A Mouse Cytoplasmic Exoribonuclease (mXRN1p) with Preference for G4 Tetraplex Substrates (original) (raw)
Abstract
Exoribonucleases are important enzymes for the turnover of cellular RNA species. We have isolated the first mammalian cDNA from mouse demonstrated to encode a 5′–3′ exoribonuclease. The structural conservation of the predicted protein and complementation data in Saccharomyces cerevisiae suggest a role in cytoplasmic mRNA turnover and pre-rRNA processing similar to that of the major cytoplasmic exoribonuclease Xrn1p in yeast. Therefore, a key component of the mRNA decay system in S. cerevisiae has been conserved in evolution from yeasts to mammals. The purified mouse protein (mXRN1p) exhibited a novel substrate preference for G4 RNA tetraplex–containing substrates demonstrated in binding and hydrolysis experiments. mXRN1p is the first RNA turnover function that has been localized in the cytoplasm of mammalian cells. mXRN1p was distributed in small granules and was highly enriched in discrete, prominent foci. The specificity of mXRN1p suggests that RNAs containing G4 tetraplex structures may occur in vivo and may have a role in RNA turnover.
Most cellular RNA species are synthesized as precursors, which are processed to yield mature RNA molecules. Those regions of the pre-RNA not found in the final molecule have to be degraded to avoid accumulation of unwanted RNA species. The turnover of RNA species, in particular mRNA, is important in determining the levels and regulation of gene expression (for review see Ross 1995, 1996; Caponigro and Parker, 1996). Furthermore, the spatial distribution of certain proteins is achieved by localized control of mRNA stability (St. Johnston, 1995). Whereas specific _cis_- and _trans_-acting factors are required for the regulated decay of specific mRNAs (Brawerman, 1993), a more general pathway for the degradation of unwanted RNA is likely to exist into which different RNA species are funneled. Seminal work (for review see Beelman and Parker, 1995; Caponigro and Parker, 1996) in the yeast Saccharomyces cerevisiae has identified deadenylation-dependent and -independent decay of mRNA, which requires several enzymatic activities including decapping, endoribonuclease, poly(A) nuclease, and 3′–5′ and 5′–3′ exoribonuclease.
Xrn1p (for review see Kearsey and Kipling, 1991; Heyer, 1994) and Rat1p (also known as Tap1p, Hke1p, Exonuclease 2; for review see Stevens, 1993) are 5′–3′ exonucleases from S. cerevisiae. These two enzymes are the only examples of purified 5′–3′ exonucleases in RNA turnover in pro- and eukaryotes (for review see Stevens, 1993) and share substantial sequence homology (see Fig. 2), yet both enzymes have functionally diverged. A primarily nuclear role for Rat1p has been suggested (Amberg et al., 1992; Kenna et al., 1993; Henry et al., 1994), whereas Xrn1p acts and is localized in the cytoplasm (Hsu and Stevens, 1993; Henry et al., 1994; Muhlrad and Parker, 1994; Muhlrad et al., 1994; Heyer et al., 1995).
Figure 2.

Structure and evolutionary conservation of mouse mXRN1p. (A). Structural relation of mXRN1p to other yeast and mammalian proteins. The domains of strongest homology are indicated as black boxes (I–III). An additional domain (IV) with intermittent but highly conserved sequence stretches between mXRN1p, Xrn1p, and ExoIIp is hatched. Numbers above the boxes represent the percentage of amino acid identity to mXRN1p. References to the sequences are found in Materials and Methods. (B) Phylogenetic tree of mXRN1p and related proteins. M.m., M. musculus; S.c., S. cerevisiae; S.p., S. pombe; aa, amino acids.
Xrn1p has been suggested to be the major 5′–3′ exoribonuclease in cytoplasmic mRNA turnover (Stevens, 1978, 1980) and is active in deadenylation-dependent and -independent pathways (Caponigro and Parker, 1996). The analysis of XRN1 mutants suggested a role in RNA turnover of pre-rRNA (Stevens et al., 1991; Henry et al., 1994) and mRNA (for review see Beelman and Parker, 1995; Caponigro and Parker, 1996; Jacobson and Peltz, 1996). In addition to molecular defects in RNA metabolism, the mutants exhibit pleiotropic phenotypes including slow growth, meiotic arrest, and defects in microtubule-related processes (for review see Heyer, 1994). Therefore, it is not surprising that this gene has been isolated in several different screens. Xrn1p (Larimer and Stevens, 1990) is also known as Sep1p (Kolodner et al., 1987; Tishkoff et al., 1991), Stpβp (Dykstra et al., 1990, 1991), Kem1p (Kim et al., 1990), Rar5p (Kipling et al., 1991), and Ski1p (for review see Wickner, 1996). It is unclear whether all mutant phenotypes are the consequence of the RNA metabolism defects.
Here we report the first isolation of mammalian cDNAs demonstrated to encode an exoribonuclease active in RNA turnover. mXRN1p is the structural and functional mouse homolog of the S. cerevisiae Xrn1p exoribonuclease. Therefore, it is likely to be involved in mRNA turnover and rRNA processing in mouse cells. mXRN1p localizes to cytoplasmic granules and is enriched in prominent foci. The purified mouse protein exhibits 5′–3′ exoribonuclease activity and a substrate preference for RNA G4 tetraplex– containing substrates in binding and hydrolysis over a monomeric RNA substrate of the same sequence. This specificity was not previously identified for S. cerevisiae Xrn1p. The mXRN1p exonuclease activity preferred RNA substrates over DNA substrates, either G4 or monomeric. This suggests that RNA G4 tetraplex structures may occur in vivo, possibly with a role in RNA turnover.
Materials and Methods
Media and Genetic Methods
The methods used for growing and constructing S. cerevisiae strains (Sherman et al., 1982) and media for S. cerevisiae (Sherman et al., 1982; Bähler et al., 1994) have been described. To test sensitivity to benomyl, S. cerevisiae cultures were grown in SD-ura medium, and the titer was adjusted to 2 × 107 cells/ml. 3 μl of cells from serial 10-fold dilutions were spotted on plates containing 0 or 15 μg/ml benomyl. Plates were incubated for 2 d at 30°C and photographed.
S. cerevisiae Strains
The S. cerevisiae strain WDHY131 (MATa ura3-52 trp1 leu2Δ1 his3Δ200 pep4::HIS3 prb1-Δ1.6R can1R xrn1Δ::LEU2) has been described (Bashkirov et al., 1995). For the meiosis experiment we used S. cerevisiae strains that are isogenic derivatives of the wild-type isolate S. cerevisiae SK-1 (Kane and Roth, 1974). The parent strain derives from a single spore, and marker systems have been developed in N. Kleckner's laboratory (Harvard University, Cambridge, MA), who kindly supplied basic strains. These strains show efficient and fast sporulation (see Table I). WDHY187 (MATa/MATα ho::LYS2/ho::LYS2 lys2/lys2 ura3/ura3 leu2::hisG/leu2:: hisG his4B/his4X XRN1/xrn1Δ::URA3), WDHY213 (MATa/MATα ho:: LYS2/ho::LYS2 lys2/lys2 ura3/ura3 leu2::hisG/leu2::hisG his4B/his4X can1R/CAN1 xrn1Δ::URA3/xrn1Δ::LEU2), and WDHY344 (MATa/MATα ho::LYS2/ho::LYS2 lys2/lys2 ura3/ura3 leu2::hisG/leu2::hisG his4B/his4X can1R/CAN1 xrn1Δ::ura3/xrn1Δ::LEU2) were constructed for this study. A spontaneous mutation inactivating the URA3 gene disrupting XRN1 in WDHY344 was isolated on medium containing 5-fluoro-orotic acid.
Table I.
Complementation of Sporulation Defect in S. cerevisiae xrn1Δ by mXrn1
| Genotype (strain) + plasmid | 3-4 Spored asci |
|---|---|
| % | |
| Wild type (WDHY187) | 82.0 |
| xrn1Δ (WDHY213) | 7.3 |
| xrn1Δ (WDHY344) + vector | 10.0 |
| xrn1Δ (WDHY344) + p_XRN1_ | 75.3 |
| xrn1Δ (WDHY344) +p_mXrn1_ | 71.5 |
| xrn1Δ (WDHY344) + p_mXrn1 Δ39_ | 64.1 |
Plasmids
The complementation plasmids are based on the S. cerevisiae CEN-ARS plasmid YCp50 (see Bashkirov et al., 1995). p_XRN1_ (pWDH276) was derived from pJI82 (Kim et al., 1990) containing a chromosomal fragment of the XRN1 region by deletion of a vector SalI site and engineering a SalI site just upstream from the ATG of the XRN1 open reading frame for efficient further recloning. p_mXrn1_ (pWDH347) and p_mXrn1Δ39_ (pWDH348) were derived from p_XRN1_ by substituting the XRN1 open reading frame with the full-length mouse cDNAs using the promoter SalI site and the vector HindIII site. pGAL_XRN1_ is pRDK249 (Johnson and Kolodner, 1991) in which the XRN1 open reading frame is expressed under the control of the GAL10 promoter. The mouse full-length cDNAs were cloned in pRDK249, replacing the resident XRN1 open reading frame and resulting in pGAL_mXrn1_ (=pWDH349) and pGAL_mXrn1Δ39_ (=pWDH350). Vector control pGAL (=pWDH181) contains no open reading frame under the control of the GAL10 promoter of pRDK249.
Cloning of Mouse mXrn1
For PCR amplification, two oligonucleotides ending with EcoRI recognition sequences at their 5′ ends (5′-GGAATTCCI(C/A)GIGCIAA(A/G) ATGAA(C/T)CA(A/G)CA-3′ and 5′-GGAATTCATIAG(G/A)TCIGC(G/ A)T CIAGICC(G/A)TA-3′, where I refers to inosine) were synthesized. These sequences correspond to highly conserved regions of S. cerevisiae XRN1 (Kim et al., 1990) and Schizosaccharomyces pombe Exo2 (Szankasi and Smith, 1992, 1996), as well as S. cerevisiae RAT1 (Amberg et al., 1992). The amino acid sequence encoded by the first oligonucleotide (PRAKMNQQ) corresponds to amino acid residues 90–97, and the sequence encoded by the second oligonucleotide (YGLDADLI) corresponds to residues 203–210 of the S. cerevisiae Xrn1 protein. PCR was performed with 5 ng of mouse testis cDNA as a template for 30 cycles of 5 s at 94°C, 15 s at 37°C, and 2 min at 72°C. The PCR products were analyzed on a 4% agarose gel (NuSieve®; FMC BioProducts, Rockland, ME), eluted from the gel, and subcloned in M13mp19 for sequence analysis. A 363-bp PCR product was used as a probe for the first round screening of a mouse testis cDNA bank. Recombinant phages λ511, λ14, and λ1620 were isolated from a mouse testis cDNA library in λgt10 (BALB/c; Clontech, Palo Alto, CA), λ2100 from a mouse testis cDNA library in λgt11 (BALB/C; Clontech), and λ10, λ1, λ6, λ4, λ9, and λ3 from a mouse thymus cDNA library in λZAP (B6/CBAF1J; Stratagene Inc., La Jolla, CA) using the probes indicated in Fig. 1 A. All cDNA clones displayed in Fig. 1 were sequenced on both strands. Full-length cDNA constructs were reconstructed by joining inserts from λ511 and λ10 at their unique HpaI sites for p_mXRN1_ and joining inserts from λ511 and λ1 for p_mXrn1Δ39._ Standard procedures were used for DNA library screening, DNA hybridization, and sequencing. These sequence data are available from EMBL/GenBank/DDBJ under accession number X91617.
Figure 1.

Cloning of mouse mXrn1. Summary of mXrn1 cDNA cloning. The reconstructed 5,497-bp cDNA is shown as a thin line. Positions of the translation start (ATG) and the stop (TAA) codon are indicated. Halfarrows show the position of the PCR primers. The open box represents the PCR-generated probe for the firstround cDNA library screening by DNA hybridization. The cloned cDNA inserts of recombinant phages are presented as thick lines, some of them with hatched boxes indicating the DNA regions used as hybridization probes for cDNA walking. Short poly (T) stretches at the 3′ end of some cDNA derived from the 3′ poly(A) are shown. The 39-bp insert in the cDNA of phages λ10, λ6, λ9, and λ3 at position 4806 is designated by an open triangle.
Purification of Mouse mXRN1p
S. cerevisiae strain WDHY131 (xrn1Δ) bearing plasmid pGAL_mXrn1_ was induced by galactose for overproduction of mouse mXRN1p as described (Johnson and Kolodner, 1991). After harvesting and washing in 20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 1 mM EDTA, and 1 mM PMSF, the cells were frozen in liquid nitrogen and stored at −70°C. 24 g of cells was thawed on ice. All the steps were performed at 4°C. mXRN1p purification was monitored by Western blot with a rabbit polyclonal anti-mXRN1p antibody. The cell slurry was diluted to 1.25 ml of buffer A containing 300 mM NaCl per gram of cells. Buffer A is 20 mM Tris-HCl, pH 7.5, 1 mM EDTA, 10% (wt/vol) glycerol, 10 mM β-mercaptoethanol, 0.2 mM PMSF, 2 mM benzamidine, 2 μM pepstatin A, and 2 μM leupeptin. Cells were disrupted in a bead beater (Biospec Products, Bartlesville, OK) with six 30-s pulses with 2 min of cooling between pulses. The glass beads were washed twice with 10 ml buffer A/300 mM NaCl. Cell lysate and wash were combined and centrifuged in a Ti45 rotor (45 min, 40 krpm at 4°C) resulting in fraction I (15.7 mg/ml, 75 ml). Fraction I was loaded on a phosphocellulose column (P11; Whatman Inc., Clifton, NJ) equilibrated with buffer A/300 mM NaCl at a flow rate of 70 ml/h. The flowthrough was collected as fraction II (100 ml, 10 mg/ml) and centrifuged at 8 krpm for 20 min to collect the precipitate. The supernatant (fraction III, 4 mg/ml) was reapplied on a new P11 column preequilibrated in buffer A/300 mM NaCl. After washing with buffer A/300 mM NaCl, proteins were eluted with 150 ml of buffer A/1 M NaCl. The eluting fractions containing mXRN1p were pooled (35 ml) and concentrated on an ultra filtration unit (Centriprep100; Amicon, Beverly, MA) to result in fraction IV (0.9 mg/ml, 3.5 ml). The conductivity of fraction IV was adjusted to 1 M NaCl and loaded on a gel filtration column (Sephacryl S-200; Pharmacia, Piscataway, NJ; 2 cm2 × 70 cm) at a flow rate of 19 ml/h. mXRN1p-containing fractions were dialyzed against buffer B containing 20 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 1.0 mM DTT, 500 mM NaCl, and 60% (wt/vol) glycerol and stored at −20°C (fraction V; 0.02 mg/ml, 0.32 ml). All experiments were performed using fraction V.
Protein Methods and Antibodies
Glass bead extracts of total S. cerevisiae protein were done as described (Johnson and Kolodner, 1991). Protein extracts and fractionation from adult mouse testis cells was performed as described (Dignam, 1990). Proteins were separated on 8% SDS-PAGE and either stained with Coomassie brilliant blue or analyzed by Western blotting. As first antibodies, we used affinity-purified rabbit anti-mXRN1p antibodies (0.2 μg/ml) or the Ig fraction of rat anti-mXRN1p antibodies (1:5,000 dilution), which recognize mouse mXRN1p but not S. cerevisiae Xrn1p. Signals were detected using HRP-conjugated anti–rabbit or anti–rat IgG antibodies (1: 4,000 or 1:7,500 dilution, respectively) as secondary antibodies in the ECL system (Amersham Corp., Arlington Heights, IL) according to manufacturer's instructions. To visualize the S. cerevisiae Xrn1p on the same blot, the anti–S. cerevisiae Xrn1p mouse mAb H8 (Heyer et al., 1995) (1 μg/ml) was used after detecting mouse mXRN1p. Xrn1p signals were generated using HRP-conjugated rabbit anti–mouse Ig antibodies (1:4,000 dilution; ECL system; Amersham Corp.).
The anti–mouse mXRN1p antibodies were generated against a His(6) fusion to amino acids 1,132–1,719 of mXRN1p, which was overexpressed in Escherichia coli using the pT7-7 system (Tabor and Richardson, 1985). The protein was purified by Ni++ chelate affinity chromatography (Qiagen, Inc., Chatsworth, CA) according to the manufacturer's instructions followed by preparative SDS-PAGE and electroelution of the protein band from the gel slice. 400 and 100 μg of purified antigen were injected into rabbits or rats, respectively, and the immune response was boosted three times. Antibodies were affinity purified from serum on His(6)–mXRN1p(aa 1,132–1,719) containing nitrocellulose strips as described (Pringle et al., 1991).
Biochemical Methods
Exoribonuclease activity was assayed essentially as described (Stevens, 1978, 1980; Käslin and Heyer, 1994). 3.72 pmol (2.6 × 104 cpm/pmol) of [32P]GTP-labeled T7 in vitro transcript of 367 nt mouse β-actin was used as a substrate in reactions containing 100 fmol of mXRN1p.
G4 tetraplex DNA or RNA was prepared from deoxy-oligonucleotide (5′-TATGGGGGAGCTGGGGAAGGTGGGATTT-3′; called GL) or the same sequence of ribo-oligonucleotide (rGL) and 5′ end-labeled with T4 polynucleotide kinase using [γ-32P]ATP essentially as described (Frantz and Gilbert, 1995). For binding assays (20 μl), 0.3 fmol of 5′ 32P-labeled tetraplex RNA (rGL[G4]), tetraplex DNA (GL[G4]), or single-stranded oligo (rGL[SS]) were incubated with 0.05–200 fmol protein at 4°C for 20 min in binding buffer (20 mM Hepes, pH 7.5, 100 mM KCl, 10% glycerol, 1 pmol unlabeled oligonucleotide TP-S 5′-TGGACCAGACCTAGCA3′, and 1 μg poly [dI-dC]: poly[dI-dC]) and run on 6% polyacrylamide gel (Sen and Gilbert, 1988; Liu and Gilbert, 1994). The dried gels were quantified using a PhosphorImager. For nuclease assays, 20 fmol of the GL(G4), rGL(G4), GL(SS), and rGL(SS) substrates and 2.5 fmol of mXRN1p unless otherwise specified were used in binding buffer supplemented with 3 mM MgCl2 without the unlabeled TP-S oligonucleotide. The reaction was carried out at 37°C. The extent of hydrolysis was determined by quantification of the dinucleotide first cleavage product on a PhosphorImager expressed as fmol of cleaved substrate.
mXRN1p was immunoprecipitated from fraction V using magnetic beads coated with sheep anti–mouse IgG1 (Fc) (30 mg/ml; Dynabeads M-450; Dynal Inc., Great Neck, NY) coupled to 20 μg of the anti-Xrn1p mAbs H8 or B4 (Holler et al., 1994) by overnight rocking at 4°C in 500 μl PBS (0.15 M NaCl and 0.01 Na-phosphate, pH 7.4) containing 0.1% BSA. Both anti-Xrn1p mAbs are of the IgG1 subtype; mAb-H8 recognizes both S. cerevisiae Xrn1p and mouse mXRN1p, whereas mAb-B4 is specific for the S. cerevisiae Xrn1 protein. After coupling, the beads were washed three times in PBS containing BSA and once in PBS containing BSA, 0.5 M NaCl, and 5% (wt/vol) glycerol. Each sample of Dynabeads loaded with primary and secondary antibodies, or only with primary antibody (control), was divided on two parts and separately incubated with 40 ng of mXRN1p (fraction V) in 500 μl of PBS containing BSA, 0.5 M NaCl, and 5% (wt/vol) glycerol for 12 h at 4°C. After washing the Dynabeads four times in the same buffer and once in 10 mM Tris-HCl, pH 8.8, the binding between primary and secondary antibodies was broken by treatment with 25 μl of freshly prepared 100 mM triethylamine, pH 11.5, for 10 min at 4°C. The immunoprecipitate was neutralized with 2 μl of 1 M Tris-HCl, pH 7.5. 5 μl of the immunoprecipitate was used for the binding reaction with 32P-labeled G4 DNA as substrate, and another 5 μl was used for immunoblotting for detection of mXrn1p. Under these conditions the exonuclease function of mXRN1p lost its activity (data not shown).
Immunofluorescence Methods
Cells were grown in DME (Life Technologies, Gaithersburg, MD) and 15% FCS on clean glass slides to 80% confluence. Benomyl was added to the culture medium at 40 μg/ml for 5 h, while cold-treated cells were kept at 4°C for 10 h. The efficacy of the drug treatment or the cold treatment was demonstrated by the reduced immunofluorescence using anti-tubulin antibodies (see Fig. 9). Cells were washed in PBS and fixed for 10 min in 4% formaldehyde, 0.02% glutaraldehyde, and 0.2% Triton X-100/PBS. After subsequent washing, cells were simultaneously immunostained (Giese et al., 1995) with rat anti–tubulin antibodies (Serotec Ltd., Oxford, UK; second antibody was an FITC conjugate, Sigma Chemical Co., St. Louis, MO) and rabbit anti–mouse mXRN1p antibodies (second antibody was a Cy3 conjugate, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Immunostaining of the actin cytoskeleton was performed with coumarin–phaloidin (Sigma Chemical Co.). Antibodies against vimentin were kindly provided by G. Giese (MPI Cell Biology, Ladenburg, Germany). The anti-mXRN1p antibodies used for immunofluorescence were the same affinity-purified antibodies used in Western blot analysis (see Fig. 6). Fig. 6 demonstrates their specificity for mXRN1p. Finally, preparations were embedded in antifade solution (Vector Laboratories, Inc., Burlingame, CA) and analyzed using a fluorescence microscope (Axioskop; Carl Zeiss, Inc., Thornwood, NY) equipped with single-band pass filters for excitation of red, green, and blue fluorescence (Chroma Technologies Corp., Brattleboro, VT). Images of high magnification and resolution were obtained using a black and white CCD camera controlled by ISIS fluorescence image analysis software (METASystems, Altlussheim, Germany). The number of foci was counted in 50 cells using a size cutoff of 300 nm; given are mean numbers ± 1 SD. The diameter of foci was determined in at least 27 foci from two independent experiments; given is the mean diameter ± 1 SD. Measurements were performed on digitized and enhanced images using the measurement option of the ISIS fluorescence image analysis software package.
Figure 9.
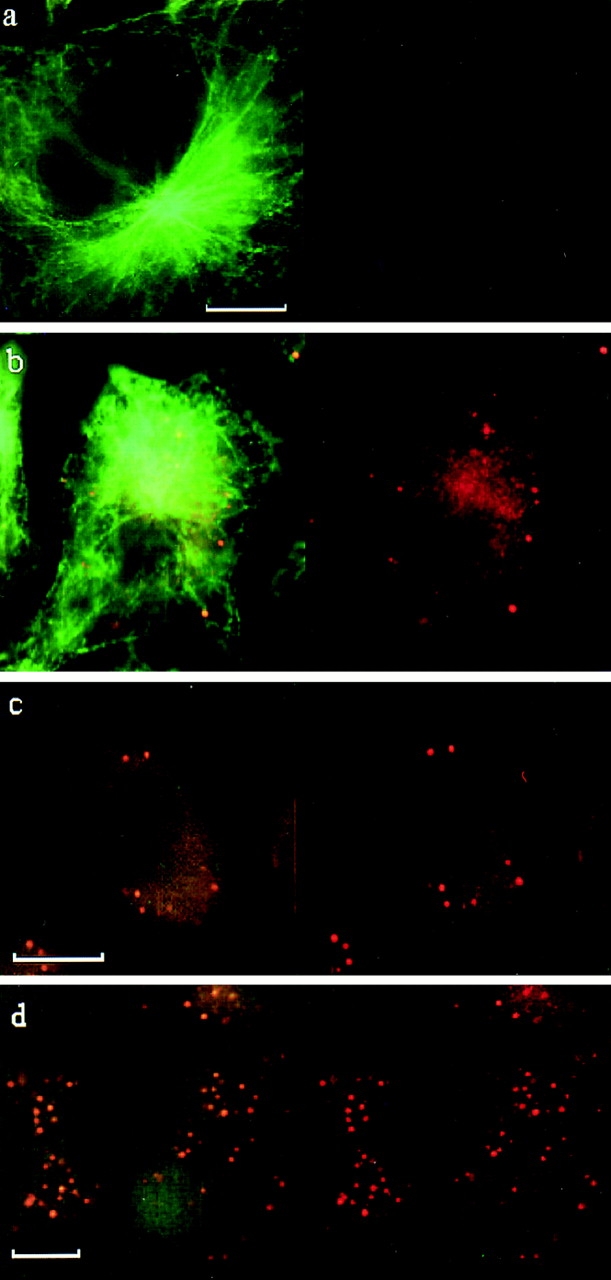
Localization of mouse mXRN1p in cytoplasmic foci. Indirect immunofluorescence of mXRN1p (red, right column) and superimposed to tubulin immunofluorescence (green, left column) in the mouse E10 fibroblast cell line. (a) Control with preimmune serum (right column) and anti-tubulin antibodies (left column). (b–d) Anti–mouse mXRN1p antibodies (right column) and anti-tubulin antibodies (left column). a and b show untreated cells; c shows cells kept at 4°C; d shows cells treated with benomyl. The position of the nucleus is seen as a negative imprint in the immunofluorescence; note the absence of mXRN1p signal in this region. Bars: 10 μm.
Figure 6.
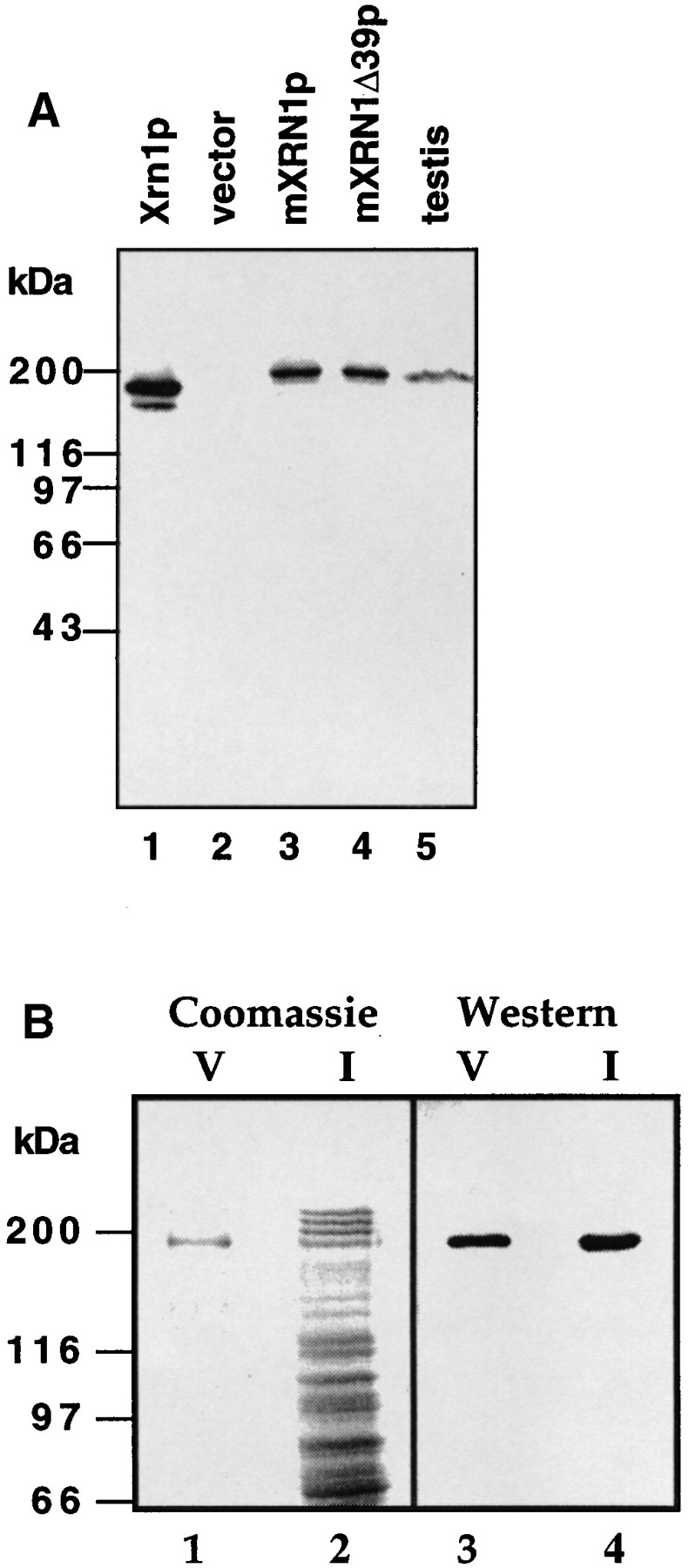
Purification and Western blot analysis of mXRN1p. (A) Analysis of total protein after induction from strain WDHY131 (xrn1Δ) transformed with pGAL_XRN1_ (Xrn1p; lane 1, 0.3 μg), with vector (pGAL; lane 2, 5 μg), with pGAL_mXrn1_ (mXRN1p; lane 3, 5 μg), and with pGal_mXrn1Δ39_ (mXRN1Δ39p; lane 4, 5 μg) as well as of cytoplasmic extract (90 μg) of adult mouse testis cells (lane 5). The size difference between the proteins encoded by the m_Xrn1_ and mXrn1Δ39 cDNAs could not have been resolved on this gel system. (B) Analysis of protein fractions from mouse mXrn1p purification. 42 μg of fraction I (lanes 2 and 4) and 0.2 μg of fraction V (lanes 1 and 3) were analyzed by Coomassie staining (lanes 1 and 2) and by Western blot analysis (lanes 3 and 4). Positions of the molecular mass markers are given on the left.
Computer Analysis
Homologies of mXRN1p to other proteins were detected in a FASTA search. The sequences were aligned, and the phylogenetic tree was generated with the Growtree program using the Kimura protein distance algorithm. The identity between individual homology boxes was scored in GAP alignments (gap weight = 3, length weight = 0.1). The sequences were S. cerevisiae Xrn1p (Kim et al., 1990) and Rat1p (Amberg et al., 1992), S. pombe Dhp1p (Sugano et al., 1994), ExoIIp (Szankasi and Smith, 1996), and mouse Dhm1p (Shobuike et al., 1995). All programs were implemented in the GCG software package (version 8.0; Genetics Computer Group, Inc., Madison, WI) (Devereux et al., 1984).
Results
Mouse cDNAs Encoding an Exoribonuclease
Mouse cDNAs deriving from the mammalian homolog of S. cerevisiae XRN1 (Fig. 1) have been identified by a PCRbased approach. A full-length cDNA of 5,497 nt was assembled, and an RNA consistent with this predicted size was detected in Northern blots of RNA isolated from mouse tissues (data not shown). Two cDNA variants were recovered, mXrn1 and mXrn1Δ39. mXrn1Δ39 carried an in-frame 39-bp deletion leading to a predicted protein product smaller by 13 amino acids (Fig. 1). PCR analysis of genomic DNA and cDNA derived from testis suggests that both cDNAs represent splice variants of a single-copy gene and that the deleted sequence in mXrn1Δ39 corresponds to an optional exon (data not shown). The DNA sequence (accession number X91617, not shown) predicts a 5,157-bp open reading frame for mXrn1 with a coding potential for a 194.2-kD protein composed of 1,719 amino acid residues and, for mXrn1Δ39, a 5,118-bp open reading frame potentially encoding a 192.9-kD protein. The putative initiation codon for both cDNAs is located in a region with highly significant homology to the NH2-terminal region of the homologous genes (Kim et al., 1990; Amberg et al., 1992) (see Fig. 2 A). In addition, the sequence around this ATG initiation codon (AAAATGGGA) fits well to the consensus proposed by Kozak (1991) with a purine and a guanosine at positions −3 and +4, respectively. Given the additional fact that the cDNAs exhibit biological activity (Figs. 3–5 and below), we conclude that the indicated ATG is highly likely to be the start codon of the mXrn1 gene. The 3′ end of both cDNAs is characterized by a relatively short oligo(A) tail of between 8 and 22 residues in six different clones (Fig. 1). Short poly(A) tails have been suggested to be of importance for the regulation of expression of several eukaryotic genes (Baker, 1993).
Figure 3.

Complementation of the slow growth and benomyl hypersensitivity of a S. cerevisiae xrn1Δ mutation by mouse mXrn1. Strain WDHY131 (xrn1Δ) was transformed with either vector (YCp50, row 1), p_XRN1_ (row 2), p_mXrn1_ (row 3), or p_mXrn1Δ39_ (row 4). Serial dilutions of cultures were spotted on medium and incubated as described in Materials and Methods.
Figure 5.

Complementation of a molecular mRNA turnover defect in xrn1 cells by mouse mXrn1. Equal amounts of poly (A)+ (3 μg) and poly(A)− (15 μg) RNA from the four strains described in Fig. 3 were fractionated on a gel and analyzed by hybridization with specific probes as described (Hsu and Stevens, 1993). Given are the relative amounts of rRNA loaded as quantified from scanning of the ethidium bromide–stained gel. The distribution of the specific mRNAs between the poly(A)+ and poly(A)− fractions were quantified by PhosphorImager.
Database searches revealed homologies to the protein sequences of the genes used for the PCR cloning strategy (see Materials and Methods), S. cerevisiae XRN1 and S. pombe exo2, and to a group of related proteins and predicted proteins from S. cerevisiae, S. pombe, and mouse (Fig. 2). No other significant homologies were identified. The phylogenetic relationship between the individual sequences (Fig. 2 B) suggests the existence of two subfamilies of related proteins with demonstrated or suspected exonuclease activity. In both subfamilies, the two yeast sequences show a slightly closer relationship to each other than to the mouse sequence.
Mouse mXRN1p Is Functional in S. cerevisiae
The sequence data suggested that the mouse cDNAs derive from a homolog of the yeast genes, and we sought functional evidence to support this notion. To this end, we constructed plasmids placing the two mouse cDNA variants under the control of the cognate S. cerevisiae XRN1 promoter to yield p_mXrn1_ and p_mXrn1Δ39_ for the long and short variants, respectively. These plasmids together with the equivalent S. cerevisiae construct (p_XRN1_) were used to assay for complementation of defects caused by a deletion of the XRN1 gene in S. cerevisiae (see Figs. 3–5).
xrn1Δ cells exhibit pleiotropic phenotypes caused by defects in RNA turnover (for review see Stevens, 1993; Caponigro and Parker, 1996) and in the cytoskeleton (Kim et al., 1990; Interthal et al., 1995). Both mouse cDNAs complemented the slow-growth phenotype almost as efficiently as the cognate plasmid–borne gene (doubling times: vector control, 209 min; p_XRN1_, 148 min; p_mXrn1_, 166 min; p_mXrn1Δ39_, 170 min; see also Fig. 3 for a semiquantitative test). Moreover, the benomyl hypersensitivity of the S. cerevisiae mutant was also complemented to a large extent, but not as efficiently as by the S. cerevisiae XRN1 gene (Fig. 3).
A striking phenotype of xrn1Δ cells is a quantitative meiotic prophase arrest in pachytene leading to highly reduced sporulation (Table I) (Bähler et al., 1994; Tishkoff et al., 1995). This phenotype is complemented by both mouse cDNAs essentially to the same extent as by the S. cerevisiae gene (Table I).
One RNA turnover defect in xrn1Δ cells is the accumulation of a fragment of the internal transcribed spacer of pre-rRNA, which is usually degraded during cytoplasmic processing of the 20S precursor to the 18S RNA (Stevens et al., 1991) (Fig. 4 A). The accumulation of this fragment can be visualized by Northern blot analysis in total RNA of xrn1Δ cells (Fig. 4 B, lane 1) but not in xrn1Δ cells containing the wild-type gene on a plasmid (lane 2). Both mouse cDNAs complemented this molecular defect of the S. cerevisiae mutant to the same extent as the cognate gene (lanes 3 and 4).
Figure 4.
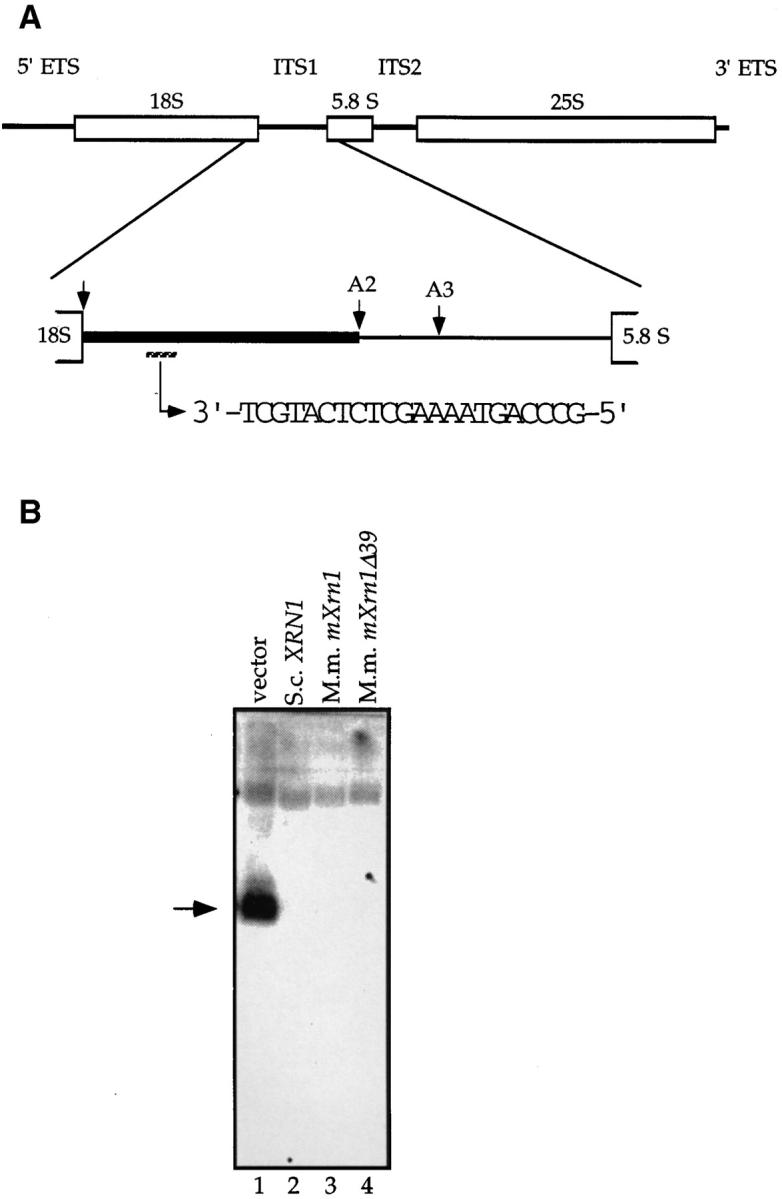
Complementation of the pre-rRNA processing defect of a S. cerevisiae xrn1Δ mutation by mouse mXrn1. (A) Structure of the 35S pre-rRNA in S. cerevisiae with the relevant processing sites of ITS1 (Eichler and Craig, 1994; Venema and Tollervey, 1995). The fragment of ITS1 accumulating in xrn1 cells but usually degraded in wild-type cells is indicated as a bold line in the blow-up of ITS1. Sequence and position of the oligonucleotide used for Northern blot analysis of total RNA is indicated. (B) Complementation of a molecular rRNA turnover defect in xrn1 cells by mXrn1. Northern blot analysis of total RNA from strain WDHY131 (xrn1Δ) transformed with either vector (lane 1), p_XRN1_ (lane 2), p_mXrn1_ (lane 3), or p_mXrn1Δ39_ (lane 4) was done as described (Stevens et al., 1991). The arrow indicates the location of the ∼200-nt band accumulating in the xrn1Δ strain.
mRNA turnover defects in xrn1Δ cells are signaled, for example, by the accumulation of certain mRNAs as poly(A)− species (Hsu and Stevens, 1993). A large _XRN1_dependent effect can be observed for the RP51A mRNA, whereas only a small effect can be documented for ACT1 mRNA (Fig. 5), consistent with previous observations (Hsu and Stevens, 1993). Both defects can be complemented by the cognate wild-type gene or by both mouse cDNAs, essentially to the same extent (Fig. 5).
Purification and Characterization of mXrn1p
The open reading frame discovered in the mouse cDNA predicted a protein of 194 kD, and a polypeptide of this size can be visualized by immunoblot analysis in S. cerevisiae cells overexpressing the cDNAs from the regulated GAL10 promoter (Fig. 6 A, lanes 3 and 4). Moreover, a polypeptide of identical size can be detected in cytoplasmic protein extracts from mouse testis (lane 5). This demonstrates that the mouse gene from which we derived the cDNAs encodes the predicted protein in vivo. No immunoreactive bands of this size were detected in high salt extracts of nuclei (data not shown), consistent with the localization of the protein in the cytoplasm (see below). In addition, this analysis demonstrated the specificity of the anti-mXRN1p antibodies because they recognize only the mXRN1p band in protein extracts from mouse testis (Fig. 6 A) and other tissues as well as from mouse E10 cells (data not shown). To obtain direct biochemical evidence that mouse mXRN1p exhibits exoribonuclease activity, we purified the protein (Fig. 6 B) and demonstrated in vitro exoribonuclease activity (Fig. 7 A). Because both mouse cDNA variants (mXrn1 and mXrn1Δ39) were found to have identical biological activity in all interspecies complementation assays (Figs. 3–5), we decided to purify and characterize only the longer version of mouse mXRN1p.
Figure 7.




Characterization of mXRN1p. (A) Time course of RNA hydrolysis by mXRN1p. Shown are the means ± SD of three independent experiments. (B) Binding to G4 tetraplex RNA and DNA substrates. Tetraplex RNA (rGL[G4], solid circle), tetraplex DNA (GL[G4], open circle), or a monomeric single-stranded RNA oligonucleotide of the same sequence (rGL[SS], solid square) were used in binding assays as described in Materials and Methods. The amount of free nucleic acid was plotted against mXRN1p concentration. Shown are the means ± SD of three independent experiments. For some values the error bars do not exceed the limits of symbol. (C) Hydrolysis of G4 tetraplex substrates. The cleavage activity of mXRN1p for the substrates used in _B_—tetraplex RNA (rGL[G4], solid circle), tetraplex DNA (GL[G4], open circle), monomeric single-stranded RNA oligonucleotide of the same sequence (rGL[SS], solid square), and monomeric single-stranded DNA oligonucleotide of the same sequence (GL[SS], open square)—was determined and is expressed as fmoles of cleaved input substrate. Time courses were from 0 to 30 min. 0 min was defined as the fmoles of cleaved substrate before addition of enzyme because the reaction was too fast to sample after addition of enzyme. Shown are the means ± SD of three independent determinations. For some values the error bars do not exceed the limits of symbol. (D) Protein titration of mXRN1p for hydrolysis of G4 tetraplex substrates. The cleavage activity of mXRN1p for two substrates used in _B_—tetraplex DNA (GL[G4], left) and tetraplex RNA (rGL[G4], right), 15 fmol each—was determined in reactions containing 0–62.5 fmol of mXRN1p for 15 min at 37°C (lanes 1 and 6, 0 fmol; lanes 2 and 7, 0.5 fmol; lanes 3 and 8, 2.5 fmol; lanes 4 and 9, 12.5 fmol; lanes 5 and 10, 62.5 fmol). Shown is the autoradiograph of a gel resolving the substrate (GL[G4] or rGL[G4]), the enzyme–substrate complex (C), and the cleavage product (CP). Monomeric substrate (GL[ss] or rGL[ss]) is also present in minor quantities because of spontaneous decomposition of the substrate.
A nuclear role for Xrn1p has been proposed based on the specificity of the enzyme for G4 tetraplex–containing DNA substrates (Liu and Gilbert, 1994; Liu et al., 1995). mXRN1p exhibited a substrate binding specificity for G4 tetraplex–containing DNA and RNA substrates (Fig. 7 B). In these experiments (Fig. 7, B–D) we used the same sequence oligonucleotides either as RNA (rGL) or as DNA (GL) in their monomeric form (rGL[ss], GL[ss]) or in their G4 tetraplex form (rGL[G4], GL[G4]) after forming the tetraplex in vitro and purifying the tetraplex substrate as described in Materials and Methods. In binding experiments, the equilibrium constants derived from Scatchard analysis of the data shown in Fig. 7 B showed a higher affinity of mXRN1p for the G4 RNA substrate (Keq = 2.95 ± 0.32 × 1010 M−1) than for the G4 DNA substrate (Keq = 1.26 ± 0.24 × 1010 M−1) or the monomeric RNA oligonucleotide of the same sequence (Keq = 2.57 ± 0.34 × 109 M−1). These differences are small but significant, as indicated by the nonoverlapping standard deviations. Kinetic analysis of the exonuclease activity using G4 tetraplex and monomeric RNA as well as DNA substrates revealed more striking differences (Fig. 7 C). mXRN1p hydrolyzed the first cleavage in the RNA substrates (G4 tetraplex and monomeric) with a biphasic kinetics, exhibiting a fast early and a slower late component. Both DNA substrates were hydrolyzed with a more uniform and significantly slower kinetics than the RNA substrates. In particular, a large difference was apparent between the RNA and DNA G4 tetraplex substrates. During the first reaction phase, hydrolysis of the RNA G4 substrate was at least 14 times faster than that of G4 DNA (Fig. 7 C). The kinetics data of the hydrolysis also demonstrate a significant preference of mXRN1p for the G4 tetraplex versus monomeric RNA substrate (Fig. 7 C) consistent with the quantitative binding studies (Fig. 7 B).
A protein titration of mXRN1p using G4 tetraplex DNA and RNA substrates is shown in Fig. 7 D. These data visualize again the preference of mXRN1p for RNA substrates. In addition, the gel shows the position of the first cleavage product (CP in Fig. 7 D), a radiolabeled two-nucleotide product. This was also shown by further high resolution gel electrophoresis using appropriate standards (data not shown). Because the substrate has been labeled at its 5′ end, this analysis also demonstrates that mXRN1p is a 5′– 3′ exonuclease.
Several lines of evidence suggest that the exonuclease activity and the substrate specificity for G4 tetraplex substrates is intrinsic to mXRN1p and not due to a minor contaminant. First, the 5′–3′ exonuclease activity of mXRN1p and its enzymatic characteristics is consistent with the biochemical analysis of the homologous enzymes from S. cerevisiae (Stevens, 1980; Johnson and Kolodner, 1991, 1994) and S. pombe (Szankasi and Smith, 1992; Käslin and Heyer, 1994), which also possess 5′–3′ exonuclease activity. Second, the mouse enzyme has been purified from a S. cerevisiae strain deleted for the endogenous XRN1 gene, which encodes Xrn1p, the major 5′–3′ exonuclease activity in S. cerevisiae cells. Third, the amount of protein added in the nuclease experiments (Fig. 7, A and C; see Materials and Methods) was small, working at excess of substrate. Any contaminant would have to be an unusually active nuclease, presently unknown in S. cerevisiae. In side-by-side experiments using mXRN1p and S. cerevisiae Xrn1p, the mouse enzyme exhibited higher specific activity than the yeast enzyme (Bashkirov, V.I., and W.-D. Heyer, unpublished data). Fourth, the amount of protein added in the binding experiments (Fig. 7 B) was low, showing 50% binding of 0.3 fmol of substrate at only 2.3fold excess of mXRN1p (0.68 fmol as calculated from Fig. 7 B). This quantitatively excludes a minor contaminant being responsible for the G4 tetraplex binding activity. Fifth, an immunoprecipitation experiment (Fig. 8) demonstrated that the G4 tetraplex specificity was intrinsic to mXRN1p by an independent method. Using two anti-Xrn1p mAbs, one cross-reacting with mXRN1p (H8) and the other (B4) not cross-reacting with the mouse protein, we demonstrated that only H8, the antibody recognizing the mouse protein, could immunoprecipitate mXRN1p (Fig. 8 A) and that the immunoprecipitate formed a specific complex with G4 tetraplex DNA (Fig. 8 B). Sixth, the observed coupling of the binding and the cleavage reaction on several G4-containing substrates (Fig. 7, B and C, and data not shown) argues that both activities were mediated by one protein. Finally, the temperature optimum of the mXRN1p nuclease activity was determined to be 37°C, typical for a mammalian but atypical for a yeast enzyme. The S. cerevisiae Xrn1p nuclease activity showed an optimum at 30°C.
Figure 8.

mXRN1p is responsible for G4 tetraplex–specific binding. mXrn1p was immunoprecipitated using anti-Xrn1p mAb H8, which cross-reacts with mXrn1p (lanes 1 and 2), with control antibody B4, which does not recognize mXRN1p (lanes 3 and 4) or with a control lacking secondary antibody (lanes 5 and 6) as described in Materials and Methods. 5 μl of the precipitate was analyzed by immunoblotting (A) using a rat anti–mXRN1p antibody. Positions of the molecular mass markers are given on the right. In B, 5 μl of the precipitate was analyzed for complex formation with the G4 tetraplex DNA substrate (GL[G4]). C denotes the position of the complex and G4 denotes the position of the free substrate.
mXRN1p Has Two Types of Localization in Mammalian Cytoplasm
mXRN1p immunostaining revealed a general granular signal and an enrichment in a number of discrete, prominent foci (>300 nm) in the cytoplasm of mouse E10 cells (Fig. 9) and skin fibroblast cell lines as well as in rat RBL-1 and human HeLa cells (data not shown). We focus here on mouse E10 cells because their large and well-defined cytoplasm allowed more precise definition of the sublocalization. However, essentially the same conclusions are reached from the analysis of the other cell types. The cytoplasmic foci and the general granular staining did not appear with preimmune control antibodies (Fig. 9) or with other antibodies against cytoskeletal components including tubulin (Fig. 9), actin, and vimentin (data not shown). Double and triple immunofluorescence experiments using antibodies against mXRN1p and against cytoskeletal components concomitantly demonstrated at the light microscopic level the general absence of tubulin, actin, or vimentin from the mXRN1p containing foci (data not shown). This is consistent with the immunofluorescence data shown in Fig. 9. In mouse E10 cells, an average of 10.5 ± 4.1 mXRN1p-containing foci were detected per cell, with an average diameter of 570 ± 113 nm. Similar numbers of foci were observed in mouse skin fibroblasts (11.3 ± 8.5) and in HeLa cells (11.1 ± 4.3).
The general granular staining (Fig. 9) consists of smaller foci (<250 nm) resolved at high contrast and resolution only by digital image analysis (data not shown). Addition of benomyl, a drug that inhibits microtubule polymerization and effectively destroys microtubular structures, essentially abolished the cytoplasmic microtubuli network as expected and also the general cytoplasmic localization of mXRN1p but not the localization in foci (Fig. 9 d). Similarly, cold treatment (Fig. 9 c), which is known to destroy microtubular structures, reduced the cytoplasmic microtubular immunofluorescence as well as the general granular cytoplasmic but not the localization in foci of mXRN1p. The similarity between the cytological appearance of mXRN1p staining in Fig. 9, c and d, suggests that the benomyl effect is not a drug-related artifact.
The cytoplasmic localization of mXRN1p in mammalian cells is consistent with the localization of Xrn1p in S. cerevisiae (Heyer et al., 1995), which could not provide the structural details seen here for the small size of the yeast cells. There is no evidence by immunofluorescence or cell fraction studies that Xrn1p in S. cerevisiae occurs in the nucleus (Heyer et al., 1995). Equally, mXRN1p in mouse has only been identified in the cytoplasm of tissue culture cells or in testis tissue preparations by use of immunofluorescence and cell fraction techniques (Fig. 9; Scherthan, H., V.I. Bashkirov, and W.-D. Heyer, unpublished data). Although the data presented here show no positive evidence for nuclear localization of mXRN1p, it will be very difficult to totally exclude that small amounts of mXRN1p are present in the nucleus. A detailed study using confocal laser scanning microscopy will help resolve this issue.
Discussion
mXRN1p Is the Mouse Homolog of the Major S. cerevisiae Cytoplasmic Exoribonuclease Xrn1p
S. cerevisiae Xrn1p is likely to be the major cytoplasmic exoribonuclease for RNA turnover of mRNA, the internal transcribed spacer of pre-rRNA, and possibly other RNA species (for review see Stevens, 1993; Caponigro and Parker, 1996; Jacobson and Peltz, 1996). The conservation of the molecular structure (Fig. 2) and the cellular functions (Figs. 3–5) gives compelling evidence that mXRN1p of mouse is the Xrn1p homolog of higher eukaryotes. This strongly suggests a similar function for mXRN1p in mouse cells. This is consistent with the evolutionary conservation of the general mRNA structure (cap, poly(A) tail) and of the pre-rRNA structure and processing (Eichler and Craig, 1994; Venema and Tollervey, 1995).
To date, no difference in biological activity of the two mXRN1p variants in mouse (mXrn1 and mXrn1Δ39) has been detected, and the biological significance of the two forms remains unclear. They may have variable distribution and relative abundance in different tissues.
It is presently unclear whether mXRN1p is related to a 5′ exoribonuclease partially purified from cytoplasmic mouse sarcoma cell extracts (Coutts and Brawerman, 1993). However, the hydrolysis products of both activities are somewhat different. mXRN1p and its S. cerevisiae homolog, Xrn1p (Liu and Gilbert, 1994), make a dinucleotide as a first cleavage product and mononucleotides thereafter, whereas the mouse sarcoma activity produces mono-, di-, and trinucleotides (Coutts and Brawerman, 1993). mXRN1p is obviously unrelated to the ∼37-kD 3′–5′ exoribonuclease purified from human cells (Caruccio and Ross, 1994).
Cellular Roles of Mouse mXRN1p and S. cerevisiae Xrn1p: Nuclear versus Cytoplasmic
Much attention has been given to possible biological functions of G4 tetraplex structures potentially occurring at telomeric DNA (Sen and Gilbert, 1988; for review see Williamson, 1994). In particular, it was suggested that Xrn1p of S. cerevisiae plays a role in nuclear DNA metabolism as an endonuclease acting on G4 tetraplex substrates (Liu and Gilbert, 1994). However, neither Xrn1p nor mouse mXRN1p are endonucleases, but rather are 5′–3′ exonucleases producing generally mononucleotide products with a first cleavage product of two nucleotides (Fig. 7 d; Stevens, 1980; Liu and Gilbert, 1994; Bashkirov, V.I., and W.-D. Heyer, unpublished observation).
The mutant phenotypes in S. cerevisiae (see Introduction for references) and the in situ localization in yeast (Heyer et al., 1995) and mouse (Fig. 9) suggested a cytoplasmic rather than a nuclear role for the enzymes; therefore, we tested the G4 tetraplex specificity of mouse mXRN1p on RNA and DNA substrates of the same sequence using the corresponding monomeric oligonucleotides as further controls. Previous work on S. cerevisiae Xrn1p (Liu and Gilbert, 1994) was not quantitative and examined G4 DNA substrates but did not analyze RNA substrates. The results of this analysis (Fig. 7, B–D) clearly showed a high specificity of mXRN1p for RNA over DNA substrates and a strong preference for the G4 RNA substrate versus monomeric substrate. Similar substrate preferences were found with the S. cerevisiae Xrn1 protein (Bashkirov, V.I., and W.-D. Heyer, unpublished results).
The pachytene arrest of cells lacking this protein (Bähler et al., 1994; Tishkoff et al., 1995) was interpreted as a result of a role of this protein in nuclear DNA metabolism (Liu and Gilbert, 1994). Other molecular defects manifest in xrn1 cells (see Introduction) may also indirectly lead to this meiotic arrest phenotype. Moreover, it is unclear whether G4 tetraplex DNA structures form at all during meiotic prophase. In conclusion, all available evidence regarding in situ localization, substrate specificity, and mutant phenotype in S. cerevisiae suggests that these proteins are cytoplasmic in both organisms, consistent with a role in cytoplasmic RNA turnover.
Implications of the Mouse mXRN1p Specificity for G4 RNA Tetraplex Substrates
G4 tetraplex structures were first noted in vitro by use of RNA substrates (Zimmermann et al., 1975) requiring only as few as four contiguous G residues (Cheong and Moore, 1992). They are no less likely to occur in vivo than G4 tetraplex DNA structures (Kim et al., 1991). The occurrence of cellular enzymes such as mouse mXRN1p (Fig. 7) and Xrn1p (Bashkirov, V.I., and W.-D. Heyer, unpublished observation) with high specificity for G4 tetraplex RNA substrates suggests that these structures may actually occur in vivo. Biochemical evidence suggested G4 tetraplex RNA formation as a mechanism for the dimerization of the HIV-1 genomic RNA (Sundquist and Heaphy, 1993). A G-rich region has been implicated in the endonucleolytic cleavage of the human insulin-like growth factor II mRNA (Scheper et al., 1995). However, the importance of forming a stem–loop structure with a C-rich strand (Scheper et al., 1995) makes involvement of a G4 tetraplex structure less likely.
If G4 tetraplex RNA occurs in vivo, what could be the functional significance? Given the role of Xrn1p and, by implication, of mXRN1p in RNA turnover, one might speculate on a functional role of this RNA structure in RNA metabolism, specifically RNA turnover. From the enzymological properties of mXRN1p, it is evident that it will bind to G4 tetraplex–containing RNAs with preference because the Keq shows at least a 10-fold difference, and that it will hydrolyze G4 RNA with an initial rate 15 times faster than monomeric RNA. However, the progression of the exonuclease activity is clearly slowed by the G4 tetraplex structure as shown by using 3′-end labeled substrates (Bashkirov, V.I., and W.-D. Heyer, unpublished observation). In vivo, G-rich sequences have been found to stabilize RNA sequences 3′ but not 5′ of the G stretch (Vreken and Raué, 1992; Decker and Parker, 1993; Muhlrad et al., 1995), consistent with Xrn1p being the relevant 5′–3′ exoribonuclease in RNA turnover. However, G4 tetraplex formation might not be required for this effect because the Xrn1p already pauses at G-rich sites on monomeric (i.e., non-G4) substrates (Johnson and Kolodner, 1994). Addition of a G stretch upstream of the AUG initiation codon greatly destabilized the PGK mRNA, reducing the half-life from 35 to 7 min (Muhlrad et al., 1995). This effect has not been found for the MFA2 mRNA or with an insertion of a G-rich sequence downstream of the stop codon in the PGK mRNA (Decker and Parker, 1993; Muhlrad et al., 1994, 1995). This differential effect could be explained by the enzymatic properties of Xrn1p/ mXRN1p by suggesting that, in the former case (general destabilization by G stretch insertion), G tetraplex formation occurs. This would attract the nuclease activity to hydrolyze the sequence 5′ to the G4 tetraplex. In the latter case (no general destabilization), no G4 tetraplex formation occurs, leaving the overall half-life of the full-length mRNA unchanged but stabilizing, as in the former case, the sequence 3′ of the G insertion.
It has been proposed that decapping of mRNA is the major control point for mRNA decay (Caponigro and Parker, 1996). Apparently the only gene encoding such an activity has been identified in S. cerevisiae, and strains lacking this activity exhibit a growth impairment (Beelman et al., 1996), as do S. cerevisiae xrn1Δ cells. Besides a control point by a decapping enzyme, a second control point exerted by the S. cerevisiae Xrn1p or correspondingly by the mouse mXRN1p exoribonuclease is suggested here. Although Xrn1p has been shown to be more active on decapped RNA, the residual activity on capped RNAs (Stevens, 1978) paired with the G4 specificity provides substance for such a control point. RNA G4 tetraplex formation is a slow process compared with most other nucleic acid annealing processes (Sundquist and Heaphy, 1993). Therefore, occurrence of G4 tetraplex structures is correlated to the lifetime of an RNA. A possible model for the role of G4 tetraplex RNA structures is that they attract the degradation of the RNA by Xrn1p/mXRN1p to ensure turnover of an old RNA. mXRN1p itself does not contribute to G4 tetraplex formation because it does not catalyze the formation of this structure (Bashkirov, V.I., and W.-D. Heyer, unpublished observation), unlike, for example, the nuclear protein Rap1p of S. cerevisiae, which is involved in telomere metabolism (Giraldo and Rhodes, 1994). Alternatively, G4 RNA structures, which may be formed quickly from short G-rich sequences (Kim et al., 1991), may be used in the cell to squelch the activity of Xrn1p/ mXRN1p to achieve overall regulation of the exoribonuclease activity itself.
Implications of the mXRN1p Localization
A major distinction between eukaryotic and prokaryotic organisms is the subcellular compartmentalization of the eukaryotic cell in membrane-bound compartments including nucleus, cytoplasm, mitochondria, ER, and Golgi apparatus. In recent years this general organizational picture of the eukaryotic cell was further refined when it was realized that molecular processes like RNA splicing (Fu and Maniatis, 1990; for review see Spector, 1993; Lamond and Carmo-Fonseca, 1993) or DNA replication (Mills et al., 1989; for review see Spector, 1993) are confined to subcompartments within the nucleus. Spatial isolation of lytic activities is of advantage for the cell as demonstrated by the existence of the lysosome compartment. Concentration and confinement of RNA hydrolytic activity in the cytosol is similarly advantageous to minimize a possible interference of the exonuclease with normal cellular processes. The mXRN1p-containing foci are possible sites of RNA turnover in the cytoplasm. Alternatively, the mXRN1p-containing foci may represent storage sites for mXRN1p. However, this is unlikely because the cDNAs do not encode an inactive precursor enzyme but rather an active exoribonuclease (Fig. 7). It can be expected that not only RNA turnover but also other molecular processes are confined to specific sites in the eukaryotic cytosol. Because mXRN1p is the first RNA turnover protein to be localized in mammalian cells, colocalization studies with other RNA turnover functions that would support the subcompartmentalization model are unfortunately not possible.
The majority of translatable mRNA in fibroblasts is associated with the cytoskeleton (Taneja et al., 1992). In developing Drosophila embryos and in nerve cells, localized translation of specific mRNAs has been linked to cytoplasmic transport of mRNA along microtubules (for review see St. Johnston, 1995), implying the presence of microtubule-associated proteins that link mRNA to transport along microtubules. The codiscovery of the S. cerevisiae XRN1 gene as KEM1 (Kim et al., 1990) has suggested that, in addition to being an exoribonuclease, the protein is a microtubule-associated protein (Interthal et al., 1995). The majority of mouse mXRN1p is localized in foci, which do not contain tubulin. However, the mXRN1p molecules localized more generally in the cytoplasm show general colocalization with the cytoplasmic tubulin network (Fig. 9 b). Moreover, this general sublocalization of mXRN1p but not the foci was abolished when the cells were incubated with a microtubular inhibitor (Fig. 9 d). Cold treatment mimicked this effect (Fig. 9 c), arguing against a drug artifact. This suggests that this subpopulation of mXRN1p molecules is associated with microtubules, possibly relating RNA turnover to the cytoskeleton.
Acknowledgments
We thank Drs. W. Filipowicz, J. Kohli, and Y. Nagamine for critically reading the manuscript, O. Bezzubova for kind help in the cDNA cloning, and Dr. R. Jessberger for providing the thymus library. Drs. P. Szankasi and G.R. Smith kindly communicated data before their publication. Drs. A. Johnson and R. Kolodner kindly supplied their overexpression vector. Drs. G. Giese and P. Traub kindly provided antivimentin antibody and mouse skin fibroblasts. Dr. H. Neitzel kindly provided the E10 cell line.
This work was supported by a career development award (Swiss Talents in Academic Research and Teaching) and a research grant of the Swiss National Science Foundation to W.-D. Heyer, and East-European collaborative grants of the Swiss National Science Foundation and an International Research Scholar's award from the Howard Hughes Medical Institute to V.I. Bashkirov and W.-D. Heyer. H. Scherthan was supported in part by the Deutsche Forschungsgemeinschaft. The Basel Institute of Immunology was founded and is supported by F. Hoffmann-La Roche & Co. Ltd.
Footnotes
Please address all correspondence to Wolf-Dietrich Heyer, Institute of General Microbiology, Baltzer-Str. 4, CH-3012 Bern, Switzerland. Tel.: 41 31 631 46 56. Fax: 41 31 631 46 84. e-mail: heyer@imb.unibe.ch
References
- Amberg DC, Goldstein AL, Cole CN. Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiaerequired for the efficient nucleocytoplasmic trafficking of mRNA. Genes & Dev. 1992;6:1173–1189. doi: 10.1101/gad.6.7.1173. [DOI] [PubMed] [Google Scholar]
- Bähler J, Hagens G, Holzinger G, Scherthan H, Heyer W-D. Saccharomyces cerevisiae cells lacking the homologous pairing protein p175SEP1arrest at pachytene during meiotic prophase. Chromosoma. 1994;103:129–141. doi: 10.1007/BF00352322. [DOI] [PubMed] [Google Scholar]
- Baker, E.J. 1993. Control of poly(A) length. In Control of Messenger RNA Stability. J.G. Belasco and G. Brawerman, editors. Academic Press, San Diego, CA. 367–415.
- Bashkirov VI, Solinger JA, Heyer W-D. Identification of functional domains in the Sep1 protein (=Kem1, Xrn1), which is required for transition through meiotic prophase in Saccharomyces cerevisiae. . Chromosoma. 1995;104:215–222. doi: 10.1007/BF00352186. [DOI] [PubMed] [Google Scholar]
- Beelman CA, Parker R. Degradation of mRNA in eukaryotes. Cell. 1995;81:179–183. doi: 10.1016/0092-8674(95)90326-7. [DOI] [PubMed] [Google Scholar]
- Beelman CA, Stevens A, Caponigro G, LaGrandeur TE, Hatfield L, Fortner DM, Parker R. An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature (Lond) 1996;382:642–646. doi: 10.1038/382642a0. [DOI] [PubMed] [Google Scholar]
- Brawerman, G. 1993. mRNA degradation in eukaryotic cells: an overview. In Control of Messenger RNA Stability. J.G. Belasco and G. Brawerman, editors. Academic Press, San Diego, CA. 149–159.
- Caponigro G, Parker R. Mechanisms and control of mRNA turnover in Saccharomyces cerevisiae. . Microbiol Rev. 1996;60:233–249. doi: 10.1128/mr.60.1.233-249.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruccio N, Ross J. Purification of a human polyribosome-associated 3′ to 5′ exoribonuclease. J Biol Chem. 1994;269:31814–31821. [PubMed] [Google Scholar]
- Cheong C, Moore PB. Solution structure of an unusually stable RNA tetraplex containing G- and U-quartet structures. Biochemistry. 1992;31:8406–8414. doi: 10.1021/bi00151a003. [DOI] [PubMed] [Google Scholar]
- Coutts M, Brawerman G. A 5′ exoribonuclease from cytoplasmic extracts of mouse sarcoma 180 ascites cells. Biochim Biophys Acta. 1993;1173:57–62. doi: 10.1016/0167-4781(93)90242-6. [DOI] [PubMed] [Google Scholar]
- Decker CJ, Parker R. A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes & Dev. 1993;7:1632–1643. doi: 10.1101/gad.7.8.1632. [DOI] [PubMed] [Google Scholar]
- Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD. Preparation of extracts from higher eukaryotes. Methods Enzymol. 1990;182:194–203. doi: 10.1016/0076-6879(90)82017-v. [DOI] [PubMed] [Google Scholar]
- Dykstra CC, Hamatake RK, Sugino A. DNA strand transfer protein β from yeast mitotic cells differs from strand transfer protein a from meiotic cells. J Biol Chem. 1990;265:10968–10973. [PubMed] [Google Scholar]
- Dykstra CC, Kitada K, Clark AB, Hamatake RK, Sugino A. Cloning and characterization of DST2, the gene for DNA strand transfer protein β from Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:2583–2592. doi: 10.1128/mcb.11.5.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler DC, Craig N. Processing of eukaryotic ribosomal RNA. Prog Nucleic Acid Res Mol Biol. 1994;47:197–239. doi: 10.1016/s0079-6603(08)60051-3. [DOI] [PubMed] [Google Scholar]
- Frantz JD, Gilbert W. A yeast gene product, G4p2, with a specific affinity for quadruplex nucleic acids. J Biol Chem. 1995;270:9413–9419. doi: 10.1074/jbc.270.16.9413. [DOI] [PubMed] [Google Scholar]
- Fu X-D, Maniatis T. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature (Lond) 1990;343:437–441. doi: 10.1038/343437a0. [DOI] [PubMed] [Google Scholar]
- Giese G, Wiegers W, Kubbes M, Traub P. Okadaic acid co-induces vimentin expression and cell cycle arrest in MPC-11 mouse plasmacytoma cells. J Cell Physiol. 1995;163:145–154. doi: 10.1002/jcp.1041630117. [DOI] [PubMed] [Google Scholar]
- Giraldo R, Rhodes D. The yeast telomere-binding protein RAP1 binds to and promotes the formation of DNA quadruplexes in telomeric DNA. EMBO (Eur Mol Biol Organ) J. 1994;13:2411–2420. doi: 10.1002/j.1460-2075.1994.tb06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer W-D. The search for the right partner: homologous pairing and DNA strand exchange proteins in eukaryotes. Experientia. 1994;50:223–233. doi: 10.1007/BF01924005. [DOI] [PubMed] [Google Scholar]
- Heyer W-D, Johnson AW, Reinhart U, Kolodner RD. Regulation and intracellular localization of Saccharomyces cerevisiaestrand exchange protein 1 (Sep1/Xrn1/Kem1), a multifunctional exonuclease. Mol Cell Biol. 1995;15:2728–2736. doi: 10.1128/mcb.15.5.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Y, Wood H, Morrissey JP, Petfalski E, Kearsey S, Tollervey D. The 5′ end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO (Eur Mol Biol Organ) J. 1994;13:2452–2463. doi: 10.1002/j.1460-2075.1994.tb06530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler A, Bashkirov VI, Solinger JA, Reinhart U, Heyer W-D. Use of monoclonal antibodies in the functional characterization of the Saccharomyces cerevisiaeSep1 protein. Eur J Biochem. 1995;231:329–336. doi: 10.1111/j.1432-1033.1995.tb20704.x. [DOI] [PubMed] [Google Scholar]
- Hsu CL, Stevens A. Yeast cells lacking 5′–3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structures. Mol Cell Biol. 1993;13:4826–4835. doi: 10.1128/mcb.13.8.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interthal H, Bellocq C, Bähler J, Bashkirov VI, Edelstein S, Heyer W-D. A role of Sep1 (=Kem1, Xrn1) as a microtubule-associated protein in Saccharomyces cerevisiae. . EMBO (Eur Mol Biol Organ) J. 1995;14:1057–1066. doi: 10.1002/j.1460-2075.1995.tb07088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A, Peltz SW. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu Rev Biochem. 1996;65:693–739. doi: 10.1146/annurev.bi.65.070196.003401. [DOI] [PubMed] [Google Scholar]
- Johnson AW, Kolodner RD. Strand exchange protein 1 from Saccharomyces cerevisiae.A novel multifunctional protein that contains DNA strand exchange and exonuclease activities. J Biol Chem. 1991;266:14046–14054. [PubMed] [Google Scholar]
- Johnson AW, Kolodner RD. The activity of the Saccharomyces cerevisiaestrand exchange protein 1 intrinsic exonuclease during joint molecule formation. J Biol Chem. 1994;269:3664–3672. [PubMed] [Google Scholar]
- Kane SM, Roth R. Carbohydrate metabolism during ascospore development in yeast. J Bacteriol. 1974;118:8–14. doi: 10.1128/jb.118.1.8-14.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käslin E, Heyer W-D. A multifunctional exonuclease from vegetative Schizosaccharomyces pombecells exhibiting in vitro strand exchange activity. J Biol Chem. 1994;269:14094–14102. [PubMed] [Google Scholar]
- Kearsey S, Kipling D. Recombination and RNA processing: a common strand? . Trends Cell Biol. 1991;1:110–112. doi: 10.1016/0962-8924(91)90101-e. [DOI] [PubMed] [Google Scholar]
- Kenna M, Stevens A, McCammon M, Douglas MG. An essential yeast gene with homology to the exonuclease-encoding XRN1/KEM1gene also encodes a protein with exoribonuclease activity. Mol Cell Biol. 1993;13:341–350. doi: 10.1128/mcb.13.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ljungdahl PO, Fink GR. Kem mutations affect nuclear fusion in Saccharomyces cerevisiae. . Genetics. 1990;126:799–812. doi: 10.1093/genetics/126.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Cheong C, Moore PB. Tetramerization of an RNA oligonucleotide containing a GGGG sequence. Nature (Lond) 1991;351:331–332. doi: 10.1038/351331a0. [DOI] [PubMed] [Google Scholar]
- Kipling D, Tambini C, Kearsey SE. rar mutations which increase artificial chromosome stability in Saccharomyces cerevisiaeidentify transcription and recombination proteins. Nucleic Acids Res. 1991;19:1385–1391. doi: 10.1093/nar/19.7.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodner R, Evans DH, Morrison PT. Purification and characterization of an activity from Saccharomyces cerevisiaethat catalyzes homologous pairing and strand exchange. Proc Natl Acad Sci USA. 1987;84:5560–5564. doi: 10.1073/pnas.84.16.5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Structural features in eukaryotic mRNAs that modulate initiation of translation. J Biol Chem. 1991;266:19867–19870. [PubMed] [Google Scholar]
- Lamond AI, Carmo-Fonseca M. Localisation of splicing snRNPs in mammalian cells. Mol Biol Rep. 1993;18:127–133. doi: 10.1007/BF00986767. [DOI] [PubMed] [Google Scholar]
- Larimer FW, Stevens A. Disruption of the gene XRN1, coding for a 5′–3′ exoribonuclease, restricts yeast cell growth. Gene. 1990;95:85–90. doi: 10.1016/0378-1119(90)90417-p. [DOI] [PubMed] [Google Scholar]
- Liu Z, Gilbert W. The yeast KEM1gene encodes a nuclease specific for G4 tetraplex DNA: implications of in vivo functions for this novel DNA structure. Cell. 1994;77:1083–1092. doi: 10.1016/0092-8674(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Liu Z, Lee A, Gilbert W. Gene disruption of a G4-DNA-dependent nuclease in yeast leads to cellular senescence and telomere shortening. Proc Natl Acad Sci USA. 1995;92:6002–6006. doi: 10.1073/pnas.92.13.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills AD, Blow JJ, White JG, Amos WB, Wilcock D, Laskey RA. Replication occurs at discrete foci spaced throughout nuclei replicating in vitro. J Cell Sci. 1989;94:471–477. doi: 10.1242/jcs.94.3.471. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Parker R. Premature translational termination triggers mRNA decapping. Nature (Lond) 1994;370:578–581. doi: 10.1038/370578a0. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Decker CJ, Parker R. Deadenylation of the unstable mRNA encoded by the yeast MFA2gene leads to decapping followed by 5′– 3′ digestion of the transcript. Genes & Dev. 1994;8:855–866. doi: 10.1101/gad.8.7.855. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Decker CJ, Parker R. Turnover mechanisms of the unstable yeast PGK1mRNA. Mol Cell Biol. 1995;15:2145–2156. doi: 10.1128/mcb.15.4.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR, Adams AEM, Drubin DG, Haarer BK. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–602. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Ross J. mRNA stability in mammalian cells. Microbiol Rev. 1995;59:423–450. doi: 10.1128/mr.59.3.423-450.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J. Control of messenger RNA stability in higher eukaryotes. Trends Genet. 1996;12:171–175. doi: 10.1016/0168-9525(96)10016-0. [DOI] [PubMed] [Google Scholar]
- Scheper W, Meinsma D, Holthuizen PE, Sussenbach JS. Longrange RNA interaction of two sequence elements required for endonucleolytic cleavage of human insulin-like growth factor II mRNAs. Mol Cell Biol. 1995;15:235–245. doi: 10.1128/mcb.15.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature (Lond) 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and J.B. Hicks. 1982. Methods in Yeast Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 120 pp.
- Shobuike T, Sugano S, Yamashita T, Ikeda H. Characterization of cDNA encoding mouse homolog of fission yeast dhp1+gene: structural and functional conservation. Nucleic Acids Res. 1995;23:357–361. doi: 10.1093/nar/23.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL. Macromolecular domains within the cell nucleus. Annu Rev Cell Biol. 1993;9:265–315. doi: 10.1146/annurev.cb.09.110193.001405. [DOI] [PubMed] [Google Scholar]
- Stevens A. An exoribonuclease from Saccharomyces cerevisiae: effect of modifications of 5′ end groups in the hydrolysis of substrates to 5′-nucleotides. Biochem Biophys Res Commun. 1978;81:656–661. doi: 10.1016/0006-291x(78)91586-3. [DOI] [PubMed] [Google Scholar]
- Stevens A. Purification and characterization of a Saccharomyces cerevisiaeexoribonuclease which yields 5′-mononucleotides by a 5′–3′ mode of hydrolysis. J Biol Chem. 1980;255:3080–3085. [PubMed] [Google Scholar]
- Stevens, A. 1993. Eukaryotic nucleases and mRNA turnover. In Control of Messenger RNA Stability. J.G. Belasco and G. Brawerman, editors. Academic Press, San Diego, CA. 449–471.
- Stevens A, Hsu CL, Isham KR, Larimer FW. Fragments of the internal transcribed spacer I of pre-rRNA accumulate in Saccharomyces cerevisiaelacking 5′–3′ exoribonuclease 1. J Bacteriol. 1991;173:7024–7028. doi: 10.1128/jb.173.21.7024-7028.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Johnston D. The intracellular localization of messenger RNAs. Cell. 1995;81:161–170. doi: 10.1016/0092-8674(95)90324-0. [DOI] [PubMed] [Google Scholar]
- Sugano S, Shobuike T, Takeda T, Sugino A, Ikeda H. Molecular analysis of the dhp1 + gene of Schizosaccharomyces pombe: an essential gene that has homology to the DST2 and RAT1 genes of Saccharomyces cerevisiae. . Mol Gen Genet. 1994;243:1–8. doi: 10.1007/BF00283869. [DOI] [PubMed] [Google Scholar]
- Sundquist WI, Heaphy S. Evidence for interstrand quadruplex formation in the dimerization of human immunodeficiency virus 1 genomic RNA. Proc Natl Acad Sci USA. 1993;90:3393–3397. doi: 10.1073/pnas.90.8.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szankasi P, Smith GR. A single-stranded DNA exonuclease from Schizosaccharomyces pombe. . Biochemistry. 1992;31:6769–6773. doi: 10.1021/bi00144a017. [DOI] [PubMed] [Google Scholar]
- Szankasi P, Smith GR. Requirement of S. pombe exonuclease II, a homologue of S. cerevisiaeSep1, for normal mitotic growth and viability. Curr Genet. 1996;30:284–293. doi: 10.1007/s002940050134. [DOI] [PubMed] [Google Scholar]
- Tabor S, Richardson CC. A bacteriophage T7 RNA polymerase/ promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci USA. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja KL, Lifshitz LM, Fay FS, Singer RS. Poly(A) RNA codistribution with microfilaments: evaluation by in situ hybridization and quantitative digital imaging microscopy. J Cell Biol. 1992;119:1245–1260. doi: 10.1083/jcb.119.5.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff D, Johnson AW, Kolodner RD. Molecular and genetic analysis of the gene encoding the Saccharomyces cerevisiaestrand exchange protein SEP1. Mol Cell Biol. 1991;11:2593–2608. doi: 10.1128/mcb.11.5.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff D, Rockmill B, Roeder GS, Kolodner RD. The sep1 mutant of Saccharomyces cerevisiaearrests in pachytene and is deficient in meiotic recombination. Genetics. 1995;139:495–509. doi: 10.1093/genetics/139.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema J, Tollervey D. Processing of pre-ribosomal RNA in Saccharomyces cerevisiae. . Yeast. 1995;11:1629–1650. doi: 10.1002/yea.320111607. [DOI] [PubMed] [Google Scholar]
- Vreken P, Raué HA. The rate-limiting step in yeast PGK1mRNA degradation is an endonucleolytic cleavage in the 3′-terminal part of the coding region. Mol Cell Biol. 1992;12:2986–2996. doi: 10.1128/mcb.12.7.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB. Double-stranded RNA viruses of Saccharomyces cerevisiae. . Microbiol Rev. 1996;60:250–265. doi: 10.1128/mr.60.1.250-265.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson JR. G-quartet structures in telomeric DNA. Annu Rev Biophys Biomol Struct. 1994;23:703–730. doi: 10.1146/annurev.bb.23.060194.003415. [DOI] [PubMed] [Google Scholar]
- Zimmermann SB, Cohen GH, Davies DR. X-ray diffraction and model-building study of polyguanylic and polyinosinic acid. J Mol Biol. 1975;92:181–192. doi: 10.1016/0022-2836(75)90222-3. [DOI] [PubMed] [Google Scholar]