Alanine Scanning of the Hepatitis C Virus Core Protein Reveals Numerous Residues Essential for Production of Infectious Virus (original) (raw)
Abstract
Hepatitis C virus (HCV) is an important human pathogen affecting an estimated 3% of the world's population. Recent advances have enabled in vitro propagation of the virus and allow assembly and egress to be investigated for the first time. As a component of the virion, the HCV core protein likely functions primarily in infectious virus production, although little is known about the determinants of this activity. To investigate the roles of core in the viral life cycle, we performed a comprehensive deletion and alanine scanning mutagenesis study of this protein in the context of a genotype 2a reporter virus. We have confirmed that core protein is essential for infectious virion production and have identified numerous residues required for this role. The infectivity of several assembly-defective core mutants could be rescued by compensatory mutations identified in p7 and NS2, suggesting genetic interactions with core and highlighting the importance of these nonstructural proteins in infectious virion morphogenesis.
Hepatitis C virus (HCV) is a major etiological agent of liver disease, with an estimated 170 million people infected worldwide (53). Along with flaviviruses and pestiviruses, HCV constitutes the family Flaviviridae, a group of enveloped, positive-sense RNA viruses with immense human and agricultural impact. The HCV genome is approximately 9,600 nucleotides in length and, on introduction into the cell, is translated via an internal ribosome entry site to yield a polyprotein of about 3,000 residues (reviewed in reference 30). The polyprotein is processed co- and posttranslationally by viral and cellular proteases to liberate the individual viral proteins, sequentially designated C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B. Core (C) and the envelope glycoproteins (E1 and E2) make up the structural components of the virion. p7 and NS2 are nonstructural (NS) proteins thought to be involved in infectious virus production (20, 45). NS2 also has protease activity responsible for the cleavage of its own carboxy terminus from the polyprotein. NS3, along with its cofactor NS4A, is the protease responsible for the remainder of the NS protein processing; it also possesses helicase and nucleotide triphosphatase activities required for RNA replication. NS5B is the viral RNA-dependent RNA polymerase. NS4B and NS5A have as-yet-undefined but essential roles in RNA replication (30).
Core, a highly conserved 21-kDa protein, is the initial translation product of the viral genome. By analogy to the related flaviviruses and pestiviruses, as well as its detection in both patient sera and cell culture-derived virions, core is presumed to function primarily as a component of the infectious virus (7, 55, 29). Consistent with this structural role, core has been shown to interact homotypically to form dimers and higher-order complexes both in vitro and in cellular expression systems (6, 34, 26, 23). Core has also been reported to interact with the envelope glycoproteins E1 and E2 (38, 31, 3). The amino-terminal region of core is highly basic and has been shown mediate association of the protein with RNA (46, 12, 48). The carboxy terminus of core acts as a signal sequence to target E1 to the endoplasmic reticulum (ER) membrane (46). This signal sequence is cleaved by the host enzymes signal peptidase and signal peptide peptidase (SPP) (36, 46), with the later cleavage responsible for liberating core from the ER membrane and directing it to lipid droplets (36). Recent studies of infected cells suggest that lipid droplets and their associated membranes are the only subcellular compartments in which core accumulates, leading to speculation that these cholesterol-rich deposits may be sites of particle assembly (44). In addition to its presumed roles in assembly, core has been implicated in numerous interactions with cellular proteins and is proposed to modulate cellular functions (reviewed in reference 43). RNA structures within the 5′ end of the core-coding sequence have been shown to be important for infectivity in both cell cultures and chimpanzees (37).
Recently, systems supporting a full infectious life cycle of HCV in cell culture have been reported (29, 52, 57). These systems (HCVcc) take advantage of a genotype 2a patient isolate, JFH-1, which both replicates robustly in the absence of adaptive mutations and supports the release of infectious particles (21, 52, 57). JFH-1 infectivity could be enhanced by substitution of its structural proteins with those of another genotype 2a isolate, J6, leading to a chimeric genome termed J6/JFH (29). The advent of the cell culture infectious systems allows the role of core in the viral life cycle to be studied for the first time. Here we have performed a comprehensive deletion and alanine scanning mutagenesis study of the core protein in the context of an infectious J6/JFH reporter virus. We have confirmed that core protein is essential for infectious virion production in the HCVcc system and identified numerous residues critical for this activity. Passage of assembly-defective core mutant genomes identified compensatory mutations in p7 and NS2, indicating genetic interactions with core and adding to the growing evidence of the importance of these NS proteins in virion morphogenesis.
MATERIALS AND METHODS
Cell culture.
Huh-7.5 cells were propagated in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA) containing sodium pyruvate, 10 μM nonessential amino acids, and 10% fetal bovine serum (FBS). Cells were grown at 37°C in 5% CO2.
Plasmid constructs.
Plasmids were constructed by standard methods. Constructs were verified by restriction enzyme digestion and sequencing of PCR amplified segments. Descriptions of the cloning strategies are provided below, and plasmid and primer sequences are available upon request. These genomes are based on the J6/JFH chimeric genotype 2a sequence (29), or its derivative J6/JFH(p7-Rluc2A), in which a Renilla luciferase-2A (Rluc2A) sequence is inserted between p7 and NS2 (20). For J6/JFH(p7-Rluc2A) the 995-bp Rluc2A coding sequence begins after nucleotide 2791 and is followed by nucleotide 2782 and the remainder of the genome, duplicating three amino acids of NS2 (J6/JFH genome numbering). Rluc2A is predicted to be translocated into the ER lumen and released from p7 by signal peptidase; the autoproteolytic peptide 2A mediates cleavage of its own carboxy terminus, leaving a non-native proline at the amino terminus of NS2.
J6/JFH and J6/JFH(p7-Rluc2A) with deletions and alanine substitutions.
Deletions or alanine substitutions were introduced by PCR amplification of the core-coding region of J6/JFH with primers containing the desired changes. Primary PCR products containing the engineered mutations were assembled by amplification with the flanking primers RU-6009 (5′-CGACGGCCAGTGAATTCTAATACG-3′) and RU-5743 (5′-ATGCCATGCGGTGTCCAG-3′). PCR products were digested with EcoRI and KpnI and ligated to the 12,073-bp fragment of J6/JFH(p7-Rluc2A) or the 11,074-bp fragment of J6/JFH digested with the same enzymes.
J6/JFH with p7, NS2, NS3, and core mutations.
Isolated compensatory mutations were reengineered into wild-type J6/JFH and the parental core mutants. To facilitate cloning, the wild-type J6/JFH sequence was modified to include unique, silent restriction sites at positions 2392 (BglII) and 2955 (NotI) (termed J6/JFH1.1). Compensatory mutations were engineered by PCR amplification of the appropriate J6/JFH1.1 sequences with primers containing the desired changes. Primary PCR products were assembled by amplification with the flanking primers RU-6020 (5′-TATGTGGGAGGGGTTGAG-3′) and RU-5721 (5′-GCTACCGAGGGGTTAAGCACT-3′). PCR products were digested with BglII and NotI (p7 mutants) or NotI and SpeI (NS2 and NS3 mutants) and ligated to the 11,804-bp BglII-NotI fragment or the 11,219-bp NotI-SpeI fragment of J6/JFH1.1. To engineer the compensatory changes into the parental core mutants, the 11,074-bp EcoRI-KpnI fragment of the NS protein mutant was ligated to the 1,290-bp EcoRI-KpnI fragment of the appropriate core protein mutant.
RNA transcription.
In vitro transcripts were generated as previously described (29). Briefly, plasmids were linearized by digestion with XbaI, templates were purified over Minelute column (QIAGEN, Valencia, CA), and 1 μg was transcribed in a 10-μl reaction by using the T7 Megascript kit (Ambion, Austin, TX). Reactions were incubated at 37°C for 3 h, followed by a 15-min digestion with 3 U of DNase I (Ambion). RNA was cleaned up by using an RNeasy kit (QIAGEN) with an additional on-column DNase treatment for samples that would subsequently be analyzed by quantitative reverse transcription-PCR (qRT-PCR). RNA was quantified by determining the absorbance at 260 nm, and its integrity was verified by agarose gel electrophoresis.
RNA transfection and growth curves.
RNA was transfected into Huh-7.5 cells by electroporation as previously described (29). Briefly, Huh-7.5 cells were treated with trypsin, washed twice with ice-cold RNase-free phosphate-buffered saline (AccuGene PBS; BioWhittaker, Rockland, ME), and resuspended at 1.5 × 107 cells/ml in PBS. Then, 2 μg of each RNA to be electroporated was mixed with 0.4 ml of cell suspension and immediately pulsed with an ElectroSquare Porator ECM 830 (BTX, Holliston, MA) (820 V, 99 μs, five pulses). Electroporated cells were diluted in 30 ml of DMEM-10 mM nonessential amino acids-10% FBS (Invitrogen) and plated into 24-well and P100 tissue culture dishes.
At 8, 24, 48, and 72 h postelectroporation cells in 24-well plates were washed with Dulbecco PBS and lysed with Renilla lysis buffer (Promega, Madison, WI) or with RLT buffer (QIAGEN) containing 0.14 M β-mercaptoethanol (RLT/β-ME) for assay of replication by luciferase activity or qRT-PCR, respectively. At the same time points, cell culture supernatants from the P100 dishes were completely removed, clarified with a 0.22-μm-pore-size filter, divided into aliquots, and frozen at −80°C for analysis of infectivity; fresh DMEM-10 μM nonessential amino acids-10% FBS was then added to the cells. For reporter viruses, infectivity was assayed by infection of naive cells with 600 μl of clarified supernatant and incubation for 48 h, with subsequent lysis in Renilla lysis buffer (Promega) and analysis of luciferase activity. Each infection was done in triplicate.
Western blot.
Cells were lysed at 48 h postelectroporation with Renilla lysis buffer (Promega). Lysates were separated by sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis (SDS-12% PAGE) for analysis of core and β-actin, and by SDS-10% PAGE for analysis of NS5A. After transfer to nitrocellulose membrane, blots were blocked for 1 h with 5% milk-TBST (0.02 M Tris [pH 7.4], 0.2 M NaCl, 0.1% Tween 20). For core detection, monoclonal antibody (MAb) HCM-071-5 (Austral Biologicals, San Ramon, CA), which recognizes amino acids 10 to 53, was diluted 1:750 to 1:1,000 in TBST. For NS5A detection, MAb 9E10 (29) was diluted 1:100 in TBST. For β-actin detection, mouse anti-β-actin MAb (Sigma, St. Louis, MO) was diluted 1:5,000 in TBST. After overnight incubation at 4°C (C and NS5A) or 2 h of incubation at room temperature (β-actin) and extensive washing with TBST, rabbit anti-mouse immunoglobulin-horseradish peroxidase secondary antibody (Pierce, Rockford, IL) was added at a 1:5,000 dilution in 5% milk-TBST for 30 min at room temperature. After additional washing, blots were developed with SuperSignal West Femto chemiluminescent substrate (Pierce).
RNA extraction and qRT-PCR.
For analysis of replication, total RNA was extracted from cells lysed with RLT/β-ME (QIAGEN) and homogenized by centrifugation through a QiaShredder column (QIAGEN) for 2 min at 14,000 × g. RNA was isolated by using an RNeasy kit (QIAGEN) and quantified by determining the absorbance at 260 nm. Fifty nanograms of total cellular RNA was used per qRT-PCR. For analysis of released viral genomes, RNA was isolated from 1 ml of cell culture supernatant, harvested at 48 h postelectroporation, by using the QiaAmp UltraSens kit (QIAGEN). Then, 2 μl of isolated RNA was used per qRT-PCR.
qRT-PCRs were performed by using a LightCycler amplification kit (Roche, Basel, Switzerland) with primers directed against the viral 5′ untranslated region. Reactions (20 μl) were assembled according to the manufacturer's instructions. qRT-PCR amplification and analysis were performed by using a LightCycler 480 (Roche) and associated software.
Core ELISA.
HCV core antigen enzyme-linked immunosorbent assay (ELISA; Ortho Clinical Diagnostics, Raritan, NJ) was performed according to the manufacturer's instructions. Briefly, cell culture supernatants harvested at 48 h postelectroporation were diluted 1:10 [wild-type J6/JFH(p7-Rluc2A)] or undiluted (mutants) and applied to plates coated with a mixture of anti-core mouse MAbs. Antigen was detected by addition of a second MAb cocktail, conjugated to horseradish peroxidase. Use of multiple MAbs with nonoverlapping epitopes ensured detection of all of the core mutants. After a washing step, wells were developed by the addition of substrate and subsequent stop solution; the absorbance at 490 nm was then measured. Core was quantified by using Softmax software and compared to a standard curve assayed in parallel.
RT-PCR.
For analysis of revertants and verification of infectious viruses, total cellular RNA was isolated from cells 48 h postinfection by using an RNeasy kit as described above. Approximately 0.2 ng of RNA was converted to first-strand DNA by using SuperScript III first-strand synthesis kit (Invitrogen) and the primers RU-5716 (5′-CTGCGGAACCGGTGAGTACAC-3′), RU-5927 (5′-CAAGGTCTCCCACTTTCAG-3′), RU-5999 (5′-GTGTACCTAGTGTGTGCCGCTCTACC-3′), and RU-6000 (5′-ATGCCGCTAATGAAGTTCCAC-3′). After digestion with 1 U of RNase H for 20 min at 37°C, one-quarter of the RT reaction was PCR amplified with appropriate primers. The PCR products were gel purified using GFX columns (GE, Piscataway, NJ) and used directly for sequencing.
Immunohistochemistry.
Immunohistochemical detection of NS5A was performed as described previously (29). Briefly, cells were fixed with 100% methanol at 48 h postelectroporation, blocked for 1 h with PBS-0.2% milk-1.0% bovine serum albumin-0.1% saponin, and incubated for 5 min with 3% hydrogen peroxide. After being washed with PBS and PBS-0.1% saponin, cells were incubated for 1 h at room temperature with a 1:200 dilution of anti-NS5A MAb (9E10) (29). The secondary antibody, ImmunPress goat anti-mouse peroxidase (Vector, Burlingame, CA), was diluted 1:10 and incubated for 30 min at room temperature. After washing, staining was developed with DAB substrate (Vector) for 5 min. Nuclei were counterstained with Hemotoxylin-2 (Sigma).
Titer by limiting dilution.
Limiting dilution assay was performed as described previously (29). Briefly, clarified cell culture supernatants harvested at various time points postelectroporation were serially diluted, and 50 μl of each dilution used to infect approximately 3 × 103 cells. At 72 h postinfection, cells were fixed, and infected cells were detected by immunohistostaining for NS5A. The 50% tissue culture infective dose (TCID50) was calculated (29).
Intracellular infectivity assay.
Cells were collected at 72 h postelectroporation by treatment with trypsin and pelleting at 500 × g for 3 min. Pellets were washed and resuspended in 500 μl of DMEM-10 μM nonessential amino acids-10% FBS. Cells were lysed by four freeze-thaw cycles, and debris were pelleted by two centrifugations at 1,500 × g for 5 min. The supernatant was collected and used to infect naive cells in the presence of 10 μg/μl of anti-CD81 MAb (JS81; Pharmingen, San Diego, CA) or isotype control MAb (anti-mouse immunoglobulin G1; Pharmingen). After 24 h of infection, the medium was replaced, and after an additional 24 h the cells were lysed in RLT/β-ME, and the total cellular RNA was harvested for qRT-PCR analysis as described above.
RESULTS
Core protein is essential for infectious virus production in HCVcc.
Core protein is thought to be an integral part of the HCV virion. Whether core is actually required for infectious virus production in the HCVcc system, however, is not known. To determine the contributions of various regions of the core protein to infectivity, we engineered in-frame deletions of core between amino acids 57 and 160 into the J6/JFH(p7-Rluc2A) genome, a Renilla luciferase-containing reporter virus that replicates and produces high titers of virus in Huh-7.5 cells (20) (Fig. 1A). Residues before amino acid 57 were not included in the deletion analysis because of conserved RNA structures in this region that are important for RNA replication (37). Similarly, amino acids 161 to 191 were left intact to ensure proper membrane targeting of E1 (46). In vitro-generated RNA transcripts of each genome were electroporated into Huh-7.5 cells. Viral RNA replication was measured by assay of cell lysates for luciferase activity after 8 and 48 h. None of the seven partial deletions of core significantly affected RNA replication compared to wild-type J6/JFH(p7-Rluc2A) (Fig. 1B). In contrast, a genome containing a lethal mutation in the NS5B RNA-dependent RNA polymerase motif GDD to GND, J6/JFH(p7-Rluc2A)/GND, did not replicate.
FIG. 1.
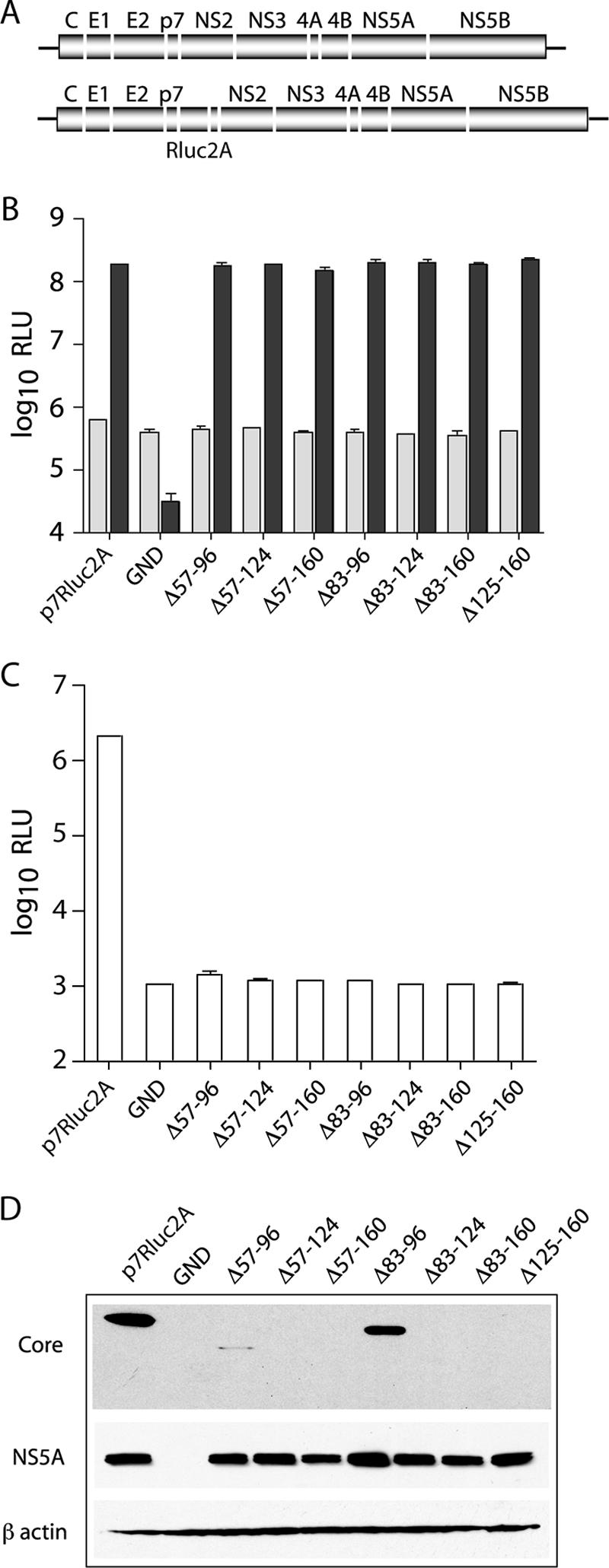
Deletions in core abolish infectious virus production. (A) Schematic representation of the full-length J6/JFH genome (top) and the J6/JFH(p7-Rluc2A) reporter virus (bottom). The J6/JFH(p7-Rluc2A) genome has an insertion of Renilla luciferase and the Foot-and-mouth disease virus 2A peptide between p7 and NS2. (B) Replication of J6/JFH(p7-Rluc2A) genomes with deletions in core. Replication was assayed by luciferase activity at 8 h (light gray) and 48 h (dark gray) postelectroporation. (C) Infectious virus production at 48 h postelectroporation. Means and standard errors of the mean (SEM) of duplicate electroporations are shown. (D) Western blot analysis of lysates harvested at 48 h postelectroporation. Results for core (top panel), NS5A (middle panel), and β-actin (lower panel) are shown.
Infectious particle production was assayed at 48 h postelectroporation by inoculation of naive Huh-7.5 cells with filtered cell culture supernatants. Infectivity was quantified by luciferase assay of the infected cell lysates after 48 h. In contrast to the robust infectious virus production of wild-type J6/JFH(p7-Rluc2A), none of the genomes containing deletions in core produced detectable levels of infectious virus (Fig. 1C). Since the engineered deletions may have led to the degradation of core, we analyzed protein expression levels in transfected cells at 48 h postelectroporation. Core was detected by using MAb HCM-071-5, which recognizes an epitope outside the region of mutagenesis. NS5A was detected by using MAb 9E10 (29). Whereas NS5A expression levels were similar for all replicating genomes, robust core expression was detected only in cells transfected with wild-type J6/JFH(p7-Rluc2A) or with a genome encoding the smallest core deletion, J6/JFH(p7-Rluc2A)/Δ83-96 (Fig. 1D). Extremely low levels of core were also detected in cells transfected with J6/JFH(p7-Rluc2A)/Δ57-96 (Fig. 1D). These data indicated that core protein is essential for infectious virus production and that it is destabilized by large deletions within its central region.
An alanine scan of core amino acids 57 to 191 reveals numerous residues critical for infectious virus production.
To more precisely map regions of core essential for infectious virus production in HCVcc, we performed a comprehensive mutagenesis of amino acids 57 to 191 to alanine. Residues 1 to 56 were again omitted to avoid complicating RNA effects; amino acids 161 to 191, however, were included in this analysis. Core residues were mutated to alanine in blocks of four amino acids in the context of the J6/JFH(p7-Rluc2A) genome. Huh-7.5 cells were electroporated with in vitro-transcribed RNA generated from each construct. RNA replication was measured at various time points postelectroporation by quantification of the luciferase activity in the cell lysates; replication levels at 8 and 48 h are shown (Fig. 2A). In accordance with the results of the deletion analysis, none of the alanine mutations tested significantly affected RNA replication compared to wild-type J6/JFH(p7-Rluc2A), whereas J6/JFH(p7-Rluc2A)/GND did not replicate (Fig. 2A).
FIG. 2.

Alanine scanning mutagenesis of core. (A) Replication of J6/JFH(p7-Rluc2A) genomes with quadruple alanine substitutions in core at 8 h (light gray) and 48 h (dark gray) postelectroporation. (B) Infectious virus production at 48 h postelectroporation. Residues mutated to alanine are identified. Dotted line indicates background luciferase activity. Means and SEM of at least duplicate electroporations are shown.
To assay for infectious virus production, naive Huh-7.5 cells were infected with filtered supernatants harvested at 48 h postelectroporation. Cell-associated luciferase activity was measured at 48 h postinoculation to quantify infectivity. Despite near wild-type levels of replication, the majority of the mutants were significantly impaired in infectious virus production, producing less than 1% of wild-type J6/JFH(p7-Rluc2A) infectivity (Fig. 2B). Exceptions were J6/JFH(p7-Rluc2A)/C181-184A, J6/JFH(p7-Rluc2A)/C185-188A, and J6/JFH(p7-Rluc2A)/C189-191A, which produced close to wild-type levels of infectious virus. RT-PCR and sequencing of viral RNA isolated from cells infected with these viruses verified the presence of the engineered mutations in these genomes (data not shown). These results indicated that numerous residues within the carboxy-terminal two-thirds for the core protein are dispensable for RNA replication but essential for efficient infectious virus production.
Some alanine mutations disrupt core stability.
We hypothesized that the alanine substitutions may have destabilized the core protein, leading to the observed defects in infectious virus production. Cell lysates were analyzed at 48 h postelectroporation for the presence of core and NS5A by Western blotting (Fig. 3). Although all replicating constructs expressed similar levels of NS5A, the stability of core protein varied among the alanine scanning mutants. In mutants that stably expressed core, core levels were much the same as for the wild type. Exceptions were constructs J6/JFH(p7-Rluc2A)/C161-164A and J6/JFH(p7-Rluc2A)/C169-172A, in which core could be detected but was significantly decreased compared to wild-type (Fig. 3, lower panel). With the exception of a possible unprocessed form visible on overexposure of the J6/JFH(p7-Rluc2A)/C169-172A blot (Fig. 3, lower panel), no differences in core processing were detected, although the ability of our gel system to resolve the processing products was not known. It was not surprising that J6/JFH(p7-Rluc2A)/C189-191A did not affect processing by signal peptidase since the native terminal residue of J6 core is an alanine and was therefore left unchanged. None of the mutants with undetectable core protein were among those that produced low levels of infectious virus (Fig. 2B), in keeping with the finding that core expression is essential for infectious virus production. These data indicated that, while about half of the alanine-substitution mutants were blocked at the level of stable core expression, many were severely impaired in infectious virus production despite wild-type levels of core protein.
FIG. 3.

Stability of mutant core proteins. Western blot analysis of lysates harvested at 48 h postelectroporation. Results for core (top panels), NS5A (middle panels), and β-actin (lower panels) are shown. Residues mutated to alanine are identified. The lowest panel is an overexposure of the core blot demonstrating low levels of protein expression. Blots are representative of at least duplicate analyses from independent electroporations.
Alanine mutants do not produce noninfectious particles.
It was possible that mutations in core might lead to defects in entry, such as particle disassembly. Mutants that stably expressed intracellular core were further analyzed to determine whether they produced noninfectious virions. To assess HCV RNA release, total RNA purified from cell culture supernatants at 48 h postelectroporation was analyzed by qRT-PCR (Fig. 4A). Although RNA was detected in the supernatants of all cells harboring replicating genomes, the levels of RNA released by the core mutants were not significantly higher than those secreted by an assembly-defective genome encoding an in-frame deletion of the envelope proteins, J6/JFH(p7-Rluc2A)/ΔE1E2 (20). This release of RNA by replication-competent genomes, regardless of structural protein expression and infectious virus production, has been reported previously (42, 52).
FIG. 4.
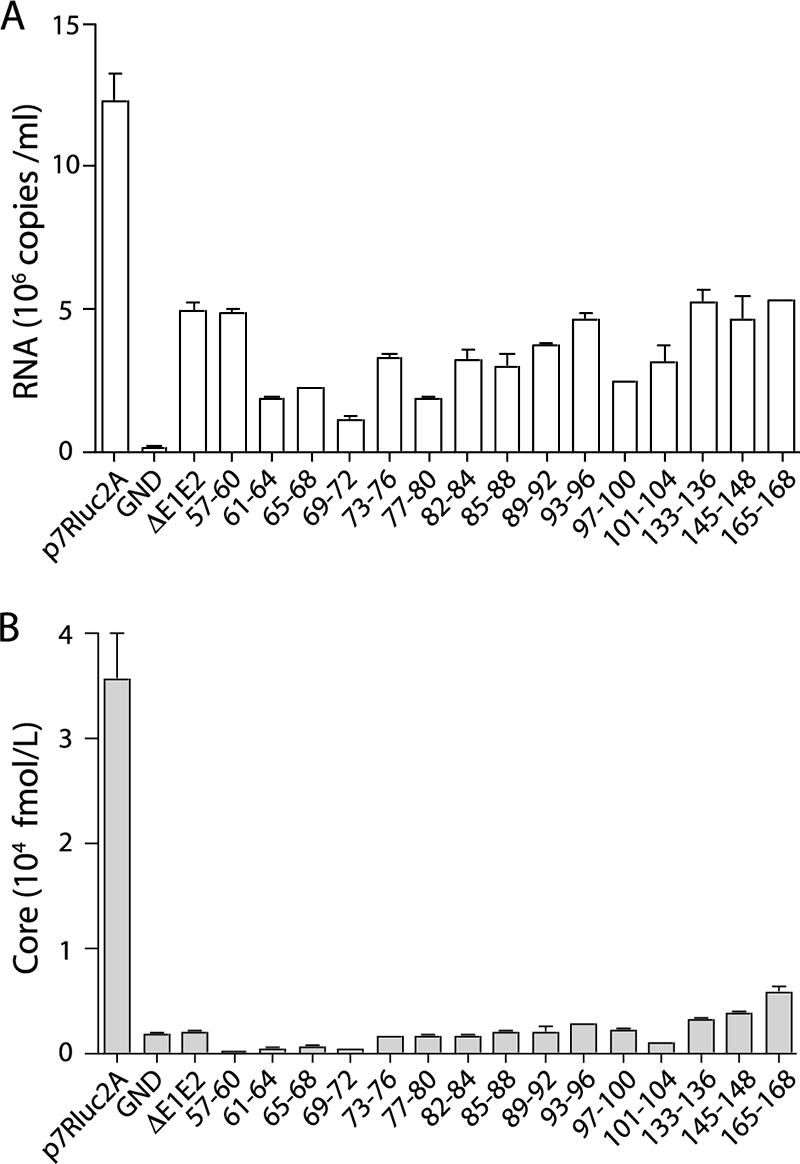
Noninfectious particles are not released by core alanine-substitution mutants. Total viral RNA release (A) and total core protein release (B) at 48 h postelectroporation are shown. Only mutants with stable intracellular core protein were tested; residues mutated to alanine are designated. Means and SEM of at least duplicate electroporations are shown.
To investigate whether the core mutations led to the production of RNA-deficient particles, core secretion was quantified. Cell culture supernatants harvested at 48 h postelectroporation were assayed for core by ELISA (Fig. 4B). None of the core alanine-substitution mutants produced drastically higher levels of secreted core than did J6/JFH(p7-Rluc2A)/ΔE1E2, suggesting that RNA-deficient noninfectious particles were not produced. Taken together, these data indicate that the majority of the core alanine-substitution mutants are defective at a step after the accumulation of core protein and before the release of infectious or noninfectious particles.
Serine 99, but not serines 53 or 116, is essential for infectious virus production.
The alanine scanning mutagenesis included serine residues previously reported to be substrates for phosphorylation (47, 32). To investigate whether the mutation of these serines was responsible for the loss of infectivity of J6/JFH(p7-Rluc2A)/C97-100A and J6/JFH(p7-Rluc2A)/C113-116A, we individually mutated serine 99, serine 116, and, for completeness, serine 53 to alanine. Whereas mutation of these residues did not affect RNA replication (Fig. 5A), mutation of serine 99 to alanine severely impaired infectious virus production (Fig. 5B). S53A and S116A mutations had less significant effects on infectivity and were reduced approximately fivefold compared to the wild type (Fig. 5B). Mutation of each combination of two serines to alanine confirmed that serine 99 was the only residue absolutely required for infectious virus production (data not shown). Mutation of each serine to aspartic acid to mimic constitutive phosphorylation abolished the production of infectious virus in all three cases (data not shown).
FIG. 5.
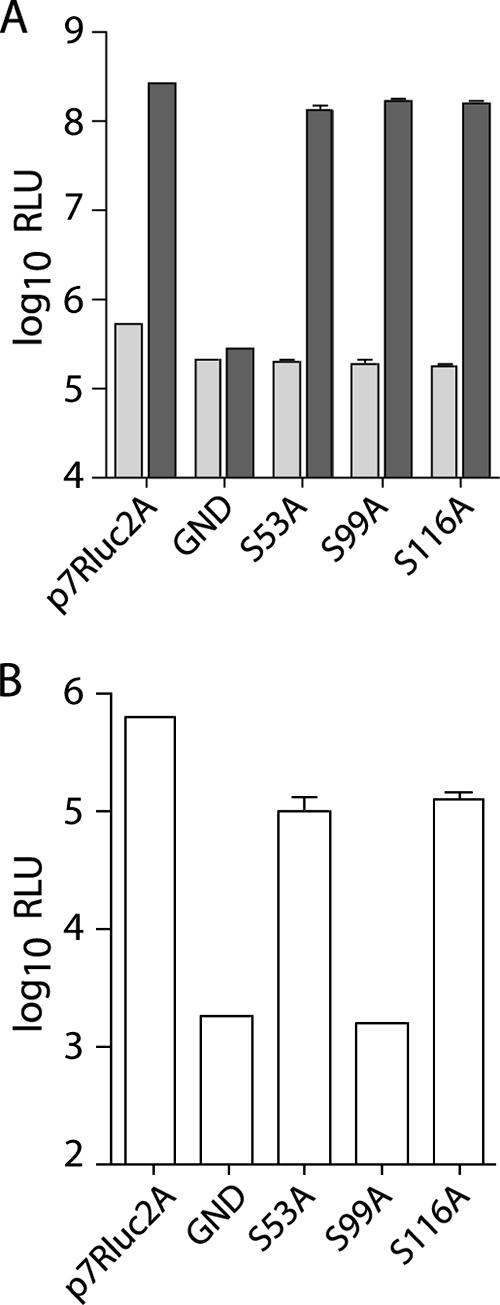
Serine 99 is essential for infectious virus production. (A) Replication of J6/JFH(p7-Rluc2A) genomes with serine-to-alanine substitutions in core at 8 h (light gray) and 48 h postelectroporation (dark gray). (B) Infectious virus production at 48 h postelectroporation. Means and SEM are shown for serine mutants; wild-type and polymerase-defective controls represent single electroporations.
These results suggest that the mutation of serine 99 alone could account for the loss of infectivity seen in J6/JFH(p7-Rluc2A)/C97-100A but that the other two serines are not absolutely required for infectivity. The robust detection of core protein in cells transfected with J6/JFH(p7-Rluc2A)/C97-100A suggests that the S99A mutation does not affect protein stability (Fig. 3). Whether S99 is phosphorylated, and the relevance of this to infectivity, remains to be investigated.
Core alanine-substitution mutants can be rescued by compensatory mutations in the NS proteins.
To identify second site changes that could compensate for the defective core proteins, cells harboring the mutant genomes were serially passaged so as to select for the emergence of spreading virus. Genomes encompassing alanine substitutions between amino acids 57 and 104 were selected for this analysis, since they appeared to be in a region of core in which protein stability was not compromised by the mutations (Fig. 3). In order to avoid the emergence of second site mutations related to the presence of the luciferase reporter gene, each of these quadruple alanine mutations was cloned into the nonreporter J6/JFH genome. For each construct the passaging experiment was conducted in duplicate.
Because initial levels of virus production from these genomes were very low or undetectable, electroporated cells, rather than culture supernatants, were serially passaged (37). Replication at each passage was monitored by immunohistostaining for NS5A. Although J6/JFH electroporated cells maintained a high proportion of NS5A-positive cells, those harboring genomes that could replicate but not produce infectious virus had diminishing quantities of NS5A-positive cells over time (data not shown). For several of the genomes (J6/JFH/C65-68A, J6/JFH/C69-72A, and J6/JFH/C93-96A) the number of NS5A-positive cells either did not diminish as rapidly or else markedly increased during passage, indicating that infectious virus may have emerged. Subsequently, cell-free supernatants from these populations were passaged three times on naive cells. RNA harvested from the infected cells was then RT-PCR amplified, and the population was sequenced over the core to NS5A region. For each of the mutants, the originally engineered alanine substitutions were maintained, and a number of additional mutations were present; those that conveyed an infectious phenotype when individually reengineered into the parental alanine mutant are shown (Fig. 6A).
FIG. 6.

Compensatory mutations in NS proteins are isolated by passaging. (A) Putative topology diagram of the p7 to NS3 region of the HCV polyprotein. Positions of isolated mutations are designated. (B) Infectious virus production at 48 h postelectroporation of core alanine-substitution mutants with or without compensatory mutations. Infectious virus was detected by immunohistostaining for NS5A at 48 h postinoculation of naive cells. Nuclei were counterstained with hemotoxylin-2. Core mutations are designated on the left; additional mutations are described above. All mutations are according to J6/JFH polyprotein numbering.
For J6/JFH/C65-68A, second site mutations emerged at nucleotides U2666C (J6/JFH amino acid F776L, p7 F26L) and A3965G (J6/JFH amino acid T1209A, NS3 T178A). The presence of the F776L mutation significantly increased infectious virus production of J6/JFH/C65-68A from undetectable levels. The NS3 change, T1209A, while allowing detectable infectivity, very weakly rescued the core mutant and was not analyzed further (Fig. 6B). The J6/JFH/C69-72A genome produced very low titers of infectious virus in the context of J6/JFH(p7-Rluc2A) (Fig. 2B). These titers were drastically increased by the presence of either of two independent emergent mutations, U2667C (J6/JFH amino acid F776S, p7 F26S) or G2978C (J6/JFH amino acid A880P, NS2 A67P). Unlike the other core mutants, J6/JFH/C69-72A was the only genome in which both replicate experiments produced revertant viruses, with F776S isolated from one population and A880P from the other. This genome also reverted the most quickly, in three passages as opposed to six (data not shown). The J6/JFH/C93-96A genome also produced very low titers in the J6/JFH(p7-Rluc2A) background (Fig. 2B). Infectious virus production was slightly increased by the emergence of the U2667C (J6/JFH amino acid F776S, p7 F26S) mutation (Fig. 6B).
These data suggest that mutations in the NS proteins are capable of overcoming severely inhibitory mutations in the core protein. It is interesting that a mutation of p7 (F776) emerged independently in each of the passaged genomes. This mutation maps to within the first transmembrane domain (TMD) of p7; the NS2 mutation also maps to a putative transmembrane region (Fig. 6A).
Compensatory changes in p7 and NS2 overcome an infectious particle assembly defect of the parental core mutants.
We next quantitatively analyzed the effects of the emergent NS protein changes on the parental core mutants. Replication was monitored by qRT-PCR analysis of total cellular RNA at various time points after electroporation (Fig. 7A). No significant differences in RNA replication were seen for the core alanine-substitution mutants, in the presence or absence of the compensatory mutations, compared to wild-type J6/JFH or J6/JFH/ΔE1E2; J6/JFH/GND did not replicate. These data suggested that the NS mutations did not act by increasing the replication of the core mutants.
FIG. 7.
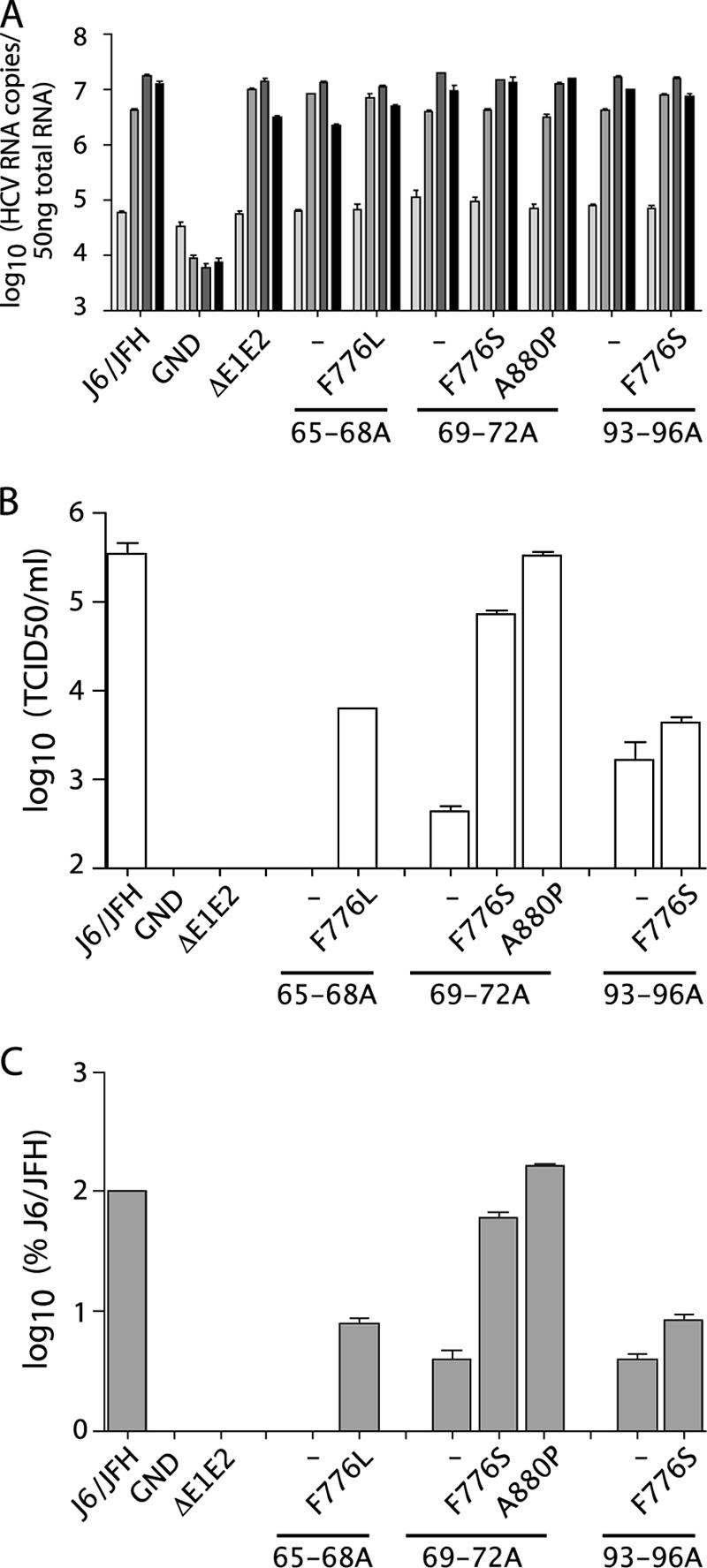
Compensatory mutations rescue core alanine-substitution mutants. (A) Replication of J6/JFH genomes with core alanine substitutions and the presence or absence of compensatory mutations at 8, 24, 48, and 72 h postelectroporation (light gray to black, respectively). Replication was measured by qRT-PCR analysis of RNA accumulation. (B) Infectious virus production at 48 h postelectroporation as measured by limiting dilution assay (TCID50). (C) Intracellular infectious virus accumulation at 72 h postelectroporation as measured by quantification of RNA replication in infected cells by qRT-PCR at 48 h postinoculation. Means and SEM of duplicate electroporations are shown.
Infectious titers of the core alanine-substitution mutants, with or without the compensatory mutations, were quantified by limiting dilution assay. As seen in the context of J6/JFH(p7-Rluc2A), the core mutants produced less than 1% of wild-type titers. J6/JFH/C65-68A was especially impaired, with no detectable infectious virus produced (Fig. 7B). The presence of the second site changes drastically increased the titers of J6/JFH/C65-68A and J6/JFH/C69-72A, with J6/JFH/C69-72A/A880P titers reaching almost wild-type levels. J6/JFH/C93-96A was not as efficiently rescued by the presence of the F776S change, indicating that the same p7 mutation had disparate effects on two different core mutations. J6/JFH/ΔE1E2 and J6/JFH/GND did not produce infectious virus. RNA release correlated with infectious virus titers, suggesting that increased secretion of noninfectious particles did not account for rescue of the core defects (data not shown).
Although the core alanine-substitution mutants were defective in the release of infectious particles, it was possible that they supported assembly of intracellular infectious particles but were defective in egress. To determine whether the compensatory mutations acted at the level of secretion, cells were washed and lysed by multiple freeze-thaw cycles at 72 h postelectroporation. Cleared lysate was used to infect naive cells and infectivity was quantified by analysis of RNA levels in the infected cells after 48 h. Core mutants J6/JFH/C65-68A, J6/JFH/C69-72A, and J6/JFH/C93-96A did not produce significant levels of intracellular infectivity, indicating that these mutants were impaired before infectious particle assembly (Fig. 7C). The presence of the compensatory mutations increased intracellular infectious virus production in proportion to the increased titers.
We further investigated the defect imposed by the C65-68A, C69-72A, and C93-96A mutations by determining the subcellular localization of the mutant core proteins in transfected cells. Similar to wild-type core, the mutant proteins were found to colocalize with lipid droplets (data not shown). These data suggest that the core mutants are blocked at an early step of infectious virion production and that the p7 and NS2 second site changes can overcome this defect. The more drastic effects of the F776L/S mutations on J6/JFH/C65-68A and J6/JFH/C69-72A compared to the slight enhancement of J6/JFH/C93-96A infectivity also suggest that a core-specific mechanism may account for this rescue of infectivity.
Characterization of p7 and NS2 mutations in wild-type virus.
It was possible that the NS protein changes might considerably increase the fitness of wild-type virus and that this enhancement could account for the suppression of the core defects. To test this hypothesis, we engineered the F776L, F776S, and A880P mutations into the wild-type J6/JFH genome and analyzed replication at various times postelectroporation by qRT-PCR (Fig. 8A). None of the NS protein mutants differed significantly in replication kinetics from J6/JFH or J6/JFH/ΔE1E2; J6/JFH/GND did not accumulate viral RNA. This again indicated that the compensatory mutations did not act by increasing RNA replication.
FIG. 8.
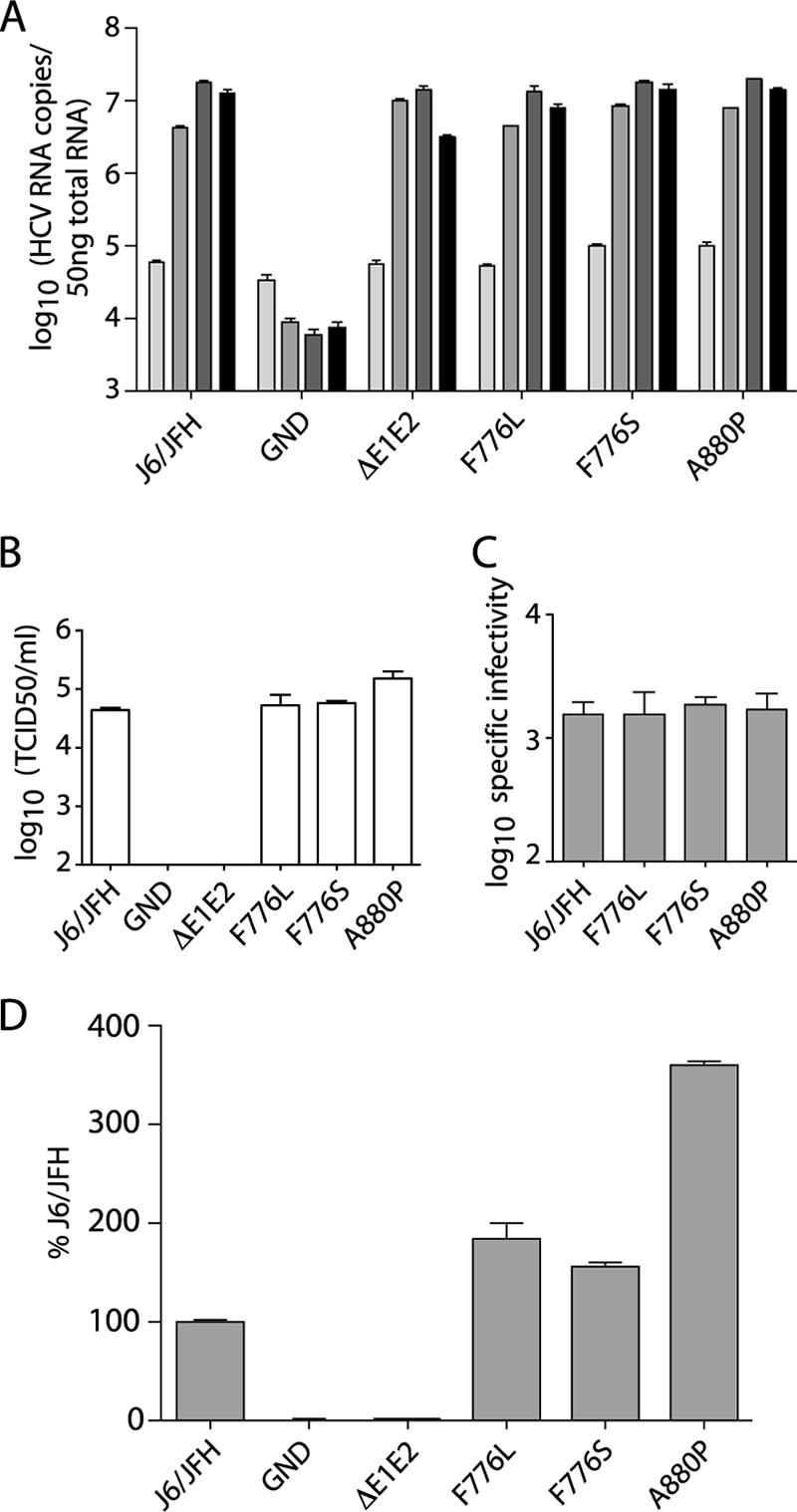
Characterization of compensatory mutations in wild-type virus. (A) Replication of J6/JFH genomes with NS protein mutations at 8, 24, 48, and 72 h postelectroporation; light gray to black, respectively. Replication was measured by qRT-PCR analysis of RNA accumulation. (B) Infectious virus production at 72 h postelectroporation measured by limiting dilution assay (TCID50). (C) Specific infectivity of particles released 72 h postelectroporation as calculated by division of released RNA copies/ml by the TCID50/ml. (D) Intracellular infectious virus accumulation at 72 h postelectroporation as measured by quantification of RNA replication in infected cells by qRT-PCR at 48 h postinoculation. Means and SEM of duplicate electroporations are shown.
Infectious virus titers were determined by limiting dilution assay of cell culture supernatants harvested at 72 h postelectroporation. J6/JFH/F776L and J6/JFH/F776S had titers nearly identical to those of the wild-type virus, whereas J6/JFH/A880P produced titers approximately half a log higher than those of J6/JFH (Fig. 8B). The specific infectivity of the particles was determined by relating released RNA copies/ml to the infectious titer. The specific infectivity of wild-type J6/JFH was similar to what has been previously reported (29) and did not differ significantly from genomes containing the NS protein changes (Fig. 8C). These data indicated that the presence of the p7 and NS2 mutations led to minimal enhancement of infectious virus production in the wild-type genetic background.
We next tested whether the presence of the NS protein changes affected the accumulation of intracellular infectious virus. At 72 h postelectroporation, cells were washed and lysed by multiple freeze-thaw cycles, and the clarified lysates were used to infect naive cells. Infectivity was measured by qRT-PCR analysis of the infected cells after 48 h. The presence of the p7 or NS2 mutations resulted in slight increases in intracellular infectious virus compared to J6/JFH, with J6/JFH/A880P producing almost fourfold more than the wild-type levels (Fig. 8D). The levels of intracellular infectivity correlated well with the released infectious titers.
The minimal enhancement of J6/JFH fitness in the presence of the F776S or F776L changes again suggests that these mutations may specifically compensate for defects in core amino acids 65 to 72 and, to a lesser extent, core 93-96. The A880P mutation, while capable of more significant enhancement of wild-type infectious virus production, may also be involved in a specific genetic interaction with core protein, since the effect of this mutation on J6/JFH/C69-72 is drastically greater than the effect on J6/JFH.
DISCUSSION
The advent of a tissue culture infectious system for HCV has allowed the role of the core protein in authentic virion production to be studied for the first time. Here, we have performed a comprehensive deletion and mutational analysis of core and identified numerous sequences essential for infectivity (summarized in Fig. 9). Consistent with the ability of subgenomes lacking the structural proteins to replicate autonomously (4), we did not identify any core mutants with RNA replication defects. Although core is not required for replication, mutations in conserved RNA structural elements at the 5′ end of the core-coding region can affect replication in a full-length genome (37); these sequences were therefore excluded from our analysis. The possible phenotypic contribution of RNA structure to mutants in the remainder of the core protein was not assessed.
FIG. 9.

Summary of core functional sequences. (A) J6 core amino acid sequence. (B) Previously reported core functional regions (reviewed in reference 35). (C) Summary of results of alanine scanning mutagenesis of residues 57 to 191. Infectious virus production is indicated by shading: black, 50 to 100% J6/JFH(p7-Rluc2A); and gray, 0.3 to 1% wild-type. Mutation of residues highlighted by dashed boxes destabilized the core protein. Isolation of compensatory mutations indicated by stars. Residues mutated to alanine are numbered above.
Despite efficient RNA replication, the majority of core mutants showed severely impaired virion production. In contrast, mutation of the carboxy-terminal residues 181 to 190 did not significantly impact infectious titers. These residues contribute to the E1 signal sequence, a known substrate of SPP (36). The inability of mutations in this region to impact infectivity suggested that SPP cleavage might occur amino terminal to these residues or that the alanine substitutions in this region did not affect the efficiency of SPP processing. Previously, mutation of A180/S183/C184 to V/L/V has been shown to abolish SPP processing of core (36, 2, 16). It is possible that substitution of these amino acids with alanine, rather than with the bulkier leucine or valine, did not disrupt SPP cleavage. Consistent with this hypothesis, transmembrane regions containing helix-disrupting residues with small side chains have been shown to be optimal substrates for SPP (28), with the small side chain deemed sufficient in some reports (50). Alternately, residues 180 to 184 may not play a role in SPP processing in the HCVcc system. Interestingly, we found that the mutation of residues 169 to 172 to alanine led to the production of very low levels of core that appeared to be incompletely processed, although the ability of our gel system to accurately resolve the processed and unprocessed forms of core has not been investigated (16). Notably, this sequence contains three conserved small or helix-disrupting residues (PGC) (7). It is also possible that SPP processing is disrupted by the carboxy-terminal alanine substitutions but that this cleavage is not required for infectious particle production. Although studies of related viruses and analysis of core from HCV-infected patient sera have strongly suggested an essential role for SPP cleavage (15, 50, 55), some studies of nucleocapsid-like particle assembly systems have found SPP processing to be dispensable for budding (33, 51). Also unknown is whether the core signal sequence performs additional roles after its liberation by SPP. The insensitivity of this region to alanine substitutions suggests it likely acts solely as a signal sequence, although it is possible that additional functions can tolerate these mutations.
A number of the deletion or alanine substitution mutations severely affected the expression or stability of the core protein. These low or undetectable levels of core presumably led to the inability of these mutants to produce infectious virus. Several studies have reported that mutations, deletions, or truncations of the central hydrophobic domain of core lead to proteosome-mediated degradation of the protein (17, 49, 5, 36, 19). This instability has been suggested to result from impaired association of these mutants with membranes and lipid droplets (36). The amino-terminal domain of core, recognized as the first 117 to 120 amino acids (6, 35), is highly basic, flexible, and likely functions in the binding and packaging the genomic RNA (6, 27, 26). Not surprisingly, the majority of mutations in this region did not lead to protein destabilization. Interestingly, the sensitivity of residues following amino acid 105 to mutation suggested that the highly flexible domain might be shorter than previously predicted.
Regardless of stable core expression, the majority of alanine-substitution mutants were severely impaired in infectivity. Low levels of infectious virus production were achieved by mutation of some residues between amino acids 69 and 104, although a small deletion in this region (Δ83-96) abolished released infectivity. These residues encompass a putative homotypic interaction domain (residues 86 to 106) (39), which has been shown to be essential for the budding of nucleocapsid-like particles in mammalian cells (19) but not required in a cell-free assembly system (22). Mutation of amino acids 145 to 148 to alanine also permitted a low level of infectious virus production. These amino acids are in a predicted flexible region between two helices (5). Mutation of neighboring residues in this putative loop region resulted in core degradation, possibly because of the replacement of two prolines implicated in lipid droplet association (18).
Interestingly, second site mutations in the NS proteins p7, NS2, and NS3 were found to rescue the infectivity of several core alanine-substitution mutants by overcoming an early defect in virus morphogenesis. p7 is an integral membrane protein with two helical TMDs and a short basic cytosolic loop (9). p7 has been shown to oligomerize, to form ion conductive channels in vitro (14), and to be essential for infectious virus production (20). Here, mutation of p7 residue F776 to L or S was found to dramatically enhance infectious titers of J6/JFH/C65-68A and J6/JFH/C69-72A, respectively, while an independently isolated F776S mutation had a much less significant ability to rescue infectivity of J6/JFH/C93-96A. These observations, along with the meager effects of F776 mutations on wild-type virus, suggested that F776 might be involved in a specific genetic interaction with core amino acids 65 to 72. This p7 residue is well conserved among HCV genotypes, with L, V, or I also represented (24). Interestingly, a sequence encoding the F776S mutation has recently been isolated from a patient infected with a genotype 1a virus (10). Structural modeling of p7 has suggested that F776 resides on the phenylalanine-rich face of TMD1 (9, 11). Both hexameric and heptameric models of p7 oligomeric ion channels predict that F776 faces out of the pore and possibly stabilizes interactions with TMD2 (11, 40). This positioning of F776 could leave it accessible for interactions with other membrane-associated proteins, including core. Interaction of p7 with the structural proteins has recently been suggested by studies of intergenotypic chimeras in which the homology of p7 and upstream sequences enhanced infectious virus production (41). Although a direct interaction of p7 and core might be envisioned for the hydrophobic amino acids 93 to 96, the basic nature of wild-type core amino acids 65 to 72 argues against their accessibility to p7 intramembrane sequences.
Alternately, compensatory mutations in p7 might act to increase interactions of core with any of the numerous factors involved in the production of an infectious particle. Determinants in TMD1 have been shown to influence the delayed or incomplete processing of p7 from both E2 and NS2 (8). Putative disruption of the TMD1 helix or destabilization of interactions between the TMDs might be envisioned to increase processing of p7 intermediates, thereby increasing concentrations of E2 and NS2 available for assembly functions (8). A recent study of core binding to E1 has suggested that this interaction depends on core amino acids 72 to 91 (38), a region closely apposed to the alanine-substitution mutations in all three of the rescued genomes described here. The effect of these core mutations on E1 binding and a possible role for p7 in modulation of this process remains to be investigated.
An independent compensatory mutation in NS2 was isolated by passage of the J6/JFH/C69-72 genome. NS2 is a membrane-associated cysteine protease and immediately follows p7 in the viral polyprotein (13). Although the exact topology of NS2 is disputed, the A880P mutation would be predicted to lie within the third transmembrane or membrane-associated helix (54). NS2 residue 880 is predominantly L, with V also represented and A prevalent in genotype 2 sequences. Proline in this position has not been reported, a finding consistent with its predicted helical environment. Unlike the p7 changes, the A880P mutation significantly increased infectious virus production in the context of a wild-type genome. Although this increase was much less dramatic than the corresponding enhancement of J6/JFH/C69-72 infectivity, it suggested that this adaptive mutation might act solely by overcoming a threshold of viral fitness, thereby suppressing the defective core phenotype. Alternately, the A880 might mediate specific genetic interactions with core amino acids 69 to 72 in addition to a general enhancement of infectivity. Recent studies of intergenotypic chimeras have suggested that optimal infectivity requires NS2 TMD1 and the structural proteins to be of the same genotype, as well as homology between the remainder of NS2 and the replicase proteins (41). Although the normal RNA accumulation of J6/JFH/A880P suggested that interactions required for replication were not affected, association of NS2 with NS3 could be envisioned to impact infectious virus production, as found in the related pestiviruses (1).
A compensatory mutation in NS3 was found to very slightly improve infectivity of J6/JFH/C65-68A, elevating it to detectable levels without affecting RNA replication (Fig. 6B and data not shown). This T1209A change resides between the protease and helicase domains of NS3. A role for NS3 in HCV infectious virus production has recently been alluded to by the isolation of a Q1251L change that enhanced infectivity of an intergenotypic chimera (56). Involvement of NS3 in infectious virus production has also been documented in the related flaviviruses and pestiviruses (1, 25). Interestingly, recent microscopy studies have suggested that NS3 colocalizes with core on lipid droplets (44).
In conclusion, a comprehensive mutagenesis study of the HCV core protein has identified numerous residues critical for its role in virion production. The identification of compensatory mutations in p7 and NS2 that efficiently overcome several defects imposed by core protein mutation illustrates the central role of these NS proteins in virion morphogenesis.
Acknowledgments
We thank Cristian Cruz, Patricia Holst, Michelle Hunter, Erica Machlin, Michelle Moh, Maryline Panis, Merna Torres, and Anesta Webson for laboratory support and technical assistance. We are grateful to Matthew Evans and Martina Kopp for helpful discussions and for critical reading of the manuscript.
This study was supported by The Greenberg Medical Research Institute and the Gates Foundation (FNIH/GCGH 6-37980-G03-07). C.T.V. was supported by NIDDK grant DK081193.
Footnotes
▿
Published ahead of print on 18 July 2007.
REFERENCES
- 1.Agapov, E. V., C. L. Murray, I. Frolov, L. Qu, T. M. Myers, and C. M. Rice. 2004. Uncleaved NS2-3 is required for production of infectious bovine viral diarrhea virus. J. Virol. 78**:**2414-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ait-Goughoulte, M., C. Hourioux, R. Patient, S. Trassard, D. Brand, and P. Roingeard. 2006. Core protein cleavage by signal peptide peptidase is required for hepatitis C virus-like particle assembly. J. Gen. Virol. 87**:**855-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumert, T. F., S. Ito, D. T. Wong, and T. J. Liang. 1998. Hepatitis C virus structural proteins assemble into viruslike particles in insect cells. J. Virol. 72**:**3827-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290**:**1972-1974. [DOI] [PubMed] [Google Scholar]
- 5.Boulant, S., R. Montserret, R. G. Hope, M. Ratinier, P. Targett-Adams, J. P. Lavergne, F. Penin, and J. McLauchlan. 2006. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 281**:**22236-22247. [DOI] [PubMed] [Google Scholar]
- 6.Boulant, S., C. Vanbelle, C. Ebel, F. Penin, and J. P. Lavergne. 2005. Hepatitis C virus core protein is a dimeric alpha-helical protein exhibiting membrane protein features. J. Virol. 79**:**11353-11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bukh, J., R. H. Purcell, and R. H. Miller. 1994. Sequence analysis of the core gene of 14 hepatitis C virus genotypes. Proc. Natl. Acad. Sci. USA 91**:**8239-8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrere-Kremer, S., C. Montpellier, L. Lorenzo, B. Brulin, L. Cocquerel, S. Belouzard, F. Penin, and J. Dubuisson. 2004. Regulation of hepatitis C virus polyprotein processing by signal peptidase involves structural determinants at the p7 sequence junctions. J. Biol. Chem. 279**:**41384-41392. [DOI] [PubMed] [Google Scholar]
- 9.Carrere-Kremer, S., C. Montpellier-Pala, L. Cocquerel, C. Wychowski, F. Penin, and J. Dubuisson. 2002. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J. Virol. 76**:**3720-3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castelain, S., D. Bonte, F. Penin, C. Francois, D. Capron, S. Dedeurwaerder, P. Zawadzki, V. Morel, C. Wychowski, and G. Duverlie. 2007. Hepatitis C Virus p7 membrane protein quasispecies variability in chronically infected patients treated with interferon and ribavirin, with or without amantadine. J. Med. Virol. 79**:**144-154. [DOI] [PubMed] [Google Scholar]
- 11.Clarke, D., S. Griffin, L. Beales, C. S. Gelais, S. Burgess, M. Harris, and D. Rowlands. 2006. Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro. J. Biol. Chem. 281**:**37057-37068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cristofari, G., R. Ivanyi-Nagy, C. Gabus, S. Boulant, J. P. Lavergne, F. Penin, and J. L. Darlix. 2004. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral positive-strand RNA in vitro. Nucleic Acids Res. 32**:**2623-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grakoui, A., D. W. McCourt, C. Wychowski, S. M. Feinstone, and C. M. Rice. 1993. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. USA 90**:**10583-10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffin, S. D., L. P. Beales, D. S. Clarke, O. Worsfold, S. D. Evans, J. Jaeger, M. P. Harris, and D. J. Rowlands. 2003. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 535**:**34-38. [DOI] [PubMed] [Google Scholar]
- 15.Heimann, M., G. Roman-Sosa, B. Martoglio, H. J. Thiel, and T. Rumenapf. 2006. Core protein of pestiviruses is processed at the C terminus by signal peptide peptidase. J. Virol. 80**:**1915-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hope, R. G., M. J. McElwee, and J. McLauchlan. 2006. Efficient cleavage by signal peptide peptidase requires residues within the signal peptide between the core and E1 proteins of hepatitis C virus strain J1. J. Gen. Virol. 87**:**623-627. [DOI] [PubMed] [Google Scholar]
- 17.Hope, R. G., and J. McLauchlan. 2000. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J. Gen. Virol. 81**:**1913-1925. [DOI] [PubMed] [Google Scholar]
- 18.Hope, R. G., D. J. Murphy, and J. McLauchlan. 2002. The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J. Biol. Chem. 277**:**4261-4270. [DOI] [PubMed] [Google Scholar]
- 19.Hourioux, C., M. Ait-Goughoulte, R. Patient, D. Fouquenet, F. Arcanger-Doudet, D. Brand, A. Martin, and P. Roingeard. 2007. Core protein domains involved in hepatitis C virus-like particle assembly and budding at the endoplasmic reticulum membrane. Cell Microbiol. 9**:**1014-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones, C. T., C. L. Murray, D. K. Eastman, J. Tassello, and C. M. Rice. 2007. Hepatitis C virus p7 and NS2 proteins are essential for infectious virus production. J. Virol. 81**:**8374-8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato, T., T. Date, M. Miyamoto, A. Furusaka, K. Tokushige, M. Mizokami, and T. Wakita. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125**:**1808-1817. [DOI] [PubMed] [Google Scholar]
- 22.Klein, K. C., S. R. Dellos, and J. R. Lingappa. 2005. Identification of residues in the hepatitis C virus core protein that are critical for capsid assembly in a cell-free system. J. Virol. 79**:**6814-6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein, K. C., S. J. Polyak, and J. R. Lingappa. 2004. Unique features of hepatitis C virus capsid formation revealed by de novo cell-free assembly. J. Virol. 78**:**9257-9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuiken, C., K. Yusim, L. Boykin, and R. Richardson. 2005. The Los Alamos hepatitis C sequence database. Bioinformatics 21**:**379-384. [DOI] [PubMed] [Google Scholar]
- 25.Kümmerer, B., and C. M. Rice. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious virus. J. Virol. 76**:**4773-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kunkel, M., M. Lorinczi, R. Rijnbrand, S. M. Lemon, and S. J. Watowich. 2001. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J. Virol. 75**:**2119-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunkel, M., and S. J. Watowich. 2004. Biophysical characterization of hepatitis C virus core protein: implications for interactions within the virus and host. FEBS Lett. 557**:**174-180. [DOI] [PubMed] [Google Scholar]
- 28.Lemberg, M. K., and B. Martoglio. 2002. Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol. Cell 10**:**735-744. [DOI] [PubMed] [Google Scholar]
- 29.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309**:**623-626. [DOI] [PubMed] [Google Scholar]
- 30.Lindenbach, B. D., H. J. Thiel, and C. M. Rice. 2007. Flaviviridae: viruses and their replication, p. 1101-1152. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 1. Lippincott-Raven Publishers, Philadelphia, PA. [Google Scholar]
- 31.Lo, S.-Y., M. J. Selby, and J.-H. Ou. 1996. Interaction between hepatitis C virus core protein and E1 envelope protein. J. Virol. 70**:**5177-5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu, W., and J. H. Ou. 2002. Phosphorylation of hepatitis C virus core protein by protein kinase A and protein kinase C. Virology 300**:**20-30. [DOI] [PubMed] [Google Scholar]
- 33.Majeau, N., V. Gagne, M. Bolduc, and D. Leclerc. 2005. Signal peptide peptidase promotes the formation of hepatitis C virus non-enveloped particles and is captured on the viral membrane during assembly. J. Gen. Virol. 86**:**3055-3064. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto, M., S. B. Hwang, K.-S. Jeng, N. Zhu, and M. M. C. Lai. 1996. Homotypic interaction and multimerization of hepatitis C virus core protein. Virology 218**:**43-51. [DOI] [PubMed] [Google Scholar]
- 35.McLauchlan, J. 2000. Properties of the hepatitis C virus core protein: a structural protein that modulates cellular processes. J. Viral Hepat. 7**:**2-14. [DOI] [PubMed] [Google Scholar]
- 36.McLauchlan, J., M. K. Lemberg, G. Hope, and B. Martoglio. 2002. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 21**:**3980-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McMullan, L. K., A. Grakoui, M. J. Evans, K. Mihalik, M. Puig, A. D. Branch, S. M. Feinstone, and C. M. Rice. 2007. Evidence for a functional RNA element in the hepatitis C virus core gene. Proc. Natl. Acad. Sci. USA 104**:**2879-2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakai, K., T. Okamoto, T. Kimura-Someya, K. Ishii, C. K. Lim, H. Tani, E. Matsuo, T. Abe, Y. Mori, T. Suzuki, T. Miyamura, J. H. Nunberg, K. Moriishi, and Y. Matsuura. 2006. Oligomerization of hepatitis C virus core protein is crucial for interaction with the cytoplasmic domain of E1 envelope protein. J. Virol. 80**:**11265-11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nolandt, O., V. Kern, H. Muller, E. Pfaff, L. Theilmann, R. Welker, and H. G. Krausslich. 1997. Analysis of hepatitis C virus core protein interaction domains. J. Gen. Virol. 78**:**1331-1340. [DOI] [PubMed] [Google Scholar]
- 40.Patargias, G., N. Zitzmann, R. Dwek, and W. B. Fischer. 2006. Protein-protein interactions: modeling the hepatitis C virus ion channel p7. J. Med. Chem. 49**:**648-655. [DOI] [PubMed] [Google Scholar]
- 41.Pietschmann, T., A. Kaul, G. Koutsoudakis, A. Shavinskaya, S. Kallis, E. Steinmann, K. Abid, F. Negro, M. Dreux, F. L. Cosset, and R. Bartenschlager. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 103**:**7408-7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pietschmann, T., V. Lohmann, A. Kaul, N. Krieger, G. Rinck, G. Rutter, D. Strand, and R. Bartenschlager. 2002. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 76**:**4008-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ray, R. B., and R. Ray. 2001. Hepatitis C virus core protein: intriguing properties and functional relevance. FEMS Microbiol. Lett. 202**:**149-156. [DOI] [PubMed] [Google Scholar]
- 44.Rouille, Y., F. Helle, D. Delgrange, P. Roingeard, C. Voisset, E. Blanchard, S. Belouzard, J. McKeating, A. H. Patel, G. Maertens, T. Wakita, C. Wychowski, and J. Dubuisson. 2006. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 80**:**2832-2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakai, A., M. S. Claire, K. Faulk, S. Govindarajan, S. U. Emerson, R. H. Purcell, and J. Bukh. 2003. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl. Acad. Sci. USA 100**:**11646-11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santolini, E., G. Migliaccio, and N. La Monica. 1994. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 68**:**3631-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shih, C.-M., C.-M. Chen, S.-Y. Chen, and Y.-H. W. Lee. 1995. Modulation of the _trans_-suppression activity of hepatitis C virus core protein by phosphorylation. J. Virol. 69**:**1160-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimoike, T., S. Mimori, H. Tani, Y. Matsuura, and T. Miyamura. 1999. Interaction of hepatitis C virus core protein with viral sense RNA and suppression of its translation. J. Virol. 73**:**9718-9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki, R., K. Tamura, J. Li, K. Ishii, Y. Matsuura, T. Miyamura, and T. Suzuki. 2001. Ubiquitin-mediated degradation of hepatitis C virus core protein is regulated by processing at its carboxyl terminus. Virology 280**:**301-309. [DOI] [PubMed] [Google Scholar]
- 50.Targett-Adams, P., T. Schaller, G. Hope, R. E. Lanford, S. M. Lemon, A. Martin, and J. McLauchlan. 2006. Signal peptide peptidase cleavage of GB virus B core protein is required for productive infection in vivo. J. Biol. Chem. 281**:**29221-29227. [DOI] [PubMed] [Google Scholar]
- 51.Vauloup-Fellous, C., V. Pene, J. Garaud-Aunis, F. Harper, S. Bardin, Y. Suire, E. Pichard, A. Schmitt, P. Sogni, G. Pierron, P. Briand, and A. R. Rosenberg. 2006. Signal peptide peptidase-catalyzed cleavage of hepatitis C virus core protein is dispensable for virus budding but destabilizes the viral capsid. J. Biol. Chem. 281**:**27679-27692. [DOI] [PubMed] [Google Scholar]
- 52.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11**:**791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.World Health Organization. 1997. Hepatitis C: global prevalence. Wkly. Epidemiol. Rec. 72**:**341-344. [PubMed] [Google Scholar]
- 54.Yamaga, A. K., and J. H. Ou. 2002. Membrane topology of the hepatitis C virus NS2 protein. J. Biol. Chem. 277**:**33228-33234. [DOI] [PubMed] [Google Scholar]
- 55.Yasui, K., T. Wakita, K. Tsukiyama-Kohara, S. I. Funahashi, M. Ichikawa, T. Kajita, D. Moradpour, J. R. Wands, and M. Kohara. 1998. The native form and maturation process of hepatitis C virus core protein. J. Virol. 72**:**6048-6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yi, M., Y. Ma, J. Yates, and S. M. Lemon. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81**:**629-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102**:**9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]