The role of ATP and adenosine in the brain under normoxic and ischemic conditions (original) (raw)
Abstract
By taking advantage of some recently synthesized compounds that are able to block ecto-ATPase activity, we demonstrated that adenosine triphosphate (ATP) in the hippocampus exerts an inhibitory action independent of its degradation to adenosine. In addition, tonic activation of P2 receptors contributes to the normally recorded excitatory neurotransmission. The role of P2 receptors becomes critical during ischemia when extracellular ATP concentrations increase. Under such conditions, P2 antagonism is protective. Although ATP exerts a detrimental role under ischemia, it also exerts a trophic role in terms of cell division and differentiation. We recently reported that ATP is spontaneously released from human mesenchymal stem cells (hMSCs) in culture. Moreover, it decreases hMSC proliferation rate at early stages of culture. Increased hMSC differentiation could account for an ATP-induced decrease in cell proliferation. ATP as a homeostatic regulator might exert a different effect on cell trophism according to the rate of its efflux and receptor expression during the cell life cycle. During ischemia, adenosine formed by intracellular ATP escapes from cells through the equilibrative transporter. The protective role of adenosine A1 receptors during ischemia is well accepted. However, the use of selective A1 agonists is hampered by unwanted peripheral effects, thus attention has been focused on A2A and A3 receptors. The protective effects of A2A antagonists in brain ischemia may be largely due to reduced glutamate outflow from neurones and glial cells. Reduced activation of p38 mitogen-activated protein kinases that are involved in neuronal death through transcriptional mechanisms may also contribute to protection by A2A antagonism. Evidence that A3 receptor antagonism may be protective after ischemia is also reported.
Keywords: Adenosine, Adenosine A2A receptors, ATP, Ecto-ATPase inhibitors, Ischemia, P2 purinergic receptors
Historical overview
The physiological roles of adenosine triphosphate (ATP) and its metabolite adenosine have been historically linked to cell metabolism since ATP is an ubiquitous intracellular energy source in a number of enzymatic processes. However, over the years both metabolites have emerged as very versatile molecules of biological systems, being implicated in a variety of cell processes, from platelet aggregation to neurotransmission. The term ‘purinergic signalling’ was first introduced in the scientific literature by Burnstock [1]. An implicit concept for sustaining the hypothesis of purinergic neurotransmission proposed in the 1970s was the existence of purinergic receptors. The first evidence in this direction suggested the existence of two different subfamilies of such ‘purinoceptors’, identified as P1 and P2 receptors, selective for adenosine and ATP respectively [2]. Four different subtypes of P1-G-protein-coupled receptors: A1, A2A, A2B and A3 are known at present [3], whereas P2 purinoceptors belong to two major families: P2X ligand-gated ion channel receptors and P2Y G-protein-coupled receptors [4]. Cloning experiments supported this classification and helped to subdivide P2 receptors into seven P2X and eight P2Y subtypes [5] (plus the recently deorphanized GPR17 receptor [6]).
Roles of ATP in neurotransmission under normoxic conditions
The first studies on the role of extracellular ATP in hippocampal neurotransmission indicated prominent inhibitory action of this purinergic nucleotide on synaptic activity [7, 8]. However, several lines of evidence led to the hypothesis that ATP-mediated inhibition of hippocampal neurotransmission was probably mediated by adenosine acting on A1 receptors. In fact, this response was theophylline-sensitive [7–9] and absent in A1 knockout mice [10]. This assumption was supported by the fact that, in the hippocampus, extracellular ATP is rapidly converted into adenosine by ecto-ATPases and ecto-nucleotidases [11] and that adenosine acting on A1 receptors exerts a well-described inhibitory role on CA1 excitatory neurotransmission [3]. In recent years, a more detailed observation of ATP-evoked effects, supported by the synthesis of new pharmacological tools, has helped to clarify the effective role of purinergic nucleotides in the hippocampus. In particular, it emerged that ATP mediates inhibitory effects by P2 receptor activation [12–14].
In a recent paper, we contributed to elucidating the role of ATP on CA1 hippocampal neurotransmission, and we reinforced the concept that ATP-mediated effects are not necessarily linked to adenosine formation in this brain region [15]. ATP is hydrolysed by ecto-NTPDase, enzymes located on the cell surface in the CNS that limit ATP, ADP and AMP spatio-temporal activity [11, 16]. There are three different known NTPDases: NTPDase1 hydrolyzes ATP and ADP equally well, NTPDase2 has a high preference for ATP, NTPDase3 is a functional intermediate, preferably hydrolyzing ATP [17]. In our study, we took advantage of some recently synthesized compounds: ARL 67156, which, at micromolar concentrations, inhibits rat NTPDase1 and 3 transiently transfected in Chinese hamster ovary cells, showing negligible activity on NTPDase2 [18]; BGO 136, a new inhibitor described as a selective NTPDase1 and 2 blocker with Ki values in the high micromolar range [19]; and the recently synthesized PV4, which strongly inhibits rat NTPDase1, 2 and 3 with Ki values in the nanomolar range [20]. By using these inhibitors that are able to block NTPDase activity without interfering with P2 receptor activation, we demonstrated that ATP exerts an inhibitory action, independent of its degradation to adenosine. As shown in Fig. 1, the application of ATP during a NTPDase activity blockade still elicits a decrease in evoked synaptic responses which is even more pronounced than that evoked by ATP alone.
Fig. 1a, b.
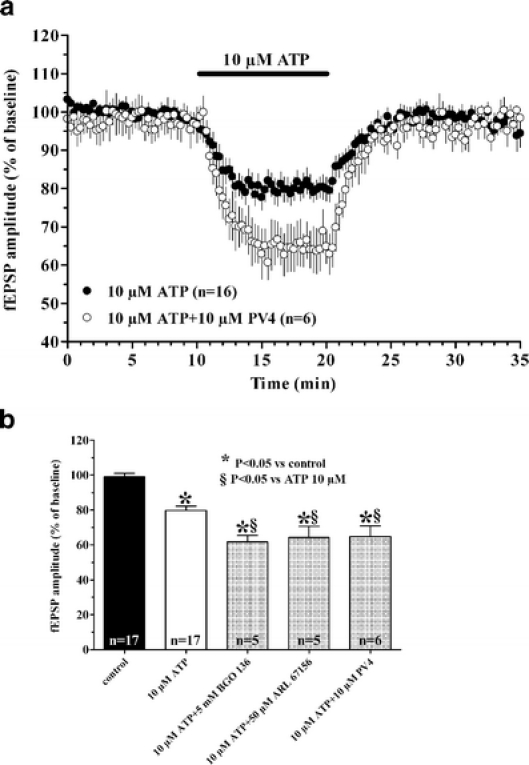
The inhibitory effect induced by ATP on fEPSP amplitude is potentiated in the presence of different NTPDase inhibitors. a Time-course of fEPSP amplitude before, during and after the application of ATP in the absence or in the presence of the NTPDase1,2,3 inhibitor PV4. Each point in the graph represents the mean ± SE of fEPSP value measured as percent of baseline, pre-drug level. b Columns in the graph summarize the average amplitude (mean ± SE) of evoked fEPSP recorded from CA1 hippocampal region in control conditions, 5 min after superfusion of ATP alone and 5 min after ATP superfusion in the presence of different ecto-ATPases inhibitors. Note that the inhibitory effect of ATP on fEPSP amplitude is potentiated by BGO 136, PV4 and ARL 67156. *P < 0.05 one-way ANOVA, Newman-Keuls multiple comparison post-hoc test versus pre-drug value. §P < 0.05, one-way ANOVA, Newman-Keuls multiple comparison post-hoc test versus 10 μM ATP treated slices. (Modified from [15])
An excitatory effect of exogenous ATP on hippocampal neurotransmission has also been reported by different authors. This effect, which was observed after drug removal, was described to persist in in vitro preparations for a relatively prolonged period, up to 1 h [21–25]. This ‘long-lasting’ potentiation of synaptic responses was compared to electrically evoked LTP, firstly described in the hippocampus by Abrams and Kandel [26] and called ‘ATP-induced LTP’. In agreement, we observed a potentiation of the synaptic responses after drug removal when slices were superfused in the presence of the metabolically stable ATP-analogue ATPγS (Fig. 2) [15]. In addition to this ‘long-lasting’ excitatory effect evoked by the exogenous application of P2 agonists, we also demonstrated an excitatory tone exerted by endogenous ATP. Slices superfused with P2 antagonists show a small but significant reduction in synaptic transmission (Fig. 3). These data demonstrate that tonic activation of P2 receptors contributes to glutamatergic excitatory neurotransmission in the hippocampus, an observation that is in line with previous work [27].
Fig. 2.
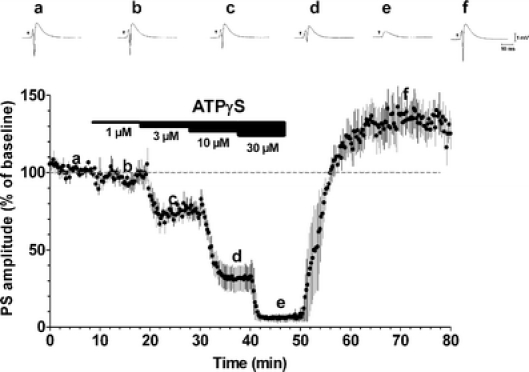
Inhibitory and excitatory effects of the stable ATP analogue ATPγS. Averaged time-course (n = 4) of PS amplitude before, during and after the application of different concentrations of ATPγS. PS amplitude (mean ± SE) is measured as percent of baseline level. Upper panels represent single traces recorded in a typical experiment before, during and after ATPγS application at different concentrations
Fig. 3.
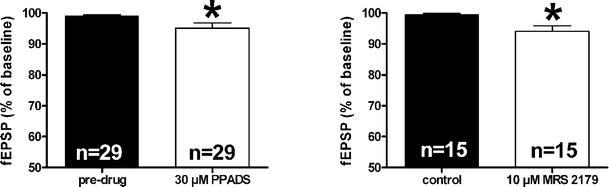
Excitatory effects of endogenous ATP. Bars in the graphs represent the average of fEPSP amplitude in the presence of P2 antagonists: PPADS (30 μM) and MRS 2179 (10 μM). *P < 0.05, paired Student’s _t_-test. (Modified from [15])
Roles of ATP in ischemic conditions
The role of ATP may become critical during pathological conditions such as ischemia, when extracellular ATP concentrations increase. An enhanced outflow of radioactive ATP from hippocampal slices during in vitro ischemic-like insults was first reported by Juranyi and co-workers [28], and the first demonstration that ATP outflow increases in vivo during the induction of focal ischemia in the rat was reported by Melani and colleagues [29]. Thus, at the ischemic site, levels of extracellular nucleotides may remain elevated for long periods of time after injury. Evidence supports the idea that, under such pathological conditions, released ATP may exert an excitotoxic role by acting on its receptors, thus enhancing Ca2+ inward currents and altering synaptic activity and cellular plasticity. Non-selective antagonists of P2 receptors, suramin and PPADS, and the selective antagonists, BBG and MRS 2179 of P2X7 and P2Y1 receptors respectively, prevent the irreversible failure of neurotransmission induced by a prolonged period of OGD in hippocampal slices [15]. Moreover, they protect from development of anoxic depolarization (AD), which is a rapid and regenerative wave of depolarization that propagates in tissue and represents an unequivocal sign of sufferance [15]. Antagonists of P2 receptors have been proved protective against cell death induced by either ATP itself, hypoglycemia or glutamate exposure in primary cultures of brain-derived neurones [30–33]. Intrastriatal ATP injection in rats induced, 24 h later, a clearly lesioned area [34]. The ATP-induced damage was concentration-dependent, mimicked by ATPγS and α,β meATP (but not by ADP or adenosine) and blocked by RB2, a non-specific P2 antagonist [34]. Suramin, another non-specific P2 receptor antagonist, administered 30 min before occlusion of the middle cerebral artery, resulted in a significant decrease in infarct and oedema volume 6 h after brain injury [35]. In agreement, it was demonstrated that RB2 [36] and PPADS [37] improve neurological deficit and reduce the damage induced in rats in a model of focal ischemia in vivo. Moreover, RB2 induces the expression of P2X7 receptors on reactive microglia in the remote ipsi and contralateral cingulate and medial frontal cortex and striatum. Although a pro-apoptotic role has been attributed to the P2X7 receptor, results suggest that microglial cells expressing the P2X7 receptor can be implicated in tissue damage as well as in the defence and repairing processes in the remote ipsi and contralateral undamaged areas [36].
In considering the effect of ATP during ischemia, it must be taken into account that ATP itself is involved in control of cerebrovascular regulation [38] and that the metabolite of ATP, ADP, that interacts with P2Y1/P2Y12 receptors, is a potent platelet aggregator. However, clinical studies that checked the antiplatelet therapy of the selective P2Y12 receptor antagonist clopidogrel reported a response variability [39, 40].
Although evidence suggests that ATP exerts a detrimental role under ischemia, there are reports that it may exert a trophic role in terms of cell division and differentiation in both differentiated and undifferentiated cells [41–43], including adult neural stem cells [44]. Interesting data correlate these trophic effects with the occurrence of spontaneous Ca2+ waves that propagate among adjacent cells in a self-renewing manner. Kawano et al. [45] reported that ATP-induced initiation and propagation of intracellular Ca2+ waves in human mesenchymal stem cells (hMSCs) promote activation of transcription factors (e.g. NFAT) that are involved in cell differentiation. In the same study, they demonstrated that ATP-induced Ca2+ waves disappear in the fully differentiated adipogenic phenotype. We recently reported that ATP is spontaneously released from hMSCs during the early stages of culture (P0-P5). Moreover, we reported that ATP decreases proliferation rate (Fig. 4) and modulates specific ionic current hMSCs [46]. A high extracellular ATP concentration at earlier cell culture passages suggests an important role of ATP in regulating cell differentiation. Increased hMSC differentiation may account for an ATP-induced decrease in cell proliferation.
Fig. 4a, b.
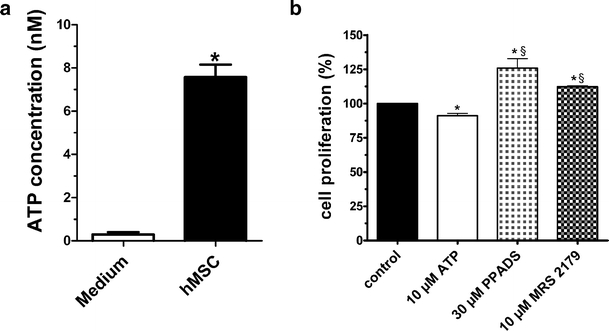
Human mesenchymal stem cells in culture spontaneously release ATP that modulates cell proliferation. a Extracellular concentrations of ATP were measured in the medium containing hMSCs and in control medium not containing cells. Data are expressed as mean ± SE, n = 11, unpaired Student’s _t_-test: *P < 0.0001 vs medium alone. b Effect on hMSC proliferation after daily application of ATP (10 μM) and P2 antagonists, PPADS (30 μM) and MRS 2179 (10 μM). Data are expressed as percentage of proliferation. Proliferation of untreated cells was assumed as 100%. The cell number was determined after 5 days of culture by a culture counter. Each column bar represents the mean ± SE of n = 4 for each experimental condition. Paired Student’s t_-test: *P < 0.05 vs respective control; one-way ANOVA, Newman-Keuls post-test: §_P < 0.05 vs 10 μM ATP-treated cells. (Modified from [46])
Therefore, it can be envisaged that ATP, as an autocrine/paracrine homeostatic regulator, exerts different effects on cell trophism according to its extracellular concentrations, distinct cell populations involved, differential expression and recruitment of P2 receptors and of more or less sustained stimulation of the same receptor. Balancing these effects may be relevant in the post-ischemic brain, when a neuroregenerative process could promote tissue repair [47]. On this basis, determination of ATP extracellular concentrations at various times after ischemia induction may help to identify which types of receptors may be stimulated under ischemia and explain the role of detrimental versus trophic ATP during ischemia. ATP present in the extracellular space is rapidly metabolised by membrane-bound NTPDase. Extracellular ATP concentrations, evaluated in the brain to date, have been underestimated, since it has not been possible to selectively inhibit ecto-NTPDase. In fact, the selective inhibitor ARL 67156, tentatively used thus far to inhibit ecto-NTPDase, interferes with the ATP assay method [29].
Role of adenosine in cerebral transmission under normoxic conditions
Adenosine exerts an important tonic modulation of synaptic transmission in the brain. This tonic inhibition of synaptic transmission is evoked by stimulation of A1 receptors, as demonstrated in several brain regions, such as the hippocampus, striatum and olfactory cortex [48, 49]. The inhibitory effect of adenosine A1 receptor stimulation has a pre- and postsynaptic component. Activation of the presynaptic A1 receptors reduces Ca2+ influx through the preferential inhibition of N-type and, probably, Q-type channels [50, 51]. Inhibition of presynaptic calcium currents decreases transmitter release [52], and adenosine, by stimulation of A1 receptors, has been found to inhibit the release of virtually all classical neurotransmitters: glutamate, acetylcholine, dopamine, noradrenaline and serotonin (see in [53]). In particular, powerful suppression of glutamate release from presynaptic terminals has been described in the hippocampus [54, 55], where adenosine A1 receptor activation reduces the number of quanta released (but not the size of individual quanta nor postsynaptic glutamate receptor sensitivity) in the Schaffer collateral-commissural pathway [56]. The postsynaptic effect of A1 receptors consists of direct hyperpolarisation of neurones via activation of GIRK channels (Kir 3.2 and 3.4 channels: potassium inward rectifiers) [57, 58]. Endogenous adenosine exerts tonic inhibition of excitatory neurotransmission. The selective A1 antagonist, DPCPX, causes a 15% increase in synaptic potential amplitude in in vitro brain slices [59]. This is an expected result in a brain region where the adenosine concentration at a receptor level was calculated around 200 nM [59, 60] and A1 receptors, whose affinity for adenosine is in the low nanomolar range, are highly expressed. These data are confirmed by the fact that, in slices taken from homozygous A1 receptor knockout mice, no evidence was found for an endogenous inhibitory action by adenosine in the Schaffer collateral pathway in the CA1 region of the hippocampus or at the mossy fibre synapses in the CA3 region [61].
Opposite effects from A1-mediated synaptic inhibition are elicited by A2A receptor activation, which has been shown to mediate excitatory actions in synaptic function [62–64]. In the hippocampus in vitro, A2A receptor stimulation results in a Ca2+-dependent release of acetylcholine [65, 66]. Furthermore, the application of CGS 21680, a selective A2A receptor agonist, decreases the ability of A1 receptor agonists to inhibit excitatory neurotransmission [9, 67]. This effect suggests that A2A receptor stimulation increases synaptic transmission through A1 receptor desensitisation [68, 69]. However, there is also evidence that A2A receptors increase excitatory amino acid release. In fact, the selective stimulation of adenosine A2A receptors augments the amount of glutamate released in the hippocampus and striatum of young rats [70–72].
In spite of the excitatory role in neurotransmission brought about by A2A receptors, the net effect of adenosine is an inhibitory tonus on neurotransmission, in accordance with observations suggesting that activation of A2A receptors requires protracted stimulation to induce evident effects on synaptic transmission [59]. It is worth noticing that the role of A2A receptors in the striatum is recently gaining interest in light of their heterodimerisation with D2 dopamine receptors. The association between A2A and D2 receptors results in an antagonistic interaction that provides a rationale for evaluating A2A-selective antagonists in Parkinson’s disease, supported by epidemiological evidence indicating an inverse relationship between caffeine consumption and the risk of developing this pathology [73, 74]. It was suggested that A2A antagonists not only provide symptomatic relief but also decelerate dopaminergic neurone degeneration in patients.
Discrepancies about the role of adenosine A3 receptors in the brain are present in the literature. Activation of this receptor subtype has been associated with both excitatory and inhibitory effects, even in the same brain region. An excitatory role of A3 receptors has been supported by evidence indicating that, in the rat hippocampus, their activation attenuates LTD and allows induction of LTP elicited by a subliminal weak-burst protocol [75]. In addition, in the same brain area, A3 receptor activation through a selective adenosine A3 agonist has been shown to antagonize the adenosine A1 receptor-mediated inhibition of excitatory neurotransmission [8]. Moreover, A3 receptor stimulation always attenuates the inhibition of hippocampal slice neurotransmission, in a PKC-dependent manner, caused by presynaptic metabotropic glutamate receptors [76]. Whole-cell patch clamp recordings in CA3 hippocampal pyramidal neurones demonstrate that A3 receptor activation results in a significant potentiation of high threshold hippocampal Ca2+ currents by a PKA-dependent mechanism [77]. Finally, facilitation of the onset of epileptiform discharge has been observed in the presence of the selective A3 receptor agonist Cl-IB-MECA [78], and a reduction in such epileptic activity was observed when A3 receptors, activated by endogenously released adenosine during seizures, were blocked by the selective antagonist MRS 1191 [79].
Contrary to previous results, an inhibitory action has been attributed to A3 receptors by Brand and colleagues [80], who demonstrated that, in rat cortical neurones, the selective activation of this adenosine receptor subtype is involved in inhibition of excitatory neurotransmission, suggesting a synergistic action with the inhibitory effect of adenosine brought about by A1 receptor activation. Despite results obtained by A3 receptor stimulation, evidence that a selective block of A3 receptors does not affect neurotransmission in the CA1 region of the hippocampus under normoxic conditions indicates that endogenous adenosine at physiological concentrations does not exert tonic activation of A3 receptors [8, 81]. This is in line with evidence that A3 receptor activation requires micromolar levels of extracellular adenosine, which can be reached only during pathological conditions of impairment in energy supply (for example during hypoxia or ischemia [82–85]).
Role of adenosine in ischemic conditions
Extracellular adenosine concentrations increase dramatically during ischemia [82, 86, 87]. During ischemia, following the imbalance between ATP degradation and resynthesis, the intracellular concentration of adenosine increases. Therefore, increased adenosine concentrations in the extracellular space during ischemia are likely due to the equilibrative transporter that carries adenosine out of cells. Adenosine formation can also take place at the extracellular level, through the hydrolysis of extracellular ATP operated by NTPDases and ecto-5′-nucleotidase (e5′-NTs) (see Fig. 5). Hence, these enzymes have a dual function in modulating purinergic neurotransmission: (1) they rapidly interrupt ATP-mediated signalling by degrading extracellular nucleotides and (2) they give rise to extracellular adenosine, which activates P1 receptors. However, recent evidence in vitro suggests that released ATP does not substantially contribute to the adenosine concentration in the extracellular milieu during ischemia [88].
Fig. 5.
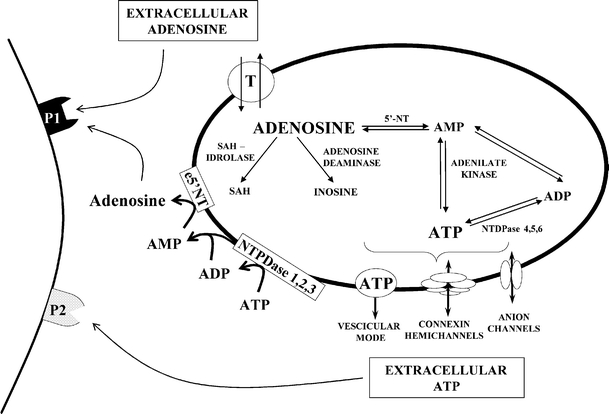
Schematic drawing of intracellular and extracellular adenosine formation. In the extracellular space, adenosine and ATP act on own purinergic receptor subtypes: P1 and P2 receptors, respectively. ADP adenosine diphosphate, AMP adenosine monophosphate, ATP adenosine triphosphate, e5′-NT ecto-5′-nucleotidase, 5′-NT 5′-nucleotidase, NTDPase ecto-nucleoside triphosphate diphosphohydrolases, P1 adenosine receptor, P2 ATP receptor, SAH S-adenosylhomocysteine, T bidirectional nucleoside transporter. (Modified from [142])
Adenosine-potentiating agents, which elevate endogenous adenosine levels by either inhibiting its metabolism by adenosine deaminase or kinase [89, 90] or preventing its transport [91, 92], offer protection against ischemic neuronal damage in different in vivo ischemia models. Moreover, adenosine infusion into the ischemic striatum during transient focal ischemia proves to significantly ameliorate the neurological outcome and reduce infarct volume [93].
A temporal correlation exists between adenosine outflow and synaptic potential inhibition in rat hippocampal slices during ischemia-like conditions [84, 88]. Synaptic inhibition during ischemia is greatly dependent on adenosine, which, by stimulating A1 receptors, exerts a protective role by reducing the Ca2+ influx, thus counteracting the presynaptic release of excitatory neurotransmitters [55, 94] and, in particular, glutamate, which exerts an excitotoxic role during ischemia mainly by overstimulation of NMDA receptors [95]. By directly increasing the K+ and Cl− ion conductances, adenosine stabilises the neuronal membrane potentials, thus reducing neuronal excitability [96]. Consequent reductions in cellular metabolism and energy consumption [97] and moderate lowering of the body/brain temperature [98] are protective in ischemia.
A1 receptor agonists are shown to attenuate ischemic or excitotoxic neuronal damage in both in vitro and in vivo models of cerebral ischemia (for review, see [99, 100]). In accordance, adenosine A1 antagonists given acutely exacerbate the damage induced by ischemia in different animal models of ischemia. An unselective A1 receptor antagonist, theophylline, increased mortality [101, 102]. Unlike acute treatment, chronic administration of A1 agonists worsened survival and increased neuronal loss [103], a phenomenon thought to depend on A1 receptor desensitization.
Although data converge in demonstrating a neuroprotective effect of adenosine through A1 receptors during ischemia, the use of selective A1 agonists is hampered by unwanted peripheral effects, e.g. sedation, bradycardia, hypotension [104]. Von Lubitz and co-workers [105] have reported that post-ischemic administration of the A1 receptor agonist adenosine amine congener (ADAC), which induces fewer undesirable effects, increases survival in gerbils. Moreover, we may consider that administration of agents that elevate the local concentration of adenosine at the injury site, by inhibiting its metabolism to inosine or rephosphorylation to AMP or reuptake, may have the advantage of restricting the effect of such inhibitors to areas of injury-induced adenosine release [106].
More recently, the role of A2A receptors in ischemic neuroprotection has been studied. Gao and Phillis [107] demonstrated for the first time that the non-selective A2A receptor antagonist CGS 15943 reduces cerebral ischemic injury in the gerbil following global forebrain ischemia. Subsequently, many reports have confirmed the neuroprotective role of A2A receptor antagonists in different models of ischemia. The selective A2A receptor antagonist SCH 58261 reduced ischemic brain damage in neonatal [108] and adult [83, 109] rat models of focal cerebral ischaemia. The same antagonist, subchronically administered, was protective against both brain damage and neurological deficit in the adult rat model of focal cerebral ischemia [110, 111]. Studies in A2A receptor knockout mice supported the neuroprotective role of A2A receptor antagonists on ischemic brain damage [112]. The beneficial effects of A2A antagonists in stroke were mainly attributed to reduced glutamate outflow [83, 110, 113]. Prolonged application of the A2A selective agonist CGS 21680 significantly reduced synaptic depression brought about by OGD in the hippocampus [59] and the same agonist promotes glutamate release under normoxic and ischemic conditions [70, 71, 114, 115]. In addition to neurones, adenosine A2A receptors are located on microglia [116, 117] and astrocytes [118]. On glial cells, A2A receptors mediate inhibition of the glutamate uptake transporter, GLT-1, and stimulate glutamate outflow [119]. A2A antagonists prevent the increase in glutamate levels induced by glutamate uptake inhibitors [120]. Therefore, the protective effects of A2A antagonists in brain ischemia may be largely due to reduced glutamate outflow from neurones and glial cells.
Further support of a major role for glia in the neuroprotective effect of A2A antagonism in ischemia comes from the observation that subchronic administration of the A2A receptor antagonist SCH 58261 reduces p38 mitogen-activated protein kinase (MAPK) activation in striatal and cortical microglia 24 h after permanent focal ischemia [111]. Evidence indicates that p38 MAPK is activated in glia up to 24 h after ischemia [121, 122] and is involved, through transcriptional mechanisms, in neuronal death [123, 124]. Since SCH 58261 reduces glutamate outflow in the first hours after ischemia [83], reduced p38 MAPK activation may be due to a direct effect on glial A2A receptors or may be secondary to a reduction in the excitotoxic cascade that primes p38 activation [125]. The importance of A2A receptors under ischemia is highlighted by the observation that A2A receptor expression increases on neurones and microglia after ischemia [126]. Selective inactivation of A2A receptors on bone-marrow-derived cells (wild-type mice transplanted with A2A receptor knockout bone marrow cells) attenuates infarct volumes and ischemia-induced expression of several proinflammatory cytokines in the brain [127]. Therefore, protective effects of A2A antagonists may be attributed to inhibition of inflammation product production.
In several studies, A2A receptor agonists have been found to be protective in the global ischemia model in the gerbil [128, 129]. Jones and co-workers [130] show that peripheral administration of the A2A receptor agonist CGS 21680 protects the hippocampus against kainate-induced excitotoxicity. However, the direct injection of CGS 21680 into the hippocampus failed to afford protection, while the A2A antagonist ZM 241385, when injected directly into the hippocampus, reduced kainate-induced neuronal damage [131]. These data suggest that the neuroprotective properties of A2A agonists are mainly due to peripherally mediated effects. Major mechanisms that may account for A2A-mediated protection include inhibition of platelet aggregation and vasodilation [89] and anti-inflammatory actions. A2A receptors on neutrophils may account for inhibition of adhesion to endothelial cells and ensuing production of free radicals [132, 133].
On this basis, adenosine and its receptors are considered targets for therapeutic implementation in the treatment of stroke. At the moment, a possible adenosynergic therapeutic strategy after ischaemia that is worth consideration is that of increasing adenosine concentrations at the ischemic sites by inhibitors of adenosine metabolism or reuptake in association with adenosine A2A antagonists. When considering the possible use of adenosine kinase inhibitors, it should be taken into account that adenosine represents only a small percentage of nucleotide content [134], therefore inhibition of its rephosphorylation to ATP by adenosine kinase inhibitors does not weigh upon the ATP content.
The few studies present in the literature concerning the role of A3 receptors in the pathophysiology of cerebral ischemia are rather contradictory. We have demonstrated that selective antagonism of A3 receptors facilitates the recovery of synaptic activity induced by ischemic preconditioning in rat hippocampal slices [81]. A harmful role of A3 receptors during in vitro OGD was confirmed by our observation that blocking the A3 adenosine receptor consistently abolishes or delays the occurrence of anoxic depolarization (AD) and significantly protects from the irreversible disruption of excitatory neurotransmission caused by a severe ischemic episode [135]. These results are in agreement with the observation that acute administration of a selective adenosine A3 agonist exacerbates the damage elicited by global ischemia in the gerbil [136]. On the contrary, it was demonstrated that chronic pre-ischemic administration of an A3 agonist protects against ischemic neuronal damage [136]. This effect may be attributed to desensitisation of A3 receptors. In fact, both human and rat A3 receptors are desensitised within a few minutes after agonist exposure [137, 138].
Contrary to the above information, Hentschel and colleagues [139] demonstrated that under hypoxic conditions, selective activation of A3 adenosine receptors brings about an inhibition of excitatory neurotransmission on cortical neurones, indicating that A3 receptors may sustain the neuroprotective action of adenosine induced by A1 receptors. Consistent with these reports, mice lacking A3 adenosine receptors show increased neurodegeneration in response to repeated episodes of moderate hypoxia [140] or an increase in cerebral infarction after transient ligation of the middle cerebral artery [141]. These opposite results regarding an excitatory or inhibitory role of A3 receptors on synaptic activity under hypoxia/ischemia may be reconciled by our recent data [135], suggesting that in a first phase of ischemia, A3 receptors play a protective synergistic role with A1 receptors. Severe ischemia would transform the A3 receptor-mediated effects from protective to injurious.
Taken together, these data suggest that the outcome of A3 receptor stimulation on synaptic transmission during hypoxic/ischemic phenomena depends on the intensity and duration of stimulation.
Concluding remarks
Purinergic signalling, e.g. adenosine and ATP, by activating specific membrane receptors (P1 and P2 respectively), is strictly correlated in orchestrating brain cell functions either under physiological normoxic or ischemic circumstances. However, the respective contribution of each single element is hard to discern from the total outcome, due to the rapid and ubiquitous enzymatic interconversion of these molecules.
In the brain, both compounds are likely to play a role in neuronal activity, and the inhibitory action of endogenous adenosine in excitatory neurotransmission (mainly due to A1 receptor activation) is well documented. Conversely our data, in line with previous observations in the literature, suggest that tonic activation of P2 receptors contributes to excitatory transmission in the hippocampus. Such an effect is modest, but it is unmasked by the application of P2 purinergic antagonists that reduce evoked synaptic responses.
An inhibitory effect elicited by exogenous ATP application has been frequently described and is mostly due to enzymatic degradation to adenosine and the subsequent activation of A1 receptors. However, our evidence demonstrates that ATP itself may inhibit hippocampal synaptic transmission since an even more pronounced effect is observed when enzymatic ATP degradation is blocked. Moreover, in our studies in the hippocampus, a potentiation of neurotransmission by the ATP analogue, ATPγS, develops a few minutes after the inhibitory effect has been washed out, and it persists for 45 min to 1 h. Within the brain, ATP is involved in the synaptic plasticity phenomenon and may be an important mediator of long-lasting effects in synaptic plasticity.
Either adenosine or ATP seems to become a particularly important signalling molecule under pathological conditions, such as ischemia, when the extracellular concentration of either compound drastically rises. It is well known that adenosine exerts important neuroprotective effects during brain ischemic insults by activating adenosine A1 receptors, which profoundly inhibit synaptic transmission and in particular the release of glutamate, known to contribute to ischemic damage. Under severe ischemia, a prolonged stimulation of both A2A and A3 receptors may be deleterious. The role of endogenously released ATP during cerebral ischemia is mainly deleterious, as found in our work and in the literature, since the application of P2 antagonists always results in a reduction in ischemic damage. The mechanisms underlying these effects are still unknown, but they can be correlated to the tonic excitatory action of endogenous ATP found in many brain regions (for example in the hippocampus and prefrontal cortex).
Purinergic signalling has an ancient phylogenetic origin. Expression of P2 receptors on cell membrane and responses to extracellular ATP are found in primitive prokaryotic species up to evolved animals and plants, suggesting an important and highly conserved role of extracellular purine nucleotides during evolution. Similarly, ontogenetic development in several species seems to involve purinergic signalling, especially during its first stages. Our observation that hMSCs, an undifferentiated line of cells able to originate a number of different cell lineages (adipocytes, chondrocytes, osteoblasts, neurones), spontaneously release ATP into the extracellular space and express functional purinergic P2 receptors, which modulate different kinds of membrane currents, and that ATP, during the early stages of culture, inhibits proliferation indicates an autocrine/paracrine mechanism of action for extracellular ATP in modulating cell functions of undifferentiated stem cells at early developmental stages. Such effects may be important in brain neurogenesis during development and in responses to neurodegenerative stimuli.
Acknowledgements
The present work was supported by Fondazione Monte dei Paschi of Siena, Siena, Italy.
References
- 1.Burnstock G, Dumsday B, Smythe A (1972) Atropine resistant excitation of the urinary bladder: the possibility of transmission via nerves releasing a purine nucleotide. Br J Pharmacol 44:451–461 [DOI] [PMC free article] [PubMed]
- 2.Burnstock G, Cocks T, Kasakov L et al (1978) Direct evidence for ATP release from non-adrenergic, non-cholinergic (“purinergic”) nerves in the guinea-pig taenia coli and bladder. Eur J Pharmacol 49:145–149 [DOI] [PubMed]
- 3.Fredholm BB, IJzerman AP, Jacobson KA et al (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–552 [PMC free article] [PubMed]
- 4.Abbracchio MP, Burnstock G (1994) Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol Ther 64:445–475 [DOI] [PubMed]
- 5.North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82:1013–1067 [DOI] [PubMed]
- 6.Ciana P, Fumagalli M, Trincavelli ML et al (2006) The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J 25:4615–4627 [DOI] [PMC free article] [PubMed]
- 7.Cunha RA, Sebastiao AM, Ribeiro JA (1998) Inhibition by ATP of hippocampal synaptic transmission requires localized extracellular catabolism by ecto-nucleotidases into adenosine and channeling to adenosine A1 receptors. J Neurosci 18:1987–1995 [DOI] [PMC free article] [PubMed]
- 8.Dunwiddie TV, Diao L, Proctor WR (1997) Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci 17:7673–7682 [DOI] [PMC free article] [PubMed]
- 9.O’Kane EM, Stone TW (1998) Interaction between adenosine A1 and A2 receptor-mediated responses in the rat hippocampus in vitro. Eur J Pharmacol 362:17–25 [DOI] [PubMed]
- 10.Masino SA, Diao L, Illes P et al (2002) Modulation of hippocampal glutamatergic transmission by ATP is dependent on adenosine A1 receptors. J Pharmacol Exp Ther 303:356–363 [DOI] [PubMed]
- 11.Zimmermann H, Braun N (1996) Extracellular metabolism of nucleotides in the nervous system. J Auton Pharmacol 16:397–400 [DOI] [PubMed]
- 12.Rodrigues RJ, Almeida T, Richardson PJ et al (2005) Dual presynaptic control by ATP of glutamate release via facilitatory P2X1, P2X2/3, and P2X3 and inhibitory P2Y1, P2Y2, and/or P2Y4 receptors in the rat hippocampus. J Neurosci 25:6286–6295 [DOI] [PMC free article] [PubMed]
- 13.Mendoza-Fernandez V, Andrew RD, Barajas-Lopez C (2000) ATP inhibits glutamate synaptic release by acting at P2Y receptors in pyramidal neurons of hippocampal slices. J Pharmacol Exp Ther 293:172–179 [PubMed]
- 14.Luthardt J, Borvendeg SJ, Sperlagh B et al (2003) P2Y(1) receptor activation inhibits NMDA receptor-channels in layer V pyramidal neurons of the rat prefrontal and parietal cortex. Neurochem Int 42:161–172 [DOI] [PubMed]
- 15.Coppi E, Pugliese AM, Stephan H et al (2007) Role of P2 purinergic receptors in synaptic transmission under normoxic and ischaemic conditions in the CA1 region of rat hippocampal slices. Purinergic Signalling 3:203 [DOI] [PMC free article] [PubMed]
- 16.Zimmermann H, Braun N, Kegel B et al (1998) New insights into molecular structure and function of ectonucleotidases in the nervous system. Neurochem Int 32:421–425 [DOI] [PubMed]
- 17.Zimmermann H (2000) Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol 362:299–309 [DOI] [PubMed]
- 18.Iqbal J, Vollmayer P, Braun N et al (2007) A capillary electrophoresis method for the characterization of ecto-nucleoside triphosphate diphosphohydrolases (NTPDase) and the analysis of inhibitors by in-capillary enzymatic microreaction. Purinergic Signalling 1:349–358 [DOI] [PMC free article] [PubMed]
- 19.Kukulski F, Komoszynski M (2003) Purification and characterization of NTPDase1 (ecto-apyrase) and NTPDase2 (ecto-ATPase) from porcine brain cortex synaptosomes. Eur J Biochem 270:3447–3454 [DOI] [PubMed]
- 20.Muller CE, Iqbal J, Baqi Y et al (2006) Polyoxometalates-a new class of potent ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors. Bioorg Med Chem Lett 16:5943–5947 [DOI] [PubMed]
- 21.Wieraszko A, Seyfried TN (1989) ATP-induced synaptic potentiation in hippocampal slices. Brain Res 491:356–359 [DOI] [PubMed]
- 22.Fujii S, Kato H, Furuse H et al (1995) The mechanism of ATP-induced long-term potentiation involves extracellular phosphorylation of membrane proteins in guinea-pig hippocampal CA1 neurons. Neurosci Lett 187:130–132 [DOI] [PubMed]
- 23.Fujii S, Kato H, Kuroda Y (1999) Extracellular adenosine 5′-triphosphate plus activation of glutamatergic receptors induces long-term potentiation in CA1 neurons of guinea pig hippocampal slices. Neurosci Lett 276:21–24 [DOI] [PubMed]
- 24.Fujii S, Kato H, Kuroda Y (2002) Cooperativity between extracellular adenosine 5′-triphosphate and activation of N-methyl-D-aspartate receptors in long-term potentiation induction in hippocampal CA1 neurons. Neuroscience 113:617–628 [DOI] [PubMed]
- 25.O’Kane EM, Stone TW (2000) Characterisation of ATP-induced facilitation of transmission in rat hippocampus. Eur J Pharmacol 409:159–166 [DOI] [PubMed]
- 26.Abrams TW, Kandel ER (1988) Is contiguity detection in classical conditioning a system or a cellular property? Learning in Aplysia suggests a possible molecular site. Trends Neurosci 11:128–135 [DOI] [PubMed]
- 27.Pankratov Y, Castro E, Miras-Portugal MT et al (1998) A purinergic component of the excitatory postsynaptic current mediated by P2X receptors in the CA1 neurons of the rat hippocampus. Eur J Neurosci 10:3898–3902 [DOI] [PubMed]
- 28.Juranyi Z, Sperlagh B, Vizi ES (1999) Involvement of P2 purinoceptors and the nitric oxide pathway in [3H]purine outflow evoked by short-term hypoxia and hypoglycemia in rat hippocampal slices. Brain Res 823:183–190 [DOI] [PubMed]
- 29.Melani A, Turchi D, Vannucchi MG et al (2005) ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem Int 47:442–448 [DOI] [PubMed]
- 30.Amadio S, D’Ambrosi N, Cavaliere F et al (2002) P2 receptor modulation and cytotoxic function in cultured CNS neurons. Neuropharmacology 42:489–501 [DOI] [PubMed]
- 31.Cavaliere F, D’Ambrosi N, Ciotti MT et al (2001) Glucose deprivation and chemical hypoxia: neuroprotection by P2 receptor antagonists. Neurochem Int 38:189–197 [DOI] [PubMed]
- 32.Volonte C, Merlo D (1996) Selected P2 purinoceptor modulators prevent glutamate-evoked cytotoxicity in cultured cerebellar granule neurons. J Neurosci Res 45:183–193 [DOI] [PubMed]
- 33.Volonte C, Ciotti MT, D’Ambrosi N et al (1999) Neuroprotective effects of modulators of P2 receptors in primary culture of CNS neurones. Neuropharmacology 38:1335–1342 [DOI] [PubMed]
- 34.Ryu JK, Kim J, Choi SH et al (2002) ATP-induced in vivo neurotoxicity in the rat striatum via P2 receptors. Neuroreport 13:1611–1615 [DOI] [PubMed]
- 35.Kharlamov A, Jones SC, Kim DK (2002) Suramin reduces infarct volume in a model of focal brain ischemia in rats. Exp Brain Res 147:353–359 [DOI] [PubMed]
- 36.Melani A, Amadio S, Gianfriddo M et al (2006) P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab 26:974–982 [DOI] [PubMed]
- 37.Lammer A, Gunther A, Beck A et al (2006) Neuroprotective effects of the P2 receptor antagonist PPADS on focal cerebral ischaemia-induced injury in rats. Eur J Neurosci 23:2824–2828 [DOI] [PubMed]
- 38.Marrelli SP, Khorovets A, Johnson TD et al (1999) P2 purinoceptor-mediated dilations in the rat middle cerebral artery after ischemia-reperfusion. Am J Physiol 276:H33–H41 [DOI] [PubMed]
- 39.Lahiri P, Chaudhuri U, Chattopadhyay A et al (2005) Structural insights in platelet receptor synergism-antiplatelet therapy in post-ischemic cerebrovascular events. Blood Cells Mol Dis 34:248–256 [DOI] [PubMed]
- 40.Ziegler S, Schillinger M, Funk M et al (2005) Association of a functional polymorphism in the clopidogrel target receptor gene, P2Y12, and the risk for ischemic cerebrovascular events in patients with peripheral artery disease. Stroke 36:1394–1399 [DOI] [PubMed]
- 41.Neary JT (2000) Trophic actions of extracellular ATP: gene expression profiling by DNA array analysis. J Auton Nerv Syst 81:200–204 [DOI] [PubMed]
- 42.Rathbone MP, DeForge S, Deluca B et al (1992) Purinergic stimulation of cell division and differentiation: mechanisms and pharmacological implications. Med Hypotheses 37:213–219 [DOI] [PubMed]
- 43.Franke H, Illes P (2006) Involvement of P2 receptors in the growth and survival of neurons in the CNS. Pharmacol Ther 109:297–324 [DOI] [PubMed]
- 44.Mishrda SK, Braun N, Shukla V et al (2006) Extracellular nucleotide signaling in adult neural stem cells: synergism with growth factor-mediated cellular proliferation. Development 133:675–684 [DOI] [PubMed]
- 45.Kawano S, Otsu K, Kuruma A et al (2006) ATP autocrine/paracrine signaling induces calcium oscillations and NFAT activation in human mesenchymal stem cells. Cell Calcium 39:313–324 [DOI] [PubMed]
- 46.Coppi E, Pugliese AM, Urbani S et al (2007) ATP modulates cell proliferation and elicits two different electrophysiological responses in human mesenchymal stem cells. Stem Cells 25:1840–1849 [DOI] [PubMed]
- 47.Scholzke MN, Schwaninger M (2007) Transcriptional regulation of neurogenesis: potential mechanisms in cerebral ischemia. J Mol Med 85:577–588 [DOI] [PubMed]
- 48.Latini S, Pedata F (2001) Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem 79:463–484 [DOI] [PubMed]
- 49.von Lubitz DK, Lin RC, Bischofberger N et al (1999) Protection against ischemic damage by adenosine amine congener, a potent and selective adenosine A1 receptor agonist. Eur J Pharmacol 369:313–317 [DOI] [PMC free article] [PubMed]
- 50.Yawo H, Chuhma N (1993) Preferential inhibition of omega-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature 365:256–258 [DOI] [PubMed]
- 51.Wu LG, Saggau P (1994) Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron 12:1139–1148 [DOI] [PubMed]
- 52.Prince DA, Stevens CF (1992) Adenosine decreases neurotransmitter release at central synapses. Proc Natl Acad Sci USA 89:8586–8590 [DOI] [PMC free article] [PubMed]
- 53.Fredholm BB, Dunwiddie TV (1988) How does adenosine inhibit transmitter release? Trends Pharmacol Sci 9:130–134 [DOI] [PubMed]
- 54.Burke SP, Nadler JV (1988) Regulation of glutamate and aspartate release from slices of the hippocampal CA1 area: effects of adenosine and baclofen. J Neurochem 51:1541–1551 [DOI] [PubMed]
- 55.Corradetti R, Lo CG, Moroni F et al (1984) Adenosine decreases aspartate and glutamate release from rat hippocampal slices. Eur J Pharmacol 104:19–26 [DOI] [PubMed]
- 56.Lupica CR, Proctor WR, Dunwiddie TV (1992) Presynaptic inhibition of excitatory synaptic transmission by adenosine in rat hippocampus: analysis of unitary EPSP variance measured by whole-cell recording. J Neurosci 12:3753–3764 [DOI] [PMC free article] [PubMed]
- 57.Takigawa T, Alzheimer C (1999) G protein-activated inwardly rectifying K+ (GIRK) currents in dendrites of rat neocortical pyramidal cells. J Physiol 517(Pt 2):385–390 [DOI] [PMC free article] [PubMed]
- 58.Takigawa T, Alzheimer C (2002) Phasic and tonic attenuation of EPSPs by inward rectifier K+ channels in rat hippocampal pyramidal cells. J Physiol 539:67–75 [DOI] [PMC free article] [PubMed]
- 59.Latini S, Bordoni F, Corradetti R et al (1999) Effect of A2A adenosine receptor stimulation and antagonism on synaptic depression induced by in vitro ischaemia in rat hippocampal slices. Br J Pharmacol 128:1035–1044 [DOI] [PMC free article] [PubMed]
- 60.Latini S, Bordoni F, Pedata F et al (1999) Extracellular adenosine concentrations during in vitro ischaemia in rat hippocampal slices. Br J Pharmacol 127:729–739 [DOI] [PMC free article] [PubMed]
- 61.Moore KA, Nicoll RA, Schmitz D (2003) Adenosine gates synaptic plasticity at hippocampal mossy fiber synapses. Proc Natl Acad Sci USA 100:14397–14402 [DOI] [PMC free article] [PubMed]
- 62.Pedata F, Pepeu G, Spignoli G (1984) Biphasic effect of methylxanthines on acetylcholine release from electrically-stimulated brain slices. Br J Pharmacol 83:69–73 [DOI] [PMC free article] [PubMed]
- 63.Latini S, Pazzagli M, Pepeu G et al (1996) A2 adenosine receptors: their presence and neuromodulatory role in the central nervous system. Gen Pharmacol 27:925–933 [DOI] [PubMed]
- 64.Sebastiao AM, Ribeiro JA (1996) Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog Neurobiol 48:167–189 [DOI] [PubMed]
- 65.Spignoli G, Pedata F, Pepeu G (1984) A1 and A2 adenosine receptors modulate acetylcholine release from brain slices. Eur J Pharmacol 97:341–342 [DOI] [PubMed]
- 66.Cunha RA, Johansson B, Fredholm BB et al (1995) Adenosine A2A receptors stimulate acetylcholine release from nerve terminals of the rat hippocampus. Neurosci Lett 196:41–44 [DOI] [PubMed]
- 67.Cunha RA, Johansson B, van der Ploeg I et al (1994) Evidence for functionally important adenosine A2a receptors in the rat hippocampus. Brain Res 649:208–216 [DOI] [PubMed]
- 68.Dixon AK, Widdowson L, Richardson PJ (1997) Desensitisation of the adenosine A1 receptor by the A2A receptor in the rat striatum. J Neurochem 69:315–321 [DOI] [PubMed]
- 69.Stone TW, Nikbakth M-R, O’Kane EM (2004) Adenosine and purines. In: Riedel G, Platt B (eds) From messengers to molecules: memories are made of these. Kluwer Academic/Plenum, New York, p 196–223
- 70.Popoli P, Betto P, Reggio R et al (1995) Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol 287:215–217 [DOI] [PubMed]
- 71.Corsi C, Melani A, Bianchi L et al (1999) Striatal A2A adenosine receptor differentially regulate spontaneous and K+-evoked glutamate release in vivo in young and aged rats. Neuroreport 10:687–691 [DOI] [PubMed]
- 72.Corsi C, Melani A, Bianchi L et al (2000) Striatal A2A adenosine receptor antagonism differentially modifies striatal glutamate outflow in vivo in young and aged rats. Neuroreport 11:2591–2595 [DOI] [PubMed]
- 73.Ross GW, Abbott RD, Petrovitch H et al (2000) Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 283:2674–2679 [DOI] [PubMed]
- 74.Ascherio A, Zhang SM, Hernan MA et al (2001) Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann Neurol 50:56–63 [DOI] [PubMed]
- 75.Costenla AR, Lopes LV, de Mendonca A et al (2001) A functional role for adenosine A3 receptors: modulation of synaptic plasticity in the rat hippocampus. Neurosci Lett 302:53–57 [DOI] [PubMed]
- 76.Macek TA, Schaffhauser H, Conn PJ (1998) Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci 18:6138–6146 [DOI] [PMC free article] [PubMed]
- 77.Fleming KM, Mogul DJ (1997) Adenosine A3 receptors potentiate hippocampal calcium current by a PKA-dependent/PKC-independent pathway. Neuropharmacology 36:353–362 [DOI] [PubMed]
- 78.Laudadio MA, Psarropoulou C (2004) The A3 adenosine receptor agonist 2-Cl-IB-MECA facilitates epileptiform discharges in the CA3 area of immature rat hippocampal slices. Epilepsy Res 59:83–94 [DOI] [PubMed]
- 79.Etherington LA, Frenguelli BG (2004) Endogenous adenosine modulates epileptiform activity in rat hippocampus in a receptor subtype-dependent manner. Eur J Neurosci 19:2539–2550 [DOI] [PubMed]
- 80.Brand A, Vissiennon Z, Eschke D et al (2001) Adenosine A(1) and A(3) receptors mediate inhibition of synaptic transmission in rat cortical neurons. Neuropharmacology 40:85–95 [DOI] [PubMed]
- 81.Pugliese AM, Latini S, Corradetti R et al (2003) Brief, repeated, oxygen-glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus: role of adenosine receptors. Br J Pharmacol 140:305–314 [DOI] [PMC free article] [PubMed]
- 82.Melani A, Pantoni L, Corsi C et al (1999) Striatal outflow of adenosine, excitatory amino acids, gamma-aminobutyric acid, and taurine in awake freely moving rats after middle cerebral artery occlusion: correlations with neurological deficit and histopathological damage. Stroke 30:2448–2454 [DOI] [PubMed]
- 83.Melani A, Pantoni L, Bordoni F et al (2003) The selective A2A receptor antagonist SCH 58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Res 959:243–250 [DOI] [PubMed]
- 84.Latini S, Bordoni F, Corradetti R et al (1998) Temporal correlation between adenosine outflow and synaptic potential inhibition in rat hippocampal slices during ischemia-like conditions. Brain Res 794:325–328 [DOI] [PubMed]
- 85.Pearson T, Damian K, Lynas RE et al (2006) Sustained elevation of extracellular adenosine and activation of A1 receptors underlie the post-ischaemic inhibition of neuronal function in rat hippocampus in vitro. J Neurochem 97:1357–1368 [DOI] [PubMed]
- 86.Dux E, Fastbom J, Ungerstedt U et al (1990) Protective effect of adenosine and a novel xanthine derivative propentofylline on the cell damage after bilateral carotid occlusion in the gerbil hippocampus. Brain Res 516:248–256 [DOI] [PubMed]
- 87.Phillis JW, Smith-Barbour M, O’Regan MH et al (1994) Amino acid and purine release in rat brain following temporary middle cerebral artery occlusion. Neurochem Res 19:1125–1130 [DOI] [PubMed]
- 88.Frenguelli BG, Wigmore G, Llaudet E et al (2007) Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus. J Neurochem 101:1400–1413 [DOI] [PMC free article] [PubMed]
- 89.Phillis JW, O’Regan MH (1989) Deoxycoformycin antagonizes ischemia-induced neuronal degeneration. Brain Res Bull 22:537–540 [DOI] [PubMed]
- 90.Lin Y, Phillis JW (1992) Deoxycoformycin and oxypurinol: protection against focal ischemic brain injury in the rat. Brain Res 571:272–280 [DOI] [PubMed]
- 91.DeLeo J, Schubert P, Kreutzberg GW (1988) Protection against ischemic brain damage using propentofylline in gerbils. Stroke 19:1535–1539 [DOI] [PubMed]
- 92.Parkinson FE, Xiong W, Zamzow CR (2005) Astrocytes and neurons: different roles in regulating adenosine levels. Neurol Res 27:153–160 [DOI] [PubMed]
- 93.Kitagawa H, Mori A, Shimada J et al (2002) Intracerebral adenosine infusion improves neurological outcome after transient focal ischemia in rats. Neurol Res 24:317–323 [DOI] [PubMed]
- 94.Pedata F, Latini S, Pugliese AM et al (1993) Investigations into the adenosine outflow from hippocampal slices evoked by ischemia-like conditions. J Neurochem 61:284–289 [DOI] [PubMed]
- 95.Choi DW (1990) Possible mechanisms limiting N-methyl-D-aspartate receptor overactivation and the therapeutic efficacy of N-methyl-D-aspartate antagonists. Stroke 21:III20–III22 [PubMed]
- 96.Greene RW, Haas HL (1991) The electrophysiology of adenosine in the mammalian central nervous system. Prog Neurobiol 36:329–341 [DOI] [PubMed]
- 97.Tominaga K, Shibata S, Watanabe S (1992) A neuroprotective effect of adenosine A1-receptor agonists on ischemia-induced decrease in 2-deoxyglucose uptake in rat hippocampal slices. Neurosci Lett 145:67–70 [DOI] [PubMed]
- 98.Gourine AV, Dale N, Gourine VN et al (2004) Fever in systemic inflammation: roles of purines. Front Biosci 9:1011–1022 [DOI] [PubMed]
- 99.von Lubitz DK (2001) Adenosine in the treatment of stroke: yes, maybe, or absolutely not? Expert Opin Invest Drugs 10:619–632 [DOI] [PubMed]
- 100.Pearson T, Currie AJ, Etherington LA et al (2003) Plasticity of purine release during cerebral ischemia: clinical implications? J Cell Mol Med 7:362–375 [DOI] [PMC free article] [PubMed]
- 101.Jarrott DM, Domer FR (1980) A gerbil model of cerebral ischemia suitable for drug evaluation. Stroke 11:203–209 [DOI] [PubMed]
- 102.Rudolphi KA, Keil M, Fastbom J et al (1989) Ischaemic damage in gerbil hippocampus is reduced following upregulation of adenosine (A1) receptors by caffeine treatment. Neurosci Lett 103:275–280 [DOI] [PubMed]
- 103.Jacobson KA, von Lubitz DK, Daly JW et al (1996) Adenosine receptor ligands: differences with acute versus chronic treatment. Trends Pharmacol Sci 17:108–113 [DOI] [PMC free article] [PubMed]
- 104.Williams M (1993) Purinergic drugs: opportunities in the 1990s. Drug Dev Res 28:438
- 105.von Lubitz DK, Lin RC, Paul IA et al (1996) Postischemic administration of adenosine amine congener (ADAC): analysis of recovery in gerbils. Eur J Pharmacol 316:171–179 [DOI] [PMC free article] [PubMed]
- 106.Phillis JW, Goshgarian HG (2001) Adenosine and neurotrauma: therapeutic perspectives. Neurol Res 23:183–189 [DOI] [PubMed]
- 107.Gao Y, Phillis JW (1994) CGS 15943, an adenosine A2 receptor antagonist, reduces cerebral ischemic injury in the Mongolian gerbil. Life Sci 55:L61–L65 [DOI] [PubMed]
- 108.Bona E, Aden U, Gilland E et al (1997) Neonatal cerebral hypoxia-ischemia: the effect of adenosine receptor antagonists. Neuropharmacology 36:1327–1338 [DOI] [PubMed]
- 109.Monopoli A, Lozza G, Forlani A et al (1998) Blockade of adenosine A2A receptors by SCH 58261 results in neuroprotective effects in cerebral ischaemia in rats. Neuroreport 9:3955–3959 [DOI] [PubMed]
- 110.Pedata F, Gianfriddo M, Turchi D et al (2005) The protective effect of adenosine A2A receptor antagonism in cerebral ischemia. Neurol Res 27:169–174 [DOI] [PubMed]
- 111.Melani A, Gianfriddo M, Vannucchi MG et al (2006) The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res 1073–1074:470–480 [DOI] [PubMed]
- 112.Chen JF, Huang Z, Ma J et al (1999) A2A adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci 19:9192–9200 [DOI] [PMC free article] [PubMed]
- 113.Marcoli M, Raiteri L, Bonfanti A et al (2003) Sensitivity to selective adenosine A1 and A2A receptor antagonists of the release of glutamate induced by ischemia in rat cerebrocortical slices. Neuropharmacology 45:201–210 [DOI] [PubMed]
- 114.O’Regan MH, Simpson RE, Perkins LM et al (1992) The selective A2 adenosine receptor agonist CGS 21680 enhances excitatory transmitter amino acid release from the ischemic rat cerebral cortex. Neurosci Lett 138:169–172 [DOI] [PubMed]
- 115.Simpson RE, O’Regan MH, Perkins LM et al (1992) Excitatory transmitter amino acid release from the ischemic rat cerebral cortex: effects of adenosine receptor agonists and antagonists. J Neurochem 58:1683–1690 [DOI] [PubMed]
- 116.Fiebich BL, Biber K, Lieb K et al (1996) Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2A-receptors. Glia 18:152–160 [DOI] [PubMed]
- 117.Saura J, Angulo E, Ejarque A et al (2005) Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J Neurochem 95:919–929 [DOI] [PubMed]
- 118.Lee YC, Chien CL, Sun CN et al (2003) Characterization of the rat A2A adenosine receptor gene: a 4.8-kb promoter-proximal DNA fragment confers selective expression in the central nervous system. Eur J Neurosci 18:1786–1796 [DOI] [PubMed]
- 119.Nishizaki T, Nagai K, Nomura T et al (2002) A new neuromodulatory pathway with a glial contribution mediated via A2A adenosine receptors. Glia 39:133–147 [DOI] [PubMed]
- 120.Pintor A, Galluzzo M, Grieco R et al (2004) Adenosine A2A receptor antagonists prevent the increase in striatal glutamate levels induced by glutamate uptake inhibitors. J Neurochem 89:152–156 [DOI] [PubMed]
- 121.Irving EA, Barone FC, Reith AD et al (2000) Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res Mol Brain Res 77:65–75 [DOI] [PubMed]
- 122.Wu DC, Ye W, Che XM et al (2000) Activation of mitogen-activated protein kinases after permanent cerebral artery occlusion in mouse brain. J Cereb Blood Flow Metab 20:1320–1330 [DOI] [PubMed]
- 123.Barone FC, Irving EA, Ray AM et al (2001) Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev 21:129–145 [DOI] [PubMed]
- 124.Gao Y, Signore AP, Yin W et al (2005) Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab 25:694–712 [DOI] [PubMed]
- 125.Kawasaki H, Morooka T, Shimohama S et al (1997) Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem 272:18518–18521 [DOI] [PubMed]
- 126.Trincavelli ML, Melani A, Guidi S et al (2007) Regulation of A2A adenosine receptor expression and functioning following permanent focal ischemia in rat brain. J Neurochem. DOI 10.1111/j.1471-4159.2007.04990.x [DOI] [PubMed]
- 127.Yu L, Huang Z, Mariani J et al (2004) Selective inactivation or reconstitution of adenosine A2A receptors in bone marrow cells reveals their significant contribution to the development of ischemic brain injury. Nat Med 10:1081–1087 [DOI] [PubMed]
- 128.Sheardown MJ, Knutsen LJS (1996) Unexpected neuroprotection observed with the adenosine A2A receptor agonist cgs 21680. Drug Dev Res 39:108
- 129.von Lubitz DK, Lin RC, Jacobson KA (1995) Cerebral ischemia in gerbils: effects of acute and chronic treatment with adenosine A2A receptor agonist and antagonist. Eur J Pharmacol 287:295–302 [DOI] [PMC free article] [PubMed]
- 130.Jones PA, Smith RA, Stone TW (1998) Protection against kainate-induced excitotoxicity by adenosine A2A receptor agonists and antagonists. Neuroscience 85:229–237 [DOI] [PubMed]
- 131.Jones PA, Smith RA, Stone TW (1998) Protection against hippocampal kainate excitotoxicity by intracerebral administration of an adenosine A2A receptor antagonist. Brain Res 800:328–335 [DOI] [PubMed]
- 132.Sitkovsky MV, Ohta A (2005) The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol 26:299–304 [DOI] [PubMed]
- 133.Mayne M, Fotheringham J, Yan HJ et al (2001) Adenosine A2A receptor activation reduces proinflammatory events and decreases cell death following intracerebral hemorrhage. Ann Neurol 49:727–735 [DOI] [PubMed]
- 134.Latini S, Corsi C, Pedata F et al (1995) The source of brain adenosine outflow during ischemia and electrical stimulation. Neurochem Int 27:239–244 [DOI] [PubMed]
- 135.Pugliese AM, Coppi E, Spalluto G et al (2006) A3 adenosine receptor antagonists delay irreversible synaptic failure caused by oxygen and glucose deprivation in the rat CA1 hippocampus in vitro. Br J Pharmacol 147:524–532 [DOI] [PMC free article] [PubMed]
- 136.von Lubitz DK, Lin RC, Popik P et al (1994) Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol 263:59–67 [DOI] [PMC free article] [PubMed]
- 137.Palmer TM, Benovic JL, Stiles GL (1995) Agonist-dependent phosphorylation and desensitization of the rat A3 adenosine receptor. Evidence for a G-protein-coupled receptor kinase-mediated mechanism. J Biol Chem 270:29607–29613 [DOI] [PubMed]
- 138.Trincavelli ML, Tuscano D, Marroni M et al (2002) A3 adenosine receptors in human astrocytoma cells: agonist-mediated desensitization, internalization, and down-regulation. Mol Pharmacol 62:1373–1384 [DOI] [PMC free article] [PubMed]
- 139.Hentschel S, Lewerenz A, Nieber K (2003) Activation of A(3) receptors by endogenous adenosine inhibits synaptic transmission during hypoxia in rat cortical neurons. Restor Neurol Neurosci 21:55–63 [PubMed]
- 140.Fedorova IM, Jacobson MA, Basile A et al (2003) Behavioral characterization of mice lacking the A3 adenosine receptor: sensitivity to hypoxic neurodegeneration. Cell Mol Neurobiol 23:431–447 [DOI] [PMC free article] [PubMed]
- 141.Chen GJ, Harvey BK, Shen H et al (2006) Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res 84:1848–1855 [DOI] [PubMed]
- 142.Pedata F, Pugliese AM, Coppi E et al (2007) Adenosine in the central nervous system: effects on neurotransmission and neuroprotection. Immunol Endocr Metab Agents Med Chem 7:304–321