Microtubule-mediated Transport of Incoming Herpes Simplex Virus 1 Capsids to the Nucleus (original) (raw)
Abstract
Herpes simplex virus 1 fuses with the plasma membrane of a host cell, and the incoming capsids are efficiently and rapidly transported across the cytosol to the nuclear pore complexes, where the viral DNA genomes are released into the nucleoplasm. Using biochemical assays, immunofluorescence, and immunoelectron microscopy in the presence and absence of microtubule depolymerizing agents, it was shown that the cytosolic capsid transport in Vero cells was mediated by microtubules. Antibody labeling revealed the attachment of dynein, a minus end–directed, microtubule-dependent motor, to the viral capsids. We propose that the incoming capsids bind to microtubules and use dynein to propel them from the cell periphery to the nucleus.
The entry of animal viruses into their host cells involves adsorption to cell surface receptors, penetration into the cytosol, and uncoating of the viral genome. For viruses that replicate in the nucleus, entry also entails extensive movement of viruses and viral capsids through the cytoplasm. Transport over long distances is particularly critical for viruses that infect neurons in which the site of entry can be located far from the cell body and the nucleus.
Since movement of virus-sized particles through the cytosol is not likely to occur by free diffusion due to high viscosity and steric obstacles (Bray, 1992; Luby-Phelps, 1994), viruses most likely exploit the cell's motile functions for transport. The limited information available suggests that viruses can make use of both microtubules (MT)1 and actin filaments. EM analysis has shown MT binding of certain viral capsids, such as adenovirus and reovirus in vivo and in vitro (for review see Luftig, 1982) as well as herpes simplex virus 1 (HSV-1) in neurons (Lycke et al., 1984, 1988; Penfold et al., 1994). Way and co-workers have described actin filament–dependent intra- and intercellular transport of vaccinia virus during egress from cells (Cudmore et al., 1995). Moreover, many viruses take advantage of the retrograde movement of endocytic organelles during entry. They are internalized by receptor-mediated endocytosis and carried within coated vesicles and endosomes from the cell periphery toward the perinuclear area of the cell (see Greber et al., 1994; Marsh and Helenius, 1989).
In this study we have focused on the intracytosolic transport of incoming HSV-1 capsids in Vero cells. HSV-1 has three structural components: capsid, tegument, and envelope. The capsid consists of the viral DNA of 152 kbp and a proteinaceous shell comprising six different proteins; it has a diameter of 125 nm, and the proteins are arranged in an icosahedron formed by 12 pentons and 150 hexons (Booy et al., 1991; Cohen et al., 1980; Heine et al., 1974; Spear and Roizman, 1972). VP5 constitutes the major protein component of both hexons and pentons (Newcomb et al., 1992; Trus et al., 1992). The tegument is a protein layer between the envelope and the capsid and contains ∼12 different viral polypeptides (Roizman and Furlong, 1974; Roizman and Sears, 1996). While the functions of most of these remain to be identified, one is a protein kinase (LeMaster and Roizman, 1980), one induces shut off of host cell protein synthesis (Read and Frenkel, 1983), and some are known to be activators and modulators of viral gene expression (Batterson and Roizman, 1983; Campbell et al., 1984; McLean et al., 1990). The viral envelope contains at least 12 different membrane proteins, some of which are involved in receptor binding and fusion.
The infectious cycle begins with virus binding to heparan sulfate receptors on the cell surface followed by glycoprotein-mediated fusion of the envelope with the plasma membrane (for review see Spear, 1993). After release into the cytosol and dissociation from some of the tegument components, the capsids move through the cytosol to the nucleus and bind to nuclear pores (Batterson et al., 1983; Lycke et al., 1988), whereafter the genome is released into the nucleus. Transcription, replication of viral DNA, and assembly of progeny capsids take place within the host nucleus (for review see Roizman and Sears, 1996; Steven and Spear, 1996). The synthesis of early viral proteins peaks after 4–6 h of virus entry, and the first progeny viruses are produced after 8 h (Honess and Roizman, 1973).
We analyzed the transport of incoming HSV-1 capsids to the nucleus in fibroblasts and found that it occurs rapidly and efficiently by an MT-mediated mechanism. The incoming capsids shed most of the associated tegument proteins and bind dynein, an MT-dependent, minus end– directed motor in the peripheral cytosol, whereafter they are transported along MT toward the cell center. The viral capsids thus make use of the machinery responsible for retrograde organelle transport in animal cells.
Materials and Methods
Cells and Antibodies
BHK-21 cells were grown in Glasgow's MEM with 5% FCS and 10% tryptose phosphate broth, and Vero cells were grown in MEM with 7.5% FCS and nonessential amino acids. Both media contained 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine. All tissue-culture reagents were obtained from GIBCO BRL (Gaithersburg, MD). Both cell lines were maintained as adherent cultures in a 5% CO2 humid incubator at 37°C and passaged twice a week. We used rabbit polyclonal antibodies against VP-5 (NC-1), VP-19c (NC-2), DNA-containing capsids (anti-HC), empty capsids (anti-LC; all provided by Roselyn Eisenberg and Gary Cohen, University of Pennsylvania, Philadelphia), and a mouse mAb 8F5 against VP5 (provided by William Newcomb and Jay Brown, University of Virginia, Charlottesville). All rabbit polyclonal antibodies to viral proteins were preadsorbed against paraformaldehyde (PFA)–fixed, Triton X-100 (TX-100)–permeabilized Vero cells. They were used at the highest possible concentration, giving no signal in noninfected cells. An mAb against ICP4, an early herpes protein, was obtained from Advanced Biotechnologies, Inc. (Columbia, MD). We used a mouse monoclonal anti-tubulin (1A2; provided by Dr. Thomas Kreis, University of Geneva, Switzerland; Kreis, 1987), a rabbit antipeptide anti–calnexin (Hammond and Helenius, 1994), and a rabbit polyclonal, affinity-purified antibody directed against a bacterially expressed fragment of dynein heavy chain (provided by Dr. Eugeni Vaisberg, University of Colorado at Boulder; Vaisberg et al., 1993). Fluorescently labeled secondary antibodies (Texas red– or fluorescein-conjugated goat anti–rabbit or goat anti–mouse) were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA), and rabbit anti-mouse was obtained from Organon Teknika-Cappel (Durham, NC).
Virological Techniques
Preparation of Stock Virus.
HSV-1, (strain F), designated passage 1, was obtained from American Type Culture Collection (Rockville, MD). BHK-21 cells grown to confluency at 37°C for 3 d were infected with an 8 ml/175 cm2 flask of MEM containing 0.2% (wt/vol) BSA and <0.01 plaque-forming units (PFU) per cell. After 2 h, 25 ml per flask of normal medium was added to the cells, and after 3 d, the medium containing secreted extracellular virus was collected. All the following steps were performed at 4°C. Cells and debris were removed by spinning at 2,000 rpm for 10 min, and the virus was collected by centrifugation from the medium using a JA-14 rotor (Beckman Instruments, Inc.. Palo Alto, CA) at 12,000 rpm for 90 min. The resulting pellets were resuspended by gentle shaking overnight in MNT buffer (30 mM MES, 100 mM NaCl, 20 mM Tris, pH 7.4). After waterbath sonification (4°C; three times for 30 s each), the suspension derived from 30 flasks was layered on top of one linear 10–40% (wt/vol) sucrose gradient in MNT buffer and centrifuged in a Beckman SW28 at 15,000 rpm for 45 min. The virus-containing band in the middle of the gradient was detected by light scattering, collected (∼3 ml per gradient), aliquoted, snap frozen in liquid nitrogen, and stored at −80°C. All experiments described here were carried out with gradient-purified virus from passage three.
Plaque Assay.
Virus was diluted in 10-fold steps into MEM with 0.2% BSA and incubated in 24-well dishes with just confluent Vero cells for 1–2 h at 37°C in a humidified 5% CO2 incubator. Unbound virus was removed by three washes in MEM-BSA, and the cells were further incubated in normal medium for 30 h. Plaques were counted using a dissecting microscope after fixation in absolute methanol (−20°C) and Giemsa staining. Titers of 109–1010 PFU/ml and protein concentrations of 0.5–2 mg/ml were obtained using this protocol.
Preparation of [3H]Thymidine-labeled virus.
BHK cells grown in an 850cm2 roller bottle to ∼70% confluency were infected with a multiplicity of infection (MOI) of 0.1 PFU per cell in MEM-BSA (0.2% wt/vol) containing 20 mM Hepes, pH 7.4. At 2 h postinfection (PI), the inoculum was removed and normal growth medium containing 5 mCi [methyl-3H]thymidine (Amersham Lifescience, Amersham, UK) was added. After 2 d, the cells and the medium were collected and transferred to 4°C, the cells were pelleted by a low speed spin, and the resulting supernatant was spun in a Beckman SW40Ti at 13,700 rpm for 2 h. The pellet was resuspended by gentle shaking overnight in MNT buffer, layered onto a linear 10–40% (wt/vol) sucrose gradient in MNT buffer, and spun in a Beckman SW40Ti at 15,800 rpm for 1 h. The gradient was fractionated into 1-ml fractions, and the radioactivity in each fraction was determined by scintillation counting. The peak fractions were pooled, aliquoted, snap frozen, and stored at −80°C. This preparation had a titer of 1 × 107 PFU/ml and contained 0.1 mg/ml protein.
Infection and Drug Treatments.
Cells were inoculated with virus for 2 h at 4°C with constant rocking in RPMI medium containing 0.2% (wt/vol) BSA, 20 mM Hepes, pH 6.8, to allow virus binding. Viral infection was initiated by shifting the cells to growth medium and 37°C in a humidified 5% CO2 incubator. In those experiments analyzing incoming viral particles, 0.5 mM cycloheximide was added to the medium to prevent synthesis of new viral proteins and progeny virus. In some experiments, the cells were incubated for 1 h before infection in normal medium containing 2 μM nocodazole, 10 μM vinblastine, 2 μM colchicine, 20 μM taxol, or 0.5 μM cytochalasin D and were maintained in these drugs during the infection. Nocodazole was stored as a stock at 20 mM, vinblastine at 10 mM, colchicine at 20 mM, taxol at 20 mM, and cytochalasin D at 10 mM, all in DMSO and at −20°C.
Electron Microscopy
Vero cells grown on culture dishes or glass coverslips (for flat embedding) for 1 d to just confluency were infected with HSV-1 at an MOI of 500 PFU per cell (or 150 PFU/cell for the nocodazole experiment) in the presence of cycloheximide. The virus was dialyzed before infection against RPMI containing 0.2% BSA for 2 h (75 kD cut off) to remove sucrose. The infected cells were analyzed by three different protocols.
Conventional Epon Embedding.
The cells were fixed with 1% glutaraldehyde (GA) in 200 mM cacodylate, pH 7.4, or with 2% GA in PBS containing 0.5 mM CaCl2 and 1 mM MgCl2 for 30 min at room temperature (for MT analysis), treated with 1% OsO4 for 1 h (or 30 min for MT), treated with 2% uranyl acetate in 50 mM maleate buffer, pH 5.2, for 1 h, dehydrated using a graded ethanol series and propylenoxid, and pelleted before embedding in Epon and cutting, or flat-embedded in Epon and cut parallel to the substrate (for MT).
Flat Embedding of Extracted, Immunolabeled Cells.
Cells were extracted with 0.5% Triton X-100 in MT stabilizing buffer (MT buffer: 80 mM Pipes-KOH, pH 6.8, 5 mM EDTA, 1 mM MgSO4, 10 μM taxol) for 2 min at 37°C (or 4°C for the 0-min time point) before the addition of twofold fixative (2% GA in MT buffer; Baas and Ahmad, 1992). The cells were fixed for 20 min at room temperature and rinsed with PBS, and remaining fixative was inactivated by three rounds of freshly prepared 2 mg/ml sodium borohydride in PBS, pH 7.4, for 15 min. After blocking with 10% FCS in PBS, the cells were incubated with primary antibody, with secondary rabbit anti–mouse antibody (if the primary was not a rabbit antibody), and with 9 nm protein A–gold (University of Utrecht, The Netherlands), each in 10% FCS in PBS for 30 min at room temperature with extensive washings with PBS in between. After a second fixation in 1% GA with 2 mg/ml tannic acid in 100 mM cacodylate buffer, pH 7.4, for 10 min, the cells were contrasted using 2% OsO4 for 10 min and 2% uranyl acetate in 50 mM maleate buffer, pH 5.2, for 1 h. They were embedded in Epon and ultrathin sections parallel to the substrate were prepared. All Epon sections (first and second protocols) were further contrasted using lead citrate and uranyl acetate.
Frozen Hydrated Cryosections.
The cells were transferred to 4°C and removed from the culture dish either by proteinase K treatment (25 μg/ml for 2–3 min on ice) and fixed (4% paraformaldehyde and 0.1% GA in 250 mM Hepes, pH 7.4), or fixed first for 20 min and then collected by scraping. After pelleting, the cells were fixed further overnight. The pellets were infiltrated with 20% (wt/vol) polyvinyl pyrolidone and 2.0 M sucrose in PBS and frozen in liquid nitrogen, and ultrathin sections were cut at −100° to −120°C and immunolabeled as previously described (Griffiths, 1993_a_ ; Sodeik et al., 1992).
Quantitation.
To analyze the efficiency of capsid transport to the nucleus (see Fig. 5), 25 EM negatives of infected, flat-embedded cells were taken at a magnification of 5,800 in a systematic random fashion for each time point (Griffiths, 1993_b_ ). The negatives were analyzed under a dissecting microscope, and the number and localization of cytosolic capsids was determined. Capsids within 100 nm of the plasma membrane were scored as being at the plasma membrane, and within 50 nm as being on MT and at the nuclear pore complex. The remaining capsids were scored as being apparently free in the cytosol. Virions present in endosomes were also counted. To quantify the effect of nocodazole (see Fig. 7), 50 negatives of conventionally embedded cells infected in the presence or absence of nocodazole (10 μM) were taken in a systematic random fashion. Cytosolic capsids were scored as being at the plasma membrane, in the cytosol, or at the nuclear pore complex. To quantify the dynein labeling, 25 micrographs of cryosections were taken at a magnification of 16,900. Since incoming herpes capsids were rare structures, the specimen was systematically translated in a defined direction, and a micrograph was taken every time a cytosolic capsid came into view. These were printed (× 2.7) and the area of each structure was outlined (virions, capsids, mitochondria, nuclei, and cytoplasm). A lattice grid was placed over the prints, and the number of points and gold particles falling into each structure was counted. The formula G/P*d2 gives the labeling density (gold particles per μm2), where G is the number of gold particles, P is the number of points of the lattice grid over the structure, and d is the distance between points (Griffiths, 1993_b_ ).
Figure 5.
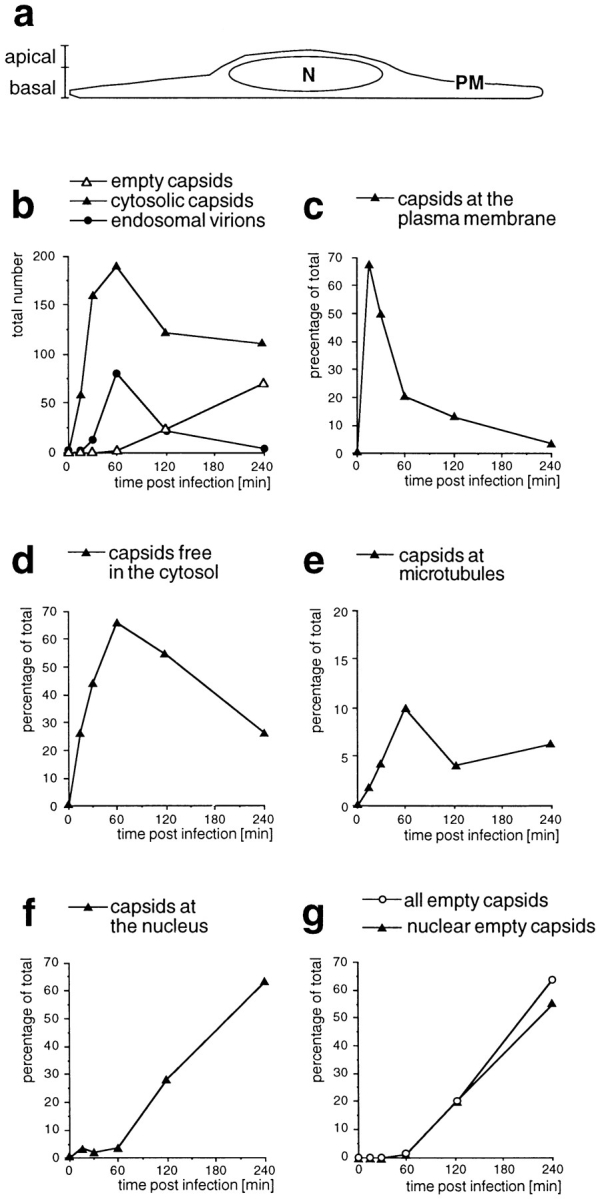
Time course of HSV-1 entry. Vero cells infected at an MOI of 500 in the presence of CH were fixed at various times postinfection with 2% glutaraldehyde in PBS and embedded in Epon for EM analysis. (a) Schematic cross-section perpendicular to the substrate through an adherent Vero cell. For quantitation, 25 random electron micrographs obtained from sections cut parallel to the substrate through the basal part of the cells were taken in a systematic fashion for each time point. “Basal” is defined as being within 2 μm distance from the substrate (30 sections of ∼50–70 nm). (b) The total numbers of cytosolic capsids (full and empty), cytosolic empty capsids, and virions in endosomes were determined for each time point. The number of cytosolic capsids increased rapidly until 1 h postinfection. At later time points, less cytosolic capsids were detected, suggesting that they were ultimately disassembled. In addition, the subcellular localization of each capsid was determined and the numbers were expressed as a percentage of the total (c–g). (c) At the earliest time point of 15 min, the majority of cytosolic capsids was localized close to the plasma membrane, from where they disappeared rapidly later. (d) Several capsids could not be localized to a particular organelle. At 1 h postinfection, there was the highest amount of those capsids, which were apparently free in the cytosol. (e) A significant portion of all cytosolic capsids, namely 10% at 1 h postinfection, colocalized transiently with microtubules. (f) Capsids began to appear at the nuclear pores at 2 h postinfection, and at 4 h, 60% of all cytosolic capsid had reached the nucleus. (g) The appearance of empty cytosolic capsids coincided with the arrival at the nucleus. Only at 4 h postinfection, there was a very small portion of empty, cytosolic capsids. N, nucleus; PM, plasma membrane.
Figure 7.
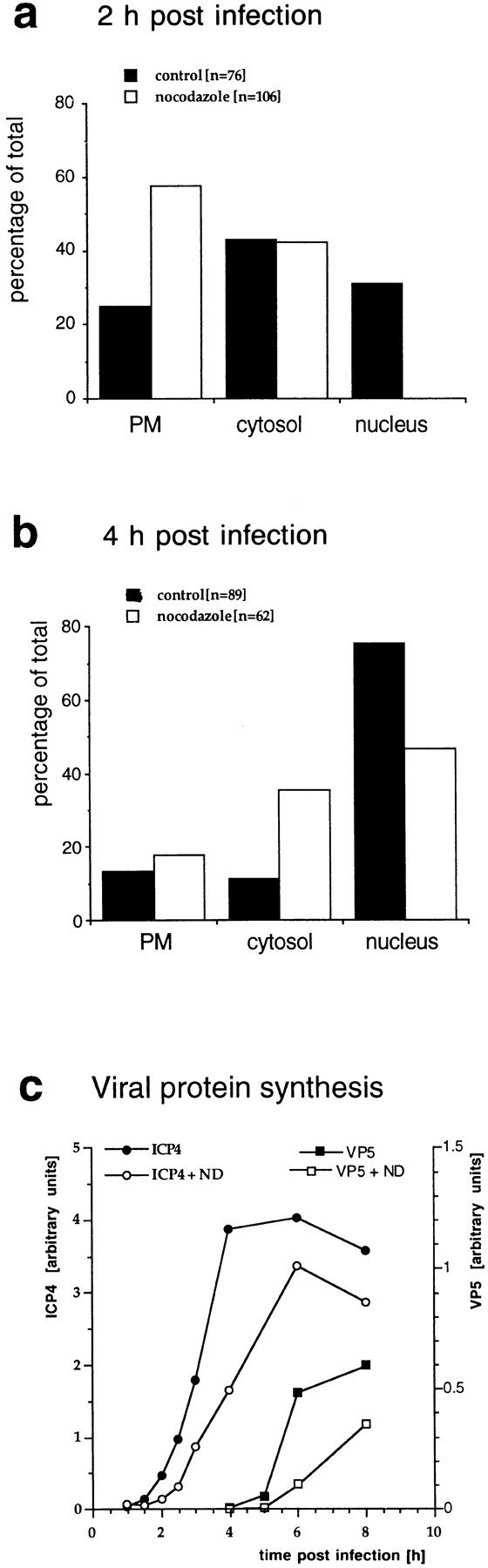
Quantitation of capsid transport and viral protein synthesis in the absence of MTs. Vero cells were infected at an MOI of 170 in the presence of CH and in the absence or presence of nocodazole to depolymerize microtubules. They were fixed at 2 (a) or 4 (b) h postinfection with 1% GA and collected by scraping and pelleting, and Epon sections were prepared. For quantitation, 50 random electron micrographs were obtained in a systematic fashion for each time point and both treatments. The subcellular localization of each cytosolic capsid was determined. (a) At 2 h postinfection in the presence of microtubules, the majority of cytosolic capsids was localized in the cytosol, and a significant fraction had reached the nucleus. In the absence of microtubules, the majority of capsids remained at the plasma membrane (PM), some were in the cytosol, and none had reached the nucleus. (b) At 4 h, >70% of the capsids had reached the nucleus. In the presence of nocodazole, a much smaller fraction was transported to the nucleus, and more capsids remained at the plasma membrane and in the cytosol. (c) Immunoblot. Vero cells were infected at an MOI of 0.5 (without CH!) in the absence or presence of 5 μM nocodazole for various times. The cells were harvested and lysed in 0.5% TX-100 in buffer, the nuclei were pelleted, and the supernatants were analyzed by immunoblotting. To monitor viral protein synthesis, antibodies to ICP4, an early viral protein, and to VP5, a late viral protein, were used. Calnexin, a membrane-bound ER protein, was used as a cellular marker protein. Equal amounts of protein were loaded per lane, and the amount of ICP4 or VP5 was normalized with the amount of calnexin. ICP4 synthesis commences at 2 h PI, whereas VP5 is first detectable at 5 h PI. The amount of ICP4 is maximal after 6 h PI. In contrast, VP5 synthesis increases during the whole time course of the experiment. At all time points, lower amounts of both, ICP4 and VP5, are synthesized in the absence of microtubules.
Quantitation of Virus Binding and Internalization
We assayed for virus binding and internalization as described previously (Helenius et al., 1980). To quantify binding, 6-well dishes containing Vero cells grown for 2 d to just confluency were set on a metal plate on an ice box and washed three times with ice cold RPMI-BSA, and [3H]thymidinelabeled virus was added (1 ml per dish). After incubation on ice for various amounts of time and intensive washings, the cells were scraped into 1 ml RPMI-BSA and pelleted, and cell-associated radioactivity was determined by scintillation counting. The data are expressed as mean values from duplicates. To assay for virus internalization, [3H]thymidine-labeled virus was bound to cells at 4°C for 2 h. The cells were washed to remove unbound virus, and then shifted to normal medium at 37°C and 5% CO2 for various lengths of time. The cells were transferred back to ice, washed, and stored at 4°C until the last time point. All the following treatments were performed at 4°C. The cells were washed with cold, protein-free PBS and incubated with 1 ml per well proteinase K (2 mg/ml in PBS; Boehringer Mannheim GmbH, Mannheim, Germany). After 1 h, 1 ml of PBS containing 1.25 mM PMSF and 3% (wt/vol) BSA was added to stop further proteolysis. The cells were collected, the wells were rinsed with wash buffer (PBS containing 0.2% BSA), and the cells along with the wash buffer were spun at 1,500 rpm for 10 min. The pellet was resuspended and pelleted again; scintillation liquid was added to the resuspended pellet (0.5 ml) and counted in a liquid scintillation counter. The amount of internalized virus was expressed as cell-associated [3H]thymidine counts (mean value from triplicates). Control cells, which were not warmed up after virus binding, showed that the proteinase K treatment removed 90–95% of cell-bound virus.
Light Microscopy
Vero cells grown on coverslips for 1–2 d to ∼70% confluency were infected with HSV-1 at an MOI of 50 PFU per cell in the presence of cycloheximide. In one protocol, cells were fixed with 3% (wt/vol) paraformaldehyde for 20 min followed by quenching remaining fixative using 50 mM NH4Cl for 10 min and permeabilization with 0.1% TX-100 for 4 min. Here, all incubations were carried out in PBS, pH 7.4, at room temperature. In an alternative protocol, the cells were preextracted with 0.5% TX100 for 10 s at 37°C in an MT stabilizing buffer (80 mM Pipes, 2 mM MgSO4, pH 6.8, 10 μM taxol), and then fixed in 100% methanol at −20°C for 4 min. Before immunolabeling at room temperature, the cells from both protocols were washed again in PBS, transferred to 10% normal goat serum for 30 min, and then labeled with the primary antibody in 10% goat serum for 20 min. The coverslips were rinsed three times for 5 min and incubated with fluorescently labeled secondary antibodies in 10% goat serum for 20 min. For double-labeling experiments, the two primary and the two secondary antibodies were applied simultaneously. After extensive washing and one short wash in water, the coverslips were mounted in Mowiol containing 2.5% (wt/vol) 1,4-diazabicyclo-[2.2.2]octane on glass slides and examined with an Axiophot fluorescence microscope (Carl Zeiss, Inc., Thornwood, NY) or a confocal fluorescence microscope (Bio Rad Laboratories, Hercules, CA). Image processing was performed using Adobe Photoshop (Adobe Systems, Inc., Mountain View, CA).
Assay for Viral Protein Synthesis
Confluent Vero cells grown in 6-cm tissue-culture dishes were infected with HSV-1 at an MOI of 0.5 PFU per cell for 30 min at 37°C and 5% CO2 in MEM containing 0.2% (wt/vol) BSA, and then incubated further in normal growth medium. At various time points, they were transferred to 4°C, washed three times with ice cold PBS, scraped, pelleted, and resuspended in 0.3 ml per dish lysis buffer (MNT with 0.5% TX-100, 2 mM DTT, 2 mM EDTA, and 10 μg/ml each of chymostatin, leupeptin, antipain, and pepstatin). The nuclei were removed by centrifugation at 5,000 rpm for 5 min. A 50-μl aliquot of the resulting supernatant was used for protein assays (bicinchoninic assay; Pierce Chemical Co., Rockford, IL), and 0.2 ml were solubilized in 5 × Laemmli sample buffer. Equal amounts of protein were loaded onto a 7.5% SDS polyacrylamide gel for electrophoresis, and the separated proteins were blotted onto nitrocellulose membranes using a semidry blotter. After blocking in 5% lowfat milk in PBS with 0.1% Tween-20, the membranes were incubated with either anti–NC-1 and calnexin or anti–ICP4 and calnexin, followed by HRP-conjugated anti–rabbit or anti–mouse secondary antibody (Pierce Chemical Co.). The secondary antibodies were visualized by enhanced chemiluminescence detection (Amersham Corp. or Pierce Chemical Co.) and Kodak film (Eastman Kodak Co., Rochester, NY). The bands were quantified by densitometry using a Visage 200 digital gel scanner (Bioimage, Ann Arbor, MI). The amount of viral protein (ICP4 or VP5) was normalized for the amount of cellular protein (calnexin).
Results
Early Events during HSV-1 Entry
Due to the large size of HSV-1 virions and capsids, early virus cell interactions can be readily visualized by EM (Campadelli-Fiume et al., 1988; Fuller et al., 1989; Lycke et al., 1988). We followed the incoming capsids at different stages of entry in Vero cells (Fig. 1). The extracellular, surface-bound virus particles had the familiar features of a herpes virus: a membrane envelope with spikes, a capsid with an electron-dense DNA core, and a tegument layer between the capsid and the envelope (Fig. 1, a and b). Images of fusion between the viral envelope and the plasma membrane were readily obtained (Fig. 1, c and d). After fusion, the capsids appeared to separate from the bulk of the tegument since an electron-dense mass remained associated with the cytoplasmic surface of the plasma membrane (Fig. 1, e and f, arrowheads; see also CampadelliFiume et al., 1988; Fuller et al., 1989).
Figure 1.

Entry and uncoating of HSV-1. Ultrathin Epon sections of Vero cells infected with HSV-1 at an MOI of 500 in the presence of cycloheximide (CH). After 2 h of virus binding at 4°C, the cells were either fixed immediately (a and b), or warmed up for 30 (c–f) or 60 min (g and h). (a and b) Binding. The main morphological features of the intact virus bound to the plasma membrane (PM) are the viral envelope (b, arrowhead) with the viral spikes (a, arrowhead), the viral capsid (arrow), and the electron-dense viral DNA genome within the capsid. (c and d) Fusion. Upon warming up, the viral envelope fuses with the plasma membrane, and the capsid (arrow) and the tegument proteins are released into the cytosol. (e and f) Release of the capsid. Preparations, which display a prominent contrast of the tegument, show that the tegument (arrowheads) stays behind at the PM. (g and h) Binding to the nuclear pore. At later time points, the capsids (arrows) have arrived at the nuclear envelope (NE), where they are exclusively located in close apposition to the nuclear pore complexes. Occasionally, fibers emanating from the pores (arrowheads) are visible, to which the capsids seem to bind. Almost all of the capsids at the nuclear pores appear empty and have lost the electron-dense DNA core. Bar, 100 nm.
After 1 h, the first capsids had reached the nucleus and attached themselves to the nuclear pore complexes (Fig. 1, g and h; see also Batterson et al., 1983; Lyke et al., 1988). They seemed to bind to cytsosolic fibers emerging from the pores (see arrowheads in Fig. 1 h). Since most of the capsids on the nuclear envelope lacked the electron-dense central mass, DNA release had apparently occurred. At later time points, some empty capsids were also seen free in the cytosol without connection to the nucleus. Occasionally, 1 h postinfection and later, intact and partially degraded virions were also seen in endosomes and lysosomes, indicating that a fraction of the inoculum was taken up by endocytosis.
The rate and efficiency of cell surface binding and subsequent internalization was assayed quantitatively using [3H]thymidine-labeled HSV-1. To monitor binding, cells were incubated with labeled virus at an MOI of 10, 50, or 100 at 4°C, and the cell-associated radioactivity was measured at different times. As previously reported, a continuous increase in virus binding was observed up to 4 h with no signs of saturation (Fig. 2 a; McClain and Fuller, 1994; Shieh et al., 1992). At 2 h—the adsorption period used in all subsequent experiments— ∼40% of the added virus was bound.
Figure 2.
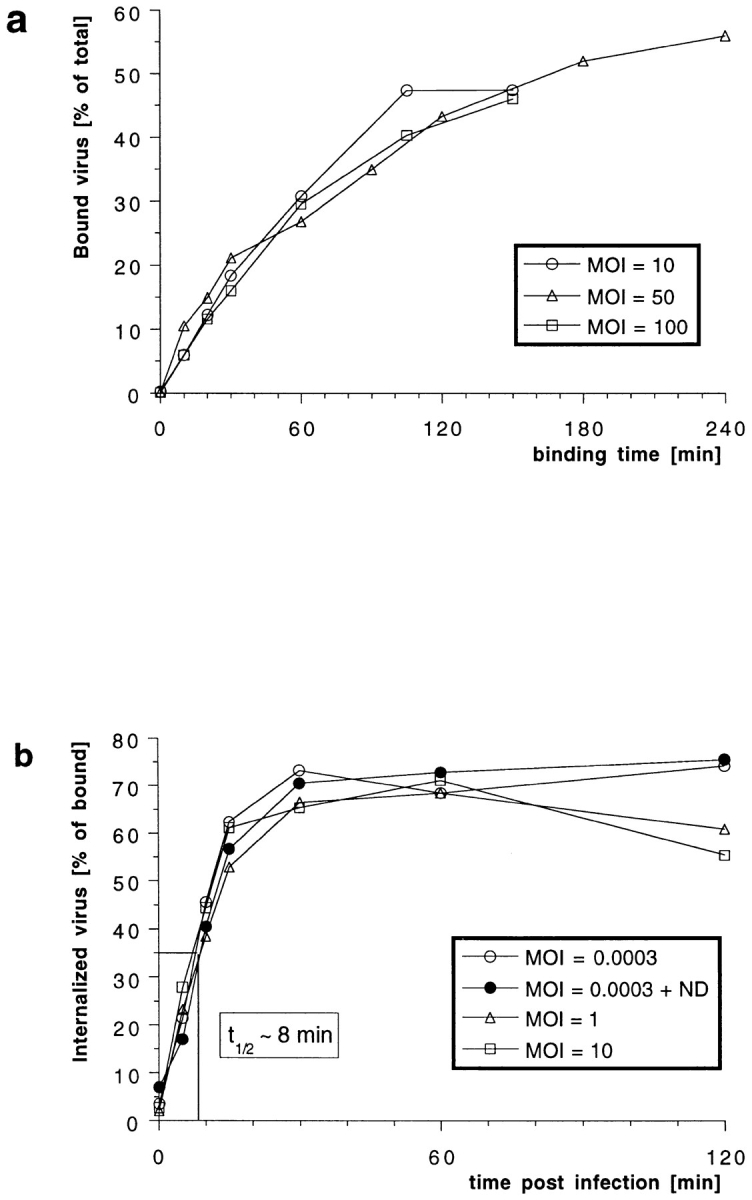
Efficiency and kinetics of HSV-1 binding and internalization. (a) Using [3H]thymidine-labeled virus at different multiplicities of infection (MOI), the amount of virus bound to cells at 4°C was quantified after various time points. There is no apparent saturation of virus binding to Vero cells. After 2 h, ∼40% of the virus added bound. (b) Using [3H]thymidine-labeled virus at different MOIs, the amount of virus internalized into cells was quantified after various time points. At time 0 min (equals 2 h of virus binding at 4°C), proteinase K treatment removes >95% of the virus bound to the plasma membrane. Upon warming up to 37°C and viral fusion, the virus is protected within the cells from the protease treatment. After 30 min, the maximal amount of virus, ∼70% of the bound virus, is internalized. The half-time (t1/2) of internalization is 8 min. Depolymerization of microtubules by nocodazole (ND) has no effect on the kinetics and the amount of internalization.
To follow internalization biochemically, a protease protection assay was used (Helenius et al., 1980). After a 2-h binding period in the cold, the cells were warmed up to 37°C for different periods of time to initiate penetration. Thereafter, they were chilled to 4°C and treated with proteinase K to detach remaining cell surface viruses. The fraction of virus protected from the protease increased rapidly after warming (Fig. 2 b) with a half-time of internalization of 8 min. The overall protease resistance reached a level of 70% within 30 min. The efficiency and rate of internalization was identical over a multiplicity range from 3 × 10−4–10 MOI. Treatment of the cells with nocodazole, a drug that depolymerizes MT (Fig. 2 b, ND), or with cytochalasin D, which depolymerizes actin filaments (not shown), had no effect, either on the efficiency of binding and internalization or on the kinetics of internalization. Consistent with previous reports using acid inactivation and plaque assays to assess virus internalization (Huang and Wagner, 1964), these results showed that HSV-1 penetration occurs rapidly and efficiently.
Transport of Capsids from the Plasma Membrane to the Nucleus
Indirect immunofluorescence microscopy was next used to analyze the transport of individual capsids from the cell periphery to the nucleus. The antibodies used were either against VP-5 (anti–NC-1) or VP19c (anti–NC-2), the two major capsid proteins, or against purified whole capsids (anti-HC and anti-LC). All the antibodies gave similar results and had in common that they stained capsids that were in the cytosol (Fig. 3, 1 h) but not capsids present in surface-bound intact viruses (Fig. 3, 0 h). Presumably, this useful selectivity was caused by antigen accessibility; the relevant capsid epitopes in the intact virus particles were probably obscured by envelope and/or tegument components. In contrast, anti-capsid antibodies labeled viral capsids on thawed cryosections (Sodeik, B., M.W. Ebersold, and A. Helenius, unpublished observations).
Figure 3.

Subcellular distribution of incoming HSV-1 capsids. Conventional immunofluorescence microscopy of Vero cells infected with HSV-1 at an MOI of 50 in the presence of CH. The cells were fixed in 3% PFA, permeabilized with 0.1% TX-100, and labeled with anti-HC, an antibody generated against purified capsids. After 2 h of virus binding at 4°C (0 h), there is no signal. At 1 h PI, numerous cytoplasmic capsids (arrows) distributed within the entire cytoplasm are detectable. At 2 h PI, the capsids begin to accumulate at the nucleus. At 4 h PI, most of the capsids decorate the nuclei. Note that at 1 h, the focus was close to the coverslip, whereas at 2 and 4 h, it was at the nuclear membrane.
The capsids in the cytoplasm appeared as small, intensely labeled spots (Fig. 3, arrows, 1 h). Using immunoelectron microscopy on thawed ultrathin cryosections, we confirmed that these spots represented single capsids (not shown). All the structures labeled in the cytosol or at the nuclear membrane by antibodies directed against the capsid or against VP5 represented single viral capsids (not shown). The number of fluorescently labeled spots increased continuously during the first hour of warming. In addition, their distribution within the cell changed with increasing time: from the cell periphery toward the nucleus where they accumulated at the nuclear rim (Fig. 3, 2 h). After 4 h, virtually all the spots were localized at the nuclear membrane (Fig. 3, 4 h) where several hundred capsids could be seen. Thus, incoming cytosolic capsids were transported efficiently from the periphery to the host nucleus.
Capsids Bind to Microtubules
Double immunofluorescence microscopy using anti-capsid and anti-tubulin antibodies showed that most of the capsids that were not bound to the nucleus were associated with MT (Fig. 4, a and b). Moreover, in many cells, capsids were seen to concentrate around the MT organizing center (MTOC) (Fig. 4 b). The close association of capsids with MT was confirmed by EM (Fig. 4, c–e). Numerous cytoplasmic capsids were seen in close proximity to cytoplasmic filaments with the expected 24-nm width and MT-like morphology. Typically, the distance between the capsid surface and the tubules was ∼50 nm, suggesting the presence of additional tethering components. The viral DNA inside the MT-associated capsids could be seen as a darkly stained material.
Figure 4.
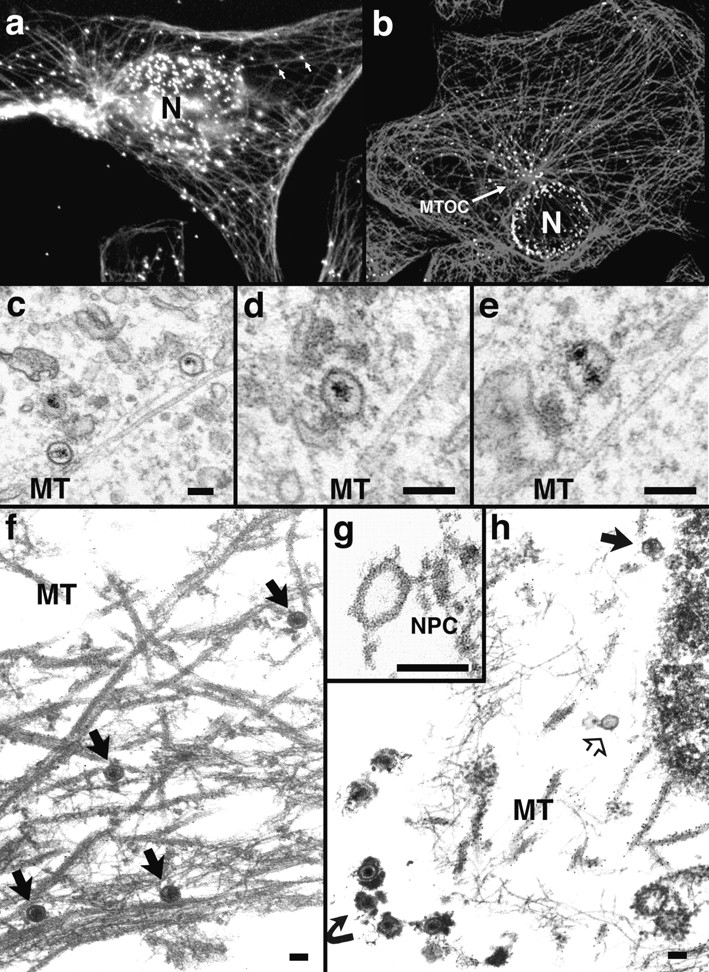
Incoming HSV-1 capsids colocalize with MTs. (a) Conventional immunofluorescence microscopy. Vero cells infected at an MOI of 50 in the presence of CH were fixed in methanol at 2 h PI, and double labeled with anti-VP5 (NC-1; FITC anti–rabbit) and antitubulin antibodies (IA2; rhodamine anti–mouse). The FITC and the rhodamine signals were documented simultaneously using the FITC filter set. Almost all capsids (arrows) not localized to the nucleus (N) colocalize with microtubules. (b) Confocal immunofluorescence microscopy. Vero cells infected at an MOI of 50 in the presence of CH were fixed in methanol 2 h PI, and double labeled with anti-capsid (HC, white) and anti-tubulin antibodies (IA2, gray). Almost all capsids not localized to the nucleus (N) colocalize with microtubules. Note that in some cells, the viral capsids accumulate at the microtubule-organizing center (MTOC). (c–e) Conventional EM. At 1 h postinfection, Vero cells infected at an MOI of 500 in the presence of CH were fixed with 2% glutaraldehyde in PBS. Epon sections were cut parallel to the substrate. Numerous incoming viral capsids are localized to microtubules (MT) as identified by their typical morphology, localization, and their 24-nm diameter. (f–h) Immunoelectron microscopy. Vero cells infected at an MOI of 500 in the presence of CH were extracted at 1 h (f) or 4 h PI (g and h) with 0.5% TX-100 in MT buffer before fixation. They were labeled with rabbit anti– tubulin followed by protein A–9 nm gold, and Epon sections were cut parallel to the substrate. The cytoskeleton, nucleus, and nuclear pore complexes (NPC), as well as cytoplasmic capsids, were preserved excellently, whereas all membranous organelles were extracted. The microtubules (MT) are easily identified, since they are heavily decorated with antibody and protein A–gold. In rather thick sections (f), the microtubules can be traced over long distances, and many capsids (filled arrows) are localized on them. These capsids still contain their electron-dense DNA core. At later time points (g and h), most of the capsids have lost their electron-dense core (h, open arrow) and are mostly located in close proximity to the nucleus. Very often the capsids are bound directly to the outer ring of the nuclear pore complex (g, NPC). Virions bound to the plasma membrane (h, curved arrowhead) have lost the trilaminar appearance of their membrane, but apparently a lot of glycoprotein and tegument remains attached to them, causing a distinct morphology easily distinguished from cytoplasmic capsids (g). Bar, 100 nm.
The tubules were unambiguously identified as MT by antitubulin labeling of specimen that had been preextracted with TX-100 before fixation. After such extraction, thicker sections could be viewed, allowing the analysis of MT over longer distances. The MT-associated capsids generally contained the viral DNA (Fig. 4 f, arrows). At later time points, most of the capsids were empty and typically located at the nuclear pores and not at the nuclear envelope membrane (Fig. 4 g). Most of these capsids did not contain DNA (Fig. 4, g and h). At 4 h postinfection, there were occasionally empty capsids localized on MT in close proximity to the nucleus (not shown). Whenever extracellular viruses were observed in these TX-100 extracted samples, they were surrounded by very-electron dense material, which probably represented the viral glycoproteins and tegument proteins that were apparently not removed during extraction (Fig. 4 h, curved arrow).
Quantitation of HSV-1 Entry and Capsid Transport
To quantify the kinetics of the cytosolic capsid transport, we determined the subcellular localization of the incoming virus by EM at various time points postinfection at an MOI of 500 (Fig. 5). To compare this experiment with the immunofluorescence microscopy, ultrathin sections were cut parallel to the substrate through the basal region of the cells (see Fig. 5 a). For each time point, 25 electron micrographs were taken in a systematic random fashion, and the number and localization of cytosolic capsids was determined. Cytosolic capsids (Fig. 5 b) were localized at the plasma membrane (Fig. 5 c), in the cytosol without obvious connection to any organelle (Fig. 5 d), at MT (Fig. 5 e), or at the nucleus (Fig. 5 f).
After warming, the total number of cytosolic capsids increased rapidly, reaching a peak after 60 min (Fig. 5 b). At later times the number of cytosolic capsids declined, suggesting that the capsids were ultimately disassembled. We also scored whether the cytosolic capsids contained the electron-dense viral DNA core or whether they appeared empty (see Fig. 1). Up to 60 min after internalization, no empty capsids were detected, whereas after 2 h 20% and after 4 h 64% had lost their electron-dense DNA core (Fig. 5 b). Virions in endosomes peaked at 1 h and sharply declined thereafter. They may have been degraded or recycled back to the cell surface, or capsids could have been released by fusion into the cytosol.
At 15 min postinfection, almost 70% of all cytosolic capsids were localized within 100 nm of the plasma membrane (see Figs. 1, c–f, and 5 c). All of the capsids close to the plasma membrane contained the viral DNA core. At 1 h postinfection, most of the cytosolic capsids (almost 80%) had an intermediate location; they were neither close to the plasma membrane or to the nucleus (Fig. 5, d and e). A significant fraction (10% of all cytosolic capsids) colocalized with MT (see Figs. 4, c–e, and 5 e). Since the capsids (125 nm) are considerably larger than the section thickness (60–70 nm), the figure for colocalization with the MT was most likely an underestimation. Of the cytosolic capsids, only one capsid (0.9% of all capsids at 4 h postinfection) was seen to colocalize with intermediate filaments (not shown).
Cytosolic capsids arrived at the nucleus at 2 h postinfection, and at 4 h, the majority of cytosolic capsids (>60%) was localized to the nucleus (Fig. 5 f). At the ealier time points (up to 1 h), <4% of the cytosolic capsids were located at the nucleus. Of the nuclear capsids, 68% at 2 h and 87% at 4 h were empty, suggesting that DNA release occurred after arrival at the nucleus. The vast majority of empty capsids were localized to the nucleus (Fig. 5 g). All capsids at the nucleus were localized to the nuclear pore complexes (see Fig. 1, g and h).
In summary, the electron microscopic experiment confirmed the immunofluorescence microscopy data. Cytosolic capsids were efficiently transported from the plasma membrane via the cytosol to the nuclear pore complexes. During transit, a significant fraction attached to MT. Empty capsids appeared soon after their arrival at the nucleus, and some of these were released into the cytosol. Ultimately, the capsids seemed to disassemble and disappear because the overall number of cytosolic capsids decreased late in infection. Interestingly, the appearance of cytosolic capsids under the plasma membrane peaked before the transport of virions into endosomes, strongly suggesting that the early cytosolic capsids were indeed derived from fusion of the virus at the plasma membrane rather than by fusion from endosomes.
Reduced Capsid Transport to the Nucleus without Microtubules
To determine whether efficient capsid transport via the cytosol depended on an intact cytoskeleton, cells were infected at an MOI of 50 for 2 h in the presence of drugs that affect MT or actin filaments. Immunofluorescence microscopy showed that neither taxol, a drug that prevents the disassembly of MT (Wilson and Jordan, 1994), nor cytochalasin D, a drug that causes depolymerization of actin filaments (Cooper, 1987), affected capsid transport to the nucleus (Fig. 6 a, upper panels). In contrast, capsid accumulation at the nuclear membrane was significantly reduced by colchicine, vinblastine, and nocodazole (Fig. 6 a, lower panels). These drugs interact with tubulin by different molecular mechanisms (Wilson and Jordan, 1994). Labeling with anti-tubulin confirmed that colchicine and nocodazole depolymerized MT, whereas vinblastine caused tubulin paracrystal formation (not shown). In the presence of these drugs, the majority of capsids remained scattered throughout the cytosol. To test whether the effect of nocodazole was reversible, the cells were analyzed 2 h after drug removal. At this time, the MT had repolymerized, and the previously dispersed capsids were concentrated on the nuclear rim (Fig. 6 b).
Figure 6.


Reduced transport of capsids to the nucleus in the absence of MTs. Immunofluorescence microscopy of Vero cells infected with HSV-1 for 2 h at an MOI of 50 in the presence of CH and drugs affecting the cytoskeleton. (a) Cytoskeletal drugs. Under control conditions, almost all capsids accumulate around the nucleus. If the cells were infected in the presence of nocodazole, colchicine, or vinblastine, respectively (lower panel), there is no accumulation of viral capsids at the nucleus; instead, the capsids are scattered throughout the entire cytoplasm. In cells infected in the presence of taxol or cytochalasin D, the capsids accumulate at the nucleus (upper panel). The cells were fixed in 3% PFA, permeabilized with 0.1% TX-100, and labeled with an antibody directed against purified capsids (HC). (b) Reversibility. In the presence of nocodazole, the amount of viral capsids localized to the nucleus is drastically reduced, whereas, after a 2-h chase in nocodazole-free medium, the capsids accumulate around the cell nuclei. Cells were infected in the presence of nocodazole for 2 h, fixed immediately (left panels), or washed and further incubated in normal medium for an additional 2 h before fixation (right panels). After extraction with 0.5% TX-100 in MT buffer and fixation in methanol, the cells were double labeled with anti-tubulin (left panels) and anti-capsid (HC, right panels).
Since we were unable to quantify the localization of the fluorescent spots representing cytosolic capsids reliably, we infected Vero cells at an MOI of 150 in the presence or absence of nocodazole and analyzed the embedded cell pellets by quantitative EM. For each experimental condition, 50 electron negatives were taken in a systematic, random fashion and the localization of cytosolic capsids was determined (Fig. 7, a and b). After 2 h of infection, 31% of the cytosolic capsids had reached the nucleus, and only 25% were still in close proximity to the plasma membrane. In contrast in the absence of MT, none had reached the nucleus, and 58% were still in the region close to the plasma membrane. After 4 h postinfection, 75% of all cytosolic capsids had reached the nucleus, 14% were at the plasma membrane, and the remaining 11% were present in the cytosol. In the presence of nocodazole, only 47% of the cytosolic capsids had reached the nucleus, 18% were still at the plasma membrane, and 36% were in the cytosol distant from both the plasma membrane and the nucleus.
As nocodazole, vinblastine, and colchicine all affect the network of MT (Wilson and Jordan, 1994), we concluded that MT are directly or indirectly involved in the rapid transport of capsids from the cell periphery to the nucleus in Vero cells. In cells devoid of functional MT, the arrival of capsids at the nucleus was significantly delayed.
Viral Infection in the Absence of Microtubules
Whether nocodazole would have an effect on productive infection was determined by monitoring the onset of viral protein synthesis in the presence or absence of the drug. Cell extracts prepared after different periods of infection were subjected to immunoblotting using antibodies against an early HSV-1 protein, ICP4, and a late structural protein, VP5. The results in Fig. 7 c show that the synthesis of both viral proteins was delayed and reduced in the absence of MT. Calnexin, an integral membrane protein of the ER (Hammond and Helenius, 1994), was used as a control and remained constant. Labeling with [35S]methionine demonstrated that nocodazole had no overall effect on protein synthesis in uninfected or HSV-1–infected Vero cells (not shown).
When immunofluorescence microscopy using ICP4 was performed, we found that, under control conditions, viral protein synthesis commenced in many cells already at 2 h postinfection. In contrast, in the presence of nocodazole, only very few cells showed ICP4 labeling. At 4 h postinfection, all control cells were strongly labeled for ICP4, whereas in the absence of MT, significantly less cells show strong labeling for ICP4 (data not shown). However, at later time points, all cells synthezised ICP4, in the presence and absence of MT.
Thus, depolymerization of MT delayed the onset of viral protein synthesis in cultured fibroblasts, but did not prevent viral infection per se.
Dynein Colocalizes with Incoming Capsids
Our results strongly suggested that incoming herpes capsids move along MT toward the nucleus. Therefore, we ask whether they would use host MT-dependent motors for their transport. Since the antibody against tubulin often obscured the morphology of putative tethering factors located between the viral capsids and the MT (see Fig. 4), we repeated these experiments without immunolabeling. Electron-dense material was usually attached to the vertices of the capsids that were associated with MT (Fig. 8). The morphology of the appendages was variable, but, in some cases (Fig. 8 d, arrow), they had the dimensions and the shape of cytoplasmic dynein, a minus end–directed MT-dependent motor. Dynein is a Y-shaped protein complex with a length of ∼50 nm. It is connected to its cargo via the stalk and to MT through two 14-nm globular domains (Gilbert and Sloboda, 1989; Vale, 1990; Vallee et al., 1988).
Figure 8.
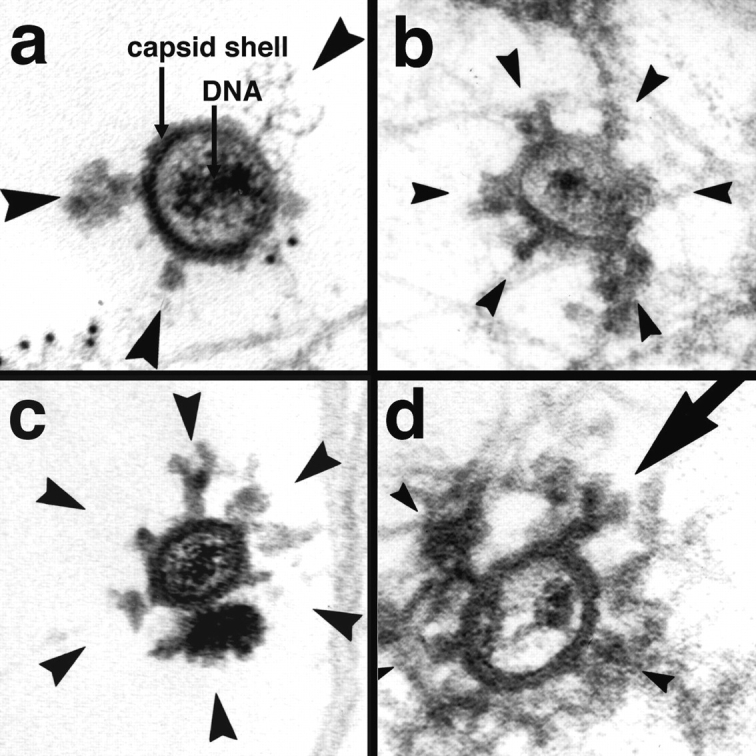
Incoming HSV-1 capsids display appendices at their vertices. At 1 h PI, Vero cells infected at an MOI of 500 in the presence of CH were extracted with 0.5% TX-100 in MT buffer for 2 min at 37°C and fixed in 1% GA in MT buffer. They were labeled with anti-tubulin followed by protein A–9 nm gold (a) or embedded directly (b–d), and Epon sections were cut parallel to the substrate. All viral capsids display some appendages on their outside (arrowheads). The appendages are visible at three, four, five, or all six corners visible in a section, which represent the pentons of a viral capsid. Some of the appendices have a morphology very similar to purified cytoplasmic dynein (large arrow in d): a large globular stalk with two smaller globular head domains. The capsid size is ∼100–110 nm; the dynein-like appendices are ∼50 nm. In thick sections of golden color, the appendices are often buried in cytoskeletal material (b and d), whereas in very thin sections of grey color (a and c), they are very prominent. The appendices have all kinds of different morphology with varying sizes and electron density, but they are always located at the pentons.
To test whether cytoplasmic dynein was, indeed, associated with the capsids, we used an affinity-purified antibody directed against the heavy chain of cytoplasmic dynein (Vaisberg et al., 1993). Immunoelectron microscopy with the anti-dynein antibody was performed on ultrathin cryosections of in situ fixed cells 1 or 2 h after initiation of virus entry. In addition to a low level of labeling of the cytoplasm, dynein was found to be localized on the surface of endosomes (Fig. 9 h), mitochondria (Fig. 9 a), and the Golgi apparatus (Fig. 9 a). Very few gold particles were seen over the nucleus and the mitochondrial matrix (Fig. 9, a, c, and d). The mitochondria, which do not contain dynein, served as a background control. Compared with the mitochondrial matrix, there was a slightly higher labeling in the nuclei, and the cytoplasm was labeled 2.3-fold higher than background (Table I).
Figure 9.
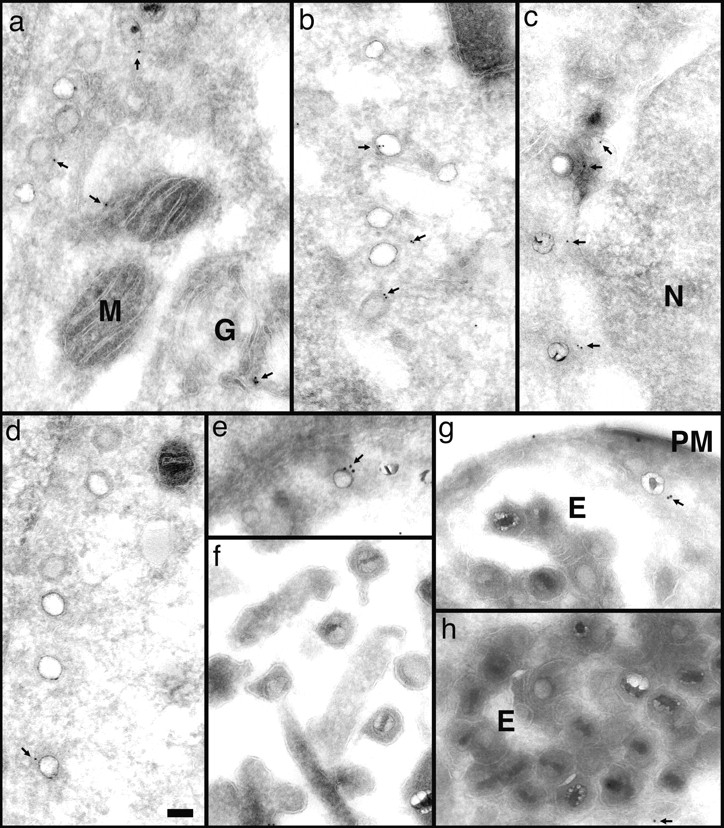
Anti-dynein antibodies label incoming viral HSV-1 capsids. Thawed cryosections of Vero cells infected for 1 (c) or 2 h (a, b, and d–h) in the presence of CH at an MOI of 500 were labeled with rabbit antibodies followed by protein A–5 nm (a–d, and g) or 10 nm gold (e, g, and h). About 5% of the incoming viral capsids (a–f, and h) are labeled by anti-dynein (arrows), an affinity-purified antibody made against a recombinant fragment of dynein heavy. Labeled capsids are localized close to the plasma membrane (g, PM), at the nuclear envelope (c and d), or within the cytoplasm without obvious connection to either one (b). Note that the capsids within the extracellular virions (f) or within the endosomes (g and h, E) are not labeled. Anti-dynein also labels the membranes of mitochondria (a, M), endosomes (h, E), and the Golgi complex (a, G) and diffusely the entire cytoplasm, whereas the mitochondrial matrix (a, b, and d) or the nucleus (d, N) show no labeling for dynein. Bar, 100 nm.
Table I.
Quantitation of Dynein Immunolabeling
| Percentage labeled* | Distance‡ | Number of points§ | Density‖ | |
|---|---|---|---|---|
| % | nm | gold/μm2 | ||
| Cystolic capsids (n = 93) | 13.0 | 48 | 32 | 3.70 |
| Viral capsids (n = 203) | 1.5 | 69 | 64 | 0.46 |
| Number of gold particles* | Number of points§ | Density‖ | ||
| gold/μm2 | ||||
| Cytosol | 469 | 3,488 | 1.21 | |
| Nucleus | 88 | 1,376 | 0.69 | |
| Mitochondria | 8 | 135 | 0.53 |
In cells containing incoming HSV-1 capsids, dynein was, in addition, found associated with many of the capsids (Fig. 9, a–f). Capsids directly underneath the plasma membrane (Fig. 9 g) and within the cytoplasm (Fig. 9 b), as well as capsids in close proximity to the nucleus (Fig. 9, a, c, and d), were labeled with anti-dynein. On average, 13% of the cytoplasmic capsids were labeled (Table I). Capsids of intact virions, either extracellular (Fig. 9 f) or within endosomes (Fig. 9, g and h), were not labeled. Only 1.5% of all capsids present in virions had colloidal gold particles in close proximity (Table I). Thus, the labeling density was eightfold higher on cytosolic capsids compared with capsids within virions, ruling out the possibility that the antidynein antibody showed cross-reactivity to capsid proteins. Given that the labeling efficiency using this technique does not exceed 10%, e.g., only 10% of the primary antibody present on the sections is detected by protein A–gold (Griffiths, 1993_a_ ), these results showed that incoming cytosolic viral capsids associated with cytoplasmic dynein.
Discussion
HSV-1 causes a range of diseases from benign common cold sores to life-threatening encephalitis. It infects epithelial cells of mucocutaneous membranes, and, as other α herpesviridae, it establishes latent infections in sensory ganglia (Whitley and Gnann, 1993). It is thought to enter neurons by fusion at the presynaptic membrane, after which the capsid is transported along the axon to the nucleus (Hill et al., 1972; Lycke et al., 1988; Roizman and Sears, 1989; Topp et al., 1994). The efficient infection of neurons and other terminally differentiated cells has made HSV-1 a useful vehicle for gene delivery with potential for gene therapy in the human brain (Glorioso et al., 1995; Karpati et al., 1996).
The biochemical and morphological assays used in our study showed that HSV-1 entry into Vero cells is both rapid and efficient. Over a broad range of multiplicities, we found that ∼70% of bound virus penetrated with a half-time of ∼8 min. More than 60% of the capsids delivered to the cytosol were transported to the nucleus within 4 h of uptake, and binding to fibers extending from the nuclear pore complexes seemed to occur. The ratio of particles to plaque-forming units for HSV-1 is between 10 and 50 (Frenkel et al., 1975; McLauchlan et al., 1992; Smith, 1964; Watson et al., 1963). Thus, if cells were infected at an MOI of 50, and 40% of these bound, the number of particles entering the cells was 140–700 according to the proteinase K assay. We determined that 60% of the internalized virus, namely 80–420 particles, uncoat their viral genome within 4 h postinfection as measured by a DNase protection assay (Ojala, P., B. Sodeik, M.W. Ebersold, and A. Helenius, manuscript in preparation).
A majority of the capsids remained associated with nuclear pores after releasing their DNA (see also Batterson et al., 1983; Lycke et al., 1988). The apparent transient stability of the empty capsids contrasts with adenovirus, another virus that delivers its DNA to the nucleus through the nuclear pore complexes, for which no empty capsids could be detected at the nuclear pore complexes (Greber et al., 1993). We cannot formally rule out that, even though the herpes capsids appeared empty at the nuclear membrane, viral DNA release had not been completed yet.
Our results demonstrate that MT are involved in capsid transport from the cell surface to the nucleus. Since the distance from the plasma membrane to the nucleus in Vero cells is not as large as in neurons, MT-assisted transport was not essential for infection, but it accelerated both the transfer of the capsids to the nucleus and the onset of viral protein synthesis. Having first detached from most of the tegument proteins at the plasma membrane, the capsids moved in minus end direction along MT to the MTOC. From there, they were transferred to the nuclear pores by unknown mechanisms. At early times (1–2 h PI), the majority of the capsids associated with the nuclear membrane proximal to the MTOC, but later the distribution became more homogenous around the nucleus.
Nocodazole and colchicine, which both depolymerize MT, and vinblastine, which causes MT paracrystal formation, reduced capsid accumulation at the nuclear envelope. These drugs were also reported to reduce the amount of infectious HSV-1 recovered from rat dorsal root ganglia infected in vitro (Kristensson et al., 1986) and from the trigeminal ganglia after inoculation of the murine cornea (Topp et al., 1994). We think that it is very likely that capsids derived from virions bound to the “apical” plasma membrane in our system (see Fig. 5 a) reached the nucleus independent of MT, since the distance between the apical plasma membrane and the nuclear membrane is only ∼200–300 nm. Thus, the larger the distance between the plasma membrane where the virion had bound and the nucleus, the more the transport to the nucleus depended on MT. In contrast with the results obtained in neurons (Kristensson et al., 1986), taxol, which stabilizes MT, did not reduce the efficiency of capsid transport, suggesting that the dynamic turnover or “treadmilling” of MT was not required for capsid transport in Vero cells. Depolymerization of actin filaments by cytochalasin D had no effect either on HSV-1 internalization or transport to the nucleus.
The presence of dynein heavy chains in the material attached to the cytosolic capsids indicated that the MT-mediated transport was powered by cytoplasmic dynein. Cytoplasmic dynein is a multisubunit protein complex of 1,270 kD consisting of two heavy chains, two to three intermediate chains, and a variable number of small subunits (Holzbaur and Vallee, 1994; Schroer, 1994). It has a length of ∼40–50 nm and a shape reminiscent of the letter Y. The two head domains contain the MT binding and ATPase sites, while the base interacts with the cargo (Gilbert and Sloboda, 1989; Vale, 1990; Vallee et al., 1988). Dynein is responsible for minus end–directed movement of chromosomes and membrane organelles along MT (Cole and LippincottSchwartz, 1995; Mitchison, 1988). Since the plus ends of MT are localized in the periphery of Vero cells close to the plasma membrane and the minus ends are anchored at the perinuclear MTOC, dynein seems a logical choice as a motor for the transport of incoming viral capsids to the nucleus. The same applies to the axons of neurons, where the MT have their plus ends toward the synapse, and the minus end anchored at the MTOC in the cell body (Black and Baas, 1989). Although some kinesin-like MT minus end–directed motors have been implicated to play a role in mitosis and meiosis, none of them has been demonstrated to be involved in interphase transport processes (Barton and Goldstein, 1996; Hirokawa, 1996). Since the MT-mediated transport that is crucial for infection of neurons also operates in cultured fibroblasts, its analysis will be greatly facilitated.
It is believed that dynein attachment to vesicles and kinetochores requires another large multimeric complex, called dynactin, which contains 10 different proteins (Holzbaur and Vallee, 1994; Schroer, 1994; Vallee and Sheetz, 1996). So far, receptor molecules for dynein and dynactin have not been identified on any cargo structures, but several studies indicate that the activity of dynein and its binding to cargo may be regulated by phosphorylation (Allen, 1995; Dillman and Pfister, 1994; Lin et al., 1994; Niclas et al., 1996). Whether the binding of dynein to HSV-1 capsids involves dynactin or other cellular components and whether it is regulated by phosphorylation remains unclear at this point. The viral protein(s) that serve as dynein receptors also remain to be identified. Since the polypeptide composition of the capsids and tegument is known, some potential candidates can, however, be singled out. The capsid itself contains six different proteins. Of these, only VP5 is known to be located at the pentons (Trus et al., 1992), which were the sites of preferential attachment of proteinaceous material onto cytosolic capsids shown here. In addition to the bona fide capsid polypeptides, components derived from the tegument are also likely to be present. For example, VP1-2, a 270-kD protein, is particularly tightly associated with the capsids (Roizman and Furlong, 1974) and may constitute part of the material seen attached to the pentons in our electron micrographs. Taking advantage of the relative simplicity of the capsids, of existing virus mutants, and of the complete genomic sequence of the virus, work is in progress to identify and to characterize the dynein receptor in HSV-1 capsids.
Abbreviations used in this paper
CH
cycloheximide
GA
glutaraldehyde
HSV-1
herpes simplex virus 1
MOI
multiplicity of infection
MT
microtubule
MTOC
microtubule organizing center
PFA
paraformaldehyde
PFU
plaque-forming unit
PI
postinfection
TX-100
Triton X-100
Footnotes
B. Sodeik thanks Drs. Gary Cohen and Roselyn Eisenberg for their generous hospitality in their labs and many helpful suggestions. Victor Stolc is acknowledged for his contribution to the immunofluorescence studies. We also thank Lisa Hartnell, Linda Iadarola, Philippe Male, and Paul Webster of the Center of Cell Imaging in the Cell Biology Department at Yale University for teaching many aspects of EM, and Henry Tan for his superb photographic work. We are grateful to Drs. Gary Whittaker, Jani Simons, Päivi Ojala, Philippe Pierre, and Urs Greber for many stimulating discussions and critical reading of the manuscript.
The work was supported by a National Institutes of Health grant to A. Helenius (AI18599) and a long-term postdoctoral EMBO fellowship to B. Sodeik (ALTF 254-1993).
Address all correspondence to Beate Sodeik, Department of Cell Biology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520-8002. Tel.: (203) 737-2612. Fax: (203) 785-7226. e-mail: sodeik@biomed.med.yale.edu
References
- Allen V. Protein phosphatase 1 regulates the cytoplasmic dynein-driven formation of endoplasmic reticulum networks in vitro. J Cell Biol. 1995;128:879–891. doi: 10.1083/jcb.128.5.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Ahmad FJ. The plus ends of microtubules are the exclusive nucleating structures for microtubules in the axon. J Cell Biol. 1992;116:1231–1241. doi: 10.1083/jcb.116.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NR, Goldstein LSB. Going mobile: microtubule motors and chromosome segregation. Proc Natl Acad Sci USA. 1996;93:1735–1742. doi: 10.1073/pnas.93.5.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterson W, Roizman B. Characterization of the HSV-associated factor responsible for the induction of α genes. J Virol. 1983;45:397–407. doi: 10.1128/jvi.46.2.371-377.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterson W, Furlong D, Roizman B. Molecular genetics of HSV. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol. 1983;45:397–407. doi: 10.1128/jvi.45.1.397-407.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MM, Baas PW. The basis of polarity in neurons. Trends Neurosci. 1989;12:211–214. doi: 10.1016/0166-2236(89)90124-0. [DOI] [PubMed] [Google Scholar]
- Booy FP, Newcomb WW, Trus BL, Brown JC, Baker TS, Steven AC. Liquid-crystalline, phage-like packing of encapsidated DNA in herpes simplex virus. Cell. 1991;64:1007–1015. doi: 10.1016/0092-8674(91)90324-r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, D. 1992. Internal movements and changes in shape. Chapter 4. In Cell Movements. Garland Publishing, Inc., New York/London. 45–49.
- Campadelli-Fiume G, Arsenakis M, Farabegole F, Roizman B. Entry of HSV-1 in BJ cells that constituitively express viral gD is by endocytosis and results in degradation of the virus. J Virol. 1988;62:159–167. doi: 10.1128/jvi.62.1.159-167.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MEM, Palfreymann JW, Preston CM. Identification of the herpes simplex virus DNA sequence which encodes a trans-acting polypeptide responsible for the stimulation of the immediate early transcription. J Mol Biol. 1984;180:1–19. doi: 10.1016/0022-2836(84)90427-3. [DOI] [PubMed] [Google Scholar]
- Cohen GH, de Leon MP, Diggelmann H, Lawrence WC, Vernon SK, Eisenberg RJ. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J Virol. 1980;34:521–531. doi: 10.1128/jvi.34.2.521-531.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Lippincott-Schwartz J. Organization of organelles and membrane traffic by microtubules. Curr Opin Cell Biol. 1995;7:55–64. doi: 10.1016/0955-0674(95)80045-x. [DOI] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudmore S, Cossart P, Griffiths G, Way M. Actin-based motility of vaccinia virus. Nature (Lond) 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- Dillman JF, Pfister KK. Differential phosphorylation in vivo of cytoplasmic dynein associated with anterogradely moving organelles. J Cell Biol. 1994;127:1671–1681. doi: 10.1083/jcb.127.6.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel N, Jacob J, Honess RW, Hayward S, Locker H, Roizman B. Anatomy of HSV DNA. III. Characterization of defective DNA molecules and biological properties of virus populations containing them. J Virol. 1975;16:153–167. doi: 10.1128/jvi.16.1.153-167.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller AO, Santos RE, Spear PG. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J Virol. 1989;63:3435–3443. doi: 10.1128/jvi.63.8.3435-3443.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SP, Sloboda RD. A squid dynein isoform promotes axoplasmic vesicle translocation. J Cell Biol. 1989;109:2379–2394. doi: 10.1083/jcb.109.5.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glorioso, J.C., M.A. Bender, W.F. Goins, D.F. Fink, and N. DeLuca. 1995. Herpes simplex virus as a gene-delivery vector for the central nervous system. In Viral vectors—Gene Therapy and Neuroscience Applications. M.G. Kapplitt and A.D. Loewy, editors. Academic Press, San Diego/New York/Boston/London/Sydney/Tokyo/Toronto. 1–23.
- Greber UF, Singh I, Helenius A. Mechanisms of virus uncoating. Trends Microbiol. 1994;2:52–56. doi: 10.1016/0966-842x(94)90126-0. [DOI] [PubMed] [Google Scholar]
- Greber UF, Willetts M, Webster P, Helenius A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 1993;75:477–486. doi: 10.1016/0092-8674(93)90382-z. [DOI] [PubMed] [Google Scholar]
- Griffiths, G. 1993_a._ Labeling reactions for immunocytochemistry. In Fine Structure Immunocytochemistry. Springer-Verlag, Berlin/Heidelberg/New York. 237–278.
- Griffiths, G. 1993_b._ Quantitative aspects of immunocytochemistry. In Fine Structure Immunocytochemistry. Springer-Verlag, Berlin/Heidelberg/New York. 371–445.
- Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine JW, Honess RW, Cassai E, Roizman B. Proteins specified by HSV. XII. The virion polypeptides of type 1 strains. J Virol. 1974;14:640–651. doi: 10.1128/jvi.14.3.640-651.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, Kartenbeck J, Simons K, Fries E. On the entry of Semliki Forest virus into BHK-21 cells. J Cell Biol. 1980;84:404–420. doi: 10.1083/jcb.84.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TJ, Field HJ, Roome APC. Intra-axonal location of HSV particles. J Gen Virol. 1972;15:253–255. doi: 10.1099/0022-1317-15-3-253. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Organelle transport along microtubules—the role of KIFs. Trends Cell Biol. 1996;6:135–141. doi: 10.1016/0962-8924(96)10003-9. [DOI] [PubMed] [Google Scholar]
- Holzbaur ELF, Vallee RB. Dyneins: molecular structure and cellular function. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Honess RW, Roizman B. Proteins specified by HSV. XI. Identification and relative molar rates of synthesis of structural and nonstructural herpes virus polypeptides in infected cells. J Virol. 1973;12:1347–1365. doi: 10.1128/jvi.12.6.1347-1365.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AS, Wagner RR. Penetration of HSV into human epidermoid cells. Proc Soc Exp Biol Med. 1964;116:863–869. doi: 10.3181/00379727-116-29392. [DOI] [PubMed] [Google Scholar]
- Karpati GHL, Nalbantoglu J, Durham H. The principles of gene therapy for the nervous system. Trends Neurosci. 1996;2:49–54. doi: 10.1016/0166-2236(96)89620-2. [DOI] [PubMed] [Google Scholar]
- Kreis TE. Microtubules containing detyrosinated tubulin are less dynamic. EMBO (Eur Mol Biol Organ) J. 1987;6:2597–2606. doi: 10.1002/j.1460-2075.1987.tb02550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensson K, Lycke E, Roeyttae M, Svennerholm B, Vahlne A. Neuritic transport of HSV in rat sensory neurons in vitro.Effects of substances interacting with microtubular function and axonal flow [nocodazole, taxol and erythro-9-3-(2-hydroxynonyl)adenine] J Gen Virol. 1986;67:2023–2028. doi: 10.1099/0022-1317-67-9-2023. [DOI] [PubMed] [Google Scholar]
- LeMaster S, Roizman B. HSV phosphoproteins. II. Characterization of the virion protein kinase and of the polypeptides phosphorylated in the virion. J Virol. 1980;35:798–811. doi: 10.1128/jvi.35.3.798-811.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SXH, Ferro KL, Collins CA. Cytoplasmic dynein undergoes intracellular redistribution concomitant with phosphorylation of the heavy chain in response to serum starvation and okadeic acid. J Cell Biol. 1994;127:1009–1019. doi: 10.1083/jcb.127.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps K. Physical properties of the cytoplasm. Curr Opin Cell Biol. 1994;6:3–9. doi: 10.1016/0955-0674(94)90109-0. [DOI] [PubMed] [Google Scholar]
- Luftig RB. Does the cytoskeleton play a significant role in animal virus replication? . J Theor Biol. 1982;99:173–191. doi: 10.1016/0022-5193(82)90397-6. [DOI] [PubMed] [Google Scholar]
- Lycke E, Hamark B, Johansson M, Krotowil A, Lycke J, Svennerholm B. Herpes simplex infection of the human sensory neuron. An electron microscopic study. Arch Virol. 1988;101:87–104. doi: 10.1007/BF01314654. [DOI] [PubMed] [Google Scholar]
- Lycke E, Kristensson K, Svennerholm B, Vahlne A, Ziegler R. Uptake and transport of herpes simplex virus in neurites of rat dorsal root ganglia cells in culture. J Gen Virol. 1984;65:55–64. doi: 10.1099/0022-1317-65-1-55. [DOI] [PubMed] [Google Scholar]
- Marsh M, Helenius A. Virus entry into animal cells. Adv Virus Res. 1989;36:107–151. doi: 10.1016/S0065-3527(08)60583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain DS, Fuller AO. Cell-specific kinetics and efficiency of HSV-1 entry are determined by two distinct phases of attachment. Virology. 1994;198:690–702. doi: 10.1006/viro.1994.1081. [DOI] [PubMed] [Google Scholar]
- McLauchlan J, Addison C, Craigie MC, Rixon FJ. Noninfectious L-particles supply functions which can facilitate infection by HSV-1. Virology. 1992;190:682–688. doi: 10.1016/0042-6822(92)90906-6. [DOI] [PubMed] [Google Scholar]
- McLean G, Rixon F, LAngeland N, Haarr L, Marsden H. Identification and characterization of the virion protein products of HSV-1 UL47. J Gen Virol. 1990;71:2953–2960. doi: 10.1099/0022-1317-71-12-2953. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ. Microtubule dynamics and kinetochore function in mitosis. Annu Rev Cell Biol. 1988;4:527–549. doi: 10.1146/annurev.cb.04.110188.002523. [DOI] [PubMed] [Google Scholar]
- Newcomb WW, Trus BL, Booy FP, Steven AC, Wall JS, Brown JC. Structure of the HSV capsid: molecular composition of the pentons and the triplexes. J Mol Biol. 1992;232:499–511. doi: 10.1006/jmbi.1993.1406. [DOI] [PubMed] [Google Scholar]
- Niclas J, Allen VJ, Vale RD. Cell cycle regulation of dynein association with membranes modulates microtubule-based organelle transport. J Cell Biol. 1996;133:585–593. doi: 10.1083/jcb.133.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfold MET, Armati P, Cunningham AL. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc Natl Acad Sci USA. 1994;91:6529–6533. doi: 10.1073/pnas.91.14.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read GS, Frenkel N. HSV mutants defective in the virion associated shutoff host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J Virol. 1983;46:498–512. doi: 10.1128/jvi.46.2.498-512.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman, B., and D. Furlong. 1974. The replication of herpes viruses. In Comprehensive Virology. H. Fraenkel-Conrat and R.R. Wagner, editors. Plenum Press, New York. 229–403.
- Roizman B, Sears AE. An inquiry into the mechanisms of herpes simplex virus latency. Annu Rev Microbiol. 1989;41:543–571. doi: 10.1146/annurev.mi.41.100187.002551. [DOI] [PubMed] [Google Scholar]
- Roizman, B., and A.E. Sears. 1996. Herpes simplex viruses and their replication. In Virology. Third edition. B.N. Fields, D.M. Knipe, and P.M. Howley, editors. Lippincott-Raven Publishers, Philadelphia. 2231–2295.
- Schroer TA. Structure, function and regulation of cytoplasmic dynein. Curr Opin Cell Biol. 1994;6:69–73. doi: 10.1016/0955-0674(94)90118-x. [DOI] [PubMed] [Google Scholar]
- Shieh MT, WuDunn D, Montgomery RI, Esko JD, Spear PG. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J Cell Biol. 1992;116:1273–1281. doi: 10.1083/jcb.116.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KO. Relationship between the envelope and the infectivity of herpes simplex virus. Proc Soc Exp Biol Med. 1964;115:814–816. doi: 10.3181/00379727-115-29045. [DOI] [PubMed] [Google Scholar]
- Sodeik B, Doms RW, Ericsson M, Hiller G, Machamer CE, van't Hof W, van Meer G, Moss B, Griffiths G. Assembly of vaccinia virus: role of the intermediate region between the endoplasmic reticulum and the Golgi complex. J Cell Biol. 1992;121:521–541. doi: 10.1083/jcb.121.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear, P.G. 1993. Membrane fusion induced by HSV. In Viral Fusion Mechanism. J. Bentz, editor. CRC Press, Boca Raton/Ann Arbor/London/Tokyo. 201–232.
- Spear PG, Roizman B. Proteins specified by HSV. V. Purification and structural proteins of the herpes virion. J Virol. 1972;9:143–159. doi: 10.1128/jvi.9.1.143-159.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven, A.C., and P.G. Spear. 1996. Herpes virus capsid assembly and envelopment. In Structural Biology of Viruses. W. Chiu, R. Burnett, and R. Garcea, editors. Oxford University Press, New York. In press.
- Topp KS, Meade LB, LaVail JH. Microtubule polarity in the peripheral processes of trigeminal ganglion cells: relevance for the retrograde transport of herpes simplex virus. J Neurosci. 1994;14:318–325. doi: 10.1523/JNEUROSCI.14-01-00318.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trus BL, Newcomb WW, Booy FP, Brown JC, Steven AC. Distinct monoclonal antibodies separately label the hexons or the pentons of herpes simplex virus capsid. Proc Natl Acad Sci USA. 1992;89:11508–11512. doi: 10.1073/pnas.89.23.11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisberg EA, Koonce MP, McIntosh JR. Cytoplasmic dynein plays a role in mammalian mitotic spindle formation. J Cell Biol. 1993;123:849–858. doi: 10.1083/jcb.123.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD. Microtubule-based motor proteins. Curr Opin Cell Biol. 1990;2:15–22. doi: 10.1016/s0955-0674(05)80025-0. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Sheetz MP. Targeting of motor proteins. Science (Wash DC) 1996;271:1539–1544. doi: 10.1126/science.271.5255.1539. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Wall JS, Paschal BM, Sheptner HS. Microtubule- associated protein 1C from brain is a two-headed cytosolic dynein. Nature (Lond) 1988;332:561–563. doi: 10.1038/332561a0. [DOI] [PubMed] [Google Scholar]
- Watson DH, Russell WC, Wildy P. Electron microscopic particle counts on Herpes virus using the phosphotungstate negative staining technique. Virology. 1963;19:250–260. doi: 10.1016/0042-6822(63)90062-x. [DOI] [PubMed] [Google Scholar]
- Whitley, R.J., and J.W. Gnann. 1993. The epidemiology and clinical manifestations of herpes simplex virus infections. In The Human Herpes Viruses. B. Roizman, R.J. Whitley, and C. Lopez, editors. Raven Press, Ltd., New York. 69–105.
- Wilson, L., and M.A. Jordan. 1994. Pharmacological probes of microtubule function. In Microtubules. J.S. Hyams and C.W. Lloyd, editors. Wiley-Liss, New York. 59–83.