Monocyte/Macrophage Suppression in CD11b Diphtheria Toxin Receptor Transgenic Mice Differentially Affects Atherogenesis and Established Plaques (original) (raw)
. Author manuscript; available in PMC: 2007 Oct 23.
Abstract
Although monocytes/macrophages are considered important in atherogenesis, their role in established plaques is unclear. For example, macrophage content is associated with plaque instability, but their loss through cell death is observed at sites of plaque rupture. To examine the role of monocytes/macrophages in atherosclerosis, we developed CD11b–diphtheria toxin (DT) receptor (DTR) transgenic mice, whereby administration of DT selectively kills monocytes/macrophages. DT treatment reduced peripheral blood monocytes and tissue macrophages and inhibited macrophage function in CD11b-DTR mice and apolipoprotein E–null (apoE−/−) mice transplanted with CD11b-DTR bone marrow. In atherogenesis experiments, DT markedly reduced plaque development and altered plaque composition, reducing collagen content and necrotic core formation. In mice with established plaques, acute DT treatment induced macrophage apoptosis and reduced macrophage content but did not induce plaque inflammation, thrombosis, or rupture. Furthermore, despite a 50% reduction in monocytes, chronic DT treatment of these mice did not alter plaque extent or composition, most likely because of ongoing recruitment/proliferation of monocytes with recovery of macrophage content. We conclude that monocytes/macrophages are critical to atherogenesis, but established plaques are more resistant to reductions in monocytes.
Keywords: apoptosis, atherosclerosis, macrophages
Macrophages are present in all stages of atherosclerosis and are considered fundamental to atherogenesis and the behavior of established plaques.1 Lipid-laden foam cells are derived from circulating monocytes that migrate into the vessel wall,2,3 and inhibition of monocyte migration, for example by disrupting a variety of chemokine/chemokine receptors, inhibits atherosclerosis development (reviewed elsewhere4). In contrast, the role of macrophages in advanced atherosclerotic plaques is more controversial. In vitro coculture studies demonstrate that low ratios of macrophages to vascular smooth muscle cells (VSMCs) induce VSMC proliferation by producing growth factors, particularly before macrophage differentiation or activation. In contrast, higher macrophage/VSMC ratios, particularly if macrophages are differentiated or activated, induce VSMC cell death by direct cell killing.5 Although macrophages can promote matrix synthesis from VSMCs,6 macrophage matrix metalloproteinases cause extracellular matrix (ECM) degradation. Macrophages also express and secrete tissue factor (particularly at sites of macrophage apoptosis7), which is responsible for much procoagulant activity of the plaque.
The direct consequences of macrophage apoptosis are also unclear. Macrophages comprise up to 50% of apoptotic cells in advanced atherosclerotic plaques.8 Macrophage apoptosis localizes to the plaque core, where it may promote core development,9 and to sites of plaque rupture in humans,10 suggesting that macrophage death itself may promote plaque instability. Although all of these potential activities of macrophages may operate in plaques, which activity predominates in plaque development or established plaques may depend on many factors, such as macrophage density and state of activation. It is thus difficult to define the role of monocyte/macrophage accumulation/activity in established lesions, a necessary prerequisite of therapies that alter macrophage numbers or function.
To examine the role of monocytes/macrophages in both established atherosclerosis and plaque formation, we developed a transgenic mouse in which diphtheria toxin (DT) conditionally ablates monocytes/macrophages. DT binds to the hbEGF receptor (heparin-binding epidermal growth factor–like growth factor)11 and, following internalization, inhibits protein synthesis. This rapidly induces apoptosis in both dividing and terminally differentiated cells. DT binds to normal mouse cells with 104 less affinity than to human cells.12 Thus, tissue-specific transgenic expression of human hbEGF confers DT sensitivity to murine cells in vivo, for example in dendritic cells, VSMCs or macrophages.13-15 Although chronic DT therapy induces a low-level antibody response, this does not impair its ability to kill cells.16 Using this system, we find that monocyte reduction by 50% profoundly affects plaque development, but has minimal effect on established plaques both acutely and with chronic suppression.
Materials and Methods
CD11b-DT Receptor Mice
All animal experiments were approved by the local Ethical Review Committee and conducted under UK Home Office licensing. The CD11b–human DT receptor (DTR) transgene was used to generate mice on the FVB/N background using conventional techniques,14 and transgene expression was detected by RT-PCR. The primer sequences used were 5′-AAGATCCGCCACAACATCG-3′ and 5′-GCAGCTCTAGGTTGGATTTCTG-3′ for the forward and reverse primers, respectively. Transplantation of apolipoprotein E (apoE) knockout mice was as previously described.17 (See the expanded Materials and Methods section in the online data supplement, available at http://circres.ahajournals.org).
Experimental Protocols
To establish the role of monocytes/macrophages in atherosclerosis we undertook three studies (Figure I in the online data supplement). ApoE−/− mice were given a high fat diet (21% fat, 0% cholate, 0.05% cholesterol - Research Diets Western Diet D12079B) and transplanted with CD11b-DTR/apoE−/− bone marrow at 8 weeks of age. Following bone marrow reconstitution, mice were administered 10 ng/g DT or saline control thrice weekly by intraperitoneal injection for the required time and euthanized 24 hours after the final injection. Serum cholesterol and triglycerides were assayed at monthly intervals, and a full lipid profile was performed at euthanasia. The experimental protocols were as follows:
- Atherogenesis study. DT was administered 2 to 3 weeks after bone marrow transplantation for a 10-week period (to 22 weeks of age).
- Acute established plaque study. Mice at 22 weeks of age were administered 2 doses of DT separated by 48 hours and euthanized 24 hours later.
- Chronic established plaque study. DT was administered at 22 weeks of age for 10 weeks.
Flow Cytometry
Mouse blood was analyzed by flow cytometry for CD3, F4/80, NK1.1, and CD11c or using Annexin V–FITC (fluorescein isothiocyanate), as described in the expanded Materials and Methods section in the online data supplement.
Peritoneal Macrophage Analysis
Peritoneal macrophage isolation, culture, immunocytochemistry, and uptake of acetylated LDL are described fully in the expanded Materials and Methods section in the online data supplement.
Lipid and Blood Cell Analysis
Lipid levels mice were measured as previously described.17 Differential white blood counts were analyzed on an automated Animal ABC Coulter Counter (Vet-ABC, Hesks) using mouse settings.
Histology and Analysis
For detailed descriptions of histology and analysis of lesions, see the expanded Materials and Methods section in the online data supplement.
Statistical Analysis
Values were expressed as mean±SEM. Groups showing similar variances were examined using 2-sample unpaired Student's t test. Where normality of distribution could not be assumed (for example buried fibrous caps), differences were examined using the nonparametric Mann–Whitney test. The significance level was set at P<0.05.
Results
Characterization of an Inducible Model of Monocyte/Macrophage Apoptosis
CD11b-DTR mice expressed human DTR–enhanced green fluorescent protein (EGFP) fusion protein from CD11b (macrophage-1) promoter sequences that direct transgene expression to monocytes/macrophages18 (supplemental Figure IIA). PCR confirmed that transgenic but not wild-type mice contained human DTR and transmitted with the expected frequency when crossed with apoE−/− mice (supplemental Figure IIB). Bone marrow–derived monocytes from wild-type and DTR mice differentiated in culture and expressed the macrophage-specific marker F4/80, indicating that DTR did not affect macrophage differentiation (data not shown). Cultured macrophages expressed the transgene mRNA by RT-PCR (supplemental Figure III) and underwent apoptosis after DT administration as assessed by time-lapse videomicroscopy and propidium iodide/Bisbenzamide staining for condensed chromatin (data not shown). In contrast, VSMCs cultured from CD11b-DTR/apoE−/− mice did not express the transgene mRNA (supplemental Figure III) and did not undergo apoptosis after DT treatment (data not shown).
We tested a range of DT doses and administration regimes for monocyte suppression in vivo. DT (10 ng/g) reduced CD3−/F4/80+ peripheral blood monocytes by 88.5±4.9% (mean±SEM, P<0.05, n=3) with no significant further reduction with 15 ng/g (Figure 1A). DT also reduced CD3−/F4/80− cells; this most likely represents F4/80-negative monocytes, as subsequent experiments revealed that chronic DT administration did not affect granulocyte counts (see below). DT treatment increased the percentage of Annexin V–positive peripheral blood cells to 7.6% by flow cytometry (Figure 1B), confirming death to be apoptosis. Flow cytometry demonstrated no change in CD3+/F4/80− (Figure 1A), NK 1.1–positive natural killer, or CD11c–positive dendritic cells (supplemental Figure IVA). DT treatment had no effect on peripheral blood populations in mice not expressing the transgene (data not shown).
Figure 1.
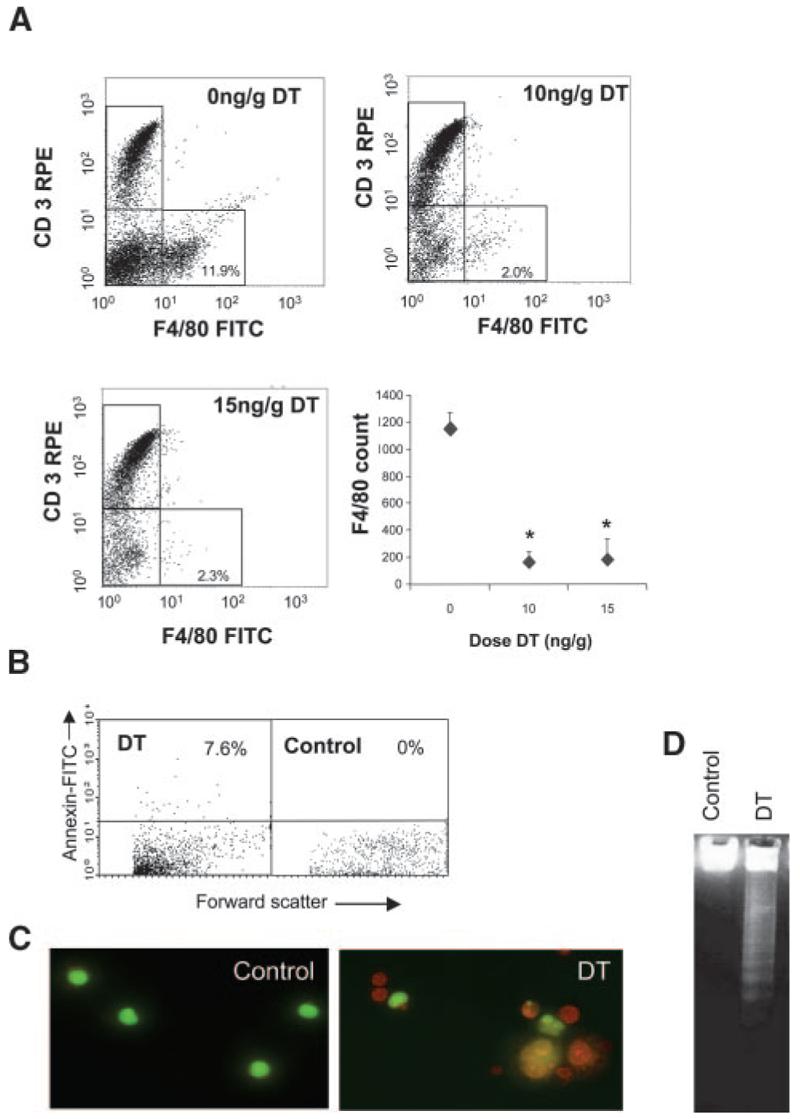
Characterization of CD11b-DTR mice. A, Representative flow-cytometric profiles from DTR-FV/B mice treated with 2 doses of 10 ng/g or 15 ng/g DT at 48-hour intervals. Blood was isolated 24 hours later, labeled with F4/80–fluorescein isothiocyanate (FITC) and CD3-R-phycoerythrin (CD3-RPE) antibodies and analyzed by flow cytometry. Absolute F4/80 counts are also shown for each dose of DT as mean±SEM. *P<0.05 (n=3) compared with controls (0 ng/g DT). B, Flow cytometry for Annexin V demonstrating increased circulating Annexin V–positive cells after DT treatment. C and D, Peritoneal macrophages isolated from these mice demonstrate apoptosis on acridine orange staining (orange cells with condensed chromatin) (C) and DNA laddering (D).
To examine whether DT administration also affected tissue macrophages, CD11b-DTR mice were administered 10 ng/g or 15 ng/g DT 48 hours apart and peritoneal macrophages isolated 24 hours later. Immunocytochemistry of parallel cultures revealed that >93% of cultured cells were F4/80 positive, and viable cells were estimated by acridine orange staining (Figure 1C). DT at 10 ng/g or 15 ng/g reduced peritoneal macrophage numbers by 84.3±2.5% and 77.4±4.1%, respectively (P<0.01, n=3). DT also induced typical DNA laddering in peritoneal macrophages, consistent with induction of apoptosis (Figure 1D). To examine whether DT inhibited function of viable macrophages, we examined uptake of acetyl LDL, an ability that requires macrophage scavenger receptors. DT at 10 ng/g or 15 ng/g reduced the percentage of viable macrophages taking up acetyl-LDL to 10.1±6.2% or 9.4±5.9%, respectively, compared with 89.7±7.6% in control mice receiving PBS (P<0.01, n=3). Previous studies have shown that DT does not affect spleen CD3+ cells or inhibit peritoneal CD3+ lymphocytes or neutrophils in CD11b-DTR mice under conditions in which peritoneal macrophages show almost complete inhibition.14
Transplantation of ApoE Knockout Mice
To study monocytes/macrophages in atherosclerosis, we backcrossed FVB CD11b-DTR mice with C57Bl6/J apoE−/− mice (supplemental Figure IIB) to generate CD11b-DTR/apoE−/− mice with >90% C57Bl6/J background and examined the ability of DT to chronically suppress monocytes/macrophages. Although thrice-weekly 10 to 15 ng/g DT administration optimally reduced peripheral blood macrophage number, chronic administration of DT induced liver inflammation and lung hemorrhage in CD11b-DTR/apoE−/− mice. Control apoE−/− mice were completely resistant to chronic DT administration (data not shown), suggesting that chronic monocyte/macrophage suppression is lethal to CD11b-DTR/apoE−/− mice and/or that the transgene is expressed at low levels outside bone marrow–derived cells.
To circumvent this problem, we irradiated and transplanted apoE−/− mice with bone marrow from CD11b-DTR/apoE−/− mice at 8 to 9 weeks of age. Irradiation suppressed total white blood cell count by 95% to 98%, with recovery of counts within 2 weeks of transplantation (supplemental Figure IVB). Two to 3 weeks after transplant, mice received saline or DT for 5 weeks and GFP-expressing cells were quantified in blood by flow cytometry. GFP could not be detected directly, as found previously,14 but could be detected using an antibody to GFP in permeabilized cells. DT reduced GFP-positive cells in whole blood by 54% compared with saline-treated mice (data not shown). Using forward/side scatter to identify monocytes, DT reduced GFP-positive monocytes from 72% to 33% of total monocyte population (Figure 2A). GFP-positive monocytes could be detected in blood of mice up to 22 weeks after CD11b-DTR transplant but were not seen in mice transplanted with control apoE−/− marrow (data not shown). In addition, GFP was not detected in control apoE−/− mice (Figure 2A).
Figure 2.
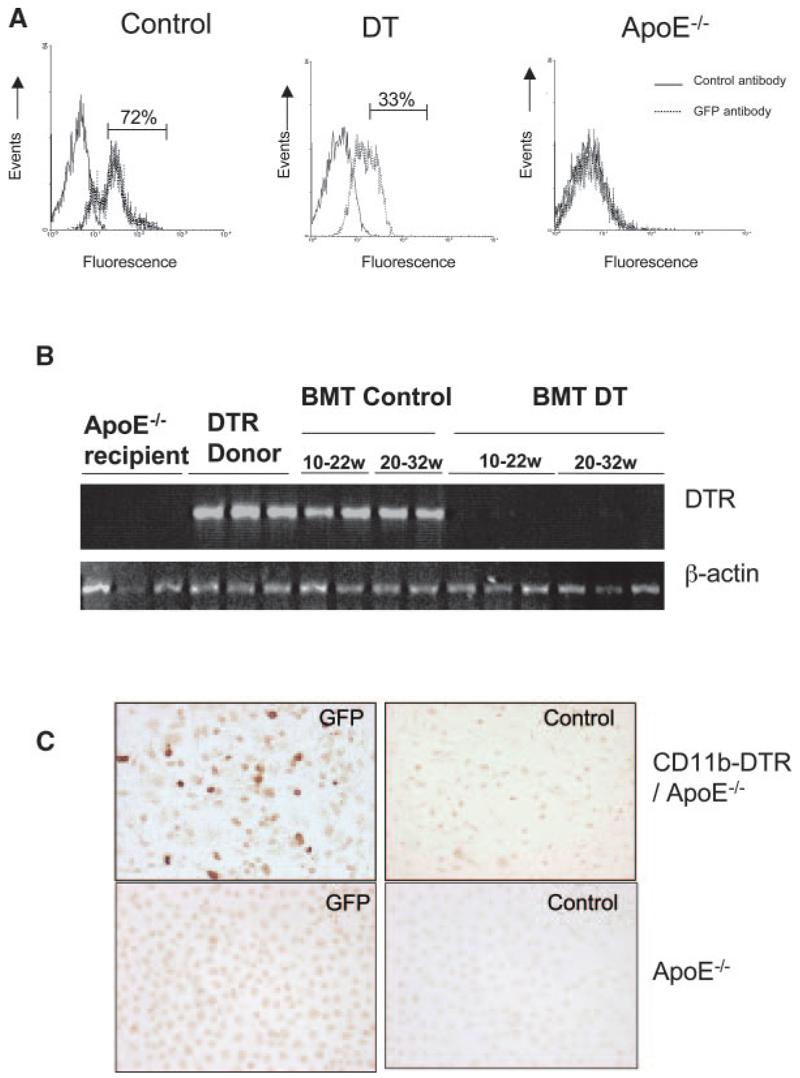
DT treatment reduces monocytes and macrophages in transplanted mice. A, ApoE−/− mice were transplanted with CD11b-DTR-EGFP/apoE−/− bone marrow at 8 weeks of age and treated with saline (control) or 10 ng/g DT 3 times per week for 5 weeks. Monocytes were separated by forward scatter/side scatter, and GFP expression was examined by flow cytometry. Nontransplanted apoE−/− mice (right) were also used as a negative control for GFP. B, PCR for DTR in blood of recipient apoE−/− mice before transplant, DTR+/+/apoE−/− donor mice and apoE−/− mice transplanted with DTR+/+/apoE−/− marrow and treated with saline control (BMT control) or DT (BMT DT) from 10 to 22 weeks or 20 to 32 weeks. C, Immunocytochemistry using anti-GFP or control antibody of peritoneal macrophages of mice transplanted with CD11b-DTR/apoE−/− marrow or control apoE−/− mice.
Previous studies demonstrated that DT treatment suppressed monocytes/macrophages for periods up to 1 week in CD11b-DTR mice.14 However, atherosclerosis studies require chronic inhibition of monocytes/macrophages. To examine whether DT could chronically reduce DTR-expressing cells in blood of transplanted mice, apoE−/− mice were transplanted with CD11b-DTR/apoE−/− marrow and DT or saline control administered from 10 to 22 or 22 to 32 weeks of age, the time points chosen for atherosclerosis studies (see below). DTR was easily detected in transplanted mice administered saline over both time courses (Figure 2B). In contrast, little or no DTR DNA was detected in blood of mice treated with DT for either time course. To examine the effect of DT on macrophage number and function, apoE−/− mice were transplanted with CD11b-DTR/apoE−/− marrow and administered saline control or 2 doses of DT 48 hours apart, 12 weeks after transplant. By immunocytochemistry, GFP was expressed in 25% to 70% (average 40%) of the total peritoneal macrophage population but was not found in control apoE−/− mice (Figure 2C).
We next examined whether peritoneal macrophages in apoE−/− mice transplanted with CD11b-DTR/apoE−/− marrow were sensitive to DT. DT induced typical apoptotic morphology within 24 hours (Figure 3A), but peritoneal macrophages of control apoE−/− mice were resistant to DT. To examine whether in vivo DT treatment of transplanted mice could reduce macrophage number and/or function, apoE−/− mice were transplanted with CD11b-DTR/apoE−/− marrow and administered saline control or 2 doses of DT 48 hours apart, 12 weeks after transplant. 10 ng/g or 15 ng/g DT reduced F4/80-positive peritoneal macrophage numbers by 31% or 40%, respectively (Figure 3B), on flow cytometry. Acridine orange staining confirmed reduction in viable macrophage numbers after 10 ng/g DT treatment (Figure 3C). In addition to reduced macrophage number, viable macrophages from mice treated with 10 ng/g DT showed reduced acetyl LDL uptake by 54.3%, compared with equal numbers of macrophages from control mice (Figure 3D and 3E). This suggests that DT could inhibit tissue macrophage number and function in transplanted mice. We also examined whether foam cell formation alters the sensitivity to DT by preincubating macrophages in oxidized LDL for 16 hours before DT administration. Oxidized LDL increased both basal and DT-induced apoptosis of peritoneal macrophages by 9% to 14% (data not shown).
Figure 3.
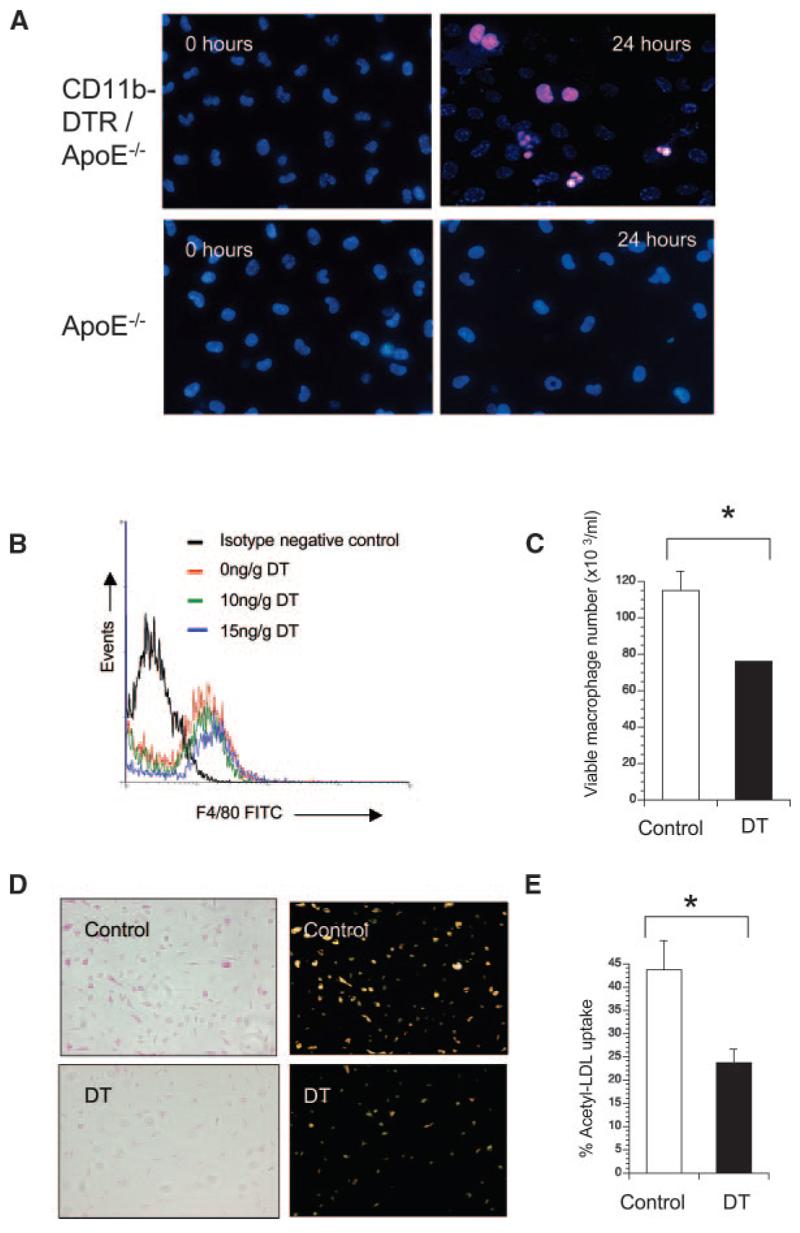
DT inhibits macrophage numbers and function in transplanted apoE−/− mice. A, Propidium iodide/Bisbenzamide staining of peritoneal macrophages cultured from mice transplanted with CD11b-DTR/apoE−/− marrow (top panels) compared with control apoE−/− mice (bottom panels) treated with DT for 24 hours. Typical apoptotic morphology (condensed, fragmented nuclei) is seen after DT treatment of mice transplanted with CD11b-DTR/apoE−/− marrow. B through E, ApoE−/− mice were transplanted with CD11b-DTR/apoE−/− marrow and treated with 2 doses of DT or saline control in vivo, before isolation of macrophages. F4/80+ peritoneal macrophages were quantified by flow cytometry (B). Viable peritoneal macrophages from mice receiving 10 ng/g DT or saline (control) were also quantified by acridine orange staining (C). D, Uptake of acetylated LDL determined by phase contrast (left) or fluorescence microscopy (right) in viable peritoneal macrophages from mice treated with 10 ng/g DT or saline (control). E, Percentage of viable macrophages demonstrating acetyl-LDL uptake. Data are means; error bars represent SEMs (n=3). *P<0.05.
Effect of Monocyte/Macrophage Ablation on Atherogenesis
We next examined the effect of 10 ng/g DT administration from 8 to 22weeks on atherosclerosis in apoE−/− mice transplanted with CD11b-DTR/apoE−/− marrow. DT administration was well tolerated, with no differences in body weight, serum cholesterol or triglycerides throughout the study, or in full lipid profiles at 22 weeks between groups (supplemental Figure VA).
At euthanasia, mice were perfusion fixed, and organs removed to examine macrophage GFP expression and plaque histology. DTR-EGFP–expressing macrophages were detected in multiple organs of transplanted mice, including lung, liver (Figure 4A and 4B), and spleen (not shown) and atherosclerotic plaques (Figure 4C and 4D), indicating successful transplant and accumulation of DTR-expressing macrophages. DT treatment reduced the percentage of plaque macrophages expressing GFP from 49.5±1.4% to 15.9±3.0% (P<0.001) (Figure 4C through 4F). VSMCs in the fibrous cap and media were GFP negative, suggesting that only macrophages within plaques expressed DTR. Histology of the liver, lungs, spleen, and pancreas was otherwise normal in DT or saline-treated mice.
Figure 4.
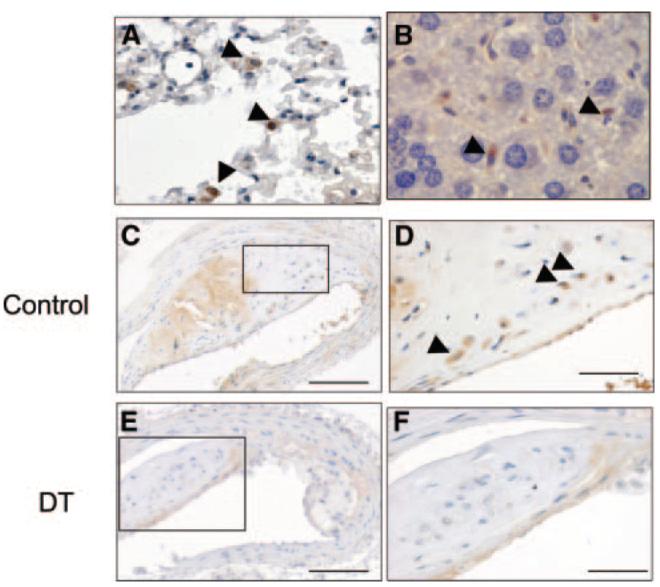
DTR-expressing macrophages are found in multiple organs. A through D, Immunohistochemistry for GFP in lung (A), liver (B), and atherosclerotic plaque (C and D) demonstrating GFP-positive macrophages in all organs (arrowheads). C through F, ApoE−/− mice were irradiated and transplanted at 8 weeks of age with CD11b-DTR/apoE−/− marrow and 10 ng/g DT or saline control injected 3 times weekly until 22 weeks. DT reduced the percentage of GFP-positive macrophages (compare F vs control [D]). D and F, Higher-power views of areas outlined in C and E. Scale bars: 200 _μ_m (C and E); and 50 _μ_m (D and F).
Descending thoracic and abdominal aortic plaque burden was analyzed using oil red O staining and en face measurements, and brachiocephalic arteries examined for plaque area, necrotic core formation, and changes in plaque composition (Picrosirius red for collagen content, immunohistochemistry for VSMC [_α_–smooth muscle actin {SMA}], macrophage [Mac3], and lymphocyte [CD3] percentages). The presence of cleaved caspase 3 (CC3) in cells with pyknotic nuclei was used to examine for apoptotic cell death and Ki67 for cell proliferation (Figure 5). We also examined plaques for loss of fibrous cap continuity, intraplaque hemorrhage, and fibrin(ogen) deposition,19 as well as counted fibrous cap-like structures below the lesion surface (“buried fibrous caps”) (Figure 5).
Figure 5.
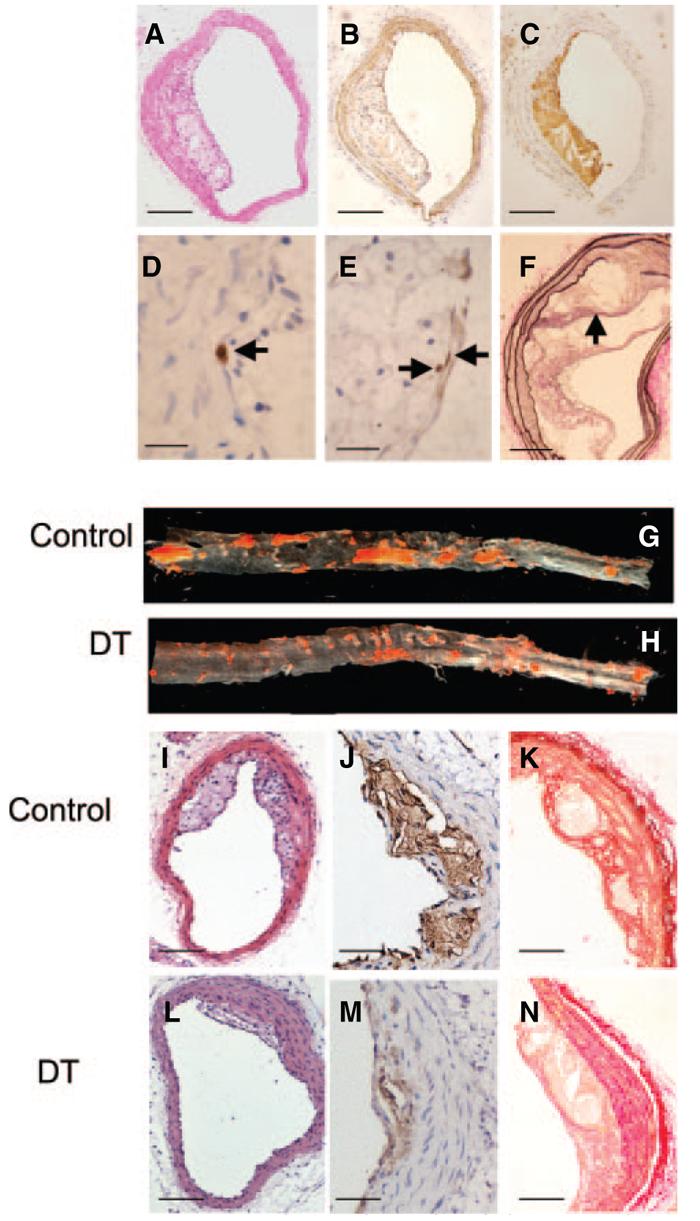
DT treatment reduces atherosclerosis in aorta and brachiocephalic arteries. Brachiocephalic artery plaques were analyzed by hematoxylin/eosin (A) and immunohistochemistry for VSMCs (SMA) (B); macrophages (Mac3) (C); cleaved caspase 3 in cells showing a pyknotic, fragmented nucleus typical of apoptosis (arrow) (D); and Ki67 (arrows) (E). F, Van Gieson stain demonstrating buried fibrous cap (arrow). G and H, Oil red O analysis of descending thoracic and abdominal aortas from mice treated with control (saline) or 10 ng/g DT. I through N, Effects of DT on plaque composition, demonstrating reduced plaque development on hematoxylin/eosin staining (I and L) and reduced necrotic cores by Mac3 staining (J and M) after DT administration. In similar-sized lesions, DT reduced plaque collagen by Picrosirius red staining (K and N). Scale bars: 100 _μ_m (A through C, I, and L); 50 _μ_m (F, K, and N); 25 _μ_m (J and M); 10 _μ_m (D and E).
DT administration markedly reduced oil red O–positive aortic lesions and brachiocephalic plaque area (Table and Figure 5). DT reduced both macrophages and VSMCs in plaques, resulting in no significant differences in their percentages. DT significantly decreased the necrotic core percentage and collagen content (Figure 5) but had no effect on total cell proliferation or apoptosis. We identified the origin of apoptotic cells by analyzing serial sections for CC3, Mac3, or SMA. In control vessels, macrophage and VSMC apoptotic frequencies were similar; in contrast, >90% apoptotic cells were macrophages in DT-treated vessels. Plaques exhibited 1 to 2 buried fibrous caps per lesion, similar to previous studies at this time point19; this was not affected by DT. Although occasional mice demonstrated intraplaque hemorrhage on Perl's staining, there was no effect of DT treatment. CD3 was expressed in <1% of cells in plaques of control or DT treated mice.
Table.
Plaque Characteristics of Mice Treated With DT
| Atherogenesis Study (10–22 weeks) | Established Plaque Study (20–32 weeks) | |||
|---|---|---|---|---|
| Experimental Protocol (DT Treatment) | Control (n=10) | DT 10 ng/g (n=10) | Control (n=8) | DT 10 ng/g (n=8) |
| Aortic Oil red O, % | 14.6±1.8 | 5.6±1.5† | 13.0±1.9 | 11.25±0.9 |
| Plaque Area _μ_m2 (×103) | 45.3±9.4 | 21.1±6.4* | 86.9±21 | 97.2±19.5 |
| Necrotic core, % | 15.4±2.8 | 9.4±2.4* | 17.6±4.1 | 21.2±4.8 |
| Picrosirius red, % | 32.4±4.3 | 12.7±4.1† | 42.2±6.6 | 60±5.6 |
| SMA, % | 11.2±1.7 | 10.2±1.3 | 25.2±4.0 | 18.9±4.7 |
| Mac3, % | 18.1±3.5 | 18.8±1.9 | 22.7±3.0 | 19.9±5.2 |
| Ki67, % | 1.6±0.7 | 1.3±0.5 | 1.4±0.3 | 2.7±0.5* |
| Cleaved caspase 3, % | 0.2±0.1 | 0.2±0.2 | 1.0±0.2 | 1.4±0.3 |
Acute Effect of DT on Established Plaques
The ability of DT treatment to significantly reduce plaque size and necrotic core percentage in atherogenesis suggests that DT inhibits monocyte numbers/migration and/or induces apoptosis in plaque macrophages. We therefore examined whether DT could induce macrophage apoptosis in established plaques in apoE−/− mice transplanted with CD11b-DTR/apoE−/− marrow treated with 2 doses of 10 ng/g DT separated by 48 hours. Plaques were examined 24 hours after the last dose of DT for morphological features of apoptosis, macrophages, CC3, TUNEL, intraplaque hemorrhage, and lumen thrombosis (Figure 6). DT treatment resulted in extensive apoptotic debris, localizing particularly to the macrophage-rich necrotic core. DT increased TUNEL staining within plaques (3.5%±0.6 versus 0.9%±0.3, P<0.01), and reduced both GFP percentage (7.9%±1.7 versus 27.5%±1.4, P<0.01) and total macrophage percentage (34.9%±2.9 versus 44.3±3.4, P<0.05). We did not see fibrous cap discontinuity, intraplaque hemorrhage, or intraluminal thrombus in DT-treated or control mice. Macrophages expressed Ki67, indicating proliferation after DT treatment. Mac387, a marker of newly migrated macrophages,20 was also found in superficial macrophages, suggesting that migration into the plaque continues after DT treatment (Figure 6J). Although DT increased apoptotic cells (indicated by TUNEL and cleaved caspase 3 staining) in liver, spleen, lymph nodes, and lung of transplanted mice, it did not significantly reduce the macrophage content of these organs nor otherwise alter their architecture.
Figure 6.
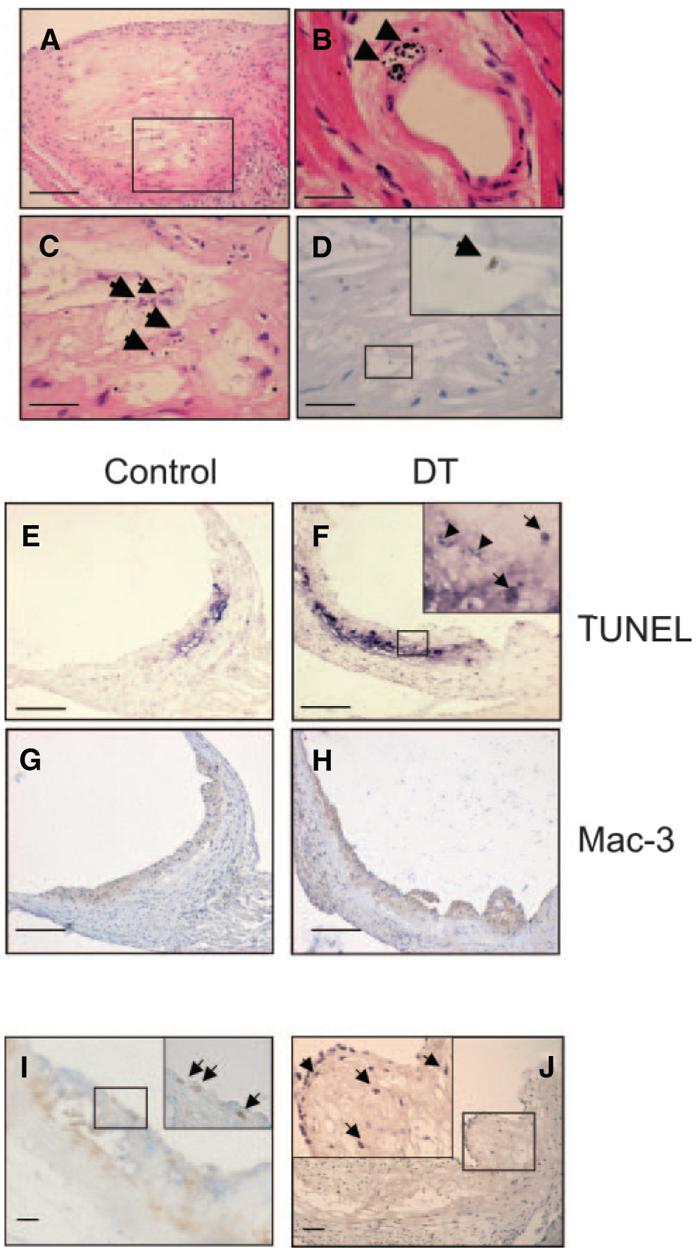
Effects of short term DT treatment on established plaques. A through D, Apoptosis in mice administered 2 doses of 10 ng/g DT or control, showing extensive apoptotic debris on hematoxylin/eosin staining of established plaque (A). B, Morphological features of apoptosis were seen in DT-treated mice, for example, in small intramyocardial artery (arrowheads). C and D, High-power views of area outlined in A showing apoptotic debris (C) and cleaved caspase 3–positive cell (D and inset). E through H, Increased TUNEL staining and reduced macrophage percentage in DT-treated vs control plaques. Inset represents high-power view of area outlined in F, showing TUNEL-positive cells (arrows) and debris (arrowheads). I, Double labeling for macrophages (blue) and Ki67 (brown) indicating superficial proliferating macrophages (arrows). J, Mac387 expression indicating presence of newly migrated macrophages (arrows). Insets represent high-power view of area outlined in (I and J). Scale bars: 200 _μ_m (A and E through H); 50 _μ_m (C, D, I, and J); 25 _μ_m (B).
Effect of Monocyte/Macrophage Ablation on Established Plaques
The effects of chronic monocyte/macrophage reduction on established plaques was examined in apoE−/− mice transplanted with CD11b-DTR/apoE−/− marrow and administered 10 ng/g DT or saline control from 22 to 32 weeks. DT treatment was well tolerated, with no difference between groups in weight gain, serum cholesterol, or triglyceride levels throughout, or full lipid profile at 32 weeks (supplemental Figure VB).
Surprisingly, DT treatment of mice with established lesions did not alter aortic oil red O or brachiocephalic plaque areas (Table). In addition, there was no significant difference observed in plaque collagen content, necrotic core percentages, VSMC or macrophage contents, or apoptosis between groups (Figure 7 and the Table). The reduction in percentage of macrophages that were GFP positive after DT did not reach statistical significance (24.4±2.1% versus 31.5%±6.5%). DT did increase numbers of proliferating cells in plaques but did not alter the frequency of buried fibrous caps, intraplaque hemorrhage, or fibrin(ogen) deposition. Fibrous cap discontinuity and luminal thrombus were not seen in any group.
Figure 7.
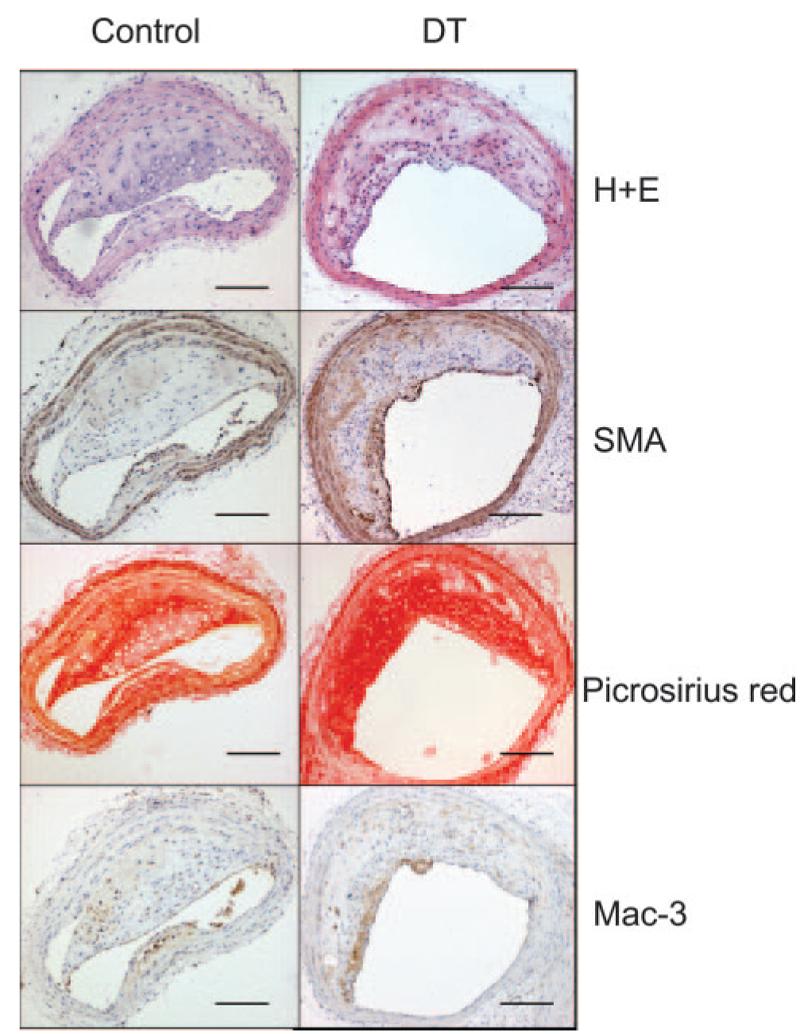
Effects of long-term DT treatment on established plaques. Effects of DT on composition of established plaques, demonstrating no effect on plaque development on hematoxylin/eosin (H+E) staining, VSMC content (SMA), collagen content (Picrosirius red), or macrophage content (Mac3). Scale bars=100 _μ_m.
To confirm that DT treatment still suppressed monocytes at 32 weeks, we additionally examined monocyte, granulocyte and lymphocyte numbers at 20 and 32 weeks by Coulter counts. Although CD11b is also expressed in neutrophils, total granulocyte count (most of which are neutrophils) was not reduced by DT. In contrast, DT reduced monocytes at 32 weeks, both in absolute terms and as a percentage of total granulocytes (supplemental Figure VI). Monocytes comprised 12.7% of the granulocyte fraction before DT administration. This was reduced to 5.7% by DT treatment. This 55% reduction correlates well with the 49% reduction in GFP-positive cells after DT administration found by flow cytometry (Figure 2) and indicates that DT suppresses monocytes throughout the experimental period. Lymphocyte counts did not change over the course of the study (not shown).
Discussion
Although numerous studies testify that monocytes/macrophages are important in atherosclerosis, the precise role of monocytes/macrophages at distinct phases of the disease is unknown. Macrophages can promote both tissue injury and repair, for example providing factors that promote VSMC proliferation and ECM synthesis but can also induce VSMC apoptosis and produce matrix metalloproteinases that degrade ECM, collagen, and elastin. Knockout of chemokine/chemokine receptors has provided evidence that monocytes/macrophages are required for plaque development. However, these mice cannot be used to define the role of monocytes/macrophages in established lesions that have developed with a normal macrophage contribution. Monocytes/macrophages can be depleted using either anti-macrophage serum or by liposomal clodronate.21-23 Although these agents suppress macrophages, and clodronate can reduce neointima formation in hypercholesterolemic rabbits,22 anti-macrophage serum has pleiotropic effects, whereas clodronate also depletes neutrophils, has systemic toxicity, and is ineffective in some tissues.23 In contrast, administration of DT to transgenic animals with tissue-specific expression of human DTR provides a potent, selective, conditional system for cellular ablation at any point in disease. In recent studies, we demonstrated that DT-induced apoptosis of VSMCs induces multiple features of plaque vulnerability in apoE−/− mice.15 In the current study, DT administration to CD11b-DTR mice reduces circulating monocyte number and could deplete tissue macrophages in some organs. DT also impaired macrophage function in the remaining cells, as demonstrated by reduction in acetylated LDL uptake of peritoneal macrophages. The complete resistance of nontransgenic mice to DT means that transplanted DTR-expressing monocytes can be selectively depleted.
We first examined the effect of monocyte/macrophage suppression in atherogenesis. DT treatment markedly reduced plaque development in both aorta and brachiocephalic arteries, most likely reflecting monocyte killing in the circulation, with subsequent reduced migration. Despite selective inhibition of monocytes/macrophages, the relative proportions of macrophages and VSMCs within plaques remained unchanged. This suggests that VSMC accumulation in plaques depends on monocytes/macrophages. For example, VSMC accumulation may be regulated by growth factors derived from macrophages, or intimal VSMCs may arise from monocytic precursor cells within blood. VSMC synthetic function may also depend on macrophages, as DT reduced collagen content of plaques. Macrophage depletion has previously been shown to reduce fibroblast synthesis of both collagen and elastin in hepatic fibrosis.14 DT treatment markedly reduced the area occupied by the necrotic core in plaques. This may be attributable to reduction in macrophage number and/or function, as DT inhibited macrophage uptake of lipid in vitro in addition to reducing macrophage numbers. Indeed, our study cannot fully discriminate the effects of reducing monocyte/macrophage numbers versus effects on their activity, as DT reduced both to an approximately similar extent. Despite monocyte suppression, markers of possible plaque rupture were unchanged. Thus, the major effect of monocyte/macrophage suppression in atherogenesis is a global reduction in plaque development. Our data also suggest that macrophage infiltration in early plaques is a major stimulus for VSMC recruitment, collagen synthesis, and necrotic core formation.
To define the role of monocytes/macrophages in established lesions, we studied the effects of DT administration in mice from 20 weeks of age, a point at which brachiocephalic plaques show advanced features,19 and plaque macrophages expressed GFP. DT treatment for 72 hours induced apoptosis of plaque macrophages, with extensive apoptotic debris in macrophage-rich parts of the plaque, and reduced GFP expression and total numbers of macrophages. Despite inducing monocyte/macrophage apoptosis in established plaques, DT treatment did not induce markers of plaque rupture. Furthermore, despite a 50% reduction in monocyte counts, chronic DT administration did not alter (1) aortic plaque burden or brachiocephalic plaque area; (2) plaque composition (including macrophage content), necrotic cores, or collagen content; or (c) markers of possible plaque rupture. These results contrast strongly with the effects of VSMC apoptosis, which induce profound changes in plaque composition.15 This suggests that selective suppression of monocytes may have minimal effects on progression or composition of established plaques, without changes in other cell populations, such as VSMCs.
The lack of effect on established plaques is unexpected. For example, macrophage death in established lesions would be predicted to enlarge the necrotic core and to produce inflammation,24 particularly as advanced plaques show defective clearance of apoptotic bodies.24 Although apoptotic debris in macrophage-rich areas of the plaque accumulated after 3 days of DT treatment, it is possible that sufficient phagocytic cells remain to engulf apoptotic macrophages to prevent secondary necrosis and inflammation in this model. Consistent with this idea, we have found recently that VSMCs are highly effective phagocytes in the vessel wall.15 Thus, unlike VSMCs,15 we cannot find evidence of significant sequelae of macrophage apoptosis in established plaques either acutely or after chronic DT treatment.
Our study has a number of limitations. First, we studied the effects of DT administration on atherosclerosis in mice that had received irradiation and bone marrow transplant. Irradiation alters plaque development in a site-specific manner, accelerating aortic root lesions and retarding thoracic aorta lesions.25 Although DT had similar effects on atherosclerosis in 2 different vascular beds, its effect might be quantitatively different at other sites, or without irradiation. Second, although VSMCs did not express the transgene, it might be expressed at low levels outside monocytes/macrophages. Although chronic DT administration did not reduce granulocytes (as seen in other studies using these mice14), lymphocytes, dendritic or NK cells, the numbers of lymphocytes present within lesions was too small to assess any change in plaque lymphocyte content or subsets. Third, chronic DT treatment of transplanted mice reduced monocyte numbers by only 50%. Although this could reflect loss of transgene in transplanted mice, PCR showed easily detectable transgene expression in control mice, with effective reduction by DT. We consider that the 50% reduction more likely reflects recovery of host monocytes, or a subset of monocytes that do not express CD11b, resulting in the monocyte population that were GFP negative in saline-treated mice. These monocytes would continue to emigrate and/or proliferate into the plaque, as demonstrated by Mac387 and Ki67 staining, respectively, such that although macrophages are depleted from the plaque after acute DT treatment, chronic DT treatment of established lesions does not reduce macrophage contents. It is also possible that some DTR-GFP–positive macrophages are resistant to DT, as demonstrated by their continued presence in the plaque despite chronic DT treatment. Finally, the DT system induces apoptosis via inhibition of protein synthesis. Death of macrophages in atherosclerosis may be both apoptotic and nonapoptotic.24 It remains possible that other forms of cell death mediate some consequences of monocyte/macrophage death in atherosclerosis.
In contrast, we consider it more likely that monocyte reduction alone is insufficient to alter composition of established lesions, without also reducing the stimuli that lead to monocyte/macrophage accumulation in plaques, such as serum cholesterol levels, cholesterol retention within lesions, and subsequent expression of adhesion molecules. Clearly, DT induced apoptosis in macrophages in established lesions and acutely reduced macrophage numbers. However, with chronic treatment, monocytes can still migrate and/or proliferate, as shown by increased proliferation in established lesions after DT treatment. Although 50% reduction in monocyte counts was sufficient to profoundly reduce atherogenesis, more aggressive ablation and/or prevention of monocyte migration/proliferation may be required to change plaque composition and reduce plaque macrophage content in established plaques. It remains to be shown whether such aggressive monocyte inhibition can be achieved without generating immune defects or other side effects.
In conclusion, we demonstrate that suppression of circulating monocytes by 50% markedly retards atherogenesis in apoE−/− mice. In contrast, similar monocyte reduction does not alter plaque development or composition in mice with established lesions, and acute macrophage apoptosis does not induce features of plaque rupture. This demonstrates that the role of monocytes/macrophages in atherosclerosis is critically dependent on extent of plaque development.
Acknowledgments
Sources of Funding
D.B. was supported by a Medical Research Council Training Fellowship. This work was also supported by British Heart Foundation grants RG98/009 and RG/04/001.
Footnotes
References
- 1.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 2.Gerrity RG, Naito HK, Richardson M, Schwartz CJ. Dietary induced atherogenesis in swine. Morphology of the intima in prelesional stages. Am J Pathol. 1979;95:775–792. [PMC free article] [PubMed] [Google Scholar]
- 3.Faggiotto A, Ross R, Harker L. Studies of hypercholesterolemia in the nonhuman primate. I. Changes that lead to fatty streak formation. Arteriosclerosis. 1984;4:323–340. doi: 10.1161/01.atv.4.4.323. [DOI] [PubMed] [Google Scholar]
- 4.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 5.Boyle J, Bowyer D, Weissberg P, Bennett M. Human blood-derived macrophages induce apoptosis in human plaque-derived vascular smooth muscle cells by Fas ligand/Fas interactions. Art Thromb Vasc Biol. 2001;21:1402–1407. doi: 10.1161/hq0901.094279. [DOI] [PubMed] [Google Scholar]
- 6.Edwards IJ, Wagner WD, Owens RT. Macrophage secretory products selectively stimulate dermatan sulfate proteoglycan production in cultured arterial smooth muscle cells. Am J Pathol. 1990;136:609–621. [PMC free article] [PubMed] [Google Scholar]
- 7.Hutter R, Valdiviezo C, Sauter BV, Savontaus M, Chereshnev I, Carrick FE, Bauriedel G, Luderitz B, Fallon JT, Fuster V, Badimon JJ. Caspase-3 and tissue factor expression in lipid-rich plaque macrophages: evidence for apoptosis as link between inflammation and atherothrombosis. Circulation. 2004;109:2001–2008. doi: 10.1161/01.CIR.0000125526.91945.AE. [DOI] [PubMed] [Google Scholar]
- 8.Lutgens E, de Muinck ED, Kitslaar PJ, Tordoir JH, Wellens HJ, Daemen MJ. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc Res. 1999;41:473–479. doi: 10.1016/s0008-6363(98)00311-3. [DOI] [PubMed] [Google Scholar]
- 9.Kockx MM. Apoptosis in the atherosclerotic plaque. Quantitative and qualitative aspects. Arterioscler Thromb Vasc Biol. 1998;18:1519–1522. doi: 10.1161/01.atv.18.10.1519. [DOI] [PubMed] [Google Scholar]
- 10.Kolodgie FD, Narula J, Burke AP, Haider N, Farb A, Hui-Liang Y, Smialek J, Virmani R. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol. 2000;157:1259–1268. doi: 10.1016/S0002-9440(10)64641-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naglich JG, Rolf JM, Eidels L. Expression of functional diphtheria toxin receptors on highly toxin-sensitive mouse cells that specifically bind radioiodinated toxin. Proc Natl Acad Sci U S A. 1992;89:2170–2174. doi: 10.1073/pnas.89.6.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pappenheimer AM, Jr, Harper AA, Moynihan M, Brockes JP. Diphtheria toxin and related proteins: effect of route of injection on toxicity and the determination of cytotoxicity for various cultured cells. J Infect Dis. 1982;145:94–102. doi: 10.1093/infdis/145.1.94. [DOI] [PubMed] [Google Scholar]
- 13.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 16.Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods. 2005;2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- 17.Mercer J, Figg N, Stoneman V, Braganza D, Bennett MR. Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ Res. 2005;96:667–674. doi: 10.1161/01.RES.0000161069.15577.ca. [DOI] [PubMed] [Google Scholar]
- 18.Dziennis S, Van Etten RA, Pahl HL, Morris DL, Rothstein TL, Blosch CM, Perlmutter RM, Tenen DG. The CD11b promoter directs high-level expression of reporter genes in macrophages in transgenic mice. Blood. 1995;85:319–329. [PubMed] [Google Scholar]
- 19.Johnson J, Carson K, Williams H, Karanam S, Newby A, Angelini G, George S, Jackson C. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation. 2005;111:1422–1430. doi: 10.1161/01.CIR.0000158435.98035.8D. [DOI] [PubMed] [Google Scholar]
- 20.Poston RN, Hussain IF. The immunohistochemical heterogeneity of atheroma macrophages: comparison with lymphoid tissues suggests that recently blood-derived macrophages can be distinguished from longer-resident cells. J Histochem Cytochem. 1993;41:1503–1512. doi: 10.1177/41.10.7504008. [DOI] [PubMed] [Google Scholar]
- 21.Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78:71–100. [PMC free article] [PubMed] [Google Scholar]
- 22.Danenberg HD, Fishbein I, Gao J, Monkkonen J, Reich R, Gati I, Moerman E, Golomb G. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation. 2002;106:599–605. doi: 10.1161/01.cir.0000023532.98469.48. [DOI] [PubMed] [Google Scholar]
- 23.Feith GW, Bogman MJ, Assmann KJ, van Gompel AP, Schalkwijk J, van Rooijen N, Koene RA. Decreased PMN accumulation and glomerular damage by clodronate liposome treatment in PMN-dependent anti-GBM nephritis in mice. Exp Nephrol. 1997;5:301–304. [PubMed] [Google Scholar]
- 24.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 25.Schiller NK, Kubo N, Boisvert WA, Curtiss LK. Effect of gamma-irradiation and bone marrow transplantation on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1674–1680. doi: 10.1161/hq1001.096724. [DOI] [PubMed] [Google Scholar]