Huntingtin–HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington's disease (original) (raw)
Abstract
The molecular mechanisms underlying the targeting of Huntingtin (Htt) to endosomes and its multifaceted role in endocytosis are poorly understood. In this study, we have identified Htt-associated protein 40 (HAP40) as a novel effector of the small guanosine triphosphatase Rab5, a key regulator of endocytosis. HAP40 mediates the recruitment of Htt by Rab5 onto early endosomes. HAP40 overexpression caused a drastic reduction of early endosomal motility through their displacement from microtubules and preferential association with actin filaments. Remarkably, endogenous HAP40 was up-regulated in fibroblasts and brain tissue from human patients affected by Huntington's disease (HD) as well as in STHdhQ111 striatal cells established from a HD mouse model. These cells consistently displayed altered endosome motility and endocytic activity, which was restored by the ablation of HAP40. In revealing an unexpected link between Rab5, HAP40, and Htt, we uncovered a new mechanism regulating cytoskeleton-dependent endosome dynamics and its dysfunction under pathological conditions.
Introduction
Huntington's disease (HD) is a neurodegenerative disorder caused by expansion of the CAG repeat in the gene encoding Huntingtin (Htt), which confers to the protein an expanded NH2-terminal polyglutamine (polyQ) stretch of >35 residues (for review see Harjes and Wanker, 2003). The function of Htt is largely unclear. It has been shown to interact with microtubules (Hoffner et al., 2002) and to display anti-apoptotic activity (Rigamonti et al., 2000, 2001). Insights into its function came from studies of Htt-interacting proteins (HIPs) and Htt-associated proteins (HAPs). For example, interactions with HIP1, HIP1R, PACSIN1, SH3GL3, and HIP14 have implicated Htt in clathrin-mediated endocytosis. Studies of HAP1 have suggested a role for Htt in axonal transport in neurons by linking vesicles to the dynein–dynactin motor complex (Block-Galarza et al., 1997; Engelender et al., 1997). The polyQ expansion confers the adjacent proline-rich sequence in Htt alterations in binding affinity to HIPs/HAPs. Thus, release from or sequestration of these molecules by mutant Htt has been implicated in the pathogenetic mechanisms.
For example, the tighter binding of HAP1 to mutant Htt is thought to impair the correct dynactin–dynein motor complex assembly and cause a trafficking defect, leading to neuronal degeneration (Block-Galarza et al., 1997; Engelender et al., 1997; Gauthier et al., 2004). Consistently, mutant Htt was recently shown to release dynein from microtubules and reduce the motility of EGFP–brain-derived neurotrophic factor–containing vesicles in vivo (Gauthier et al., 2004). However, the upstream events that target Htt and its partners to their various sites of function and the mechanisms whereby they regulate intracellular trafficking remain elusive.
In this study, we report an unexpected link between Htt and the small GTPase Rab5 via the adaptor protein HAP40. Rab5 is a key regulator of endocytosis that orchestrates the recruitment of multiple effector proteins on the early endosome membrane to regulate organelle tethering, fusion, and microtubule-dependent motility (Zerial and McBride, 2001). Our data extend the analysis of the Rab5 effector machinery by functionally implicating the interaction between Rab5 and Htt in the regulation of the differential association of early endosomes with the actin and tubulin cytoskeleton.
Results
The Htt–HAP40 complex is a novel Rab5 effector
Affinity chromatography revealed several downstream effectors of the small GTPase Rab5 (Christoforidis et al. 1999). Surprisingly, among the proteins specifically eluted from the GST-Rab5–GTPγS but not from the Rab5–guanosine diphosphate (GDP) nor GST-Rab4 affinity column, we identified Htt and HAP40 (Peters and Ross, 2001) by mass spectrometry and immunoblotting (Fig. 1, A and B). Therefore, we investigated the function of HAP40 and Htt with respect to Rab5.
Figure 1.
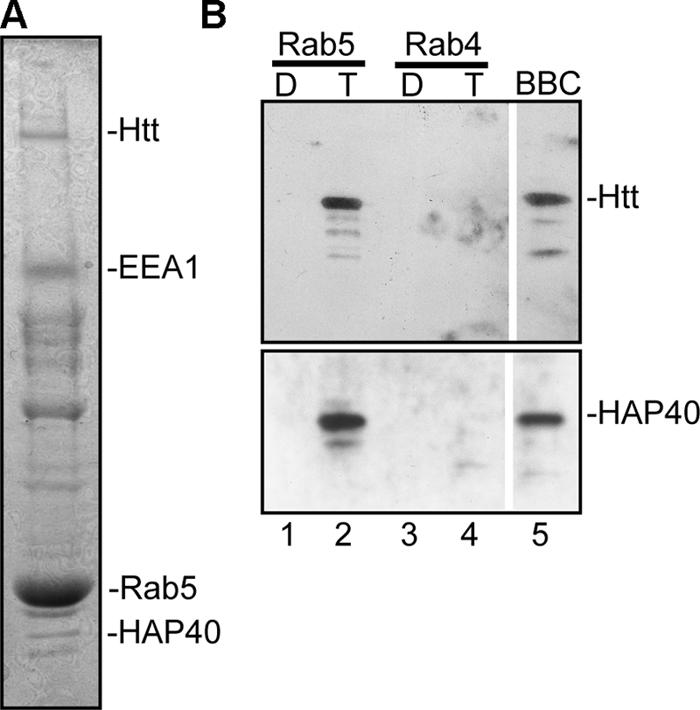
Htt and HAP40 elute from immobilized Rab5. (A) SDS-PAGE of proteins eluted from immobilized GST-Rab5 that was loaded with GTPγS. The indicated bands were found to correspond to Htt, HAP40, Rab5-GST, and EEA1 by mass spectrometry analysis. (B) Western blot analysis of chromatographic eluates from GST-Rab5 (lanes 1 and 2) and -Rab4 (lanes 3 and 4) affinity columns preloaded with GDP (D; lanes 1 and 3) or GTPγS (T; lanes 2 and 4). The bovine brain cytosol (BBC; lane 5) was used as a source of proteins. Blots were probed for Htt and HAP40 as indicated.
We first tested whether HAP40, Htt, or both interact directly and specifically with Rab5. To this end, full-length HAP40 was cloned from a rat brain cDNA library and in vitro translated. Because of its large molecular mass (348 kD; Fig. 2 A), fragments of wild-type Htt were translated to facilitate the analysis. In vitro translation yielded major products of predicted size as well as lower molecular mass bands presumably as a result of internal initiation (Fig. 2 B). Immobilized Rab5-GST fusion protein preloaded with either GDP or GTPγS were incubated with the translation products and washed, and bound proteins were eluted with glutathione analyzed by SDS-PAGE and autoradiography (Christoforidis et al., 1999). Similar to early endosome antigen 1 (EEA1), which served as a positive control, HAP40 displayed specific binding to GTPγS- versus GDP-bound Rab5 (Fig. 2 C, compare lanes 1 and 11 with lanes 2 and 12). In contrast, none of the Htt fragments exhibited significant binding to Rab5 (Fig. 2 C, lanes 3–10). Because Htt was purified on the GST-Rab5 affinity column, we next tested whether binding of Htt to Rab5 occurs indirectly and requires HAP40 as a bridge. HAP40 and Htt fragments were cotranslated in vitro (Fig. 2 B, lanes 6–9) and applied on the Rab5 columns. Indeed, the COOH-terminal part of Htt (Fig. 2 A, Htt4) was eluted together with HAP40 from GTPγS- but not GDP-bound GST-Rab5 (Fig. 2 D, lanes 7 and 8). None of the other Htt fragments displayed such binding (Fig. 2 D, lanes 1–6), which is consistent with the reported interaction map between HAP40 and Htt (Peters and Ross, 2001). Neither the Htt fragments nor HAP40 displayed binding to Rab4, Rab6, Rab7, or Rab11 (Fig. 2 D), suggesting the interaction with Rab5 is specific. Thus, we conclude that HAP40 binds to the COOH-terminal part of Htt and links the complex to active Rab5. By applying a 10-fold excess of the COOH-terminal fragment of Htt onto the Rab5 column to reduce binding of free HAP40 to Rab5 (Fig. 2 E), we estimated the stoichiometry of the Rab5/HAP40/Htt interaction in this assay to be ∼1:1:1 (see Rab5 affinity…cloning).
Figure 2.

Htt–HAP40 complex is a novel Rab5 effector. (A) Schematic 23 glutamine residues (23Q), the adjacent proline-rich sequence (P), the caspase-3 cleavage site, and the four recombinant fragments (Htt1–4) cloned in pcDNA3.1 using the indicated restriction sites. (B) Autoradiograph of in vitro–translated Htt fragments (Htt1–4) and HAP40. Full-length HAP40 cDNA and Htt fragments were in vitro translated in the presence of [35S]methionine, separated by SDS-PAGE, and autoradiographed. Given their large size, the bands of strongest intensity (arrows) and corresponding to the predicted masses for HAP40 (∼40 kD, lane 1), Htt1 (∼65 kD, lane 2), Htt2 (∼97 kD, lane 3), Htt3 (∼84 kD, lane 4), and Htt4 (∼102 kD, lane 5) were accompanied by multiple products because of either initiation of translation at internal sites or premature termination. Lanes 6–9: each Htt fragment was cotranslated with HAP40. (C) Autoradiograph of the in vitro–translated proteins in B eluted from immobilized GST-Rab5 preloaded with GTPγS (T) or GDP (D). (D) Autoradiograph of Htt fragments cotranslated in vitro with HAP40 and eluted from various immobilized Rab proteins as indicated. The experiment was performed as described in C, but Htt fragments and HAP40 were cotranslated (see B, lanes 6–9) and applied onto immobilized Rab proteins. Besides HAP40, the COOH-terminal Htt fragment (Htt4) was eluted from GST-Rab5 beads (compare lanes 7 with 8), whereas Htt1 (lanes 1 and 2), Htt2 (lanes 3 and 4), and Htt3 (lanes 5 and 6) did not show significant specific association with Rab5. Moreover, none of the Htt fragments or HAP40 displayed specific interactions with GST-Rab4, 6, 7, or 11. Positive controls (POS, lanes 9 and 10) for each Rab protein were EEA1 (∼170 kD) for Rab5, Rabenosyn-5 (∼89 kD) for Rab4 (de Renzis et al., 2002), VPS39 (∼100 kD) for Rab7 (Rink et al., 2005), and GapCenA (∼115 kD) for Rab6 and 11 (Cuif et al., 1999 and unpublished data). (E) Htt and HAP40 elute in equimolar amounts from the Rab5 column. A mixture of Htt4 and HAP40 was applied onto Rab5 columns and eluted as in D but with a 10-fold excess of Htt4 to prevent HAP40 from binding freely to Rab5. (C–E) Arrows point to correct translation products as in B.
Htt is recruited onto early endosome in a HAP40- and Rab5-dependent manner
We began testing the functional relevance of this interaction in vivo by immunofluorescence microscopy analysis of HeLa cells. First, we verified that the anti-HAP40 antibody resulted in specific staining above background levels for detection of the endogenous antigen (Fig. 3 A). Endogenous HAP40 displayed a diffuse staining in the cytoplasm and accumulated in the nucleus, whereas endogenous Htt localized to discrete cytoplasmic structures (Fig. 3 B) as reported previously (Peters and Ross, 2001; Tao and Tartakoff, 2001). Colocalization of endogenous HAP40 and Htt was hardly detectable (Fig. 3 B). Early endosomes labeled with EGFP-Rab5 displayed little colocalization with endogenous Htt (7 ± 5% overlap; n = 10; Fig. 3 C). However, the association of HAP40 with early endosomes dramatically increased upon overexpression. In cells overexpressing HAP40 (see Fig. 7 D) but not EGFP-Rab5 alone, HAP40 significantly colocalized with endogenous Htt on EGFP-Rab5–positive early endosomes (Fig. 3, compare B with D; endogenous Htt and EGFP-Rab5: 43 ± 6% overlap, n = 10; HAP40 and EGFP-Rab5: 31 ± 7% overlap, n = 10; Fig. 3, compare C with D). We experimentally verified that the HAP40 fluorescence signals (Fig. 3 D) were not caused by bleed-through of the Htt signals (because of extended AlexaFluor568 emission in the Cy5 channel) and that swapping these fluorescent dyes on the secondary antibodies resulted in similar distribution patterns of Htt and HAP40 (see Cell culture procedures). The endosomal colocalization of endogenous HAP40 and Htt was even more striking upon expression of the activated EGFP-Rab5Q79L mutant (Htt: 52 ± 7% overlap, n = 8), which caused the characteristic swelling of early endosomes (Fig. 4 A; Stenmark et al., 1994).
Figure 3.

Overexpressed HAP40 recruits Htt onto early endosomes. Immunofluorescence microscopy analysis of HeLa cells expressing EGFP-Rab5 and/or HAP40 and immunostained for HAP40 and Htt as indicated. (A) The anti-HAP40 antibody specifically recognizes its antigen (green). Cells were stained with the antibody alone (anti-HAP40) or premixed with HAP40-GST fusion (anti-HAP40 & GST-HAP40) or GST protein (anti-HAP40 & GST). The image after specific depletion of the antibody (anti-HAP40 & GST-HAP40) was obtained at sixfold prolonged exposure time compared with the others. (B) Endogenous Htt (red) and endogenous HAP40 (green); <1% overlapping (n = 10). (C) Endogenous Htt (red) and EGFP-Rab5 (green); 7% overlapping (SD ± 5%, n = 10). (D) Overexpressed HAP40 (red) recruits endogenous Htt (red) onto endosomes labeled with EGFP-Rab5WT (green); 43% overlapping of endogenous Htt with EGFP-Rab5 (merge Rab5 & Htt; SD ± 6%, n = 10) and 31% overlapping of overexpressed HAP40 with EGFP-Rab5 (merge Rab5 & HAP40; SD ± 7%, n = 10). Cells were cotransfected with HAP40 and GFP-Rab5WT expression constructs. Arrowheads in insets (magnified images of boxed areas) highlight colocalization. Bar, 10 μm. (E) Western blot analysis of early endosomes prepared from untreated (WT) or HeLa cells overexpressing HAP40 (HAP40). Blots were probed for HAP40, Htt, EEA1, transferrin receptor (Tfr), LAMP1, and GM130 as indicated.
Figure 7.

Elevated protein levels of HAP40 shift early endosomes from microtubules to actin filaments, causing a severe decrease of motility in vivo. HeLa cells and human primary fibroblasts expressing EGFP-Rab5 were imaged using time-lapse video microscopy. (A–C) Images generated by merging a stack of overlaid images collected at 300-ms intervals over 2 min, as for Fig. 5 B. Videos corresponding to A–C are available as online supplemental materials (Videos 1–5, available at http://www.jcb.org/cgi/content/full/jcb.200509091/DC1). (A) In HeLa cells, coexpression of EGFP-Rab5 and HAP40 (right) led to a drastic reduction in endosome motility compared with EGFP-Rab5 alone (WT, left). (B) In fibroblasts from HD patients (HD), such reduction was also evident from the comparison with fibroblasts from healthy individuals (WT). (C) RNAi against HAP40 in these HD fibroblasts (HAP40 RNAi) restores endosome motility. HD fibroblast cell lines were cotransfected with EGFP-Rab5 expression vector and siRNA against HAP40. (D) Similar to HeLa cells overexpressing HAP40 (compare lane 1 with lane 2), endogenous protein levels of HAP40 were elevated in five HD fibroblast cell lines (lanes 8–12) compared with fibroblasts from five healthy individuals (lanes 3–7). Blots were also probed with anti-EEA1 and anti-Htt antiserum to confirm equal loading and the identity of the HD cell lines (see Results). (E) Knockdown of HAP40 protein levels in fibroblasts from all five HD cell lines by RNAi. Western blot analysis of EEA1 and HAP40 in untransfected cells (lanes 1, 4, 7, 10, and 13), cells transfected with unrelated siRNA against GFP (lanes 2, 5, 8, 11, and 14), or HAP40 siRNA (lanes 3, 6, 9, 12, and 15). (F) EGFP-Rab5–labeled endosomes align primarily with microtubules in healthy fibroblasts. Primary human fibroblasts from healthy individuals were transfected with EGFP-Rab5 (green) and fixed and immunostained for microtubules and F-actin. The same cell is shown with its β-tubulin staining (red) on the left and for F-actin (red) on the right, as indicated. Arrowheads in the insets (magnifications of the boxed areas) point to endosomes aligned with microtubules. Overlap of EGFP-Rab5 with tubulin signals was 82% (SD ± 9%, n = 10) for healthy fibroblasts and 15% (SD ± 6%, n = 9) for HD cell lines (see G). (G) The cell processed as in F shows that EGFP-Rab5–labeled endosomes align strikingly with F-actin in fibroblasts from HD patients. Arrowheads in the insets point to endosomes (green) aligned with F-actin (red). Overlap of EGFP-Rab5 with actin was 2% (SD ± 1%, n = 10) for healthy fibroblasts (see F) and 44% (SD ± 8%, n = 10) for HD cell lines. Bar, 10 μm.
Figure 4.

Htt is recruited onto early endosomes in a Rab5- and HAP40-dependent fashion. Immunofluorescence microscopy analysis of HeLa cells transfected with expression vectors for EGFP-Rab5Q79L alone or cotransfected with siRNA duplexes against HAP40 as indicated. (A) EGFP-Rab5Q79L (green) recruits endogenous Htt (red) onto endosomes; 52% overlapping (SD ± 7%, n = 8). Cells were transfected with EGFP-Rab5Q79L expression construct alone. (B) RNAi against HAP40 (siHAP40) leads to the loss of endogenous Htt (red) from endosomes; 8% overlapping (SD ± 6%, n = 9). Cells were cotransfected with HAP40 siRNA and EGFP-Rab5Q79L (green) expression constructs. Bar, 10 μm. (C) Knockdown of HAP40 protein by RNAi as shown by Western blot analysis. EEA1 and HAP40 expression levels in untransfected cells (control) and cells transfected with unrelated (siGFP) or siRNA against HAP40 (siHAP40).
Given the difficulties detecting endogenous HAP40 and Htt on early endosomes in untreated cells (Fig. 3, B and C) we sought to verify the changes in the localization of both proteins upon the overexpression of HAP40 biochemically. To this end, we prepared early endosomes from HeLa cells for Western blot analysis. Indeed, we found that the levels of HAP40 and Htt increased on early endosomes from the HAP40 overexpressor compared with untreated cells (Fig. 3 E). Early endosome (EEA1 and transferrin receptor [Tfr]) as well as lysosomal (lysosome-associated membrane protein (LAMP); Eskelinen et al., 2003) and Golgi (GM130; Nakamura et al., 1995) markers remained unchanged, confirming equal loading and the specificity of changes through elevated HAP40 on early endosomes.
To confirm the requirement of HAP40 for the recruitment of Htt onto early endosomes, we transfected HeLa cells with short interfering RNA (siRNA) duplexes against HAP40 and unrelated siRNA (against GFP) as control. The HAP40 siRNA specifically and efficiently reduced the protein levels by ∼90% (Fig. 4 C), whereas the level of EEA1 remained unchanged. When cells were cotransfected with the expression vector for EGFP-Rab5Q79L and HAP40 siRNA, Htt was no longer detectable on the enlarged endosomes (8 ± 6% overlap, n = 9; Fig. 4 B), confirming HAP40 as a prerequisite to bridge Htt to active Rab5. Collectively, these data suggest that active Rab5 and HAP40 are rate limiting for the recruitment of Htt onto early endosomes.
The Htt–HAP40 complex inhibits the binding and motility of early endosomes on microtubules
Htt has previously been shown to bind microtubules and regulate microtubule–motor interactions (Block-Galarza et al., 1997; Engelender et al., 1997; Hoffner et al., 2002; Gauthier et al., 2004). Because Rab5 regulates endosome motility along microtubules (Nielsen et al., 1999; Hoepfner et al., 2005), we explored the role of the Htt–HAP40 complex in this process. First, by using a cell- and cytosol-free assay that recapitulates the Rab5-dependent movement of early endosomes along microtubules (Hoepfner et al., 2005), we found that Htt–HAP40 inhibited microtubule-dependent early endosome motility. Addition of the GTPγS- (containing Htt and HAP40; Fig. 1 B) but not the GDP-loaded Rab5 column eluate reduced the motility compared with control conditions (Fig. 5 A). This inhibition was specifically rescued with anti-Htt but not unrelated antibodies. The addition of 1 μM GST-HAP40 fusion protein completely blocked the in vitro motility (Fig. 5, A and B). Second, we performed a biochemical early endosome–microtubule-binding assay as described previously (Nielsen et al., 1999) with some modifications to improve the quantitative assessment. An early endosome–enriched fraction was prepared from HeLa cells pulsed with rhodamine–transferrin, incubated with taxol-stabilized microtubules, ATP, and factors to be tested, and centrifuged through a sucrose cushion. The resulting pellet of microtubule-associated material was analyzed by immunoblotting (Fig. 6 A) and fluorimetrically for the rhodamine–transferrin content (Fig. 6 D). In a dilution series for calibration, we verified that the amount of endosomes and fluorescence intensity correlated linearly (see Microtubule and actin spin-down assays). Western blotting revealed that the β-tubulin content was similar between samples, ruling out secondary effects on microtubule stability (Fig. 6 A, compare lanes 2–5 with lane 1). The addition of 1 μM GST-HAP40 protein decreased the amount of early endosomes in the pellet as revealed by EEA1 and Tfr (Fig. 6 A, compare lane 3 with lane 2). The GST-Rab5–GTPγS column eluate adding ∼0.3 μM HAP40 (Fig. 6 C) caused a similar inhibition (Fig. 6A, compare lane 4 with lane 2) that was rescued through the addition of antiserum against Htt (Fig. 6 A, compare lane 5 with lanes 4 and 2). To rule out the idea that the observed differences result from the bundling of microtubules causing unspecific cosedimentation of any membranous structure, we probed the pellets for nonendosomal contaminants in the fraction. Both the lysosomal (LAMP1) and Golgi marker (GM130) pelleted with similar efficiency in all samples. Collectively, these data indicate that Htt–HAP40 specifically lowers the binding of early endosomes to microtubules.
Figure 5.
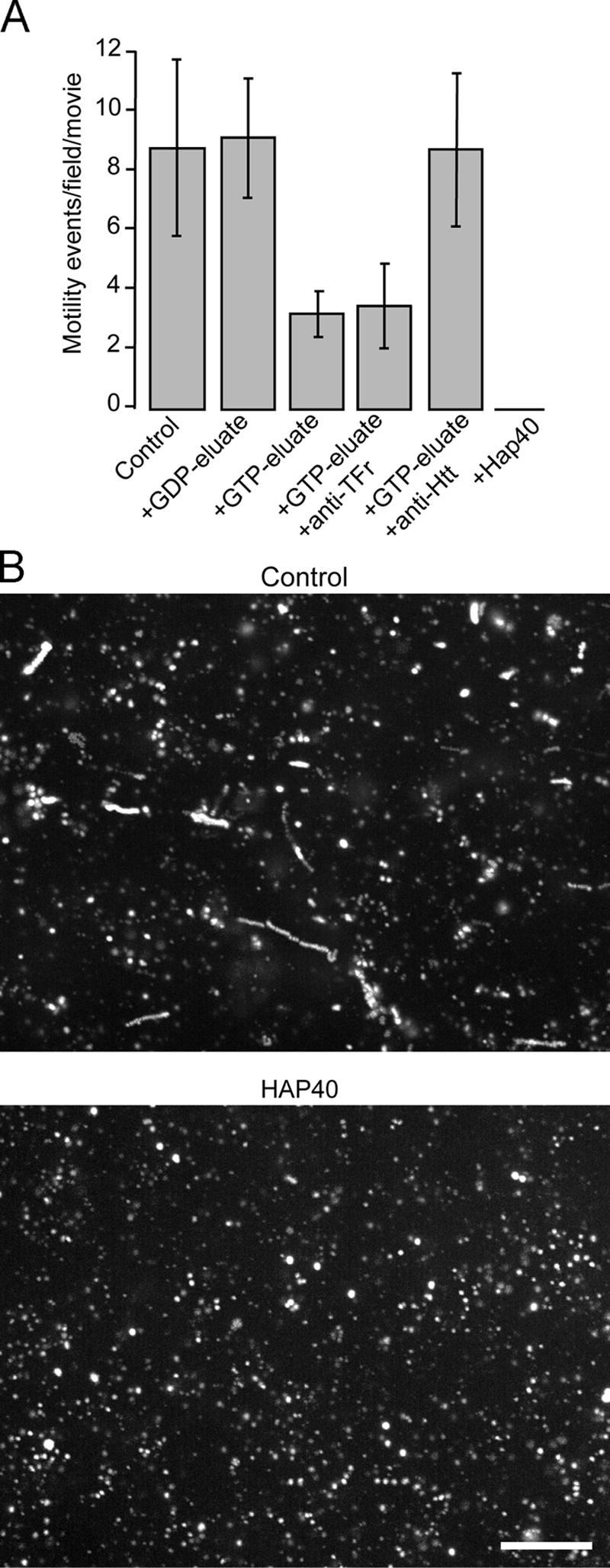
Htt and HAP40 decrease in vitro reconstituted motility of early endosomes along microtubules. (A) Purified early endosomes labeled by internalization of rhodamine–transferrin were mixed with buffer (control) alone or with various eluates of bovine brain cytosol proteins that were affinity purified on GST-Rab5 columns. Eluate obtained from columns loaded with GDP was added directly to the sample (+GDP-eluate). Eluate obtained from columns loaded with GTPγS was added directly to the sample (+GTP-eluate) or after preincubation with antiserum against the cytoplasmic domain of Tfr (+eluate + anti-Tfr), antiserum against Htt (+eluate + anti-Htt), or without eluate but with 1 μM of recombinant GST-HAP40 fusion protein (+HAP40). In vitro motility of early endosomes along microtubules recorded using time-lapse fluorescence video microscopy (see Materials and methods) was quantified by counting motility events per video. Error bars show the SD of 10 videos. (B) Videos recorded under control conditions (control) or with 1 μM GST-HAP40 fusion protein (HAP40) used for the analysis in A are displayed as merged stacks of overlaid images collected at 2-s intervals over 2 min. When represented in this manner, a moving object will generate a trajectory consisting of a linear series of overlapping spots. Bar, 10 μm.
Figure 6.

Modulation of binding of early endosomes to microtubules and F-actin by Htt and HAP40. (A) Reduction of binding of early endosomes (EE) to microtubules in vitro caused by Htt and HAP40. A spin-down assay was performed (see Materials and methods), and the resulting pellet of microtubule-associated material was analyzed by immunoblotting with antibodies against proteins indicated on the right. (B) Stimulation of binding of early endosomes to F-actin in vitro caused by Htt and HAP40. A spin-down assay was performed as described for A but with 10 μg of freshly polymerized F-actin replacing the microtubule. (A and B) Microtubules (A) or F-actin (B) spun alone (−EE, lane 1), with early endosomes (control; +EE, lane 2), with early endosomes and 1 μM GST-HAP40 fusion protein (+EE+HAP40, lane 3), with early endosomes and 10 μg GST-Rab5–GTPγS column eluate (+EE+eluate, lane 4), or eluate preincubated with anti-Htt antiserum (+EE+eluate+anti-Htt, lane 5). (C) Comparison of HAP40 protein levels in samples prepared for the early endosome–microtubule/actin-binding assays. Samples were prepared as described for A and B either with GST-HAP40 fusion (lane 1) or GST-Rab5–GTPγS column eluate (lane 2). 10-μl aliquots of each sample were separated by SDS-PAGE and immunoblotted for HAP40. Bands corresponded to the correct masses of GST-HAP40 or HAP40 as indicated on the right. Nonrelevant lanes on the same blot were sliced out in Adobe Photoshop to juxtapose the lanes shown. (D) Quantifications of early endosomes bound to microtubules. Binding was performed as described for A, but the resulting pellet of microtubule-associated material was lysed to release the rhodamine–transferrin label of early endosome. Fluorescence (arbitrary units) served as a direct measure for the amount of early endosomes bound to microtubules. Error bars represent the SD of samples in triplicate. Binding was performed in the presence of 15 μg of early endosomes and 16 μg of microtubules (control), in the absence of microtubules (−MT), in the absence of early endosomes (−EE), in the absence of ATP (−ATP), with all three components omitted (buffer), with 2 mM adenylyl-imidodiphosphate (+AMP-PNP), with 1 μM of recombinant RN-tre (+RN-tre), with 1 μM Rab-GDI (+GDI), with 1 μM HAP40-GST fusion protein (+HAP40), with 1 μM GST (+GST), with 10 μg GST-Rab5–GTPγS column eluate (+eluate), or with eluate preincubated with anti-Htt antiserum (+eluate+anti-Htt). (E) Quantifications of early endosomes bound to F-actin. Binding was performed as described for B, and quantifications were made as in D. Binding was performed in the presence of 15 μg of early endosomes, 10 μg of F-actin (control), and with the addition of various components as in D.
Having validated the assay, we next quantified the amount of endosomes bound to microtubules fluorimetrically. Omission of either microtubules or early endosomes reduced the fluorescence signal to background levels (Fig. 6 D). As reported previously (Nielsen et al., 1999), the association of early endosomes with microtubules was energy dependent and required active Rab5. Omitting ATP or substituting it with the nonhydrolyzable adenylyl-imidodiphosphate analogue resulted in an ∼50% reduction in microtubule binding. The requirement for ATP can be explained by the role of PI3-K in the assembly and maintenance of a functional Rab5 domain on endosomes (Zerial and McBride, 2001) and in recruitment of the endosomal kinesin KIF16B (Hoepfner et al., 2005). Extraction of Rab proteins from membranes by the addition of 1 μM of recombinant Rab–GDP dissociation inhibitor (GDI; Ullrich et al., 1994) or treatment with RN-tre, a GTPase-activating protein for Rab5 (Lanzetti et al., 2000), caused an ∼40% reduction of endosomes in the pellet (Fig. 6 D). As for endosome motility (Fig. 5 A), Htt and HAP40 inhibited endosome–microtubule binding. The GST-Rab5–GTPγS column eluate led to an ∼25% reduction in binding (Fig. 6 D), but supplementing the reaction with antibodies against Htt restored binding to control levels. The addition of 1 μM HAP40-GST fusion protein decreased the binding by ∼60%, whereas GST alone did not have any effect. Collectively, these data underpin the ability of HAP40 and Htt to destabilize endosome–microtubule association.
We next performed time-lapse video microscopy studies to correlate these biochemical findings in vitro with the regulation of early endosome dynamics in vivo. We detected a drastic reduction in motility of EGFP-Rab5–positive endosomes in HeLa cells overexpressing HAP40 (Fig. 7 A) compared with cells transfected with EGFP-Rab5 alone (Fig. 7 A and Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200509091/DC1). Whereas some residual motility activity was observed in the cell periphery, early endosomes in the perinuclear region appeared static, with frequent short-range movements almost completely impaired in long-range motility (Nielsen et al., 1999, Rink et al. 2005). Collectively, these data suggest that the Rab5-dependent recruitment of Htt onto endosomes by HAP40 disrupts early endosome–microtubule interactions, thus leading to a reduction in organelle motility.
HD cells display increased levels of HAP40 and are impaired in early endosome motility
We next asked whether alterations of endosome motility could occur in cells bearing the HD mutation. Primary fibroblasts from five healthy individuals and five unrelated HD patients were transfected with EGFP-Rab5 to compare the motility of early endosomes. The identity of the HD cell lines was confirmed by Western blotting to detect the polyQ-expanded Htt. Because the cells were derived from patients heterozygous for the HD gene, they express both normal and mutant Htt. The latter is known to display a lower mobility in SDS-PAGE (Trottier et al., 1995), thus causing a doublet on the blot (Fig. 7 D). Strikingly, we observed a severe reduction in early endosome motility in all HD cell lines compared with fibroblasts from healthy individuals (Fig. 7 B and Videos 3 and 4, available at http://www.jcb.org/cgi/content/full/jcb.200509091/DC1). The similarity between this phenotype and the alterations induced upon HAP40 overexpression in HeLa cells (Fig. 7 A and Videos 1 and 2) hinted to a common molecular basis. Interestingly, we discovered an ∼10-fold up-regulation of endogenous HAP40 levels by Western blotting in all HD cell lines compared with normal fibroblasts (Fig. 7 D). As a control, the levels of EEA1 remained unchanged. Moreover, we found that HAP40 protein levels were significantly elevated in striatal tissue (caudate, putamen, accumbens, and globus pallidus) from human postmortem brains affected by HD (adult onset grade) compared with control brains (Fig. 8 B). Our data suggest that the motility block in HD cells may be caused by elevated HAP40 levels, as mimicked by overexpression of HAP40 in HeLa cells. Consistently, endogenous Htt localized to EGFP-Rab5–labeled endosomes in fibroblasts from human HD patients as well as striatal STHdhQ cells from a HD mouse model (Trettel et al., 2000) but not in cells lacking the mutant Htt (Fig. 9). This phenotype is also caused by overexpressed HAP40 in HeLa cells (Fig. 3 D).
Figure 8.

Elevated endogenous HAP40 protein levels impair Rab5 dynamics in vivo in STHdhQ111/111 and STHdhQ7/111 striatal cells from a HD mouse model. (A) Cells expressing normal (STHdhQ7/7) or mutant (STHdhQ111/111 and STHdhQ7/111) Htt were transfected with EGFP-Rab5 plasmid, differentiated, and imaged using time-lapse video microscopy. Overlaid images were generated as described for Fig. 5 B. Videos corresponding to A are available as online supplemental material (Videos 6–8, available at http://www.jcb.org/cgi/content/full/jcb.200509091/DC1). Motility of Rab5 compartments was drastically reduced in STHdhQ111/111 and STHdhQ7/111 compared with STHdhQ7/7 cells. Insets show magnifications of boxed areas. Bar, 10 μm. (B) Endogenous protein levels of HAP40 are elevated in STHdhQ111/111 (lane 1) and STHdhQ7/111 (lane 2) compared with STHdhQ7/7 cells (lane 3) as well as in striatal tissue from five human postmortem brains affected by HD (lanes 4–8) compared with healthy control brains (lanes 9–13). Blots were also probed with anti-EEA1 and anti-Htt antiserum to confirm equal loading and the expression of wild-type and mutant Htt as described for Fig. 7 D.
Figure 9.

Htt and HAP40 are recruited onto Rab5 vesicles in primary fibroblasts from human HD patients and STHdhQ7/111 striatal cells from a HD mouse model. (A) Image gallery of the same fibroblast cell from a healthy individual transfected with EGFP-Rab5 (green) and fixed and immunostained for endogenous HAP40 (red) and Htt (red). Boxed areas are magnified in the top panels. (B) The same analysis of a fibroblast from a HD patient. Yellow (arrowheads) indicates colocalization of EGFP-Rab5 and Htt (merge Rab5 and Htt,) or of EGFP-Rab5 and HAP40 (merge Rab5 and HAP40). Overlapping of EGFP-Rab5 with Htt was 6% (SD ± 3%, n = 10) for fibroblasts from healthy individuals (A) and 46% (SD ± 8%, n = 10) from HD patients (B). Overlapping of EGFP-Rab5 with HAP40 was 4% (SD ± 3%, n = 10) for fibroblasts from healthy individuals (A) and 51% (SD ± 9%, n = 10) from HD patients (B). (C) Image gallery of the same differentiated STHdhQ7/7 cell (homozygous for normal Htt) transfected with EGFP-Rab5 (green) and immunostained for endogenous HAP40 (red) and Htt (red). (D) The same analysis of a differentiated STHdhQ7/111 cell (heterozygous for mutant Htt). Overlapping of EGFP-Rab5 with Htt was 5% (SD ± 4%, n = 10) for STHdhQ7/7 cells (C) and 83% (SD ± 12%, n = 10) for STHdhQ7/111 cells (D). Overlapping of EGFP-Rab5 with HAP40 was 4% (SD ± 3%, n = 10) for STHdhQ7/7 cells (C) and 86% (SD ± 15%, n = 10) for STHdhQ7/111 cells (D). The analysis was restricted to cellular outgrowths. Essentially the same results were obtained for STHdhQ111/111 cells (homozygous for mutant Htt; not depicted). Bar, 10 μm.
Our data do not exclude the possibility that the observed inhibition of early endosome motility may have other underlying causes. As a test to our hypothesis, we attempted to rescue the inhibition of early endosome motility by specifically ablating HAP40 from the HD fibroblasts by RNA interference (RNAi). Transfection of HAP40 siRNA efficiently reduced the HAP40 protein levels in these cells (Fig. 7 E) as in HeLa cells (Fig. 4 C). Indeed, endosome motility was restored by RNAi against HAP40 (Fig. 7 C and Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200509091/DC1) but not against GFP, suggesting that the up-regulation of HAP40 is indeed the underlying mechanism of the motility defect in HD fibroblasts.
Given the ability of HAP40 to reduce endosome–microtubule binding in vitro (Fig. 6, A and D), we investigated whether the observed motility block in HD cell lines was caused by a release of endosomes from microtubules in vivo. Immunofluorescence analysis on cells transfected with EGFP-Rab5 showed a considerable alignment of early endosomes to microtubules in healthy fibroblasts (82 ± 9% overlap, n = 10) but to a much lesser extent in HD cell lines (15 ± 6% overlap, n = 9; Fig. 7, compare F with G). In contrast, early endosomes were strikingly aligned with filamentous actin (F-actin) in all five HD cell lines (44 ± 8% overlap, n = 10) but not in healthy fibroblasts (2 ± 1% overlap, n = 10; Fig.7, compare G with F). A similar phenotype was obtained by the overexpression of HAP40 in HeLa cells (not depicted).
The Htt–HAP40 complex enhances the binding of early endosomes to actin
The alignment of early endosomes on F-actin in HD cell lines could be a secondary effect from the inhibition of endosome–microtubule interactions. To directly test the role of HAP40 in the association between early endosomes and actin filaments (Fig. 6, B and E), we modified the biochemical sedimentation assay used to study endosome–microtubule interactions (Fig. 6, A and D) by replacing taxol-stabilized microtubules with freshly in vitro–polymerized F-actin. Unlike for endosome–microtubule binding, depletion of active Rab5 from endosomal membranes by treatment with either Rab-GDI or RN-tre did not decrease endosome–actin interactions (Fig. 6 E). Thus, a basal level of endosome–F-actin binding activity was independent of Rab5. However, the addition of HAP40-GST fusion protein stimulated binding (∼260%) over control levels, whereas GST alone was ineffective. Evidently, endosome binding to microtubules and F-actin is reciprocally regulated through HAP40 because concentrations (1 μM) inhibiting binding to microtubules (Fig. 6 D) clearly stimulated binding to F-actin (Fig. 6 E). This effect was Rab5 dependent because when Rab-GDI or RN-tre were added together with HAP40, the stimulation was nearly abolished (Fig. 6 E). Again, immunoblotting confirmed that the amount of actin in the pellets was unaffected by any added protein and that all changes in pelletable fluorescence corresponded consistently to altered band intensities of early endosomal markers (EEA1 and TFr) but not to others (LAMP1 and GM130), indicating specific effects of Htt–HAP40 on endosome–actin binding (Fig. 6 B).
Alterations in early endosome motility in a HD mouse model of striatal cells
Next, we investigated the role of HAP40 in Rab5 dynamics in an experimental system that is more relevant for HD using immortalized STHdhQ striatal cells (Trettel et al., 2000). These cells were established from embryonic normal or HD knock-in mice and either express normal (STHdhQ7/7) or mutant Htt as a result of a CAG expansion inserted into the endogenous Htt gene (heterozygous STHdhQ7/111 and homozygous STHdhQ111/111). Thus, they reflect the closest situation to HD patients as normal, and mutant Htt are expressed at endogenous levels. Remarkably, we again found endogenous HAP40 protein levels elevated in STHdhQ7/111 and STHdhQ111/111 compared with STHdhQ7/7 cells (Fig. 8 B), which is consistent with the data on HD fibroblasts (Fig. 7 D) and brain tissue from HD patients (Fig. 8 B). Live cell imaging revealed EGFP-Rab5–positive organelles moving bidirectionally in neuronlike outgrowths as well as in the cell body of normal STHdhQ7/7 cells (Fig. 8 A and Video 6, available at http://www.jcb.org/cgi/content/full/jcb.200509091/DC1). Conversely, STHdhQ111/111 and STHdhQ7/111 cells clearly displayed a drastic reduction of endosome dynamics (Fig. 8 A and Videos 7 and 8), which is consistent with the observations on HD fibroblasts (Fig. 7 B and Videos 3 and 4). Collectively, defects of Rab5 dynamics caused by pathogenic excess of HAP40 apparently occur in peripheral tissues such as fibroblasts as well as in neuronal systems and, therefore, are of potential relevance for HD.
Increased HAP40 levels cause alterations in endocytic trafficking
To gain some insights into possible alterations of endocytic transport caused by increased levels of HAP40, we tested the uptake of transferrin in HeLa cells overexpressing HAP40 and in fibroblasts from healthy and HD patients. Fig. 10 A shows that the uptake of transferrin was reduced by 30% in HeLa cells overexpressing HAP40 compared with mock-transfected or control cells. Consistent with this result, fibroblasts from HD patients displaying higher levels of endogenous HAP40 (Fig. 7 D) displayed a similarly reduced uptake of transferrin compared with normal fibroblasts (Fig. 10 B). As for the block of endosome motility, the inhibitory effect on transferrin uptake was rescued by the depletion of HAP40 by RNAi. These data suggest that the inhibitory effects on endosome motility caused by up-regulation of HAP40 also result in defects in cargo transport through the endocytic pathway.
Figure 10.

Elevated levels of HAP40 reduces transferrin uptake. (A) HeLa cells transfected with HAP40 expression (HAP40), empty plasmid (mock), or untreated (WT) were serum starved, allowed to internalize biotinylated transferrin (Tf) for the indicated times, washed, and lysed, and internalized transferrin was quantified. The mean values of triplicate samples from one representative experiment out of three with SD (error bars, often omitted by plot symbols) are shown. (B) The same uptake for primary human fibroblasts untreated or transfected with siRNA duplexes against HAP40 (siHAP40) or unrelated GFP (siGFP). The mean values of different cell lines from five healthy persons (5× WT), five HD patients (5× HD), the same but treated with siHAP40 (5× HD siHAP40), or the mean values of triplicate sample obtained from one HD line treated with unrelated siGFP (1× HD siGFP) are shown. Error bars (mostly omitted by plot symbols) represent the SD between cell lines (5× WT, 5× HD, and 5× HD siHAP40) or of triplicate samples (1× HD siGFP).
Discussion
The key finding of this study is that the complex between HAP40 and Htt is a direct downstream effector of Rab5 that regulates the dynamics of early endosomes through a switch from microtubules to F-actin. These findings provide important new insights into how the motility of early endosomes is regulated under physiological and pathological conditions. Htt has been implicated in clathrin-mediated endocytosis, regulation of the actin cytoskeleton, and microtubule-dependent transport along the endocytic pathway via interactions with its numerous binding partners (Harjes and Wanker, 2003). Such multiplicity of roles implies that the activities of Htt in endocytic membrane trafficking need to be spatially and temporally coordinated. In showing for the first time that a complex between Htt and one of its binding partners, HAP40, can be recruited onto endosomes by interacting directly with Rab5, our data provide novel insights into the mechanisms governing the targeting of Htt to early endosomes and its regulatory activity on cytoskeleton-dependent dynamics.
Under normal physiological conditions, early endosomes undergo frequent short-range movements on actin but also long-range bidirectional movements along microtubules (Nielsen et al., 1999; Gasman et al., 2003; Rink et al. 2005). Plus end movement of early endosomes along microtubules is propelled by KIF16B (Hoepfner et al. 2005). Overexpression of HAP40, which is rate limiting for the recruitment of Htt on the membrane, caused the detachment of early endosomes from microtubules and their preferential association with actin filaments, thus limiting both their velocity and range of movements. Therefore, the Htt–HAP40–Rab5 complex is a key regulator of the switch from one type of filaments to another. Our data are consistent with previous studies documenting alterations in microtubule-dependent motility in HD model systems (Block-Galarza et al., 1997; Engelender et al., 1997; Gauthier et al., 2004). However, these studies have exclusively implicated alterations between mutant Htt and HIP/HAP effectors. For example, wild-type Htt has been shown to interact with the dynactin subunit p150_Glued_ via HAP1 and mutant Htt to disrupt the dynein motor complex in axonal transport (Gauthier et al., 2004). In this study, we have uncovered a different mechanism based on the up-regulation of an Htt adaptor, HAP40. Compelling evidence in support of this mechanism was provided by the rescue of the motility block upon depletion of HAP40 by RNAi both in HD fibroblasts and in striatal cells. Because the COOH-terminal part of Htt is responsible for the underlying interactions with HAP40 and Rab5, this endosomal recruitment affects normal as well as mutant Htt. Consistently, wild-type (overexpression of HAP40 in HeLa cells), heterozygous (STHdhQ7/111), and homozygous (STHdhQ111/111) HD cells display very similar phenotypes. In this way, functional competition with the Rab5-dependent endosomal kinesin KIF16B and disruption of the dynein–dynactin complex (Gauthier et al., 2004) could account for the compromised bidirectional microtubule-based motility. Therefore, our data suggest that multiple defects may contribute to the block of vesicular transport in HD cells, which implicates various HIP/HAPs interacting with different regions of Htt.
Previous findings have shown that Htt is also linked to Rab8 through FIP-2 (Hattula and Peranen, 2000) and that this interaction regulates cell polarization and morphogenesis. Whereas the mechanism underlying the latter process is unclear, in light of our observation, we propose that Htt is a multifunctional protein that may be recruited by different Rab GTPases through different adaptors at different intracellular locations to regulate organelle dynamics.
The primary pathological cause of HD is attributed to the abnormal activity of Htt and its interacting partners in the nucleus (Landles and Bates, 2004). The majority of HAP40 is also nuclear under normal conditions (Fig. 3; Peters and Ross, 2001), although the functional significance of this localization is unclear at present. Our data indicate that HAP40 fulfills a function in the cytoplasm. It is interesting in this respect to note that HAP40 adds to the increasing list of proteins implicated in a dual role in endocytic trafficking and nuclear signaling (Miaczynska et al., 2004). The molecular mechanism underlying the up-regulation of HAP40 unexpectedly observed in cells and brain tissue from HD patients remains to be determined. The most likely explanation is that it arises as a consequence of alterations in gene expression caused by mutated Htt in the nucleus. Messenger RNA microarray studies have revealed many transcriptional abnormalities in HD (Chan et al., 2002; Sipione et al., 2002; Sugars and Rubinsztein, 2003), although no changes for HAP40 or any other Htt-interacting partners have been reported so far. Up-regulation of HAP40 at the protein level might thus serve as a new diagnostic indicator for the disease. However, it is premature to judge to what extent this abnormality and the consequent alterations in membrane dynamics contribute to the pathogenetic mechanism of HD as compared with nuclear activities of Htt and HAP40. Although endosome motility is not perturbed in HD cells to the extent of compromising cell viability, we nevertheless uncovered defects in endocytic transport, specifically a decrease in transferrin uptake. These observations may be of particular functional relevance in neurons given the importance of long-range transport (Block-Galarza et al., 1997; Engelender et al., 1997; Gauthier et al., 2004). Therefore, it is possible that alterations in endosome motility and transport may further compromise the pathological state induced by mutated Htt on the survival and function of neurons or some selected populations of neurons where endosomal transport is particularly rate limiting in vivo. Addressing this hypothesis requires a more thorough investigation of HAP40 and Htt function in the transport of different types of cargo along the endocytic/recycling as well as degradative pathway in neuronal cells rather than fibroblasts. A deeper analysis of the role of HAP40 on the molecular level will hopefully improve our understanding of both HD pathogenesis and the mechanisms coordinating organelle–cytoskeleton interactions during membrane trafficking.
Materials and methods
Rab5 affinity chromatography and HAP40 and Htt cloning
GST-Rab5 affinity chromatography with bovine brain cytosol or in vitro–translated proteins was performed as described previously (Christoforidis et al., 1999). HAP40 was cloned from rat brain cDNA by PCR, Htt fragments were cloned by PCR on full-length Htt cDNA in pBlueScript (a gift from M. Sherman, Boston University School of Medicine, Boston, MA), and all PCR products were cloned into pcDNA3.1 (Invitrogen) and sequenced to confirm their identity. To estimate the stoichiometry of the bound complex, GST-Rab5 was incubated in the presence of a 10-fold excess of the COOH-terminal fragment of Htt over HAP40. Band intensities obtained under these conditions were quantified in ImageJ software (National Institutes of Health; Htt4/HAP40 = ∼6:1) and corrected for the number of [35S]methionine residues incorporated (Htt4/HAP40 = 22:4). The resulting ratio was ∼1:1. Assuming a 1:1 ratio of Rab protein to effector, the stoichiometry of the Rab5/HAP40/Htt interaction was estimated at ∼1:1:1.
Antibodies and recombinant proteins
Monoclonal mouse antibodies against Htt were obtained from Chemicon and Y. Trottier (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Université Louis Pasteur, Illkirch, France), and polyclonal rabbit antibody against HAP40 was obtained from Chemicon. Microtubules were revealed with mouse monoclonal anti–β-tubulin antibody (BD Biosciences), and F-actin was revealed with AlexaFluor568-conjugated phalloidin (Invitrogen). Secondary anti–mouse and rabbit IgG for immunofluorescence microscopy were conjugated to AlexaFluor568 and Cy5 (Invitrogen). Rabbit polyclonal anti-EEA1 antibody was described previously (Christoforidis et al. 1999), mouse monoclonal antibody against the cytoplasmic tail of Tfr was purchased from Invitrogen, mouse monoclonal anti-LAMP1 antibody was obtained from Becton Dickinson, and mouse monoclonal anti-GM130 antibody was obtained from Abcam. Rab-GDI and RN-tre were prepared as described previously (Ullrich et al., 1994; Lanzetti et al., 2000). GST and HAP40-GST fusion proteins were affinity purified from Escherichia coli lysates according to standard procedures.
Cell culture procedures
HeLa and human primary fibroblast cells were grown according to standard procedures. Immortalized STHdhQ striatal cell lines from control and HD knock-in mice were cultured and differentiated as described previously (Trettel et al., 2000). For transient expression studies, cells were transfected using LipofectAMINE 2000 (Invitrogen) and used 12 h after transfection for immunoblot analysis or intracellular localization studies according to standard protocols. Fixed cells were analyzed using a 100×/NA 1.40 plan-Apochromat oil immersion lens (Carl Zeiss MicroImaging, Inc.) on a microscope (Axiovert S100TV; Carl Zeiss MicroImaging, Inc.). Illumination was performed with a 100-W mercury lamp with filter sets for GFP, AlexaFluor568, DAPI, and Cy5 fluorescence. Images were acquired at 1,300 × 1,000 pixels with a digital camera (SP 1.4.4; Diagnostic Instruments) controlled by the MetaVue 6.1 software package (Universal Imaging Corp.). Raw images from various color channels were assembled and colorized in Adobe Photoshop 7.0. Brightness and contrast were adjusted for visual clarity. To validate immunofluorescence microscopy studies using AlexaFluor568- and Cy5-conjugated secondary antibodies, HAP40 was overexpressed in HeLa cells and cells stained with a mixture of anti-HAP40 and -Htt antisera. Single addition of secondary antibody against mouse IgG (for Htt) conjugated with AlexaFluor568 resulted in no detectable bleed-through into the Cy5 channel at the exposure time and filter settings used for HAP40 detection with Cy5. Moreover, signals for HAP40 that were obtained with Cy5 looked very similar in the presence or absence of AlexaFluor568. Finally, when these dyes were swapped on the secondary antibodies, very similar distribution patterns for HAP40 and Htt were obtained. Collectively, the colocalization between HAP40 and Htt revealed in Figs. 3 and 9 is not caused by bleed-through. The percentage of colocalization between immunofluorescence signals was determined as follows: raw signals from discrete membrane structures were manually counted and inspected for overlapping with signals in other channels to calculate the percentage of colocalization. Only signals above value 70 on the eight-bit tonal scale were considered specific and used for the analysis. Typically, 10 cells (n = 10) were counted to calculate the mean colocalization and SD. For overlapping studies of EGFP-labeled early endosomes with cytoskeletal filaments, these organelles were considered colocalized to either microtubules or F-actin if at least three early endosomes in a row were clearly aligned to a filament. In areas where microtubules and F-actin converged (e.g., close to the cell edge), early endosomes could often not be assigned to either type of filament. The sum of early endosomes aligned to microtubules, F-actin, and unassignable early endosomes (100%) was used to calculate percentages of colocalization. Internalized biotinylated transferrin was detected with ruthenium-labeled antitransferrin antibodies and subsequently quantified by ECL analysis as described previously (Hoepfner et al. 2005).
Cell lines and tissue samples
The primary human fibroblast cell lines GM00023, GM00024, GM00037, GM00038, and GM00041 (Apparently Healthy cell collection) and GM04281, GM04723, GM03621, GM04869, and GM04847 (Huntington Disease cell collection) were obtained from Coriell Cell Repositories. Cell lines were established from both genders from healthy, unrelated individuals by the age of 3–31 yr or from clinically affected onset HD patients by the age of 19–32 yr. Immortalized striatal cell lines established from mouse embryos (STHdhQ) were gifts from M. MacDonald (Richard B. Simches Research Center, Boston, MA; Trettel et al., 2000). Human postmortem brain samples (CAP; Globus Pallidus) from healthy and HD grades 3 and 4 donors were provided by the Harvard Brain Tissue Resource Center.
Protein identification by mass spectrometry
Gel-separated Coomassie-stained proteins were excised from the gel slab and in-gel digested with trypsin as described previously (Shevchenko et al., 1996). Tryptic peptides were sequenced by nanoelectrospray tandem mass spectrometry on hybrid quadrupole time-of-flight mass spectrometers (Q-TOF I; Micromass, Ltd. and QSTAR Pulsar I; MDS Sciex) as described previously (Shevchenko et al., 1997). Database searching was performed by Mascot software (Matrix Science, Ltd.).
In vitro motility of early endosomes
The in vitro motility assay was essentially performed as described previously (Nielsen et al., 1999) with the following modifications (Hoepfner et al., 2005). KHMG (110 mM KCl, 50 mM Hepes-KOH, pH 7.4, 2 mM MgCl2, and 10% glycerol) was used as an assay buffer. For preparation of the antifade solution, BRB80 buffer was substituted with KHMG, and the solution was further supplemented with 10% serum. The energy mix consisted of 75 mM creatine phosphate, 10 mM ATP, 10 mM GTP, and 20 mM MgCl2 in BRB80. For the assay, taxol-stabilized microtubules were perfused in a microscopy chamber and allowed to bind to the coverslip. Next, 10 μl of 10% nonspecific rabbit serum in antifade solution was perfused in the chamber followed by 10 μl of the assay mixture (2 μl of 5 mg/ml fluorescently labeled early endosome, 1 μl of energy mix, 6 μl of the antifade solution, and 1 μl of saturated hemoglobin solution in KHMG) and was incubated for 5 min at RT. At least three videos per sample were recorded (60 frames with 2-s intervals) and analyzed as described previously (Nielsen et al., 1999).
In vivo motility of early endosomes
Cells were grown on glass coverslips, transfected with EGFP-Rab5 alone or together with siRNA against HAP40, and transferred to custom-built aluminum microscope slide chambers (Nielsen et al., 1999) just before observation. Cells were analyzed in CO2-independent medium/10% FBS (Invitrogen) at 37°C using a 100×/NA 1.40 plan-Apochromat oil immersion lens (Carl Zeiss MicroImaging, Inc.) on a microscope (IX70; Olympus) placed in a temperature-controlled chamber. Illumination was performed with a 100-W Xenon lamp fitted with a monochromator to excite GFP fluorescence. Images were acquired at 512 × 512 pixels with a camera (Cascade 512B; Roper Scientific) in stream acquisition mode over 2 min in 300-ms intervals using the MetaVue 6.1 software package. Image stacks were either converted to QuickTime videos or motility events highlighted by generating Z projections of the entire stack using the ImageJ software package.
Microtubule and actin spin-down assays
Binding of early endosomes to microtubules was performed as described previously (Nielsen et al., 1999) but with some modifications. 15 μg of prepared early endosomes pulsed with rhodamine–transferrin (Sigma-Aldrich) were incubated at RT with 5 μl of energy mix (see previous section), 1 μl of nonspecific rabbit serum, and 1 μM of recombinant candidate factors (HAP40, Rab-GDI, or RN-tre) in KHMG buffer supplemented with 1 mg/ml HeLa cell cytosol. After 20 min, 16 μg of taxol-stabilized microtubules were added to a total volume of 50 μl. After 10 min more at RT, the reaction mixture was laid over 100 μl of a 35% (wt/vol) sucrose cushion. After sedimentation at 100,000 g for 20 min at 22°C in a rotor (TLA 100; Beckman Coulter), the supernatant was removed, and the resulting pellet was washed twice in cytosol-free KHMG buffer. The pellet was lysed in 150 μl KHMG supplemented with 1% sodium deoxycholate, and the released rhodamine–transferrin was quantified in a fluorimeter. Excitation was at 550 nm, and emission was at 582 nm. A calibration curve revealed a linear correlation between emission and early endosome concentration. The equation of the linear regression analysis was y = 4,548.4x − 2,653.1 with R2 = 0.9879, where y is the emission in arbitrary units and x is the concentration of early endosomes in protein mass units. For binding of early endosomes to F-actin, globular-actin (Cytoskeleton, Inc.) was polymerized in KHMG buffer at RT for 1 h and centrifuged for 30 min at 100,000 g. The resulting pellet was washed with KHMG buffer and resuspended at 10 mg/ml. The assay was then performed with 10 μg of F-actin as described for microtubules.
Online supplemental material
All videos show EGFP-Rab5 dynamics, and images were acquired as described in In vivo motility of early endosomes. Video 1 shows a wild-type HeLa cell transfected with plasmid for EGFP-Rab5 expression only. Video 2 shows a HeLa cell cotransfected with plasmids for EGFP-Rab5 and HAP40. Video 3 shows a primary human fibroblast from a healthy person transfected with plasmid for EFGP-Rab5 expression only, whereas Video 4 shows the fibroblast from a HD patient. Video 5 shows a primary human fibroblast from a HD patient cotransfected with plasmid for EGFP-Rab5 transfection and siRNA duplexes against HAP40. Video 6 shows a striatal STHdhQ7/7 cell transfected with plasmid for EGFP-Rab5 expression only, Video 7 shows the same for an STHdhQ7/111 cell, and Video 8 shows this for an STHdhQ111/111 cell. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200509091/DC1.
Supplementary Material
[Supplemental Material Index]
Acknowledgments
We thank M. Sherman for full-length Htt cDNA, M. Matyash for the rat brain cDNA library, M. MacDonald for STHdhQ striatal cell lines, and Y. Trottier for invaluable advice, discussions, and for providing anti-Htt antibodies. We thank K. Simons, J. Howard, L. Pelkman, and J. Rink for critical reading of the manuscript. The Coriell Cell Repositories is acknowledged for providing the human primary fibroblast cell lines, and the Harvard Brain Tissue Resource Center is acknowledged for providing human postmortem brain samples from healthy and HD grades 3 and 4 donors.
This work was supported by the Max Planck Society and grants from the Human Frontier Science Program (RG-0260/1999-M) and the European Union (HPRN-CT-2000-00081). A. Pal, F. Severin, and B. Lommer are long-term Max Planck fellows.
F. Severin's present address is Biotechnology Centre, University of Technology Dresden, Cellular Machines, 01307 Dresden, Germany.
Abbreviations used in this paper: EEA1, early endosome antigen 1; F-actin, filamentous actin; GDI, GDP dissociation inhibitor; GDP, guanosine diphosphate; HAP, Htt-associated protein; HD, Huntington's disease; HIP; Htt-interacting protein; Htt, Huntingtin; LAMP, lysosome-associated membrane protein; polyQ, polyglutamine; RNAi, RNA interference; siRNA, short interfering RNA; Tfr, transferrin receptor.
References
- Block-Galarza, J., K.O. Chase, E. Sapp, K.T. Vaughn, R.B. Vallee, M. DiFiglia, and N. Aronin. 1997. Fast transport and retrograde movement of huntingtin and HAP 1 in axons. Neuroreport. 8:2247–2251. [DOI] [PubMed] [Google Scholar]
- Chan, E.Y., R. Luthi-Carter, A. Strand, S.M. Solano, S.A. Hanson, M.M. DeJohn, C. Kooperberg, K.O. Chase, M. DiFiglia, A.B. Young, et al. 2002. Increased huntingtin protein length reduces the number of polyglutamine-induced gene expression changes in mouse models of Huntington's disease. Hum. Mol. Genet. 11:1939–1951. [DOI] [PubMed] [Google Scholar]
- Christoforidis, S., H.M. McBride, R.D. Burgoyne, and M. Zerial. 1999. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 397:621–625. [DOI] [PubMed] [Google Scholar]
- Cuif, M.H., F. Possmayer, H. Zander, N. Bordes, F. Jollivet, A. Couedel-Courteille, I. Janoueix-Lerosey, G. Langsley, M. Bornens, and B. Goud. 1999. Characterization of GAPCenA, a GTPase activating protein for Rab6, part of which associates with the centrosome. EMBO J. 18:1772–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Renzis, S., B. Sonnichsen, and M. Zerial. 2002. Divalent Rab effectors regulate the sub-compartmental organization and sorting of early endosomes. Nat. Cell Biol. 4:124–133. [DOI] [PubMed] [Google Scholar]
- Engelender, S., A.H. Sharp, V. Colomer, M.K. Tokito, A. Lanahan, P. Worley, E.L. Holzbaur, and C.A. Ross. 1997. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 6:2205–2212. [DOI] [PubMed] [Google Scholar]
- Eskelinen, E.L., Y. Tanaka, and P. Saftig. 2003. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13:137–145. [DOI] [PubMed] [Google Scholar]
- Gasman, S., Y. Kalaidzidis, and M. Zerial. 2003. RhoD regulates endosome dynamics through Diaphanous-related Formin and Src tyrosine kinase. Nat. Cell Biol. 5:195–204. [DOI] [PubMed] [Google Scholar]
- Gauthier, L.R., B.C. Charrin, M. Borrell-Pages, J.P. Dompierre, H. Rangone, F.P. Cordelieres, J. De Mey, M.E. MacDonald, V. Lessmann, S. Humbert, and F. Saudou. 2004. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 118:127–138. [DOI] [PubMed] [Google Scholar]
- Harjes, P., and E.E. Wanker. 2003. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem. Sci. 28:425–433. [DOI] [PubMed] [Google Scholar]
- Hattula, K., and J. Peranen. 2000. FIP-2, a coiled-coil protein, links Huntingtin to Rab8 and modulates cellular morphogenesis. Curr. Biol. 10:1603–1606. [DOI] [PubMed] [Google Scholar]
- Hoepfner, S., F. Severin, A. Cabezas, B. Habermann, A. Runge, D. Gillooly, H. Stenmark, and M. Zerial. 2005. Modulation of receptor recycling and degradation by the endosomal kinesin KIF16B. Cell. 121:437–450. [DOI] [PubMed] [Google Scholar]
- Hoffner, G., P. Kahlem, and P.J. Djian. 2002. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: relevance to Huntington's disease. J. Cell Sci. 115:941–948. [DOI] [PubMed] [Google Scholar]
- Landles, C., and G.P. Bates. 2004. Huntingtin and the molecular pathogenesis of Huntington's disease. EMBO Rep. 5:958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetti, L., V. Rybin, M.G. Malabarba, S. Christoforidis, G. Scita, M. Zerial, and P.P. Di Fiore. 2000. The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature. 408:374–377. [DOI] [PubMed] [Google Scholar]
- Miaczynska, M., L. Pelkmans, and M. Zerial. 2004. Not just a sink: endosomes in control of signal transduction. Curr. Opin. Cell Biol. 16:400–406. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., C. Rabouille, R. Watson, T. Nilsson, N. Hui, P. Slusarewicz, T.E. Kreis, and G. Warren. 1995. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131:1715–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, E., F. Severin, J.M. Backert, A.A. Hyman, and M. Zerial. 1999. Rab5 regulates motility of early endosomes on microtubules. Nat. Cell Biol. 1:376–382. [DOI] [PubMed] [Google Scholar]
- Peters, M.F., and C.A. Ross. 2001. Isolation of a 40-kDa Huntingtin-associated protein. J. Biol. Chem. 276:3188–3194. [DOI] [PubMed] [Google Scholar]
- Rigamonti, D., J.H. Bauer, C. De-Fraja, L. Conti, S. Sipione, C. Sciorati, E. Clementi, A. Hackam, M.R. Hayden, Y. Li, et al. 2000. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J. Neurosci. 20:3705–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti, D., S. Sipione, D. Goffredo, C. Zuccato, E. Fossale, and E. Cattaneo. 2001. Huntingtin's neuroprotective activity occurs via inhibition of procaspase-9 processing. J. Biol. Chem. 276:14545–14548. [DOI] [PubMed] [Google Scholar]
- Rink, J., E. Ghigo, Y. Kalaidzidis, and M. Zerial. 2005. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 122:735–749. [DOI] [PubMed] [Google Scholar]
- Shevchenko, A., M. Wilm, O. Vorm, and M. Mann. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858. [DOI] [PubMed] [Google Scholar]
- Shevchenko, A., I. Chernushevich, W. Ens, K.G. Standing, B. Thomson, M. Wilm, and M. Mann. 1997. Rapid ‘de novo’ peptide sequencing by a combination of nanoelectrospray, isotopic labeling and a quadrupole/time-of-flight mass spectrometer. Rapid Commun. Mass Spectrom. 11:1015–1024. [DOI] [PubMed] [Google Scholar]
- Sipione, S., D. Rigamonti, M. Valenza, C. Zuccato, L. Conti, J. Pritchard, C. Kooperberg, J.M. Olson, and E. Cattaneo. 2002. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum. Mol. Genet. 11:1953–1965. [DOI] [PubMed] [Google Scholar]
- Stenmark, H., R.G. Parton, O. Steele-Mortimer, A. Lutcke, J. Gruenberg, and M. Zerial. 1994. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 13:1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugars, K.L., and D.C. Rubinsztein. 2003. Transcriptional abnormalities in Huntington disease. Trends Genet. 19:233–238. [DOI] [PubMed] [Google Scholar]
- Tao, T., and A.M. Tartakoff. 2001. Nuclear relocation of normal huntingtin. Traffic. 2:385–394. [DOI] [PubMed] [Google Scholar]
- Trettel, F., D. Rigamonti, P. Hilditch-Maguire, V.C. Wheeler, A.H. Sharp, F. Persichetti, E. Cattaneo, and M.E. MacDonald. 2000. Dominant phenotypes produced by the HD mutation in STHdhQ111 striatal cells. Hum. Mol. Genet. 9:2799–2809. [DOI] [PubMed] [Google Scholar]
- Trottier, Y., D. Devys, G. Imbert, F. Saudou, I. An, Y. Lutz, C. Weber, Y. Agid, E.C. Hirsch, and J.L. Mandel. 1995. Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat. Genet. 10:104–110. [DOI] [PubMed] [Google Scholar]
- Ullrich, O., H. Horiuchi, C. Bucci, and M. Zerial. 1994. Membrane association of Rab5 mediated by GDP-dissociation inhibitor and accompanied by GDP/GTP exchange. Nature. 368:157–160. [DOI] [PubMed] [Google Scholar]
- Zerial, M., and H.M. McBride. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2:107–117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental Material Index]