βcap73-ARF6 Interactions Modulate Cell Shape and Motility after Injury In Vitro (original) (raw)
Abstract
To understand the role that ARF6 plays in regulating isoactin dynamics and cell motility, we transfected endothelial cells (EC) with HA-tagged ARF6: the wild-type form (WT), a constitutively-active form unable to hydrolyze GTP (Q67L), and two dominant-negative forms, which are either unable to release GDP (T27N) or fail to bind nucleotide (N122I). Motility was assessed by digital imaging microscopy before Western blot analysis, coimmunoprecipitation, or colocalization studies using ARF6, β-actin, or β-actin-binding protein-specific antibodies. EC expressing ARF6-Q67L spread and close in vitro wounds at twice the control rates. EC expressing dominant-negative ARF6 fail to develop a leading edge, are unable to ruffle their membranes (N122I), and possess arborized processes. Colocalization studies reveal that the Q67L and WT ARF6-HA are enriched at the leading edge with β-actin; but T27N and N122I ARF6-HA are localized on endosomes together with the β-actin capping protein, βcap73. Coimmunoprecipitation and Western blot analyses reveal the direct association of ARF6-HA with βcap73, defining a role for ARF6 in signaling cytoskeletal remodeling during motility. Knowledge of the role that ARF6 plays in orchestrating membrane and β-actin dynamics will help to reveal molecular mechanisms regulating actin-based motility during development and disease.
INTRODUCTION
Coordination of membrane and isoactin cytoskeletal dynamics represents a critical interface in orchestrating the site-specific delivery of subcellular constituents as well as directing cellular locomotion. For example, signaling through phosphoinositides and Ras family small GTPases has been implicated as pivotal in stimulating actin cytoskeletal reorganization and plasma membrane remodeling during cell motility (Qualmann and Kessels, 2002). Phosphoinositides not only regulate the ability of profilins to enhance nucleotide exchange on actin, they can cause actin dissociation from profilin (Yin and Janmey, 2002). Phosphoinositides can also cause the dissociation of capping proteins from the barbed ends of actin filaments at the membrane, permitting filament assembly and elongation. The ability of phosphoinositides to influence cytoskeletal dynamics in a significant way is conferred by their binding affinity for so many important cytoskeletal signaling molecules, such as members of the Rho GTPase family.
Despite the fact that the Rho GTPase family of signaling proteins has been shown to modulate cytoskeletal remodeling during developmental or disease-related processes (Etienne-Manneville and Hall, 2002), the molecular mechanisms regulating the interactions of these proteins with the actin cytoskeleton have not been clearly defined. The best-characterized members of this large family are Rho, Rac, and CDC42, which signal through the actin network to regulate the assembly of stress fibers (Ridley and Hall, 1992), lamellopodia (Ridley et al., 1992), and filopodia (Kozma et al., 1995; Brown et al., 2000), respectively. Recently, it has been revealed that the Rho GTPase family may signal through the ADP-ribosylation factor (ARF) family to effect cytoskeletal remodeling during cell motility (Zhang et al., 1999; Santy and Casanova, 2001; Tarricone et al., 2001). In fact, ARF6 plays a dual role in regulating both actin cytoskeletal and plasma membrane dynamics. It also colocalizes with Rac1 on endosomes, and the two are simultaneously transported to the plasma membrane during motility (Boshans et al., 2000). Both Rac1 and ARF6 have nucleotide-dependent interactions with the Arfaptin and Arfophilin proteins (Shin et al., 2001), which may play a role in their colocalization and transport linkage. The localization of ARF6 is nucleotide dependent; in its GDP-bound form, it has been localized to the cytosol and to endosomal compartments, and when bound to GTP, it becomes localized to the plasma membrane (Gaschet and Hsu, 1999) with ARNO, its specific nucleotide exchange factor (Frank et al., 1998; Santy and Casanova, 2001). ARF6, Rac1, and Rho have all been shown to activate PIP-5-kinase (Brown et al., 2001), an enzyme that generates PI-4,5 biphosphate, though only ARF6-GTP and Rac1 do so directly (Tolias et al., 2000). This enzyme, in turn, can aid actin cytoskeletal remodeling and cell motility by unmasking the barbed ends of actin filaments capped by gelsolin (Carlier, 1998; Pollard and Borisy, 2003).
The dendritic nucleation hypothesis has recently been put forward to explain the mechanisms regulating actin assembly during motility (Machesky et al., 1999; Svitkina and Borisy, 1999; Blanchoin et al., 2000; Amann and Pollard, 2001). This hypothesis states that actin assembly and branching occurs by ARP2/3 actin nucleation, which is activated by WASP, on the sides of older actin filaments. Profilin and capping proteins function to limit the length of new actin filaments, favoring a branched assembly. Along with actin-depolymerizing factor, cofilin, these proteins provide a dynamic framework to explain actin filament assembly and turnover during motility. However, this hypothesis neither explains the physical nature of the association of the terminal actin network with the plasma membrane, nor does it address the functional diversity of the cellular isoactin network itself (Herman, 1993; Khaitlina, 2001). Indeed, despite the highly conserved nature of the actin multigene family, there is considerable evidence indicating that the actin isoforms are functionally distinct. With respect to actin-based cell motility, we have recently revealed the important role of β-actin and its isoform-specific barbed end capping protein, βcap73 (Shuster, 1995; Shuster et al., 1996; Welch and Herman, 2002).
In our experiments, we have sought to elucidate the role of ARF6 in coordinating cell motility through β-actin dynamics and membrane trafficking, using an in vitro model of wound healing. Transient transfection of HA-tagged wild-type and mutant forms of ARF6 into vascular endothelial cells together with analysis of living cell migration in response to injury help to reveal the cellular behavior as a function of ARF6 expression. In turn, colocalization studies demonstrate that dominant-negative ARF6-HA proteins (T27N and N122I) colocalize with βcap73 and early endosomal antigen-1 (EEA-1) in abnormal endosomal compartments, whereas the dominant-positive and wild-type forms predominantly colocalize with β-actin at the leading edge of motile cells. Endothelial cells expressing Q67L dominantpositive ARF6-HA spread at twice the rate of untransfected cells in time-lapse wound healing assays. Most interestingly, coimmunoprecipitation and Western blotting reveal the direct association of ARF6 with βcap73. Taken together, these results support the hypothesis that the association of ARF6 with βcap73 may play a pivotal role in regulating β-actin and membrane dynamics during cell motility.
MATERIALS AND METHODS
Cell Culture
All of our experiments were performed on bovine retinal endothelial cells. For time-lapse imaging and indirect immunofluorescence, cells were plated on 10-mm2 glass coverslips in Costar 24-well plates, at a density of 100,000 cells per well, to reach ∼80% confluence 24 h after plating. All cells were grown in DMEM supplemented with 5% bovine calf serum and 0.5% each l-glutamine and penicillin-streptomycin-fungizone.
Transfection
Transfection was carried out with the Effectene reagent kit from Qiagen (Valencia, CA), using 0.2 μg of DNA per coverslip. DNA prepared with Effectene was diluted in complete media and applied to cell populations ∼24 h after plating, when cultures had reached ∼80% confluence. These cultures were incubated for 24 h in the reagent, then washed with PBS, and fed with fresh media. Cells were transfected with plasmids for expression of ARF6 proteins with HA tags. Plasmids were engineered for wild-type ARF6; a dominant-negative, GTP binding-defective mutant, T27N; and a constitutively active, GTP hydrolysis-defective mutant, Q67L. Another HA-tagged ARF6 mutant, which is completely incapable of binding nucleotide, N122I, was a generous gift of Dr. Ralph Isberg (Tufts University.)
Time-lapse Imaging
Twenty-four to 30 h after transfection, coverslips were mounted for time-lapse imaging. Transfected monolayers, with cells attached to coverslips, were injured with a razor blade to remove half of the monolayer and then mounted in the specially designed cell culture chamber, which maintains physiological pH. The cell culture chamber was placed on the stage of a Zeiss Axiovert fluorescence microscope (40× objective, NA = 0.75; Thornwood, NY) and kept at 37°C using an air curtain (Nevtek, Burnsville, VA). A target field along the wound edge was selected and phase-contrast images were captured every 5 min using the Metamorph software package (Universal Imaging Corp., Downingtown, PA) After imaging, the coverslips with cells attached were removed from the chamber and processed for immunofluorescence microscopy.
Image Analysis
Rate of cell motility was determined by analyzing multiple images from several experiments of the time-lapse series (n ≥ 30), using Adobe Photoshop 5.5 (San Jose, CA). In each image, the space devoid of cells was measured in pixels and then converted to square micrometers. The difference in empty area between each pair of images was calculated and then divided by the elapsed time to give a rate of movement:
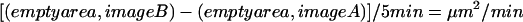
We also analyzed the behavior of single cells within and between experimental groups. Translocation of the cell was measured by selecting a feature of the nucleus and tracking its X and Y coordinates in each frame. This was converted to a linear measurement in micrometers (Metamorph, Universal Imaging Co.) Spreading was also measured, by selecting each cell around its border and converting this area measurement into square micrometers (Photoshop, Adobe.)
Indirect Immunofluorescence
Twenty-four to 36 h after transfection, cells on glass coverslips were fixed with 4% formaldehyde in DMEM and permeablized for 90 s at room temperature with buffered Triton X-100 as described (Hoock et al., 1991). Coverslips were then incubated for 45 min in the specified primary antibodies diluted in PBS/azide, followed by the corresponding secondary antibodies for 45 min. Coverslips were mounted using the SlowFade Light Antifade Kit (Molecular Probes, Eugene, OR). Slides were imaged using the same microscope setup as described above for time-lapse imaging.
Antibodies
Primary antibodies for immunofluorescence and Western blotting are anti-βcap73 (murine monoclonal IgM) and anti-β-actin (affinity-selected, rabbit polyclonal IgG), rat monoclonal anti-HA IgG (Boehringer Mannheim via Roche Molecular Biochemicals, Indianapolis, IN), and anti-ARF6 (3F8, monoclonal IgG; Yang et al., 1998). Secondary antibodies are Alexa 488– and 546–conjugated antibodies from Molecular Probes and FITC- and TRITC-conjugated antibodies from Jackson Immunofluorescence (West Grove, PA.)
For all immunofluorescence experiments, several controls were performed. Working dilutions (1:100–1:500) of labeled secondary antibodies were tested for background fluorescence and nonspecific binding, which were not detectable. For these controls, cells were fixed and permeablized under standard test conditions, but the primary antibodies were omitted. In addition, to test for cross-species and cross-isotype reactivities, all secondary antibodies were examined for their ability to combine with primary antibodies, as viewed by immunofluorescence and where applicable, Western blotting. To establish that each commercially available secondary antibody used recognized only its cognate primary antibody, secondary antibodies were tested with primary antibodies generated from a different species. For example, we confirmed that the FITC goat anti-rat IgG cross-reacted with rat IgG, but not with rabbit IgG. This was especially critical when simultaneous double immunofluorescence was performed on the same specimen. In this way, each pair of primary-secondary antibodies was tested to check for cross-species reactivity. Additionally, localization patterns obtained during colocalization studies were compared with results obtained when only a single antigen was being localized. In all cases, results from single and double immunofluorescence were consistent.
Immunoprecipitation
Cell lysis and immunoprecipitation were performed as previously described (Witczak et al., 1999; Welch and Herman, 2002). Cells were lysed in RIPA buffer containing 150 mM NaCl, 30 mM Tris-HCl, pH 8.0, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Nonidet P-40 and then incubated 10 min at room temperature in presence of protease inhibitors. Approximately 10 μg of anti-HA primary antibody was incubated with 10 μl of packed protein A/Sepharose beads (Pharmacia, Piscataway, NJ) for 1 h at room temperature with gentle rotation. Meanwhile, 250 μl of lysate (∼400 μg protein) was precleared with 10 μl of protein A/Sepharose for 1 h at room temperature. The precleared lysate was then incubated with antibody-protein A/Sepharose complex overnight at 4°C with gentle rotation. The beads were washed five times in RIPA buffer and one time in buffer containing 30 mM Tris-HCl, pH 8.0, and 50 mM NaCl. The beads were then boiled in 50 μl of 1× sample buffer for 3 min, and the supernatant was run on SDS-PAGE.
Immunoblotting
Samples (cell lysate or immunoprecipitate) were boiled in SDS sample buffer for 3 min before loading on 1.5-mm-thick polyacrylamide slab gels containing 0.1% SDS. The samples were separated by electrophoresis and were transferred to nitrocellulose (Schleicher & Schuell, Keene, NH) overnight at 200 mA in a Tris-buffered methanol/SDS solution. Western blotting was performed as described by Amersham (Piscataway, NJ). Briefly, blots were blocked with 5% nonfat dry milk in TBST (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween-20) for at least 1 h at room temperature or overnight at 4°C. For immunodetection, the blots were incubated with primary antibody at ∼5 μg/ml for at least 2 h at room temperature and incubated 1 h in ∼ 0.4 μg/ml goat anti-rabbit or anti-rat IgG HRP. Detection was performed with Supersignal Western detection reagents (Pierce, Rockford, IL). Images of stained gels were recorded with a model IS-1000 digital imager (Alpha Inotech, San Leandro, CA).
RESULTS
Expression of Mutant ARF6 in Vascular Endothelial Cells: Role in Regulating Motility
To investigate the role of ARF6 in regulating membrane and cytoskeletal dynamics, we transiently transfected microvascular endothelial cells with plasmids encoding HA-tagged wild-type and mutant ARF6. ARF6-HA expression was confirmed by Western blotting of whole cell lysates from i) untreated populations, ii) populations exposed to the Effectene transfection reagent without plasmid DNA, and iii) those transfected with each of the ARF6-HA expression plasmids (Figure 1). The blot was probed with anti-ARF6 (3F8), revealing the 20-kDa endogenous ARF6 in all cell populations and the 21-kDa HA-tagged ARF6 in the transfected populations. Results reveal that our transfection is successful and that the various ARF6 mutant proteins are being comparably expressed.
Figure 1.

Western blot of endogenous and recombinant ARF6. Bovine retinal endothelial cells were transfected with the ARF6-HA wild-type, active Q67L, and negative T27N plasmids with the Qiagen Effectene reagent for an incubation period of 48 h. Untransfected cells and cells treated with the Effectene reagent in the absence of plasmid DNA were used as negative controls. This Western blot was probed with mouse anti-ARF6 (3F8) to show both endogenous ARF6 expression (20 KD) and expression of HA-tagged ARF6 (21 KD).
To assess the influence of mutant ARF6 expression on cell motility, we quantified endothelial cell migration over a 2-h period in response to injury. We have compared the behavior of entire populations as well as individual cells. In populations transfected with the Q67L (dominant-active) ARF6 mutant, the leading edge advances at approximately twice the rate seen in untransfected control populations or those transfected with wild-type, T27N, or N122I plasmids (Figure 2). The cells at the leading edges of the Q67L-transfected populations generally display a smooth, broad lamellar fan and form a relatively contiguous leading edge. The leading edges of control and wild-type transfected populations also appear smooth and contiguous, though they do not ruffle and advance as quickly as those of the Q67L populations. Although the overall motility rates of the T27N and N122I ARF6-expressing populations are not significantly different from those of the untransfected controls or wild-type transfected populations, there are observable alterations in cell morphology and motility. Populations expressing either of the dominant-negative proteins fail to form a smooth, ruffled leading edge. Some cells do not appear to ruffle their membranes at all, producing gaps in the advancing edge, whereas other cells produce narrow, elongated pseudopodia instead of a broad membrane fan.
Figure 2.

Spreading Rates of ARF6-HA transfected populations. This chart shows the rates of movement of the leading edge of whole populations over a two hour period after monolayer injury (see MATERIALS AND METHODS). The averaged results of five experiments are shown together (±SEM) in arbitrary units, with the rate of motility of the control populations set equal to 1.
ARF6-HA Antibody Localization
To further investigate how altered motility and ability to establish membrane projections is related to the form and distribution of ARF6, we performed antibody localization in cells whose motility had been recorded in time-lapse. After transfection and time-lapse imaging, cells were fixed and processed for indirect immunofluorescence to localize ARF6-HA, in conjunction with β-actin and βcap73. In the samples analyzed by time-lapse imaging, recorded fields were relocated after fixation and staining and transfected cells along the wound edge were identified by robust anti-HA staining (Figure 3). Generally, between 15 and 50% of the cells in a transfected population highly express the HA-tagged ARF6. With the T27N mutant, efficiency is slightly lower. Individual cells expressing the Q67L ARF6 mutant spread to as much as 200% of their original size, whereas neighboring untransfected cells and cells from untransfected populations spread to a maximum of 130% of their original size, with an average maximum of 115% among the control cells examined. No significant difference was found in the cell migration rates (measured by nuclear translocation) of individual transfected cells, as compared with controls, with any of the ARF6-HA plasmids.
Figure 3.

Spreading of an individual Q67L HA-ARF6 expressing cell. (A) Live cells, 60 min after initial wounding; scale bar, 50 μm. (B) Live cells, 90 min after wounding, last frame of time-lapse. (C) Phase contrast image of fixed and stained sample. (D) β-actin staining; scale bar, 50 μm. (E) ARF6-HA staining. (F and G) β-actin and ARF6-HA staining, respectively. Scale bar, 20 μm.
Indirect immunofluorescence was used to compare the localization of the wild-type with the mutant ARF6-HA proteins (Figure 4). In the Q67L ARF6 expressing cells, ARF6-HA is distributed throughout the cytoplasm, with increased localization in membrane veils at the leading edge of crawling cells located at the in vitro wound edge. In some cells, small punctae are present, suggesting the association of some ARF6 molecules with endosomes. These Q67L-expressing cells possess large lamellar fans, consistent with the spreading seen in the individual cells analyzed in time-lapse. In cells expressing the wild-type form, cytoplasmic distribution and small endosomal pools are also seen, with increased concentration at the leading edge, though less than that seen in the Q67L-expressers. Cell morphology in wild-type transfectants is similar to that of neighboring unstained cells.
Figure 4.

Localization of HA-tagged ARF6. β-actin (red); ARF6-HA (green). In the active Q67L and Wild-type expressing cells, ARF6-HA can be seen throughout the cytoplasm and plasma membrane, and concentrated at the leading edge. In the two dominant-negative mutants, abnormal endosomal localization can be seen, and a near-absence of ARF6-HA from the periphery. Scale bar, 50 μm.
The dominant-negative forms of ARF6, T27N, and N122I show a localization pattern quite different from the wild-type and dominant-positive forms. The majority of the tagged protein is seen in abnormally large endosomes, with little or no cytoplasmic or leading edge localization. The distribution of these endosomes does not resemble that of any recognizable structure such as the Golgi complex, because they are spread throughout the central area of the cell. The morphology of these cells is clearly affected; they fail to develop a smooth lamellar fan, instead displaying narrower, branched projections or a blunted, immobile leading edge.
In all endothelial cells examined, β-actin is present at the cell periphery and is strongly concentrated at the leading edge together with βcap73 (Welch and Herman, 2002). In the wild-type and Q67L ARF6-HA–expressing cells, these concentrations of β-actin colocalize with areas of enrichment of ARF6-HA at the leading edge. The absence of dominant-negative ARF6 from the cell periphery does not appear to alter β-actin localization.
Colocalization of Dominant-Negative ARF6 with EEA-1
After our observation of the concentration of dominant-negative ARF6 on endosomes, we stained transfected endothelial cell populations for a variety of endosomal markers, in an attempt to determine if a specific type of intracellular compartment was affected. Through these experiments, we found that the endosomes in which the mutant ARF6 is seen generally also contain early endosomal antigen-1 (EEA-1, Figure 5). Other antibodies tested include those against TGN38 and Rab11, proteins associated with membrane trafficking within and from the Golgi complex. None of the antibodies tested showed colocalization with any form of ARF6 except for EEA-1.
Figure 5.

Colocalization of HA-tagged ARF6 T27N with Early Endosomal Antigen-1 and βcap73. (A) Phase contrast, (B) ARF6-HA T27N, and (C) EEA-1. Colocalization is highlighted with arrows. Scale bar, 50 μm.
βcap73 Coimmunoprecipitates with ARF6
β-actin assembly is critical for projection formation and cell motility and occurs primarily at the leading edge of motile cells. A barbed-end capping protein specific for β-actin, βcap73, has been found, and has been shown to regulate this assembly. As we have seen that β-actin colocalizes with wild-type and dominant-positive ARF6 at the leading edge, we wanted to determine whether mutant ARF6 modulation of motility might be the result of the direct association with βcap73. Indirect immunofluorescence reveals that a subpopulation of βcap73 colocalizes strongly with the dominant-negative ARF6-HA mutants within the endosomal compartment (Figure 6). This endosomal colocalization was not observed in the wild-type or Q67L-expressers. To determine whether βcap73 and ARF6 interact directly, immunoprecipitation was performed on endothelial cell populations transfected with each of the ARF6 plasmids, using the monoclonal rat anti-HA antibody to “pull down” the ectopic ARF6 proteins, together with any binding partners. The resulting product was probed via Western blot with anti-βcap73 IgM, and with the anti-HA antibody for comparison (Figure 7). Results reveal that βcap73 binds directly to all forms of HA-tagged ARF6. Levels of βcap73 bound to the wild-type protein and the Q67L and T27N mutants are comparable. Interestingly, the nucleotide binding-impaired N122I mutant precipitates four times the level of βcap73, as analyzed by spot densitometry.
Figure 6.

(A and D) Phase contrast, (B) T27N mutant ARF6-HA, (E) Q67L mutant ARF6-HA, and (C and F) βcap73. Colocalization is highlighted with arrows.
Figure 7.

Immunoprecipitation of βcap73 by ARF6-HA. (A) Western blot of total endothelial cell lysate confirming the expression of HA-tagged ARF6 proteins from vector plasmids. (B) Immunoprecipitation with Rat anti-HA mAb using the same endothelial cell lysates, Western blot for the presence of βcap73.
DISCUSSION
Our results, in conjunction with the published findings of others, support a central role for ARF6 in the coordination of plasma membrane recycling and actin cytoskeletal remodeling, but extend these observations by revealing the association of ARF6 with the β-actin network via βcap73. We find that overexpression of dominant-positive ARF6 increases endothelial cell motility, primarily by inducing increased spreading at the leading edge, where active ARF6 colocalizes with β-actin. ARF6 inactivation, studied through overexpression of dominant-negative mutants, results in impaired spreading and a blunted or arborized leading edge. Most importantly, we have shown a direct link to the β-actin cytoskeleton through βcap73, a novel, β-actin–specific barbed end capping protein.
Localization of ARF6
The information gathered by overexpression of ARF6 followed by indirect immunofluorescence labeling confirms that ARF6 is important for regulating traffic between the early and recycling endosomes and the plasma membrane. Wild-type and Q67L ARF6 appear to move efficiently from the interior of the cell to the plasma membrane; both forms are concentrated at the leading edge of motile cells. This is consistent with findings that GTP-bound ARF6 is localized to the plasma membrane in other cell types (Gaschet and Hsu, 1999). Colocalized with the dominant-positive and wild-type forms at the leading edge is β-actin, the actin isoform known to be important for cell motility (Hoock et al., 1991). In contrast, the dominant-negative T27N and N122I mutants become trapped in abnormally large early or recycling endosome pools, colocalized with βcap73. Immunoprecipitation assays reveal that βcap73 and ARF6 interact directly.
Regulation of Isoactin Dynamics during Cell Motility and Spreading
The association of ARF6 with β-actin-capping protein βcap73 reveals a direct role in signaling cytoskeletal remodeling during motility. The availability of barbed ends of β-actin filaments for assembly is clearly critical for spreading and motility. Through its binding to βcap73, ARF6 may be able to regulate the binding affinity of βcap73 for β-actin at its known β-actin binding domain, and perhaps remove it from the active peripheral zone to the recycling endosome, thus altering β-actin polymerization kinetics. Through its activation of PIP-5-kinase and the subsequent generation of PI-4,5 biphosphate, ARF6 may be able to decrease the binding affinity of βcap73 for β-actin, because PI-4,5 biphosphate has been shown dissociate capping protein gelsolin from F-actin (Schafer et al., 1996). This dissociation may be the necessary first step for ARF6 to transport βcap73 to the recycling endosome. The exact relationship among the molecules in the plasma membrane-associated actin-binding complex is unclear, but βcap73 is obviously well positioned to have a significant effect on β-actin dynamics and cell motility through its association with ARF6. In fact, cells overexpressing the calpain inhibitor calpastatin have a phenotype similar to some of the ARF6 dominant-negative–expressing cells: deficient motility, aberrant projection formation, and abnormal cell shape. Indeed, these cells have been found to have an overabundance of βcap73 that is 10 times that of their wild-type parental cells (Welch and Herman, 2002). Furthermore, overexpression of transfected βcap73 causes cellular processes that are arborized and β-actin rich, similar to the morphology seen in cells overexpressing dominant-negative ARF6.
ARF6 and Membrane Recycling
Efficient membrane recycling and timely delivery of subcellular components is necessary for cell motility. The importance of ARF6 in this process is evidenced by the increased motility seen with the Q67L dominant-positive mutant in the time-lapse assay. The same dominant-positive mutant has also been found to stimulate migration in epithelial cells (Palacios et al., 2001). Activation of ARF6 by its exchange factor, ARNO, is sufficient to promote this epithelial migration (Santy and Casanova, 2001). ARF6 has already been implicated in membrane recycling and exocytosis in epithelial (D'Souza-Schorey et al., 1998) and neural cells, evidence which, taken together, supports our model of ARF6 as a regulator of endosome recycling in endothelial cells. Clearly, understanding the role of ARF6 in regulating membrane and isoactin dynamics should prove instrumental in revealing the mechanics of cell motility.
Acknowledgments
This work was supported by National Institutes of Health Grants EY 09033 and GM 55110 to I.M.H. and in part by a grant from the from the American Heart Association to C.D.-S.
References
- Amann, K.J., and Pollard, T.D. (2001). The Arp2/3 complex nucleates actin filament branches from the sides of pre-existing filaments. Nat. Cell Biol. 3, 306–310. [DOI] [PubMed] [Google Scholar]
- Blanchoin, L., Amann, K.J., Higgs, H.N., Marchand, J.B., Kaiser, D.A., and Pollard, T.D. (2000). Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature 404, 1007–1011. [DOI] [PubMed] [Google Scholar]
- Boshans, R.L., Szanto, S., van Aelst, L., and D'Souza-Schorey, C. (2000). ADP-ribosylation factor 6 regulates actin cytoskeleton remodeling in coordination with Rac1 and RhoA. Mol. Cell. Biol. 20, 3685–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, F.D., Rozelle, A.L., Yin, H.L., Balla, T., and Donaldson, J.G. (2001). Phosphatidylinositol 4, 5-bisphosphate and Arf6-regulated membrane traffic. J. Cell Biol. 154, 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.D., Cornejo, B.J., Kuhn, T.B., and Bamburg, J.R. (2000). Cdc42 stimulates neurite outgrowth and formation of growth cone filopodia and lamellipodia. J. Neurobiol. 43, 352–364. [DOI] [PubMed] [Google Scholar]
- Carlier, M.F. (1998). Control of actin dynamics. Curr. Opin. Cell Biol. 10, 45–51. [DOI] [PubMed] [Google Scholar]
- D'Souza-Schorey, C., van Donselaar, E., Hsu, V.W., Yang, C., Stahl, P.D., and Peters, P.J. (1998). ARF6 targets recycling vesicles to the plasma membrane: insights from an ultrastructural investigation. J. Cell Biol. 140, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and Hall, A. (2002). Rho GTPases in cell biology. Nature 420, 629–635. [DOI] [PubMed] [Google Scholar]
- Frank, S., Upender, S., Hansen, S.H., and Casanova, J.E. (1998). ARNO is a guanine nucleotide exchange factor for ADP-ribosylation factor 6. J. Biol. Chem. 273, 23–27. [DOI] [PubMed] [Google Scholar]
- Gaschet, J., and Hsu, V.W. (1999). Distribution of ARF6 between membrane and cytosol is regulated by its GTPase cycle. J. Biol. Chem. 274, 20040–20045. [DOI] [PubMed] [Google Scholar]
- Herman, I.M. (1993). Actin isoforms. Curr. Opin. Cell Biol. 5, 48–55. [DOI] [PubMed] [Google Scholar]
- Hoock, T.C., Newcomb, P.M., and Herman, I.M. (1991). Beta actin and its mRNA are localized at the plasma membrane and the regions of moving cytoplasm during cellular response to injury. J. Cell Biol. 112, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitlina, S.Y. (2001). Functional specificity of actin isoforms. Int. Rev. Cytol. 202, 35–98. [DOI] [PubMed] [Google Scholar]
- Kozma, R., Ahmed, S., Best, A., and Lim, L. (1995). The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 15, 1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky, L.M., Mullins, R.D., Higgs, H.N., Kaiser, D.A., Blanchoin, L., May, R.C., Hall, M.E., and Pollard, T.D. (1999). Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc. Natl. Acad. Sci. USA 96, 3739–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios, F., Price, L., Schweitzer, J., Collard, J.G., and D'Souza-Schorey, C. (2001). An essential role for ARF6-regulated membrane traffic in adherens junction turnover and epithelial cell migration. EMBO J. 20, 4973–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard, T.D., and Borisy, G.G. (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465. [DOI] [PubMed] [Google Scholar]
- Qualmann, B., and Kessels, M.M. (2002). Endocytosis and the cytoskeleton. Int. Rev. Cytol. 220, 93–144. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., and Hall, A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., Paterson, H.F., Johnston, C.L., Diekmann, D., and Hall, A. (1992). The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401–410. [DOI] [PubMed] [Google Scholar]
- Santy, L.C., and Casanova, J.E. (2001). Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J. Cell Biol. 154, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, D.A., Jennings, P.B., and Cooper, J.A. (1996). Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J. Cell Biol. 135, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, O.H., Couvillon, A.D., and Exton, J.H. (2001). Arfophilin is a common target of both class II and class III ADP-ribosylation factors. Biochemistry 40, 10846–10852. [DOI] [PubMed] [Google Scholar]
- Shuster, C.B., Lin, A.Y., Nayak, R., and Herman, I.M. (1996). Beta cap 73, a novel beta actin-specific binding protein. Cell Motil. Cytoskel. 35, 175–187. [DOI] [PubMed] [Google Scholar]
- Shuster, C.B.H., I. M. (1995). Indirect association of ezrin with F-actin: isoform specificity and calcium sensitivity. J. Cell. Biol. 128, 837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., and Borisy, G.G. (1999). Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145, 1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarricone, C., Xiao, B., Justin, N., Walker, P.A., Rittinger, K., Gamblin, S.J., and Smerdon, S.J. (2001). The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. Nature 411, 215–219. [DOI] [PubMed] [Google Scholar]
- Tolias, K.F., Hartwig, J.H., Ishihara, H., Shibasaki, Y., Cantley, L.C., and Carpenter, C.L. (2000). Type Ialpha phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr. Biol. 10, 153–156. [DOI] [PubMed] [Google Scholar]
- Welch, A.Y., and Herman, I.M. (2002). Cloning and characterization of beta-CAP73, a novel regulator of beta-actin assembly. Int. J. Biochem. Cell Biol. 34, 864–881. [DOI] [PubMed] [Google Scholar]
- Witczak, O., Skalhegg, B.S., Keryer, G., Bornens, M., Tasken, K., Jahnsen, T., and Orstavik, S. (1999). Cloning and characterization of a cDNA encoding an A-kinase anchoring protein located in the centrosome, AKAP450. EMBO J. 18, 1858–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C.Z., Heimberg, H., D'Souza-Schorey, C., Mueckler, M.M., and Stahl, P.D. (1998). Subcellular distribution and differential expression of endogenous ADP-ribosylation factor 6 in mammalian cells. J. Biol. Chem. 273, 4006–4011. [DOI] [PubMed] [Google Scholar]
- Yin, H.L., and Janmey, P.A. (2002). Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 2, 2. [DOI] [PubMed] [Google Scholar]
- Zhang, Q., Calafat, J., Janssen, H., and Greenberg, S. (1999). ARF6 is required for growth factor- and rac-mediated membrane ruffling in macrophages at a stage distal to rac membrane targeting. Mol. Cell. Biol. 19, 8158–8168. [DOI] [PMC free article] [PubMed] [Google Scholar]