter Phys. 2005;71:021911. doi: 10.1103/PhysRevE.71.021911. [DOI] [PubMed] [Google Scholar]
- 36.Bustamante C, Smith SB, Liphardt J, Smith D. Single-molecule studies of DNA mechanics. Curr Opin Struct Biol. 2000;10:279–285. doi: 10.1016/s0959-440x(00)00085-3. [DOI] [PubMed] [Google Scholar]
- 37.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 38.McLeod SM, Xu J, Cramton SE, Gaal T, Gourse RL, Johnson RC. Localization of amino acids required for Fis to function as a class II transcriptional activator at the RpoS-dependent proP P2 promoter. J Mol Biol. 1999;294:333–346. doi: 10.1006/jmbi.1999.3262. [DOI] [PubMed] [Google Scholar]
- 39.Osuna R, Finkel SE, Johnson RC. Identification of two functional regions in Fis: the N-terminus is required to promote Hin-mediated DNA inversion but not lambda excision. EMBO J. 1991;10:1593–1603. doi: 10.1002/j.1460-2075.1991.tb07680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dame RT, Goosen N. HU: promoting or counteracting DNA compaction? FEBS Lett. 2002;529:151–6. doi: 10.1016/s0014-5793(02)03363-x. [DOI] [PubMed] [Google Scholar]
- 41.Marko JF, Siggia ED. Stretching DNA. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 42.Topping TB, Hoch DA, Gloss LM. Folding mechanism of FIS, the intertwined, dimeric factor for inversion stimulation. J Mol Biol. 2004;335:1065–1081. doi: 10.1016/j.jmb.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 43.Hobart SA, Ilin S, Moriarty DF, Osuna R, Colon W. Equilibrium denaturation studies of the Escherichia coli factor for inversion stimulation: implications for in vivo function. Protein Sci. 2002;11:1671–1680. doi: 10.1110/ps.5050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng YS, Yang WZ, Johnson RC, Yuan HS. Structural analysis of the transcriptional activation on Fis: crystal structures of six Fis mutants with different activation properties. J Mol Biol. 2000;302:1139–51. doi: 10.1006/jmbi.2000.4123. [DOI] [PubMed] [Google Scholar]
- 45.Sankararaman S, Marko JF. Entropic compression of interacting DNA loops. Phys Rev Lett. 2005;95:078104. doi: 10.1103/PhysRevLett.95.078104. [DOI] [PubMed] [Google Scholar]
- 46.Marko JF, Siggia ED. Driving proteins off DNA using applied tension. Biophys J. 1997;73:2173–2178. doi: 10.1016/S0006-3495(97)78248-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Travers A, Muskhelishvili G. DNA microloops and microdomains: a general mechanism for transcription activation by torsional transmission. J Mol Biol. 1998;279:1027–1043. doi: 10.1006/jmbi.1998.1834. [DOI] [PubMed] [Google Scholar]
- 48.Ussery D, Larsen TS, Wilkes KT, Friis C, Worning P, Krogh A, Brunak S. Genome organisation and chromatin structure in Escherichia coli. Biochimie. 2001;83:201–212. doi: 10.1016/s0300-9084(00)01225-6. [DOI] [PubMed] [Google Scholar]
- 49.Robison K, McGuire AM, Church GM. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J Mol Biol. 1998;284:241–254. doi: 10.1006/jmbi.1998.2160. [DOI] [PubMed] [Google Scholar]
- 50.Hardy CD, Cozzarelli NR. A genetic selection for supercoiling mutants of Escherichia coli reveals proteins implicated in chromosome structure. Mol Microbiol. 2005;57:1636–1652. doi: 10.1111/j.1365-2958.2005.04799.x. [DOI] [PubMed] [Google Scholar]
- 51.Bokal AJ, Ross W, Gaal T, Johnson RC, Gourse RL. Molecular anatomy of a transcriptional activation patch: Fis-RNA polymerase interactions at the Escherichia coli rrnB P1 promoter. EMBO J. 1996;16:154–162. doi: 10.1093/emboj/16.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanders ES, Johnson RC. Stepwise dissection of the Hin-catalyzed recombination reaction from synapsis to resolution. J Mol Biol. 2004;340:753–766. doi: 10.1016/j.jmb.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 53.Strick TR, Allemand JF, Bensimon D, Croquette V. Behavior of supercoiled DNA. Biophys J. 1998;74:2016–2028. doi: 10.1016/S0006-3495(98)77908-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith SB, Finzi L, Bustamante C. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science. 1992;258:1122–1126. doi: 10.1126/science.1439819. [DOI] [PubMed] [Google Scholar]
- 55.Strick TR, Allemand JF, Bensimon D, Bensimon A, Croquette V. The elasticity of a single supercoiled DNA molecule. Science. 1996;271:1835–1837. doi: 10.1126/science.271.5257.1835. [DOI] [PubMed] [Google Scholar]
- 56.Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Meth Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]