Ultraviolet radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa (original) (raw)
Abstract
To identify the mechanisms of ultraviolet radiation (UVR)–induced cell death, for which the tumor suppressor p53 is essential, we have analyzed mouse embryonic fibroblasts (MEFs) and keratinocytes in mouse skin that have specific apoptotic pathways blocked genetically. Blocking the death receptor pathway provided no protection to MEFs, whereas UVR-induced apoptosis was potently inhibited by Bcl-2 overexpression, implicating the mitochondrial pathway. Indeed, Bcl-2 overexpression boosted cell survival more than p53 loss, revealing a p53-independent pathway controlled by the Bcl-2 family. Analysis of primary MEFs lacking individual members of its BH3-only subfamily identified major initiating roles for the p53 targets Noxa and Puma. In the transformed derivatives, where Puma, unexpectedly, was not induced by UVR, Noxa had the dominant role and Bim a minor role. Furthermore, loss of Noxa suppressed the formation of apoptotic keratinocytes in the skin of UV-irradiated mice. Collectively, these results demonstrate that UVR activates the Bcl-2–regulated apoptotic pathway predominantly through activation of Noxa and, depending on cellular context, Puma.
Introduction
UV radiation (UVR) is a potent carcinogen that acts directly on DNA. Accumulated lifetime exposure to UVR is the key environmental risk factor for development of nonmelanoma skin cancers (NMSCs), such as basal and squamous cell carcinomas (Kraemer et al., 1994). The cellular response to DNA damage is centered on p53, a transcription factor that exerts its tumor-suppressive function by inducing cell cycle arrest, cell senescence, or apoptosis (Vousden and Lu, 2002). The importance of p53 in the prevention of UVR-induced skin cancer is underscored by the observation that after chronic UV irradiation, p53-deficient mice exhibit a vastly increased incidence and reduced latency of NMSC compared with wild-type (wt) animals (Li et al., 1998). Programmed cell death initiated by UVR is required to remove precancerous keratinocytes, yielding so-called sunburn cells (SBCs). Their formation appears to represent a crucial tumor-suppressive response because they arise from the cell type of origin for NMSC and their development requires functional p53 (Ziegler et al., 1994; Li et al., 1998).
Two distinct signaling pathways activate the caspases that mediate apoptosis (Strasser et al., 1995). The extrinsic pathway is initiated by “death receptors” (several members of the TNF-R family) and proceeds via caspase-8 and its adaptor FADD (Fas-associated death domain), whereas the intrinsic or mitochondrial pathway is regulated by the interacting pro- and antiapoptotic members of the Bcl-2 protein family and leads, after mitochondrial outer membrane permeabilization, to caspase-9 activation. Although UVR-induced apoptosis clearly involves the downstream effector caspases (Kuida et al., 1996), the relative roles of the extrinsic and intrinsic pathways are controversial. The extrinsic pathway is favored by evidence that membrane localization of the death receptors Fas (also called APO-1 or CD95) and TRAIL-R is up-regulated in a p53-dependent manner after UVR exposure (Bennett et al., 1998) and that UV-irradiated gld/gld (FasL-deficient) mice exhibit reduced SBC formation (Hill et al., 1999). On the other hand, UV-irradiated mice overexpressing Bcl-2 in keratinocytes exhibited fewer SBCs and more skin tumors than control animals (Rodriguez-Villanueva et al., 1998).
In the intrinsic path to cell death, the key initiators are the BH3-only members of the Bcl-2 family (Huang and Strasser, 2000). Different death stimuli activate distinct subsets of these death ligands. For example, Noxa and Puma are up-regulated during p53-mediated cell killing, and their genes are direct p53 targets (Oda et al., 2000; Nakano and Vousden, 2001; Yu et al., 2001). Gene-targeting experiments in mice have demonstrated that Puma plays a major and Noxa a more restricted role in p53-mediated apoptosis (Jeffers et al., 2003; Shibue et al., 2003; Villunger et al., 2003). Primary mouse embryonic fibroblasts (MEFs) as well as E1A oncogene transformed MEFs from Puma-deficient animals proved refractory to etoposide, and puma −/− lymphoid and myeloid cells were remarkably resistant to genotoxic damage (Jeffers et al., 2003; Villunger et al., 2003; Erlacher et al., 2005). The role of Noxa has been less clear, as its loss gave MEFs only slight, albeit significant, resistance against etoposide and did not affect any apoptotic responses in lymphoid cells (Shibue et al., 2003; Villunger et al., 2003).
Here, we have sought to delineate the pathways to cell death elicited by UV irradiation of primary MEFs, the MEFs rendered more sensitive to genotoxic damage by transformation with the adenovirus E1A and ras oncogenes (Lowe et al., 1993) and keratinocytes within whole mouse skin. We demonstrate that the Bcl-2 family regulates not only the death induced by p53 but also a p53-independent pathway in response to UVR. By exploiting MEFs that lack different BH3-only proteins, we show that in primary cells, both Noxa and Puma contribute to UVR-induced apoptosis. Unexpectedly, only Noxa plays a major role in the transformed MEFs and keratinocytes, where the noxa, but not the puma, gene proved to be induced.
Results
UV irradiation triggers both apoptotic and nonapoptotic cell death in primary MEFs but predominantly apoptosis in transformed MEFs
To examine whether UV-irradiated MEFs die by apoptosis, several well-established parameters were assessed in both primary and E1A/ras transformed MEFs (Fig. 1 and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200608070/DC1). Morphological and flow cytometric examination indicated that relatively few primary MEFs died when exposed to low-dose UVR (5–50 J/m2). Although they exhibited some morphological signs of apoptosis at these doses, at higher doses (e.g., 200 J/m2), they appeared to die predominantly by a nonapoptotic mechanism, as indicated by the prevalence of large, vacuolated cells at 6 h after irradiation (Fig. 1 A). In contrast, at both low and high doses, the MEFs sensitized to genotoxic damage by oncogenic transformation (Lowe et al., 1993) exhibited classical morphological signs of apoptosis, such as chromatin condensation and membrane blebbing (Fig. 1 A), as well as hallmark biochemical features. Notably, caspase activity was evident from the generation of the cleaved p12 and p85 fragments of the canonical caspase substrates ICAD (inhibitor of caspase-activated DNase) and PARP (poly ADP ribose polymerase), respectively (Fig. S1, A and B). The resulting release of the active DNase CAD from its inhibitor ICAD presumably accounts for the characteristic DNA fragmentation (Fig. S1 C) and the cell population with DNA content of <2C (Fig. S1 D). Consistent with the different fates of primary and E1A/ras transformed MEFs, active caspase-3 increased in E1A/ras MEFs but not in primary MEFs (Fig. 1 B). Thus, although E1A/ras transformed MEFs are highly sensitive to UVR-induced apoptosis, primary MEFs are more resistant and die at high doses, predominantly by a nonapoptotic mechanism.
Figure 1.

UVR induces apoptosis in E1A/ras transformed MEFs but predominantly nonapoptotic death in primary MEFs. (A) Cytospin preparations of untreated cells or cells exposed to 200 J/m2 UVR were stained with hematoxylin and eosin. A higher magnification of E1A/ras transformed MEFs (middle) shows an enlarged necrotic cell (arrowhead) with large cytoplasmic vesicles. Apoptotic cells (asterisks) are identifiable by cell shrinkage, dark and punctate nuclear staining, and membrane blebbing. These figures are representative of two independent lines of primary and E1A/ras transformed MEFs from wt mice. Bars: (top middle) 25 μm; (others) 50 μm. (B) Western blot to reveal active caspase-3 in primary or E1A/ras transformed wt MEFs stimulated with 100 or 10 J/m2 UVR, respectively.
Overexpression of Bcl-2 but not blockade of death receptor signaling inhibits UVR-induced apoptosis
To investigate whether the extrinsic or intrinsic signaling pathway drives UVR-induced apoptosis, two independently generated E1A/ras transformed wt MEF lines were transfected with expression vectors that encode Bcl-2 or proteins that inhibit the death receptor pathway: a well-characterized dominant-negative mutant of FADD (FADD-DN; Chinnaiyan et al., 1995) or the caspase-8 inhibitor CrmA (Strasser et al., 1995). Both inhibitors were expressed (Fig. 2, B and D) and functional, as they blocked the death of MEFs stimulated with FasL (not depicted). Nevertheless, neither FADD-DN nor CrmA notably inhibited UVR-induced apoptosis (Fig. 2, A and C). In striking contrast, Bcl-2 overexpression (Fig. 2 E) almost completely ablated UVR-induced cell death (Fig. 2 C). We conclude that the UVR-induced apoptosis of the transformed MEFs proceeds through the Bcl-2–regulated pathway.
Figure 2.
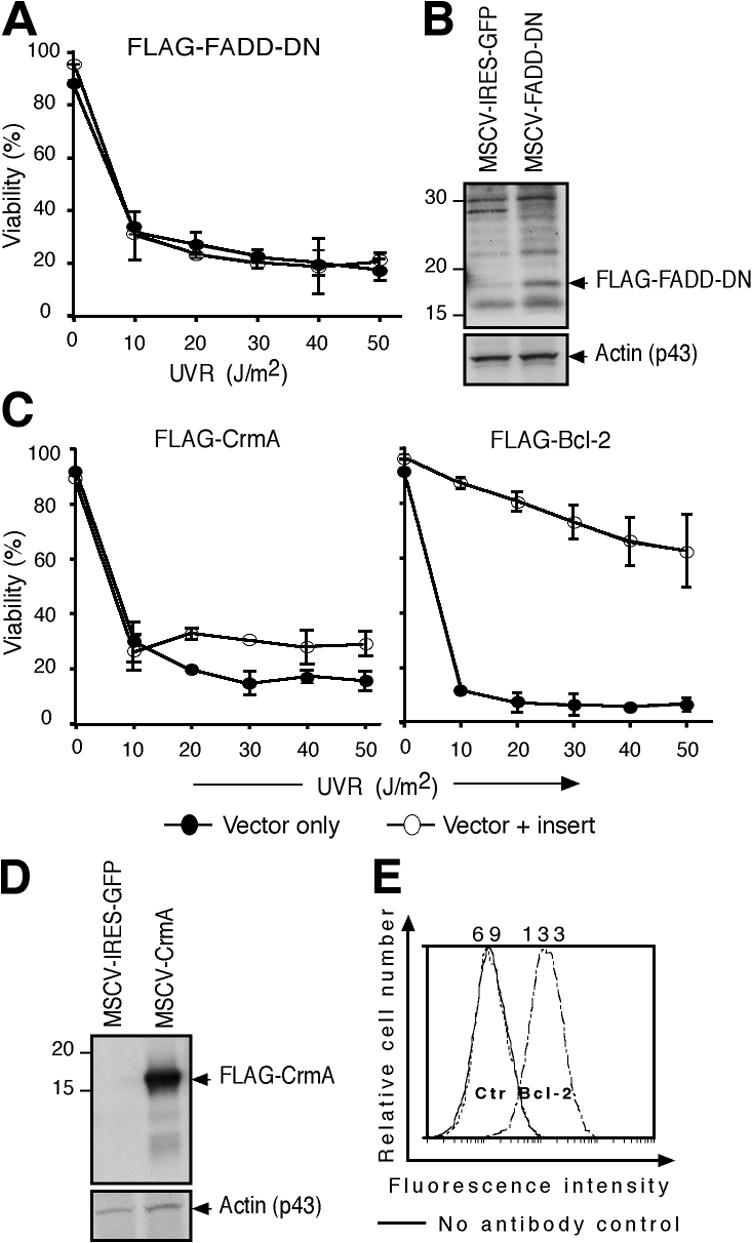
UVR-induced apoptosis of transformed MEFs is inhibited by Bcl-2 overexpression but not by blockade of the death receptor pathway. (A) E1A/ras transformed wt MEFs retrovirally transduced with a control vector or a construct encoding FLAG–FADD-DN were exposed to graded doses of UVR, and viability was analyzed at 24 h by PI staining. Data represent the mean ± SD of two independent lines. (B) Western blot analysis using anti-FLAG antibody was performed on lysates derived from FLAG–FADD-DN–expressing and control vector–infected cells. A band of the expected size (p16) was detected in FLAG–FADD-DN–expressing cells. β-Actin served as a loading control. (C) Transformed wt MEFs retrovirally transduced with a control vector, a construct encoding FLAG-tagged CrmA or Bcl-2, were exposed to graded doses of UVR, and viability was determined as in A. Data represents mean ± SD of two independent lines. (D) Western blot analysis using anti-FLAG antibody was performed on lysates derived from FLAG-CrmA–expressing and control vector–transfected cells, with β-actin as a loading control. (E) Intracellular FACS staining with a human Bcl-2–specific monoclonal antibody (Bcl-2-100).
Loss of p53, Noxa, or Puma partially protects primary MEFs against UVR-induced cell death
The strong protection conveyed by Bcl-2 (Fig. 2 C) prompted us to investigate which of its BH3-only antagonists drives this process. Primary MEFs derived from wt mice or those lacking Bim, Bad, Noxa, or Puma were exposed to graded doses of UVR, and cell viability was monitored 24 h later. Wt primary MEFs exhibited dose-dependent rates of death, which was just as extensive in MEFs lacking Bad or Bim (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200608070/DC1). In contrast, the cells lacking p53, Noxa, or Puma all displayed greater viability than wt controls at doses up to 50 J/m2 (Fig. 3 A).
Figure 3.

Significant roles for p53, Noxa, and Puma in UVR-induced killing of primary MEFs. (A) Primary MEFs from p53 −/−, noxa −/−, or puma −/− embryos were exposed to graded doses of UVR, and cell survival was determined after 24 h. (B) UVR dose response of wt, p53 −/−, noxa −/−, and noxa −/− puma −/− primary MEFs. By t test, noxa −/− puma −/− MEFs were significantly more resistant than noxa −/− MEFs to 100 J/m2 of UVR. *, P < 0.025. (C) Kinetics of cell survival for wt, p53 −/−, noxa −/−, and noxa −/− puma −/− primary MEFs subjected to 25 J/m2 UVR. Results shown represent the mean percentage of viability ± SD of MEFs from three independent embryos of each genotype.
Because many BH3-only proteins have partially redundant functions (Huang and Strasser, 2000), we also generated primary MEFs from mice lacking both Noxa and Puma, or Noxa and Bim. The primary noxa −/− puma −/− MEFs were significantly more resistant than noxa −/− cells at doses of 100 and 200 J/m2 (Fig. 3 B; P < 0.025), although the slight increases at lower doses were not significant. Remarkably, at all doses and times analyzed, the primary noxa −/− puma −/− MEFs were at least as refractory to UVR as the p53 −/− MEFs (Figs. 3, B and C), indicating that Noxa and Puma account for all p53-mediated killing of UV-irradiated primary MEFs. In contrast, the noxa −/− bim −/− primary MEFs were no more refractory than those lacking Noxa alone (Fig. S2 B). Thus, at doses up to 50 J/m2, the UVR-induced apoptosis of primary MEFs is driven mainly through p53 and its targets Noxa and Puma, whereas Bad and Bim are dispensable. At higher doses of UVR, the substantial killing of the p53 −/−, noxa −/−, puma −/−, and noxa −/− puma −/− MEFs (Fig. 3 A) probably reflects nonapoptotic cell death.
Loss of p53 or Noxa but not Puma renders transformed MEFs resistant to UVR-induced apoptosis
To circumvent the complication of nonapoptotic UVR-induced cell death, we derived independent E1A/ras transformed MEFs from three embryos of various genotypes (wt, p53 −/−, bim −/−, noxa −/−, and puma −/− E1A/ras lines 1–3). Because the E1A oncoprotein sensitizes MEFs to apoptosis by inactivating the RB tumor suppressor, we verified by intracellular FACS analysis that all transformed lines of every genotype expressed similar levels of E1A (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200608070/DC1).
When the cell lines were subjected to UVR at 10–50 J/m2 and viability analyzed 24 h later, the p53-deficient E1A/ras MEFs were consistently the most resistant, and Noxa-deficient MEFs demonstrated a marked, albeit lower, level of resistance (Fig. 4 A i and Fig. S3). In contrast, loss of Bim conveyed no protection (Fig. 4 A, i and ii). Unlike the enhanced resistance in the puma −/− primary MEFs (Fig. 3 A), their transformed derivatives behaved like wt MEFs except for slightly greater viability at 10 and 20 J/m2 (Fig. 4 A i; P < 0.025). To determine whether these protective effects persisted, viability was also analyzed at 24, 48, and 72 h after an intermediate dose of UVR (15 J/m2). The bim −/− and puma −/− cells died as rapidly as wt cells (Fig. 4 A ii). Indeed, even most of the cells deficient in p53 or Noxa had succumbed by 72 h after irradiation; only 15% of them remained viable, although this still greatly exceeded the 1% viability of the wt cells (Fig. 4 A ii). Thus, Noxa is the major initiator of UVR-induced apoptosis downstream of p53 in E1A/ras transformed MEFs.
Figure 4.

Loss of p53 or Noxa protects E1A/ras transformed MEFs from UVR-induced apoptosis. (A, i) UVR dose-response analysis of E1A/ras transformed MEFs harvested at 24 h after irradiation. *, P < 0.025. (ii) Kinetics of cell survival of E1A/ras transformed MEFs exposed to 15 J/m2 UVR. Data represent mean percentage of viability ± SD of three independent embryos (two embryos for bim −/− E1A/ras cells). (B, i) UVR dose response of E1A/ras transformed MEFs from noxa −/− bim −/− mice. (ii) Kinetics of cell survival of E1A/ras transformed noxa −/− bim −/− MEFs subjected to 15 J/m2 UVR. By t test, noxa −/− bim −/− MEFs were significantly more resistant than noxa −/− MEFs at 24 h after UVR stimulation. *, P < 0.05. (iii) UVR dose response of E1A/ras transformed noxa −/− puma −/− MEFs. Cell viability was determined at 24 h after UVR by PI staining and FACS analysis. (iv) Kinetics of cell survival of E1A/ras transformed noxa −/− puma −/− MEFs subjected to 15 J/m2 UVR. Results shown represent the mean percentage of viability ± SD of MEFs from three independent embryos of each genotype.
Loss of Noxa plus Puma or Noxa plus Bim does not render transformed MEFs as resistant to UVR as p53 loss
The observation that noxa −/− transformed MEFs were more sensitive to UVR than the corresponding p53 −/− or Bcl-2 overexpressing cells suggested that Noxa is not the sole initiator of UVR-induced apoptosis. Because our studies suggested that Puma or Bim might have an auxiliary role, we generated transformed MEFs from noxa −/− bim −/− and noxa −/− puma −/− embryos. Dose-response analysis at 24 h after UVR indicated that the noxa −/− bim −/− transformed cells appeared to survive slightly better than noxa −/− MEFs at all doses tested (Fig. 4 B i) and had a small but significant advantage at 24 h after stimulation with 15 J/m2 (Fig. 4 B ii; P < 0.05). Unexpectedly, the noxa −/− puma −/− transformed cells were no more resistant to UVR than counterparts deficient in Noxa alone (Fig. 4 B, iii and iv).
Puma mRNA is induced by UVR in primary but not E1A/ras transformed MEFs
To clarify why loss of either Noxa or Puma protected the primary MEFs (Fig. 3 A) but only loss of Noxa substantially protected the transformed MEFs (Fig. 4 A), we quantified noxa and puma transcripts by PCR at various times after treatment with UVR, or etoposide as a positive control, and normalized the values to unstimulated controls. In transformed MEFs, UVR induced noxa mRNA 2.3-fold but, surprisingly, slightly reduced the puma mRNA level (Fig. 5 A). The up-regulation of noxa mRNA was p53 dependent, as no increase occurred in p53 −/− MEFs (Fig. 5 A, top). In contrast, etoposide up-regulated both noxa and puma, albeit noxa to a greater extent (∼11- vs. 4.5-fold). The absence of puma mRNA at 6 h after etoposide treatment indicates that this transcript is rapidly induced but then degraded (Fig. 5 A).
Figure 5.
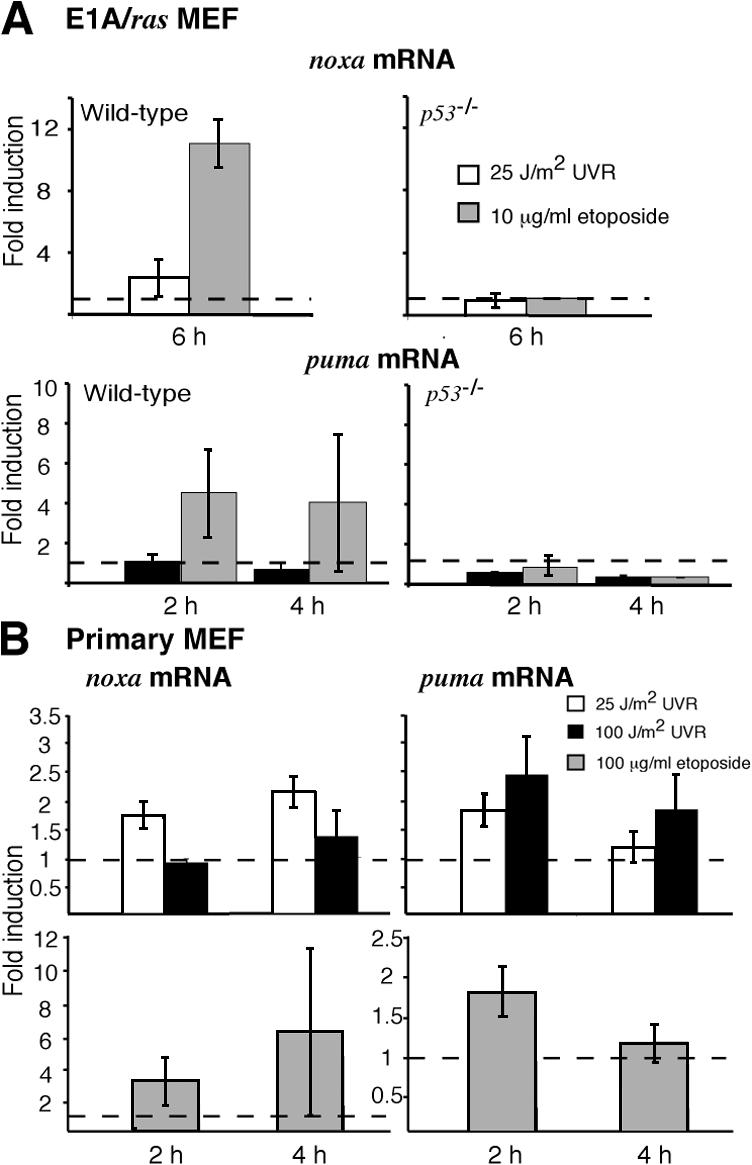
Puma mRNA is up-regulated by UVR in primary but not E1A/ras transformed MEFs. (A) Quantitative PCR analysis of noxa mRNA in E1A/ras transformed wt MEFs 6 h after treatment with 25 J/m2 UVR or 10 μg/ml etoposide. Puma mRNA levels were assessed in these MEFs at 2 and 4 h after treatment with 25 J/m2 UVR or 10 μg/ml etoposide. (B) Noxa and puma mRNA expression was assessed in wt primary MEFs after exposure to 25 or 100 J/m2 UVR or 100 μg/ml etoposide. Transcript abundance was normalized to levels observed in unstimulated control cells and represented as fold induction. Data shown represent the mean fold induction ± SD observed in MEFs from two independent wt embryos. SD is small where not visible. Dashed lines indicate a basal expression level of 1.
In primary wt MEFs, UVR, like etoposide, induced both noxa and puma mRNA (Fig. 5 B). Interestingly, puma mRNA was up-regulated slightly more by a high dose (100 J/m2) than a low dose (25 J/m2) of UVR (Fig. 5 B, top right; 2.5- vs. 1.8-fold maximum at 2 h), whereas noxa mRNA exhibited the reciprocal pattern (Fig. 5 B, top left; 1.4- vs. 2.2-fold maximum at 4 h). These results show that UVR increases puma mRNA levels in primary but not in E1A/ras transformed MEFs and that UVR dose affects the relative abundance of puma and noxa transcripts in primary cells.
In hemopoietic progenitor cells, DNA damage induces the transcriptional repressor Slug, which in turn ablates the activation of puma by p53 (Wu et al., 2005). To explore whether Slug or its close relative Snail might account for the absence of puma mRNA in UV-irradiated E1A/ras MEFs, we analyzed their activation profile by quantitative PCR. Snail mRNA levels were not affected by treatment with UVR or etoposide, in both the primary and transformed MEFs (Fig. 6, left). As we hypothesized, however, slug mRNA remained unchanged after irradiation of primary MEFs but was induced approximately threefold by UVR in wt E1A/ras MEFs (Fig. 6 A). This induction did not require p53, as slug mRNA was still up-regulated by UVR in p53 −/− E1A/ras MEFs (Fig. 6 B). In contrast, etoposide-induced up-regulation of slug in wt E1A/ras MEFs requires p53, as slug remained at basal levels in stimulated p53 −/− E1A/ras MEFs (Fig. 6 B). Thus, Slug may well be responsible for the absence of puma induction in UV-irradiated transformed MEFs (see Discussion).
Figure 6.
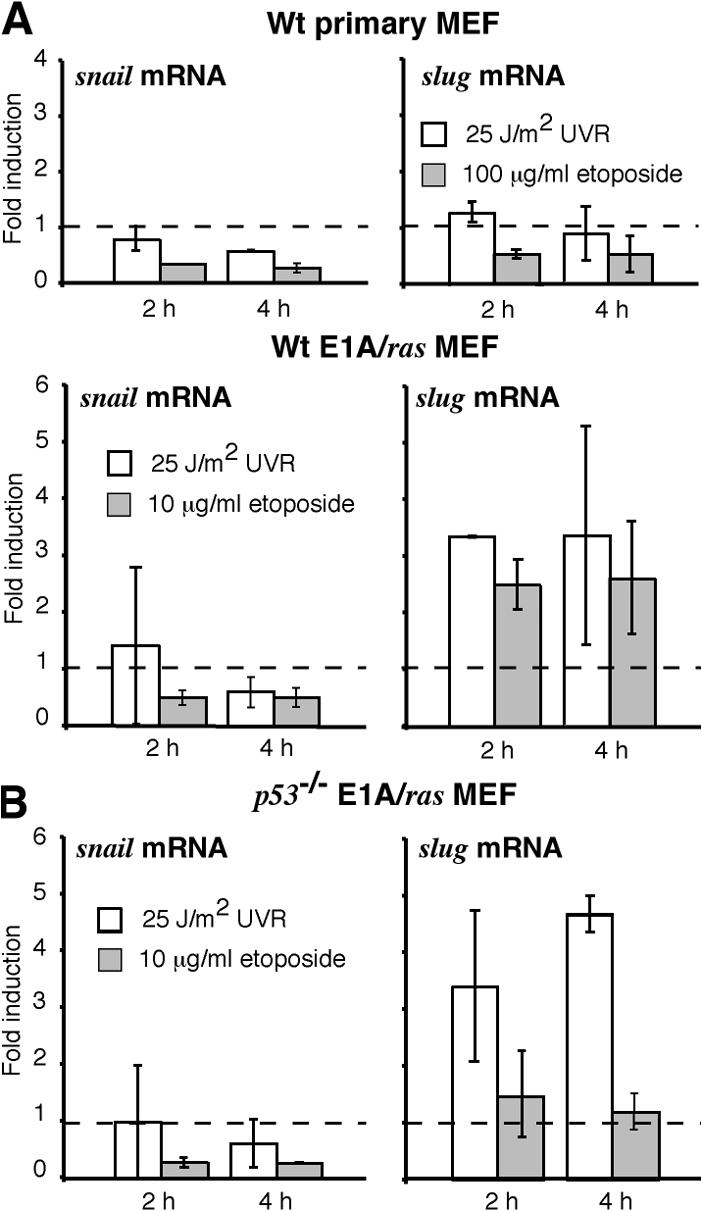
The transcriptional repressor slug is constitutively expressed in E1A/ras transformed MEFs. (A) Quantitative PCR analysis of slug and snail mRNA expression in wt primary and E1A/ras transformed MEFs. (B) Quantitative PCR analysis of slug and snail mRNA expression in p53 −/− E1A/ras transformed MEFs. Data shown represent the mean fold induction ± SD of two independent cell lines, and dashed lines indicate the basal expression level.
Bcl-2 overexpression increases the UVR resistance of transformed MEFs lacking Noxa, Puma, or even p53
To investigate the mechanism of UVR-induced death that occurs in the absence of Noxa, Puma, or p53 (Fig. 3 A), human Bcl-2 was overexpressed in E1A/ras transformed MEFs from wt, noxa −/−, puma −/−, or p53 −/− mice (Fig. 7 B). Remarkably, Bcl-2 overexpression not only enhanced resistance to UVR in wt MEFs (Fig. 2 C, right) but also in the MEFs lacking Noxa, Puma, or even p53 (Fig. 7 A). Indeed, in puma −/− MEFs exposed to 50 J/m2 UVR, Bcl-2 overexpression increased survival from <5 to 70% and in noxa −/− or p53 −/− cells, it saved ∼95% of the cells (Fig. 7 A). As a control, E1A/ras transformed noxa −/− MEFs were transfected with constructs encoding FADD-DN or CrmA, but neither of these inhibitors augmented resistance to UVR (Fig. 7 C). These results demonstrate that UVR-induced apoptosis must proceed via a Bcl-2–inhibitable pathway in addition to that orchestrated by p53 and its death effectors Noxa and Puma.
Figure 7.

Bcl-2 overexpression further inhibits UVR-induced apoptosis in Noxa- or p53-deficient transformed MEFs. (A) UVR dose-response analysis of E1A/ras transformed noxa −/−, p53 −/−, and puma −/− MEFs transfected with a human Bcl-2–GFP (open circles) expression vector or a control GFP vector (closed circles). (B) Transgenic Bcl-2 expression levels, determined by intracellular immunofluorescent staining. The numerical values represent the mean fluorescence intensity of the cell populations. The unbroken line represents negative control (staining with FITC-conjugated secondary antibody only), and the dashed line represents staining with human anti–Bcl-2 antibody plus FITC-conjugated secondary antibody. (C) Cell viability of E1A/ras transformed noxa −/− MEFs transfected with either a FADD-DN or a CrmA retroviral expression construct, was assessed as described in Fig. 2. Closed circles indicate control, and open circles indicate FADD-DN–GFP or CrmA-GFP. Data in A and C represent the mean percentage of viability ± SD of MEFs from two independent embryos.
Loss of p53 or Noxa suppresses the formation of SBCs in UV-irradiated mouse skin
To extend these results to an in vivo context, we analyzed the induction of apoptosis in the skin of UV-irradiated mice of different genotypes, first by histology and enumeration of SBCs, identified by their eosinophilic cytoplasm, pyknotic nuclei, and detachment from surrounding cells (Fig. 8 A, arrowheads), and then by TUNEL staining (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200608070/DC1), to reveal cells undergoing DNA fragmentation. Unstimulated control skin from all genotypes (Fig. 8 A, left) exhibited well-ordered stratification of keratinocytes, an intact stratum corneum, and the absence of SBCs. UV-irradiated skin from p53- or Noxa-deficient animals exhibited a relatively preserved morphology and small numbers of SBCs (Fig. 8). In contrast, UV-irradiated skin from wt and puma −/− mice were hallmarked by large vacuoles, reduced epidermal thickness, loss of the stratum corneum, and an increased number of SBCs (Fig. 8). At 72 h after UVR, wt skin exhibited substantial parakeratosis, hyperkeratosis, keratinocyte loss, and dysplasia (Fig. S5). In contrast, there were only few parakeratotic and hyperkeratotic lesions in UV-irradiated noxa −/− and p53 −/− skin at this time point, and these regions typically overlaid viable, well-ordered, and stratified keratinocytes (Fig. S5). Moreover, unstimulated control skin of each genotype displayed very few TUNEL-positive cells (Fig. S4). After stimulation with 1,000 J/m2 UVR, many more became evident in wt and puma −/− skin at 24 h, whereas considerably fewer arose in UV-irradiated skin from the p53 −/−, noxa −/−, or noxa −/− puma −/− animals (Fig. S4). These data indicate that, although loss of Puma provided no protection, the loss of p53 or Noxa significantly reduced the number of apoptotic keratinocytes within the epidermal layer (Fig. 8 B; P < 0.025).
Figure 8.

Epidermal keratinocytes are protected from UVR-induced apoptosis by the loss of p53 or Noxa. (A, left) Representative hematoxylin- and eosin-stained sections of nonirradiated skin of the indicated genotypes. (right) Representative sections of skin irradiated with 1,000 J/m2 UVR. Sc, stratum corneum; ep, epidermis; der, dermis; hf, hair follicle. Dashed lines delineate the epidermal–dermal junction. Arrowheads indicate the presence of SBCs. Bars, 100 μm. (B) Quantification of SBCs per linear centimeter of skin. The data represent the mean numbers ± SD from three to five mice of each genotype. The differences between UVR-treated wt and p53 −/− and wt and noxa −/− mice are statistically significant (P < 0.025). The difference between UVR-treated noxa −/− and p53 −/− is also statistically significant (P < 0.05).
Up-regulation of noxa mRNA in UV-irradiated whole mouse skin
Quantitative PCR analysis revealed that the mRNA expression profiles of noxa and puma observed in cultured MEFs held for irradiated whole mouse skin. In wt skin, noxa mRNA was up-regulated approximately sevenfold and puma mRNA approximately twofold at 24 h after exposure to 1,000 J/m2 UVR (Fig. 9 A). As expected, the up-regulation of noxa mRNA at this time is mediated by p53, as there was no corresponding increase in the skin of p53 −/− mice (Fig. 9 B).
Figure 9.
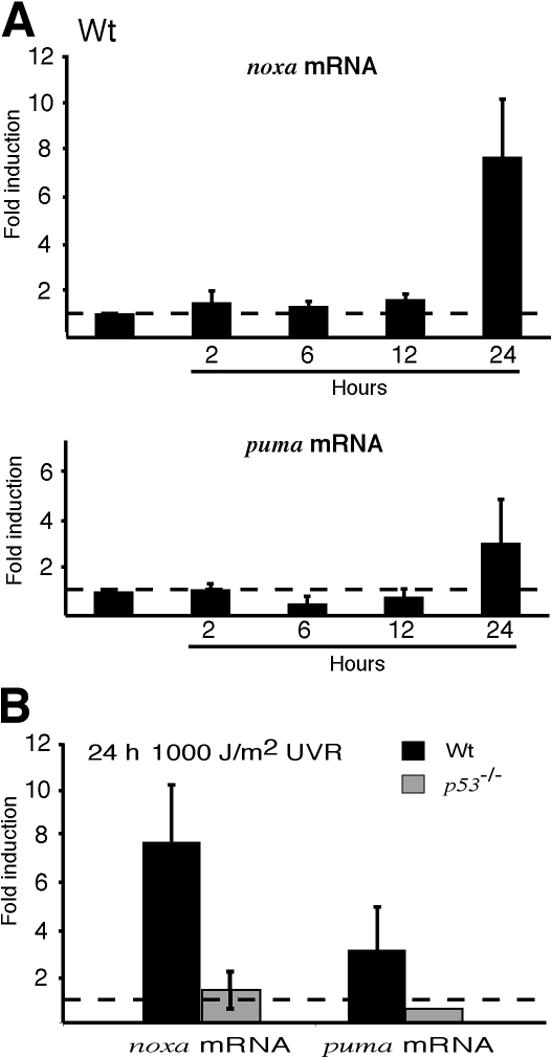
Noxa mRNA is up-regulated by UV irradiation in whole mouse skin. (A) Quantitative PCR analysis of the kinetics of noxa and puma mRNA up-regulation in UV-irradiated skin of wt animals. (B) Noxa and puma mRNA expression at 24 h after irradiation in skin of wt and p53 −/− animals.
Discussion
The death receptor pathway is dispensable for UVR-induced apoptosis of transformed MEFs
To determine whether cell surface–bound death receptors, such as Fas (Apo-1/CD95) or TNF-R1, are required for UVR-induced apoptosis, potent inhibitors of the extrinsic pathway, namely, FADD-DN and the caspase-8 inhibitor CrmA, were expressed in the transformed MEFs. As neither blocked the UVR-induced apoptosis, the death receptor pathway appears to be dispensable in these cells. Other reports, however, have implicated Fas signaling in cellular responses to UVR, such as immune suppression (Strand et al., 1996). Furthermore, cell surface expression of Fas was shown to be up-regulated by p53 after UVR exposure (Bennett et al., 1998). However, in agreement with our findings, UVR-induced apoptosis of primary MEFs and keratinocytes did not require the critical death receptor mediator caspase-8 (Varfolomeev et al., 1998; Tournier et al., 2000). How can the dispensability of death receptor signaling for UVR-induced apoptosis of keratinocytes be reconciled with the suppression of SBC formation in the skin of UV-irradiated FasL- deficient (C3H/HeJ gld/gld) mice (Hill et al., 1999)? One possible explanation is that death receptor signaling amplifies rather than initiates UVR-induced apoptosis. Alternatively, FasL-Fas signaling in the skin may represent a non–cell autonomous process, whereby FasL on leukocytes recruited by inflammatory cytokines engages Fas on UVR-damaged keratinocytes.
BH3-only proteins Noxa and, to a lesser extent, Puma initiate UVR-induced apoptosis downstream of p53
The strong inhibition of UVR-induced apoptosis by Bcl-2 overexpression implicated proapoptotic members of this family in initiating the response. Indeed, both Noxa and Puma, BH3-only proteins previously associated with commitment to γ-irradiation–induced apoptosis, proved to play critical roles. Primary MEFs lacking either Noxa or Puma exhibited substantial resistance to doses up to 50 J/m2, although little protection was evident at higher doses, where nonapoptotic death appeared to predominate. Significantly, primary MEFs lacking both Noxa and Puma proved as refractory as those lacking p53 at all doses and times studied. This finding suggests that, in these cells, Noxa and Puma are the essential mediators of all p53-induced death after UV irradiation.
Although primary MEFs lacking Bim were not resistant to UVR-induced apoptosis, a minor role for Bim appeared in the transformed noxa −/− bim −/− MEFs, which exhibited a small but statistically significant survival advantage over transformed noxa −/− MEFs. Because DNA damage is not believed to induce bim mRNA and because UVR also causes cytoplasmic damage, cytoskeletal alterations provoking Bim release (Puthalakath et al., 2001) may occur independent of the DNA damage response. In any case, the function of Bim in the UVR response probably overlaps that of BH3-only proteins that play a more prominent role, such as Noxa. Noxa alone is a weak inducer of apoptosis, because it predominantly neutralizes Mcl-1 and robust cell killing requires additional BH3-only proteins, such as Bim or Puma, which can neutralize other prosurvival members (Chen et al., 2005).
After UV irradiation, the transformed noxa −/− cells died at a rate intermediate between p53 −/− and wt counterparts. Because both Noxa and Puma are up-regulated in a p53-dependent manner in response to DNA damage in many cell types (Oda et al., 2000; Nakano and Vousden, 2001; Yu et al., 2001), it was surprising that, although the noxa −/− puma −/− primary MEFs survived high-dose UVR (100 or 200 J/m2) considerably better than noxa −/− MEFs, the transformed noxa −/− puma −/− MEFs showed no greater resistance to UVR than noxa −/− counterparts, at any dose or time examined. Examination of puma expression revealed that transformation ablated puma mRNA induction specifically in response to UVR. Because etoposide induced puma expression comparably in the primary and transformed MEFs, the puma suppression is probably mediated by a UVR-induced signal acting parallel to p53 rather than by the p53 pathway itself.
A precedent for failure of puma induction in the face of DNA damage and active p53 emerged with the discovery that in γ-irradiated hematopoietic progenitors, puma expression is silenced downstream of p53 by the transcriptional repressor Slug (Wu et al., 2005). Indeed, we found that in E1A/ras transformed MEFs, but not in primary MEFs, UVR induces transcription of Slug, which may well be responsible for the failure of p53 to induce puma expression in these cells. However, in contrast to the hematopoietic progenitors, slug was up-regulated in the transformed MEFs irrespective of whether p53 was present.
Although we show that Noxa is the principal initiator of UVR-induced apoptosis, Puma is the principal initiator of apoptosis downstream of p53 activated by other genotoxic stimuli (Jeffers et al., 2003; Villunger et al., 2003). For example, loss of Noxa did not protect lymphoid cells from any apoptotic stimulus tested and offered only minor protection to MEFs exposed to etoposide or γ-irradiation (Shibue et al., 2003; Villunger et al., 2003), whereas the corresponding puma −/− cells were markedly resistant (Jeffers et al., 2003; Villunger et al., 2003). This difference probably reflects the fact that UVR produces pyrimidine dimers, whereas γ-irradiation and etoposide produce double-strand breaks in DNA. Although all genotoxic damage leads to p53 stabilization, the activity of p53 can be altered by the different kinases (e.g., ATM and ATR) selectively activated by these two types of DNA damage (Lowndes and Murguia, 2000; Abraham, 2001). A plausible model is that the levels and/or activity of Noxa and Puma are modulated selectively by different posttranslationally modified forms of p53 and possibly also by p53-independent signals (Fig. 10 A, X or Y) that are determined by the nature of the DNA damage.
Figure 10.
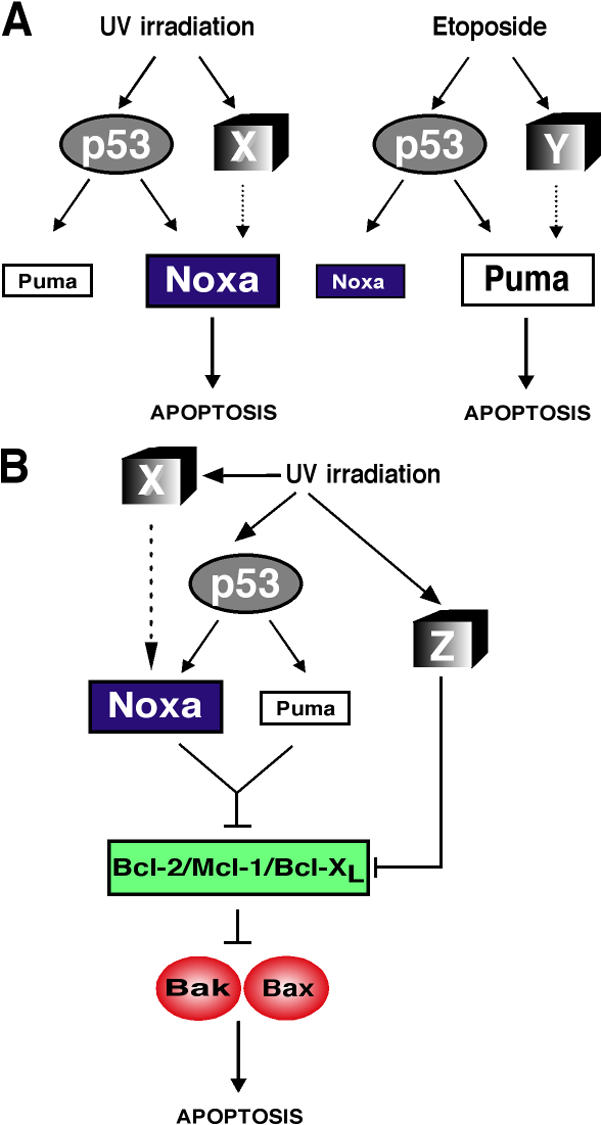
Model for the control of Noxa and Puma expression during DNA damage–induced apoptosis. (A) Model to explain why UVR-induced DNA damage triggers apoptosis in E1A/ras transformed MEFs predominantly via Noxa, whereas Puma predominates for apoptosis triggered by etoposide-induced DNA damage. X and Y represent distinct signaling pathways activated in parallel to the p53 pathway by the two different forms of DNA damage. (B) An integrated model of UVR-induced apoptosis. In addition to the p53-dependent pathway activated through Noxa and Puma, a p53-independent pathway to neutralization of antiapoptotic Bcl-2–like proteins is postulated, based on the finding that Bcl-2 overexpression provided p53 −/− E1A/ras transformed MEFs with additional protection from UVR. This pathway (Z) may involve both transcriptional and posttranslational mechanisms of regulating pro- and antiapoptotic members of the Bcl-2 family.
Bcl-2 overexpression increases the resistance of p53 −/− cells to UVR
Despite their enhanced resistance to UVR, a substantial proportion of noxa −/− and, remarkably, even p53 −/− cells still died after irradiation. The failure of FADD-DN or CrmA to inhibit the death of Noxa-deficient cells demonstrates that their UVR-induced apoptosis does not require death receptor signaling. In contrast, the substantial protection conveyed by Bcl-2 overexpression, even in the absence of p53, demonstrates that UV irradiation can trigger at least two distinct pathways to apoptosis. One of these pathways leads via p53 to induction of noxa and puma, whereas the other activates a Bcl-2–inhibitable apoptosis inducer via a p53-independent route (Fig. 10 B, Z).
What molecular mechanisms might mediate this p53-independent but Bcl-2–inhibitable pathway to UVR-induced cell death? Transformation by E1A may well have activated this pathway. By antagonizing RB, the E1A in the transformed MEFs deregulates E2F activity, including that of E2F1 (Sherr, 2001), which can enhance expression of several BH3-only proteins (Hershko and Ginsberg, 2004) and repress that of Mcl-1 (Croxton et al., 2002). Another interesting candidate is the JNK signaling pathway. In response to UVR, the JNKs phosphorylate diverse nuclear and cytoplasmic substrates, including c-Jun, a constituent of the transcription factor AP-1 that is rapidly induced upon UVR exposure (Liu et al., 1996). Notably, Jnk1 −/− Jnk2 −/− MEFs (Tournier et al., 2000) and c-Jun–deficient fibroblasts (Shaulian et al., 2000) are abnormally resistant to UVR.
Loss of p53 or Noxa protects keratinocytes from apoptosis induced by UV irradiation
To extend our in vitro findings to a relevant physiological context, we UV irradiated the skin of wt, p53 −/−, noxa −/−, puma −/−, and noxa −/− puma −/− mice. Consistent with previous findings (Ziegler et al., 1994), UV-irradiated p53 −/− animals exhibited significantly fewer SBCs than wt controls at 24 h after irradiation, and the extent of SBC formation in noxa −/− skin was comparable to that in p53 −/− skin. In contrast, the extent of SBC formation in puma −/− skin was indistinguishable from wt controls. Even at 72 h after UVR, there was enhanced survival of keratinocytes in p53 −/− and noxa −/− skin, demonstrating that loss of p53 or Noxa provided longer term protection and not only a delay in apoptosis. The greater role of Noxa in the skin may be related to our finding that noxa was increased approximately sevenfold and puma only approximately twofold at 24 h after irradiation. p53 protein is strongly up-regulated in UV-irradiated skin from 2 to 24 h after exposure (Hall et al., 1993), and accordingly, the robust induction of Noxa at 24 h required p53.
In summary, our analyses of two different cell line systems and the skin of intact mice identify Noxa as the principal mediator of UVR-induced apoptosis. Furthermore, we provide evidence of a pathway that collaborates with the p53 pathway to activate noxa, plus the activation of a p53- and Noxa-independent pathway to apoptosis that can be blocked by Bcl-2. The delineation of these pathways will provide insight into how the stimulus specificity of BH3-only protein activation is conferred downstream of p53.
Materials and methods
Mice
All experiments with animals were conducted according to the guidelines of the Melbourne Research Directorate Animals Ethics Committee. The generation of puma −/− (Villunger et al., 2003), noxa −/− (Villunger et al., 2003), bim −/− (Bouillet et al., 1999), bad −/− (Ranger et al., 2003), and p53 −/− mice (Jacks et al., 1994) has been described. The puma −/− and noxa −/− mice were generated on an inbred C57BL/6 background using C57BL/6-derived embryonic stem cells, whereas bim −/−, bad −/−, and p53 −_/_− mice were produced on a mixed C57BL/6 × 129Sv background using 129Sv-derived embryonic stem cells and were backcrossed with C57BL/6 mice for >10 generations. MEFs were derived from day 14.5 embryos and were cultured to ∼80% confluence before passage 1 (P1).
Whole skin UV irradiation, histological analysis, and microscopic imaging
A depilated region of dorsal skin was exposed to UVR supplied by a bank of six UVB lamps (FL20SE; Heraeus Amba Lamps). Spectral output was quantified with a radiometer (model IL1400A) fitted with a detector (SEL240; International Light).
All microscopy used either a Stemi SV11 (Carl Zeiss MicroImaging, Inc.) or a microscope (Axioplan 2; Carl Zeiss MicroImaging, Inc.). The latter used objective lenses (5×/NA 0.15 and 10×/NA 0.30). Images were recorded with a camera (AxioCam) and AxioVision software (Carl Zeiss MicroImaging, Inc.).
Generation of stably transformed cell lines
The Phoenix packaging line was used to produce high-titre, replication-incompetent retrovirus using the Fugene 6 Transfection method (Roche). In brief, Phoenix cells were transfected with plasmid DNA (pWZLH.12S [E1A] and pBabePuro.H-Ras; gifts from M. Schuler, Johannes Gutenberg University, Mainz, Germany; D. Green, St. Jude Children's Research Hospital, Memphis, TN; and S. Lowe, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). Primary MEFs were infected by centrifugation for 45 min at 32°C in the presence of viral supernatant. This process was performed on 2 consecutive days, and transfected cells were selected by incubation with 3 μg/ml puromycin and 100 μg/ml hygromycin B (Invitrogen) for at least 1 wk. FLAG–FADD-DN and FLAG-CrmA DNA sequences were subcloned into the MSCV-IRES-GFP vector backbone to generate retroviral expression constructs (Pellegrini et al., 2005). GFP-labeled cells were sorted using a FACStar cell sorter (Becton Dickinson) to obtain an 85–90% GFP+ population.
Cell culture and viability assays
Early passage (P2) primary MEFs and E1A/ras transformed MEFs were maintained in a high-glucose DME supplemented with 10% fetal calf serum, 10−6 M asparagine, and 50 μM 2-mercaptoethanol. Cells were seeded at 2.5 × 105 per well (6-well tissue culture plate) and cultured to ∼60–70% confluency before irradiation with UVC (UV lamp; Sankyo Denki). UVR output was quantified with a Spectroline Shortwave (254 nm) Ultraviolet meter. Cells were stimulated for 48 h with 100 ng/ml recombinant human FasL (FLAG-tagged; Qbiogene) cross-linked with 1 μg/ml anti-FLAG M2 monoclonal antibody (Sigma-Aldrich).
To assess cell viability, both floating and adherent cells were harvested, stained with 0.5 μg/ml propidium iodide (PI), and analyzed in a FACScan (Becton Dickinson). Forward and side light scatter parameters were used to exclude debris and cell viability calculated in comparison to a negative control (no PI staining).
Biochemical and morphological analyses
Cells were washed twice with ice-cold PBS before lysis with Onyx lysis buffer (20 mM Tris-HCl, pH 7.4, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10% glycerol, 2 mM sodium orthovanadate, 50 mM sodium fluoride, 1 μg/ml pepstatin, 1 μg/ml aprotinin, and 1 μg/ml leupeptin). 20 μg of total protein per sample were separated by gel electrophoresis (Tris-glycine Novex Pre-cast gels; Invitrogen) and transferred to nitrocellulose membranes (Hybond-C extra; GE Healthcare). Membranes were probed with antibodies to poly ADP ribose polymerase (Qbiogene), β-actin (Sigma-Aldrich), HSP70 (a gift from R. Anderson, Peter MacCallum Cancer Centre, East Melbourne, Australia), FLAG epitope tag (a gift from L. O'Reilly and D. Huang, Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia), ICAD (BD Biosciences), active caspase-3 (a gift from Y. Lazebnik, Cold Spring Harbor Laboratory), and Bim (clone 3C5; Qbiogene) and visualized using the ECL Western detection kit (GE Healthcare). Cytospin preparations (1 × 105 and 1 × 104 cells) were fixed in ice-cold methanol and stained with hematoxylin and eosin.
Quantitative PCR
Total RNA was extracted from ∼1 × 106 cells or ∼100 mg of whole mouse skin using Trizol (Invitrogen) and treated with DNase I (Promega). First-strand cDNA synthesis was performed using 5 μg total RNA, SuperScript II reverse transcriptase (Invitrogen), and oligo(dT)15 primer (Promega) according to the manufacturer's instructions. One tenth of the reverse-transcription reaction was subjected to quantitative PCR using Quantitect SYBR Green PCR Master Mix (QIAGEN) in 10-μl reaction volumes and the ABI PRISM system (Applied Biosystems). β-Actin was used as an internal control, and basal transcript levels were estimated from cDNA samples from puma −/− and noxa −/− cells.
Statistics
For statistical comparison, a t test was used, with P values <0.05 considered significant.
Online supplemental material
The supplemental text discusses DNA fragmentation assay, cytochrome c release assay, TUNEL staining, and confocal microscopy. Fig. S1 shows hallmarks of apoptosis in E1A/ras transformed MEFs. Fig. S2 shows viability analyses of bim −/−, bad −/−, and noxa −/− bim −/− primary MEFs. In Fig. S3, intracellular immunofluorescent staining demonstrates that all E1A/ras transformed cell lines express E1A comparably and that cyctochrome c release after UV irradiation is abrogated by the loss of Noxa and p53. In Fig. S4, TUNEL analysis confirms the induction of apoptotic cell death in UV-irradiated epidermal keratinocytes. In Fig. S5, analysis of skin at 72 h after UV irradiation demonstrates the long-term protection afforded by the loss of Noxa or p53. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200608070/DC1.
Supplemental Material
[Supplemental Material Index]
Acknowledgments
We thank Professors S. Cory, S. Lowe, and M. Schuler and Drs. A.W. Harris, P. Bouillet, D.C.S. Huang, L.A. O'Reilly, L. Coultas, V.S. Marsden, P.N. Kelly, M. Pellegrini, and M. Van Delft for gifts of knockout mice, antibodies, retroviral constructs, invaluable assistance, and helpful discussions.
This work was supported by grants and fellowships from the Cancer Council of Victoria, the National Health and Medical Research Council (Canberra; program 257502), the National Institutes of Health (CA 80188 and 43540), the Leukemia and Lymphoma Society of America (Specialized Center of Research Grant 7015), the Juvenile Diabetes Research Foundation, and the Austrian Science Fund.
The authors declare no conflicts of interest relating to this work.
Abbreviations used in this paper: FADD, Fas-associated death domain; ICAD, inhibitor of caspase-activated DNase; MEF, mouse embryonic fibroblast; PI, propidium iodide; SBC, sunburn cell; UVR, UV radiation; wt, wild-type.
References
- Abraham, R.T. 2001. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196. [DOI] [PubMed] [Google Scholar]
- Bennett, M., K. Macdonald, S.-W. Chan, J.P. Luzio, R. Simari, and P. Weissberg. 1998. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science. 282:290–293. [DOI] [PubMed] [Google Scholar]
- Bouillet, P., D. Metcalf, D.C.S. Huang, D.M. Tarlinton, T.W.H. Kay, F. Köntgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- Chen, L., S.N. Willis, A. Wei, B.J. Smith, J.I. Fletcher, M.G. Hinds, P.M. Colman, C.L. Day, J.M. Adams, and D.C.S. Huang. 2005. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 17:393–403. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan, A.M., K. O'Rourke, M. Tewari, and V.M. Dixit. 1995. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 81:505–512. [DOI] [PubMed] [Google Scholar]
- Croxton, R., Y. Ma, L. Song, E.B. Haura, and W.D. Cress. 2002. Direct repression of the Mcl-1 promoter by E2F1. Oncogene. 21:1359–1369. [DOI] [PubMed] [Google Scholar]
- Erlacher, M., E.M. Michalak, P.N. Kelly, V. Labi, H. Niederegger, L. Coultas, J.M. Adams, A. Strasser, and A. Villunger. 2005. BH3-only proteins Puma and Bim are rate-limiting for γ-radiation and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 106:4131–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, P.A., P.H. McKee, H.D. Menage, R. Dover, and D.P. Lane. 1993. High levels of p53 protein in UV-irradiated normal human skin. Oncogene. 8:203–207. [PubMed] [Google Scholar]
- Hershko, T., and D. Ginsberg. 2004. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 279:8627–8634. [DOI] [PubMed] [Google Scholar]
- Hill, L.L., A. Ouhtit, S.M. Loughlin, M.L. Kripke, H.N. Ananthaswamy, and L.B. Owen-Schaub. 1999. Fas ligand: a sensor for DNA damage critical in skin cancer etiology. Science. 285:898–900. [DOI] [PubMed] [Google Scholar]
- Huang, D.C.S., and A. Strasser. 2000. BH3-only proteins—essential initiators of apoptotic cell death. Cell. 103:839–842. [DOI] [PubMed] [Google Scholar]
- Jacks, T., L. Remington, B.O. Williams, E.M. Schmitt, S. Halachmi, R.T. Bronson, and R.A. Weinberg. 1994. Tumor spectrum analysis in _p53_-mutant mice. Curr. Biol. 4:1–7. [DOI] [PubMed] [Google Scholar]
- Jeffers, J.R., E. Parganas, Y. Lee, C. Yang, J. Wang, J. Brennan, K.H. MacLean, J. Han, T. Chittenden, J.N. Ihle, et al. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 4:321–328. [DOI] [PubMed] [Google Scholar]
- Kraemer, K.H., M.M. Lee, A.D. Andrews, and W.C. Lambert. 1994. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch. Dermatol. 130:1018–1021. [PubMed] [Google Scholar]
- Kuida, K., T.S. Zheng, S. Na, C.-Y. Kuan, D. Yang, H. Karasuyama, P. Rakic, and R.A. Flavell. 1996. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 384:368–372. [DOI] [PubMed] [Google Scholar]
- Li, G., V. Tron, and V. Ho. 1998. Induction of squamous cell carcinoma in p53-deficient mice after ultraviolet irradiation. J. Invest. Dermatol. 110:72–75. [DOI] [PubMed] [Google Scholar]
- Liu, Z.-G., R. Baskaran, E.T. Lea-Chou, L.D. Wood, Y. Chen, M. Karin, and J.Y.J. Wang. 1996. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 384:273–276. [DOI] [PubMed] [Google Scholar]
- Lowe, S.W., H.E. Ruley, T. Jacks, and D.E. Housman. 1993. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 74:957–967. [DOI] [PubMed] [Google Scholar]
- Lowndes, N.F., and J.R. Murguia. 2000. Sensing and responding to DNA damage. Curr. Opin. Genet. Dev. 10:17–25. [DOI] [PubMed] [Google Scholar]
- Nakano, K., and K.H. Vousden. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 7:683–694. [DOI] [PubMed] [Google Scholar]
- Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 288:1053–1058. [DOI] [PubMed] [Google Scholar]
- Pellegrini, M., S. Bath, V.S. Marsden, D.C. Huang, D. Metcalf, A.W. Harris, and A. Strasser. 2005. FADD and caspase-8 are required for cytokine-induced proliferation of hemopoietic progenitor cells. Blood. 106:1581–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath, H., A. Villunger, L.A. O'Reilly, J.G. Beaumont, L. Coultas, R.E. Cheney, D.C.S. Huang, and A. Strasser. 2001. Bmf: a pro-apoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 293:1829–1832. [DOI] [PubMed] [Google Scholar]
- Ranger, A.M., J. Zha, H. Harada, S.R. Datta, N.N. Danial, A.P. Gilmore, J.L. Kutok, M.M. Le Beau, M.E. Greenberg, and S.J. Korsmeyer. 2003. _Bad_-deficient mice develop diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA. 100:9324–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Villanueva, J., D. Greenhalgh, X.J. Wang, D. Bundman, S. Cho, M. Delehedde, D. Roop, and T.J. McDonnell. 1998. Human keratin-1.bcl-2 transgenic mice aberrantly express keratin 6, exhibit reduced sensitivity to keratinocyte cell death induction, and are susceptible to skin tumor formation. Oncogene. 16:853–863. [DOI] [PubMed] [Google Scholar]
- Shaulian, E., M. Schreiber, F. Piu, M. Beeche, E.F. Wagner, and M. Karin. 2000. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell. 103:897–907. [DOI] [PubMed] [Google Scholar]
- Sherr, C.J. 2001. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2:731–737. [DOI] [PubMed] [Google Scholar]
- Shibue, T., K. Takeda, E. Oda, H. Tanaka, H. Murasawa, A. Takaoka, Y. Morishita, S. Akira, T. Taniguchi, and N. Tanaka. 2003. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17:2233–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand, S., W.J. Hofmann, H. Hug, M. Müller, G. Otto, D. Strand, S.M. Mariani, W. Stremmel, P.H. Krammer, and P.R. Galle. 1996. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells— a mechanism of immune evasion? Nat. Med. 2:1361–1366. [DOI] [PubMed] [Google Scholar]
- Strasser, A., A.W. Harris, D.C.S. Huang, P.H. Krammer, and S. Cory. 1995. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 14:6136–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier, C., P. Hess, D.D. Yang, J. Xu, T.K. Turner, A. Nimnual, D. Bar-Sagi, S.N. Jones, R.A. Flavell, and R.J. Davis. 2000. Requirement of JNK for stress-induced activation of the cytochrome c–mediated death pathway. Science. 288:870–874. [DOI] [PubMed] [Google Scholar]
- Varfolomeev, E.E., M. Schuchmann, V. Luria, N. Chiannilkulchai, J.S. Beckmann, I.L. Mett, D. Rebrikov, V.M. Brodianski, O.C. Kemper, O. Kollet, et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 9:267–276. [DOI] [PubMed] [Google Scholar]
- Villunger, A., E.M. Michalak, L. Coultas, F. Müllauer, G. Böck, M.J. Ausserlechner, J.M. Adams, and A. Strasser. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science. 302:1036–1038. [DOI] [PubMed] [Google Scholar]
- Vousden, K.H., and X. Lu. 2002. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2:594–604. [DOI] [PubMed] [Google Scholar]
- Wu, W.S., S. Heinrichs, D. Xu, S.P. Garrison, G.P. Zambetti, J.M. Adams, and A.T. Look. 2005. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 123:641–653. [DOI] [PubMed] [Google Scholar]
- Yu, J., L. Zhang, P.M. Hwang, K.W. Kinzler, and B. Vogelstein. 2001. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell. 7:673–682. [DOI] [PubMed] [Google Scholar]
- Ziegler, A., A.S. Jonason, D.J. Leffell, J.A. Simon, H.W. Sharma, J. Kimmelman, L. Remington, T. Jacks, and D.E. Brash. 1994. Sunburn and p53 in the onset of skin cancer. Nature. 372:773–776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental Material Index]