Visualization of Phosphoinositides That Bind Pleckstrin Homology Domains: Calcium- and Agonist-induced Dynamic Changes and Relationship to Myo-[3H]inositol-labeled Phosphoinositide Pools (original) (raw)
Abstract
Phosphatidylinositol 4,5-bisphosphate (PtdIns[4,5]P2) pools that bind pleckstrin homology (PH) domains were visualized by cellular expression of a phospholipase C (PLC)δ PH domain–green fluorescent protein fusion construct and analysis of confocal images in living cells. Plasma membrane localization of the fluorescent probe required the presence of three basic residues within the PLCδ PH domain known to form critical contacts with PtdIns(4,5)P2. Activation of endogenous PLCs by ionophores or by receptor stimulation produced rapid redistribution of the fluorescent signal from the membrane to cytosol, which was reversed after Ca2+ chelation. In both ionomycin- and agonist-stimulated cells, fluorescent probe distribution closely correlated with changes in absolute mass of PtdIns(4,5)P2. Inhibition of PtdIns(4,5)P2 synthesis by quercetin or phenylarsine oxide prevented the relocalization of the fluorescent probe to the membranes after Ca2+ chelation in ionomycin-treated cells or during agonist stimulation. In contrast, the synthesis of the PtdIns(4,5)P2 imaged by the PH domain was not sensitive to concentrations of wortmannin that had been found inhibitory of the synthesis of myo-[3H]inositol– labeled PtdIns(4,5)P2. Identification and dynamic imaging of phosphoinositides that interact with PH domains will further our understanding of the regulation of such proteins by inositol phospholipids.
Keywords: phosphoinositides, calcium, green fluorescent protein, pleckstrin-homology domain, phospholipase C
Pleckstrin homology (PH)1 domains are ∼120 amino acid–long protein modules that were first described in pleckstrin, the major protein kinase C substrate in platelets (16). PH domains have since been identified in several key regulatory proteins with characteristic structural features that include two orthogonal β sheets that form a sandwich with an α helix at the COOH terminus, and variable loops that create a highly charged surface (11, 16). It has been generally accepted that PH domains provide a structural basis for the interaction of certain regulatory proteins with membranes (23). The search for proteins that would bind and regulate proteins via PH domains has generally been unsuccessful, although in some cases, such as the β-adrenergic receptor kinase, the region that confers regulation by G protein βγ subunits overlaps with the PH domain (28). On the other hand, evidence is accumulating to suggest that PH domains of several proteins interact with membrane phosphoinositides, and that this interaction is critical to the regulation of those proteins (11, 16, 23).
The PH domains of several proteins have been shown to bind phosphoinositides in a specific manner with micromolar affinities in vitro (29, 31). For example, the PH domain of phospholipase C (PLC)δ1 binds inositol 1,4,5-trisphosphate (Ins[1,4,5]P3) and associates with lipid vesicles containing phosphatidylinositol 4,5-bisphosphate (PtdIns- [4,5]P2), but only weakly binds other inositol phosphates and PtdIns(4)P (24). Similarly, the PH domain of the Akt protein kinase appears to bind PtdIns(3,4)P2 but not PtdIns(4,5)P2 (13). However, some PH domains (such as that of dynamin) show much weaker and less specific interactions with inositol phospholipids (31), raising the question of whether these binding forces alone are sufficient to anchor those proteins to membranes. Whereas in vitro binding studies have been valuable in determining the ability of PH domains to associate with inositol phospholipids, the dependency of the specificity of their binding on lipid micelle composition and detergent environment (21) emphasizes the need to assess these interactions in intact cells. Another question that is raised by studies on PH domain–phosphoinositide interactions is how the binding of PH domains to the inositide headgroup affects the primary signaling functions of PtdIns(4,5)P2, particularly its availability to PLC enzymes or to PtdIns 3 kinases. It is known that other proteins that bind PtdIns(4,5)P2, such as profilin, greatly impair the ability of PLC enzymes to hydrolyze this lipid (14).
The present studies were designed to examine whether specific association of the PLCδ PH domain (PHPLCδ)with PtdIns(4,5)P2 can be exploited to visualize the cellular distribution of phosphoinositides, that are capable of binding PH domains in intact cells, and to follow their changes in real time after stimulation. Our results, obtained using a fusion protein of the PHPLCδ coupled to the green fluorescent protein (GFP), provide imaging of the dynamics of membrane PtdIns(4,5)P2 pools that interact with PH domains. They also reveal a prominent difference between the regulation of these pools compared with those labeled with myo-[3H]inositol.
Materials and Methods
Materials
Angiotensin II (Ang II; human) was obtained from Pheninsula Laboratories, Inc. (San Carlos, CA) and PDGF (recombinant, AB) from Life Technologies, Inc. (Gaithersburg, MD). Thapsigargin, ionomycin, and 1,2-bis(2-aminophenoxy)ethane N,N,_N_′,_N_-tetraacetic acid (BAPTA) were purchased from Calbiochem-Novabiochem, Corp. (La Jolla, CA), and wortmannin was a gift from Kyowa Hakko Laboratories (Tokyo, Japan). 2,3-Dimercaptopropanol (BAL), phenylarsine oxide, and quercetin were obtained from Sigma Chemical Co. (St. Louis, MO). Myo-[3H]inositol (68 Ci/mmol) and [3H]inositol-1,4,5-trisphosphate (48 Ci/mmol) was from Amersham Corp. (Arlington Heights, IL). All other chemicals were of HPLC or analytical grade.
Plasmid Constructs
The PH domains of PLCδ1 (1–170), Bruton's tyrosine kinase (1–177), Akt protein kinase (1–167), and dynamin (508–652) were amplified with the Advantage Klentaq polymerase mix (CLONTECH Labs, Inc., Palo Alto, CA) from human cDNAs (marathon cDNA from brain and K562 leukemia cells; CLONTECH Labs, Inc.) with the following primer pairs:
PLCδ: 5′-GGCATGGACTCGGGCCGGGACTTCCTG-3′, 5′-AAGATCTTCCGGGCATAGCTGTCG-3′;
Btk: 5′-CCAAGTCCTGGCATCTCAATGCATCTG-3′,
5′-TGGAGACTGGTGCTGCTGCTGGCTC-3′;
Akt: 5′-GTCAGCTGGTGCATCAGAGGCTGTG-3′,
5′-CACCAGGATCACCTTGCCGAAAGTGCC-3′;
Dyn: 5′-ATGCTCAGCAGAGGAGCAACCAGATG-3′,
5′-GAGTCCACAAGATTCCGGATGGTCTC-3′.
The amplified products were subcloned into the PGEM-Easy T/A cloning vector (Promega Corp., Madison, WI) and sequenced with dideoxy sequencing (thermosequenase; Amersham Corp.). A second amplification reaction was performed from these plasmids with nested primers that contained restriction sites for appropriate cloning into the pEGFP-N1 (PLCδ, Btk, and Akt) or pEGFP-C1 (dynamin) plasmids (CLONTECH Labs, Inc.) to preserve the reading frame. Plasmids were transfected into COS-7 cells or NIH-3T3 cells and cell lysates were resolved by SDS-PAGE followed by Western blot analysis for the presence of the GFP fusion proteins using a polyclonal antibody against GFP (CLONTECH Labs, Inc.).
Mutations were created in the PHPLCδ–GFP fusion plasmid by the QuickChange™ mutagenesis kit (Stratagene, La Jolla, CA). For practical purposes, a SalI site was introduced into the PH domain sequence which changed S34 to a T but this substitution did not change any characteristic compared with the wild-type protein. All mutations were confirmed by dideoxy sequencing and the expression of the fusion protein by Western blot analysis.
Transfection of Cells for Confocal Microscopy
Cells were plated onto poly-l-lysine–coated 30-mm-diam circular cover slips at a density of 5 × 104 cells/dish and cultured for 3 d before transfection with plasmid DNAs (1 μg/ml) using the Lipofectamine reagent (10 μg/ml; Life Technologies, Inc.) and OPTI-MEM (Life Technologies, Inc.). 48 h after transfection cells were washed twice with a modified Krebs-Ringer solution, containing (mM): NaCl 120, KCl 4.7, CaCl2 1.2, MgSO4 0.7, glucose 10, Na-Hepes 10, pH 7.4, and the coverslip was placed into a chamber that was mounted on a heated stage with the medium temperature kept at 33°C. Cells were incubated in 1 ml of the Krebs-Ringer buffer and the stimuli were added in 0.5 ml prewarmed buffer after removing 0.5 ml medium from the cells. Cells were examined in an inverted microscope under a 40× oil-immersion objective (Nikon, Inc., Melville, NY) and a BioRad laser confocal microscope system (MRC-1024) with the Lasersharp acquisition software (Bio-Rad Laboratories, Hercules, CA). All pictures presented are original recordings without postacquisition enhancing.
Analysis of Inositol Lipids
Inositol phospholipids were analyzed from either COS-7 cells transfected with the AT1a Ang II receptor together with selected GFP–PH domain fusion constructs, or from untransfected NIH-3T3 cells after labeling with myo-[3H]inositol for 24 h (COS-7 cells) or 48 h (NIH-3T3 cells) as previously described (4, 17). [3H]Inositol phosphates were analyzed by HPLC (17) or, in the COS cell experiments, by Dowex minicolumns. PtdIns(4,5)P2 mass was determined from NIH-3T3 cells and bovine adrenal glomerulosa (BAG) cells that were cultured on 12-well plates. After stimulation at 33°C, reactions were terminated with 400 μl ice-cold perchloric acid (5% final) followed by an acidic lipid extraction (3). Samples were taken to dryness with N2 stream and were subjected to alkaline hydrolysis to liberate Ins(1,4,5)P3 from PtdIns(4,5)P2 for quantitation in a radioreceptor assay, essentially as described in Challis (see reference 6) using bovine adrenocortical membranes (15) and [3H]Ins(1,4,5)P3 (Amersham Corp.).
Results
Plasma Membrane Localization of the PLCδ PH Domain Requires Interaction with PtdIns(4,5)P2
Among the known PH domains reported to interact with PtdIns(4,5)P2, the PH domain of PLCδ1 has the highest affinity (12). Therefore, we chose this PH domain to create a fluorescent probe for imaging purposes and fused it to the NH2 terminus of GFP. Expression of GFP alone showed no specific cellular localization and was present in the cytosol, as expected from a protein of its size (27 kD) lacking a localization signal. Addition of the PH domain of PLCδ1 (1–170) to the GFP (PHPLCδ–GFP) was sufficient to localize the construct to the plasma membrane when transiently expressed in various cell types (Fig. 1). A small amount of fluorescence was always present in the cytosol and the nucleus, but very little in internal membranes apart from some vesicular structures that could be membrane invaginations. Although this result was consistent with binding of the construct to membrane PtdIns(4,5)P2, it did not exclude the possibility of other interaction(s) between the PH domain and some other membrane component(s). Therefore, we mutated each of three critical basic residues, K30, K32, and R40, within the PH domain that have been previously found, based on the crystal structure of the molecule, to contribute to the high affinity binding of PLCδ to PtdIns(4,5)P2 (12). Each of the three mutations (K30L, K32L, and R40L) prevented the plasma membrane localization of the construct, whereas mutation of a non-charged residue in the same region (S34T) had no effect (Fig. 1). These results indicate that the plasma membrane localization of this fluorescent probe is based on its interaction with PtdIns(4,5)P2 through the PLCδ PH domain, and that the lipid is present predominantly in the plasma membrane.
Figure 1.
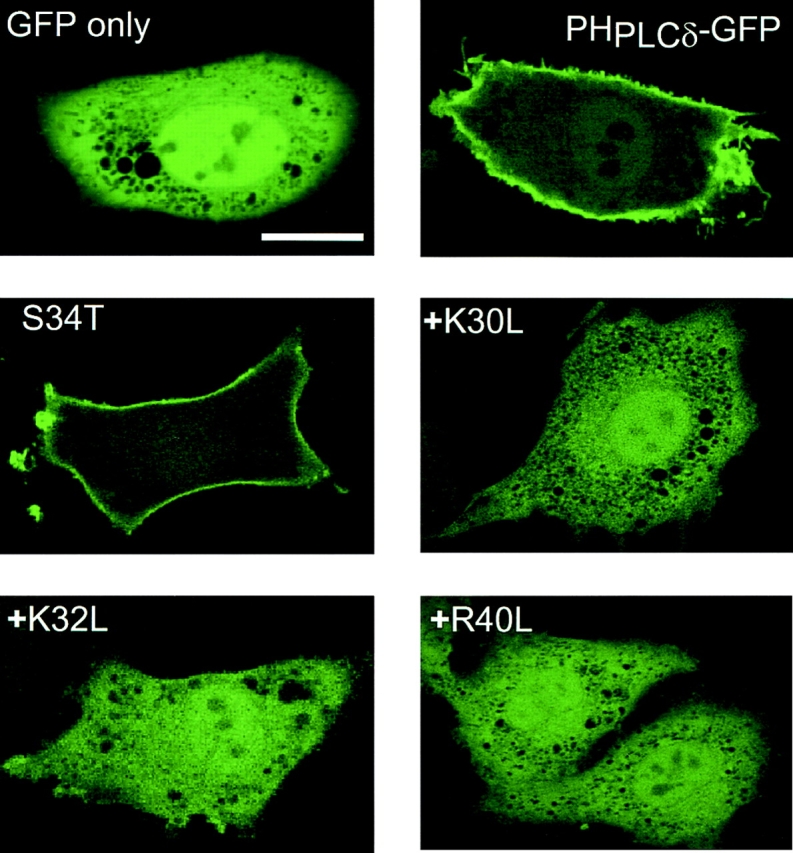

Membrane localization of PLCδ PH domain–GFP fusion protein in transfected NIH-3T3 cells. The PH domain of PLCδ (1–170) was fused to the NH2 terminus of the GFP protein and this construct or its mutants were transiently expressed in NIH-3T3 cells. Confocal images were obtained in living cells at 33°C. For technical reasons, the K30L, K32L, and R40L mutants also carried the S34T mutation but the latter alone was without effect on localization. A partial sequence of the PLCδ PH domain containing the first three β strands is shown with the mutated residues indicated by arrows. Residues that form critical contacts with Ins(1,4,5)P3 (12), are labeled by small squares. Bar, 10 μm.
PLC Activation by Ca2+ Releases the Fluorescent Probe from the Plasma Membrane into the Cytosol
If the distribution of PHPLCδ–GFP truly maps the cellular PtdIns(4,5)P2 pools, then hydrolysis of these phospholipids by endogenous PLCs should change the distribution of the fusion protein to report those phosphoinositide changes. PLC was activated in intact NIH-3T3 cells by treatment with ionomycin (10 μM) in the presence of external Ca2+ (1.2 mM) to allow a large increase in cytosolic Ca2+ concentration ([Ca2+]i) with sequential activation of PLC isozymes (30). As shown in Fig. 2, this manipulation, indeed, caused rapid disappearance of fluorescence from the plasma membrane and its simultaneous appearance in the cytosol, consistent with PLC-mediated hydrolysis of the membrane PtdIns(4,5)P2 at high [Ca2+]i. Once released into the cytosol, this 40-kD fusion construct only slowly appeared in the nucleus. Preincubation of the cells with neomycin (10 mM) for 10 min to inhibit the hydrolysis of PtdIns(4,5)P2 (8), completely prevented the release of the fluorescent signal from the membranes in the majority (>90%) of cells (not shown).
Figure 2.
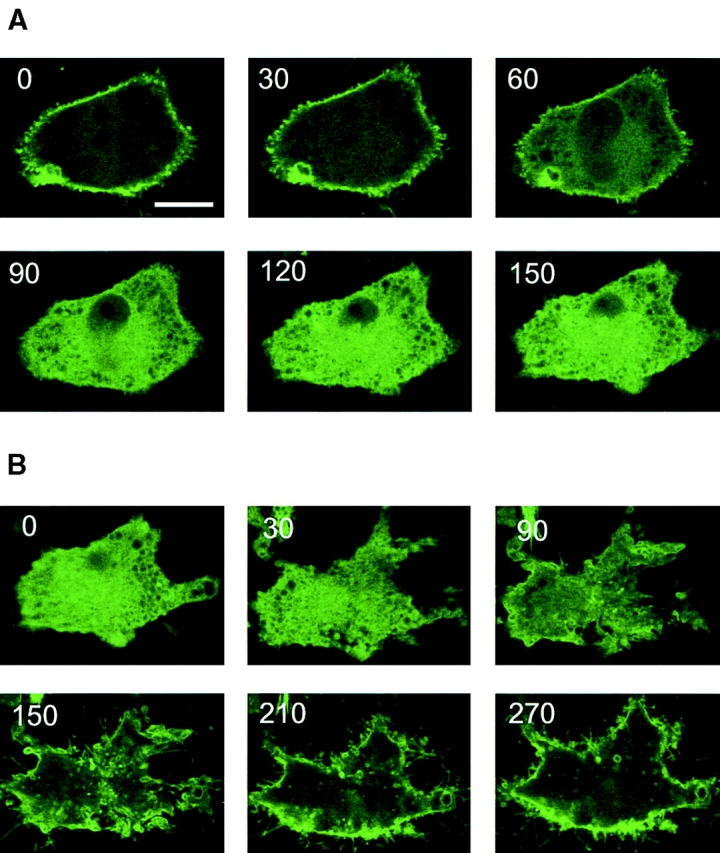

Calcium-induced changes in the membrane localization of PLCδ PH domain–GFP fusion protein and in the absolute mass of PtdIns(4,5)P2. NIH-3T3 cells were transfected with the PHPLCδ–GFP fusion construct and cells were imaged by confocal microscopy at 33°C. Ionomycin (10 μM) was added at 20 s, and images taken at 30-s intervals (A). After 5 min, 2 mM BAPTA was added to the cells and images were taken at the same interval (B). The numbers indicate the elapsed time in seconds. The changes in the absolute mass of PtdIns(4,5)P2 was measured in parallel experiments (C) and compared with the kinetics of redistribution of fluorescence. For this purpose, a fluorescence ratio was formed from the fluorescence intensities of the plasma membrane (•) or of internal bright spots (○) (Im) relative to that of cytosol (Icyt) from a line intensity plot at each of the frames from a number (n = 8) of cells and these ratios were plotted against time (D). PtdIns(4,5)P2 levels were expressed as percent of the control (i.e., before ionomycin stimulation, which was 96.5 ± 17 pmol/well, means ± SEM, n = 3 duplicate). Bar, 10 μm.
Subsequent chelation of external Ca2+ by either BAPTA or EGTA (2 mM) caused the slow reappearance of the fluorescence at the plasma membrane and, most notably, in some intracellular membrane structures (Fig. 2 B). In many cells, the first signs of relocalization were the appearance of bright foci at perinuclear membranes and in other intracellular structures that eventually translocated to the plasma membrane (Fig. 2 B). These changes were considered to reflect the resynthesis of the phosphoinositide pools as PLC activity declined in concert with the falling [Ca2+]i (see below).
To analyze whether Ca2+ release from intracellular stores without Ca2+ influx was sufficient to cause the breakdown of membrane phosphoinositides that bind PH domains, we applied ionomycin to the transfected cells in Ca2+-free medium. Under these conditions ionomycin failed to evoke a lipid signal that could be detected by the redistribution of the fluorescent construct (not shown). To calculate the concentration of Ca2+ that was required to activate PtdIns(4,5)P2 breakdown in the presence of ionomycin, we titrated extracellular Ca2+ in the presence of 2 mM BAPTA and measured [Ca2+]i under identical conditions in Fura-2–loaded NIH-3T3 cells. Based on these measurements, the concentration of Ca2+ that was required to activate PtdIns(4,5)P2 hydrolysis (assessed by the release of fluorescence from the membrane into the cytosol) was around 10 μM. The need for relatively high Ca2+ to trigger this process was also indicated by the finding that lower concentrations of ionomycin (1 μM), that evoke rapid Ca2+ release with consequential activation of the capacitive Ca2+ entry pathway, did not induce translocation of the fluorescent construct (not shown). Similarly, thapsigargin (100 nM) or PDGF (25–100 ng/ml), both of which evoked moderate [Ca2+]i increases in NIH-3T3 cells, had no effect on the distribution of the fusion protein (not shown).
Ca2+-induced Changes in Cellular PtdIns(4,5)P2 Mass Closely Correlate with Plasma Membrane Localization of PLCδ PH Domain
To examine how the cellular PtdIns(4,5)P2 pool is affected by the same manipulations that caused the redistribution of the fusion construct, we analyzed the total mass of this phospholipid from NIH-3T3 cells after ionomycin treatment and subsequent Ca2+ chelation. The total mass of PtdIns(4,5)P2 in cultured cells was measured by lipid extraction followed by alkaline hydrolysis to liberate Ins(1,4,5)P3, which was then quantitated by a radioreceptor assay (6). As shown in Fig. 2 C, ionomycin (10 μM) caused rapid breakdown of PtdIns(4,5)P2, and upon Ca2+ chelation the level of this phospholipid rapidly returned nearly to its original value within 5 min.
To analyze the correlation between PtdIns(4,5)P2 changes and the redistribution of fluorescence, we needed to quantify membrane localization. Dividing the fluorescence of the membrane with that of the adjacent cytosol (values were taken from line intensity histograms on series of images) proved to be a useful index of membrane-association of the construct. As shown in Fig. 2 D, PtdIns(4,5)P2 changes and membrane localization of the fluorescent construct correlated remarkably closely during manipulation of intracellular Ca2+, except for a clear delay observed in the plasma membrane reappearance of the fluorescence. This, however, was preceded by localization of the construct at intracellular sites (Fig. 2 D), indicating that the earliest sites of PtdIns(4,5)P2 synthesis are at intracellular membrane compartments.
Ca2+-mobilizing Hormones Can Also Initiate the Breakdown of the PtdIns(4,5)P2 Pool That Binds PLCδ PH Domain
To investigate whether the PtdIns(4,5)P2 pools that are visualized by PHPLCδ–GFP show any change after agonist stimulation, we examined the effect of the calcium mobilizing hormone, Ang II, on the redistribution of the fluorescent probe in various cells. The fluorescent construct was transfected together with the AT1a Ang II receptor into COS-7 or NIH-3T3 cells, or alone into primary cultures of adrenal glomerulosa cells that express endogenous AT1 receptors. Stimulation of the cells with Ang II caused a rapid release of the fluorescent signal from the plasma membrane to the cytosol in each of the three cell types (Figs. 3 and 4). This process was very rapid and, in contrast to ionomycin stimulation, was only transient with the construct partially relocalizing to the plasma membrane in spite of the sustained stimulation. Simultaneous changes in PtdIns(4,5)P2 mass measured in BAG cells showed again a close correlation between changes in membrane-associated fluorescence and the absolute amounts of this lipid (Fig. 4, B and C). In contrast to the effects of ionomycin, the effect of Ang II on PHPLCδ–GFP distribution was still observed in a medium nominally free of Ca2+, although this effect was very short-lived, the fluorescence being relocalized to the membrane within 2 min (not shown). These results indicate that stimulation of a G protein–coupled receptor can cause the breakdown of the PtdIns(4,5)P2 pool(s) that bind PH domains.
Figure 3.
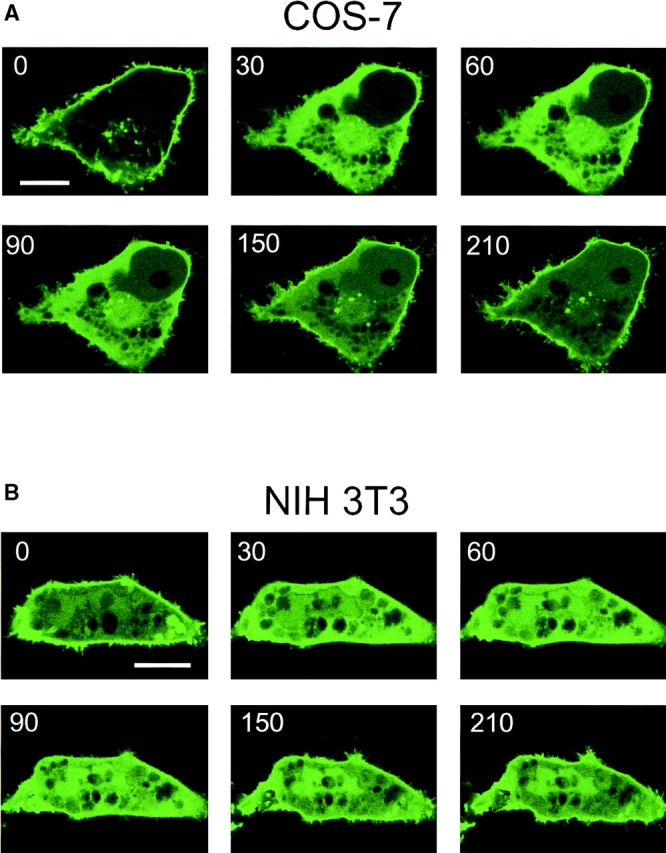
Ang II–induced changes in the membrane localization of PLCδ PH domain–GFP fusion protein in COS-7 and NIH-3T3 cells. COS-7 (A) and NIH-3T3 cells (B) were transfected with plasmids encoding the PHPLCδ–GFP fusion protein together with plasmids encoding the rat AT1a receptor. Confocal images were taken at every 15 s from cells incubated at 33°C. Stimulation by Ang II (1 μM at 20 s) was followed by a transient translocation of the fluorescent signal from the membranes to the cytosol. Bar, 10 μm.
Figure 4.


Ang II–induced changes in the membrane localization of PLCδ PH domain–GFP and absolute PtdIns(4,5)P2 mass in BAG cells. BAG were transfected with the PHPLCδ–GFP fusion construct and examined by confocal microscopy during stimulation with Ang II (10−7 M) at 33°C. The numbers indicate elapsed time in seconds. Changes in membrane localization after Ang II addition were quantitated by forming a fluorescence intensity ratio of the plasma membrane (Ipm) and the cytosol (Icyt) at various times in control cells (n = 7), and from cells pretreated with 100 μM quercetin (dashed lines, n = 5) (B). PtdIns(4,5)P2 levels were measured in parallel experiments and the values expressed as percent of control, (i.e., before Ang II stimulation, which was 149 ± 16 pmol/well). Means ± SEM are shown from three experiments each performed in duplicate. The kinetics of changes in this ratio again showed close correlation with those of the absolute lipid mass. Bar, 10 μm.
Inhibition of the Resynthesis of PtdIns(4,5)P2 Prevents Relocalization of the PLCδ PH Domain to the Plasma Membrane
To further examine the interaction between the PLCδ PH domain and the plasma membrane phosphoinositides, we used inhibitors that have been reported to inhibit the formation of PtdIns(4,5)P2 from PtdIns by inhibiting PtdIns 4 and PtdIns(4)P 5 kinases (34). These inhibitors were tested for their ability to prevent the resynthesis of PtdIns(4,5)P2 after ionomycin treatment and subsequent Ca2+ chelation. As shown in Fig. 5, both quercetin (100 μM) and phenylarsine oxide (PAO; 100 μM) were each able to prevent the resynthesis of PtdIns(4,5)P2 as measured by the total mass of this phospholipid. The inhibitory effect of PAO was completely reversed by DTT, only partially by BAL and not by β-mercaptoethanol (1 mM each). These same treatments equally affected the relocalization of the fluorescent probe to the plasma membrane upon Ca2+ chelation after ionomycin treatment: both quercetin and PAO prevented the reappearance of the fluorescent signal in membranes, and DTT but not β-mercaptoethanol, were each able to antagonize the inhibitory effect of PAO (Fig. 5 B). Interestingly, although BAL only partially restored PAO-inhibited PtdIns(4,5)P2 resynthesis, it fully restored the translocation of PHPLCδ–GFP. In bovine glomerulosa cells that were pretreated with quercetin (or with PAO), Ang II induced a more permanent release of the fluorescent construct into the cytosol instead of the transient translocation observed in control cells, indicating that once the resynthesis of the PtdIns(4,5)P2 is inhibited, the Ca2+-mobilizing agonist causes a more complete degradation of membrane PtdIns(4,5)P2 (Fig. 4, B and C).
Figure 5.


Inhibition of PtdIns(4,5)P2 synthesis prevents relocalization of the PLCδ PH domain–GFP fusion protein to the plasma membrane. NIH-3T3 cells were transfected with plasmids encoding the PLCδ PH domain–GFP fusion protein and stimulated at 33°C with 10 μM ionomycin for 5 min before addition of 2 mM BAPTA for an additional 5 min. Confocal images (A) were taken from the cells before stimulation (basal), 5 min after ionomycin addition (iono), and at the end of 5 min BAPTA treatment (+BAPTA). Cells were also preincubated with the indicated inhibitors for 5 min before ionomycin addition (WT, 10 μM; Quer [quercetin], 100 μM; PAO, 100 μM; DTT, 1 mM; BAL, 1 mM; MerOH [β-mercaptoethanol], 1 mM). PtdIns(4,5)P2 mass (B) was also measured in simultaneous experiments in non-transfected NIH-3T3 cells under identical conditions. The lipid levels were expressed as percent of the absolute control, (i.e., before ionomycin stimulation and no treatment with inhibitors, which was 91 ± 7.2 pmol/well). Means ± SEM are shown from three experiments each performed in duplicate. Bar, 10 μm.
The Resynthesis of PtdIns(4,5)P2 That Binds PH Domains Is Not Sensitive to WT Inhibition
Interestingly, high concentrations of wortmannin (WT; 10 μM) shown to inhibit type III PtdIns 4-kinases (27) did not affect relocalization of the fluorescent construct after Ca2+ chelation in ionomycin-treated cells, although it partially inhibited PtdIns(4,5)P2 resynthesis measured by the mass assay (Fig. 5). Similarly, 10 μM WT had no effect on the Ang II–induced transient translocation of the fluorescent signal (not shown). This result contrasted our previous finding that the metabolically labeled hormone-sensitive PtdIns(4,5)P2 pools are maintained by WT-sensitive PtdIns 4-kinase(s) in several agonist-stimulated cells and that stimulation of these cells in the presence of 10 μM WT leads to the depletion of these phosphoinositide pools in myo-[3H]inositol–labeled cells (27).
These findings suggest that although the myo-[3H]inositol–labeled, agonist-sensitive PtdIns(4,5)P2 pools are formed by WT-sensitive PtdIns 4-kinase(s), additional PtdIns(4,5)P2 pools that bind PH domains are synthesized by WT-insensitive mechanisms that are only inhibited by less-specific PI kinase inhibitors, such as quercetin or PAO (34).
Relationship between Ca2+- and Agonist-responsive PtdIns(4,5)P2 Pools That Bind PH Domains
The transient nature of the release of PHPLCδ–GFP from the plasma membrane after Ang II stimulation indicated the resynthesis of the membrane PtdIns(4,5)P2 during the action of the hormone. We examined whether such newly synthesized PtdIns(4,5)P2 having reassociated with PHPLCδ– GFP is available for Ca2+-induced hydrolysis. As shown in Fig. 6 A, ionomycin caused further and complete hydrolysis of PtdIns(4,5)P2 in Ang II–stimulated cells as assessed by the release of PHPLCδ–GFP from the membrane. Similar results were obtained when Ang II action was terminated by the AT1 receptor antagonist, losartan (10−5) before the addition of ionomycin (not shown). The reciprocal experiment was also performed; bovine glomerulosa cells expressing PHPLCδ–GFP were subjected to one round of treatment with ionomycin (10 μM) and Ca2+ chelation (5 mM EGTA) to allow complete hydrolysis of PtdIns(4,5)P2 and to allow its resynthesis to take place in the presence of the fluorescent construct. After the removal of ionomycin, (by several washes with a solution containing BSA), such cells still showed an Ang II–stimulated redistribution of fluorescence that did not appear to be significantly different from the changes observed in naive cells (Fig. 6 B). These results indicated that the Ca2+- and agonist-responsive pools of PtdIns(4,5)P2 that are available for binding to PH domains are largely overlapping and that the resynthesized PtdIns(4,5)P2 is still available for agonist-induced hydrolysis.
Figure 6.
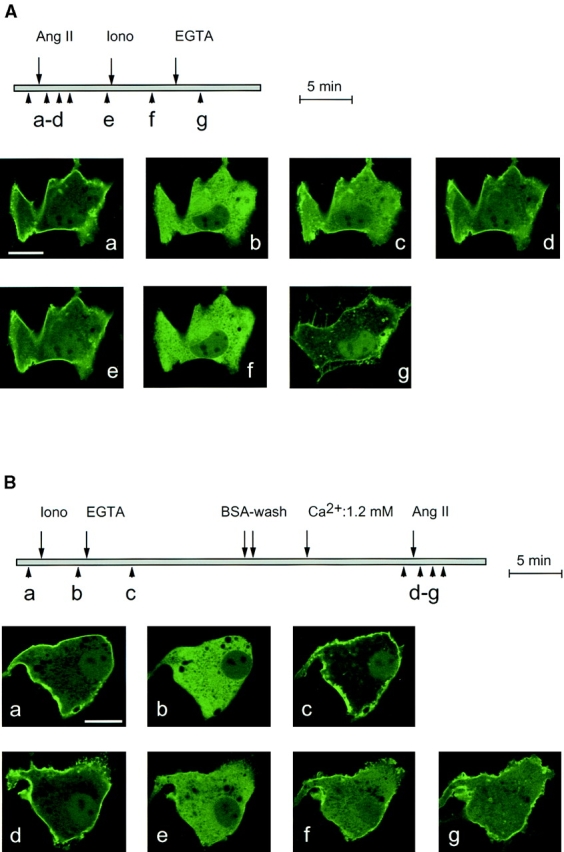
Availability of newly synthesized PtdIns(4,5)P2 to repeated stimulation of phospholipase C in BAG cells. BAG cells were transfected with plasmids encoding the PLCδ PH domain–GFP fusion protein. (A) Cells were stimulated with Ang II (10−7 M) for 5 min before addition of 10 μM ionomycin, and then 5 mM EGTA. In some experiments, losartan (10−5 M, an AT1 receptor antagonist) was added for 10 min to terminate Ang II action before the administration of ionomycin (not shown). (B) Cells were stimulated with 10 μM ionomycin and, immediately after the redistribution of fluorescence, 5 mM EGTA was added to remove Ca2+. After 10 min, cells were washed twice with incubation medium containing 5% BSA to reduce ionomycin, and then incubated in normal medium (containing 1.2 mM Ca2+) for an additional 10 min before stimulation with Ang II (10−7 M). Bar, 10 μm.
PH Domains Can Inhibit the Agonist-induced Hydrolysis of Myo-[3H]inositol–labeled PtdIns(4,5)P2
Since PH domains bind to and therefore cover the inositol phosphate headgroup of phosphoinositides, they can hinder its accessibility and hydrolysis by the agonist-regulated PLC enzymes. Alternatively, PH domains may interfere with the membrane localization of the PLC enzymes that are activated by an agonist. Although all of the known PLC isoenzymes contain a PH domain, its affinity and specificity toward inositides has not been analyzed in every case (10). To test the possibility that PH domains interfere with agonist-induced Ins(1,4,5)P3 formation from [3H]inositol-labeled PtdIns(4,5)P2, we overexpressed various PH domain–GFP constructs in COS-7 cells together with the AT1a Ang II receptor and measured Ang II–stimulated [3H]inositol phosphate formation in cells prelabeled by myo-[3H]inositol. Fig. 7 A shows that PLCδ PH domain greatly inhibited Ang II–stimulated formation of [3H]inositol phosphates and that other PH domains with low affinity for PtdIns(4,5)P2, such as that of the Bruton's tyrosine kinase or the Akt protein kinase, as well as that of dynamin (13, 18, 31), showed no similar inhibitory effect. Fluorescent constructs containing these PH domains did not show the same membrane localization as those with the PLCδ PH domain (not shown). Also, mutations within the PH domain of PLCδ that prevented its interaction with PtdIns(4,5)P2, and hence its membrane localization, failed to inhibit Ang II–induced inositol phosphate production (Fig. 7 B). The corollary of this finding is that fluorescent PH domain constructs with high enough affinity to “label” PtdIns(4,5)P2 pools are most likely to also interfere with the agonist-sensitive phosphoinositide pools, since their binding to PtdIns(4,5)P2 impedes their access to the relevant PLC enzymes.
Figure 7.
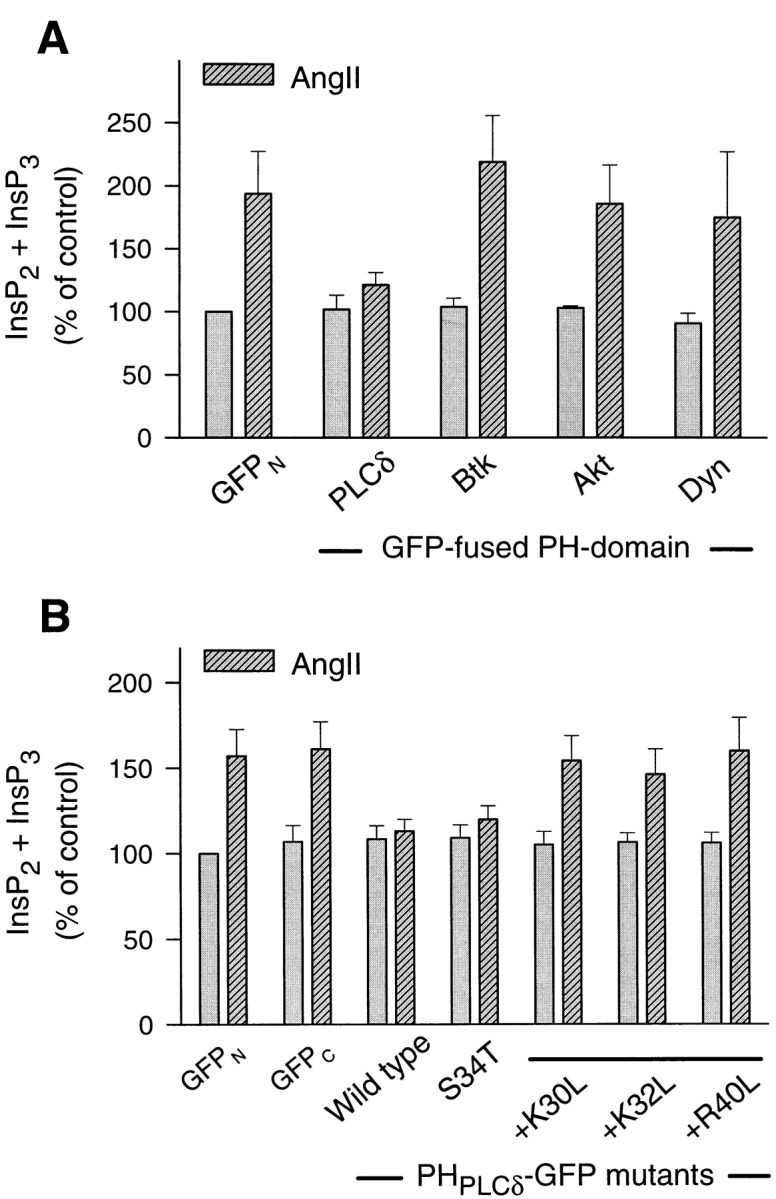
Inhibition of Ang II–induced [3H]inositol phosphate formation by the PLCδ PH domain in COS-7 cells. COS-7 cells were cotransfected with plasmids encoding the rat AT1a receptor and the indicated GFP–PH domain construct. After 24 h, cells were labeled with myo-[3H]inositol for 24 h and preincubated with 10 mM LiCl for 30 min before stimulation with 1 μM Ang II for 20 min. After extraction, inositol phosphates were separated on DOWEX columns from which InsP2 and InsP3 were eluted together. Values are expressed as percent of the unstimulated control that was transfected with the AT1a receptor and the GFP plasmid.
Discussion
The present experiments demonstrate that PH domains which bind PtdIns(4,5)P2 with high affinity and specificity, such as that of PLCδ1, can be used to visualize certain PtdIns(4,5)P2 pools in single living cells. Decreasing the affinity of the PH domain by mutation of any one of three basic residues known to form contacts with the inositol phosphate headgroup (12, 35) was sufficient to eliminate membrane localization of the construct. Also, PH domains with lower affinity for PtdIns(4,5)P2 such as that of the GTP-binding protein, dynamin, or with different specificity such as those of the Bruton's tyrosine kinase and the protein kinase, Akt, did not show the clear plasma membrane localization that was observed with the PLCδ PH domain. The high concentration of the fluorescent probe at the plasma membrane was consistent with previously established views that PtdIns(4,5)P2 is most abundant in the plasma membrane (26). The lack of localization to intracellular membranes, including the nuclear membrane where a separate inositide system has been described (7), suggests that in quiescent cells these membranes either contain only small amounts of this lipid, or that such pools are not accessible to the expressed PH domain. Since the interaction of the PH domain is formed with the inositol phosphate headgroup of PtdIns(4,5)P2, hydrolysis of the lipid by PLC is reflected in the release of the fluorescent probe from the plasma membranes to the cytosol. Such change in localization was dramatically demonstrated when endogenous PLC was activated by Ca2+ influx via Ca2+ ionophores. After Ca2+ chelation, when PLC activity subsided and PtdIns(4,5)P2 resynthesis began to take place, the appearance of bright spots at perinuclear structures preceded the localization of the fluorescence to the plasma membrane, indicating that some PtdIns(4,5)P2 synthesis also occurs in intracellular membranes.
This novel methodology, which allowed analysis of the regulation of cellular PtdIns(4,5)P2 pools from the viewpoint of PH domains showed that stimulation of a G protein–coupled Ca2+-mobilizing receptor, the AT1a Ang II receptor, was also able to activate the hydrolysis of PH domain–tagged PtdIns(4,5)P2. In a recent report, Stauffer et al. (see reference 32) reported that stimulation of PAF receptors (another G protein–coupled receptor) in a basophilic leukaemia cell line also caused a transient translocation of PLCδ PH domain using a similar methodology. Interestingly, activation of PLCγ by PDGF in NIH-3T3 cells (in this study), or by FCεRI stimulation in RBL cells (33) did not cause any visible change in the distribution of a PH domain–GFP construct (the PH domain of pleckstrin was used in reference 33). Whether this reflects the inability of PLCγ to hydrolyze PtdIns(4,5)P2 when it is covered by a PH domain remains to be determined.
Overexpression of the PLCδ PH domain was also found to inhibit agonist-induced formation of myo-[3H]inositol– labeled inositol phosphates in transfected COS-7 cells, and this effect was closely correlated with the ability of the construct to localize to the membrane. Similar finding was presented in a recent report, in which pleckstrin, via its PH domain, was shown to inhibit [3H]inositol phosphate formation in transfected COS-1 or HEK 293 cells regardless of the type of PLC that was activated (1). These results suggest that expressed PH domains also can interfere with the agonist-regulated PLC activation mechanism, either by masking the PtdIns(4,5)P2 headgroup from the enzyme's catalytic site or by competing with the PH domains of the PLC enzymes to inhibit their localization to the membranes. These results also emphasize that the PLC activity detected in cells is greatly influenced by membrane components that interact with phosphoinositides, and that the use of cell-free systems and artificial substrates cannot reveal this aspect of PLC regulation.
An important finding of the present study was the inability of WT (at μM concentrations that inhibit type III PtdIns 4 kinases) to prevent the resynthesis of the PtdIns(4,5)P2 pools available for binding of the PLCδ PH domain in both ionomycin- and agonist-stimulated cells. The same treatment completely prevented the synthesis of myo-[3H]inositol–labeled PtdIns(4,5)P2 in Ang II–stimulated adrenal glomerulosa cells (27), and also blocks the resynthesis of [3H]PtdIns(4,5)P2 after chelation of Ca2+ in ionomycin-treated NIH-3T3 cells (Varnai, P., and T. Balla, unpublished observations). This finding raised the possibility that the agonist-sensitive myo-[3H]inositol–labeled PtdIns(4,5)P2 pools are not completely identical to those that are imaged by the fluorescent PH domain of PLCδ, although both respond to agonist stimulation. Heterogeneity of phosphoinositide pools have been described earlier in a few reports. These studies indicated that some of the metabolically labeled PtdIns and PtdIns(4,5)P2 pools are not sensitive to agonist stimulation (22) and conversely, some PtdIns(4,5)P2 that is not labeled metabolically is still subject to PLC-mediated hydrolysis (20). The existence of a metabolically hyperactive hormone-sensitive PtdIns(4,5)P2 pool, however, could not be substantiated (25). Our results indicate that, unlike the [3H]inositol-labeled PtdIns(4,5)P2 pools, the PtdIns(4,5)P2 pool(s) that bind PH domains are not synthesized by WT-sensitive type III PtdIns 4 kinases. The highly abundant, tightly membrane-bound type II PtdIns 4 kinase (5), which is not sensitive to even high concentrations of WT (9) is a good candidate for synthesizing the inositides that bind PH domains. Clearly, more experiments will be required to find the explanation for the apparent discrepancy between the regulation of myo-[3H]inositol–labeled and PH domain–imaged PtdIns(4,5)P2.
An additional possibility to be considered in understanding the current results is that the Ins(1,4,5)P3 that is formed after agonist stimulation competes for the PH domain of PLCδ (and of other proteins), and contributes to the release of these proteins from the plasma membrane, especially when PtdIns(4,5)P2 levels are decreasing. Such competition by Ins(1,4,5)P3 with PtdIns(4,5)P2 for the PH domain of PLCδ has been demonstrated and also found to inhibit the catalytic efficiency of the enzyme (2, 19). Since the only known major source of Ins(1,4,5)P3 is PtdIns(4,5)P2, this competing effect of Ins(1,4,5)P3 would distort the imaging results if there were substantial amounts of Ins(1,4,5)P3 formed from PtdIns(4,5)P2 pools that did not bind PH domains in their initial steady-state before stimulation. Nevertheless, this is an open possibility and a better assessment of such a displacing effect of Ins(1,4,5)P3 requires additional studies with GFP-fused PH domains.
In summary, the present studies demonstrate that isolated PH domains fused to the GFP are capable of recognizing phosphoinositides in living cells with remarkable specificity. These interactions allow visualization of the spatiotemporal changes in phosphoinositides at the single cell level. Analysis of cellular PtdIns(4,5)P2 with this method reveals that the pools that bind PH domains my not be identical to those that can be metabolically labeled with myo-[3H]inositol. Studies on the receptor-mediated control of the PtdIns(4,5)P2 pools that bind PH domains will help to understand the manner in which phosphoinositides regulate various signaling processes via protein PH domains.
Abbreviations used in this paper
Ang II
angiotensin II
BAG
bovine adrenal glomerulosa
BAL
2,3-dimercaptopropanol
BAPTA
1,2-bis(2-aminophenoxy)ethane N,N,_N_′,_N_-tetraacetic acid
GFP
green fluorescent protein
Ins(1,4,5)P3
inositol-1,4,5-trisphosphate
PAO
phenylarsine oxide
PH
pleckstrin homology
PLC
phospholipase C
PtdIns
phosphatidylinositol
PtdIns(4)P
phosphatidylinositol 4-phosphate
PtdIns(4,5)P2
phosphatidylinositol 4,5-bisphosphate
WT
wortmannin
Footnotes
The skillful technical work of Ms. Y. Zhang is greatly appreciated.
Address all correspondence to T. Balla, National Institutes of Health, Bldg. 49, Rm. 6A35, 49 Convent Drive, Bethesda, MD 20892-4510. Tel.: (301) 496-2136. Fax: (301) 480-8010. E-mail: tambal@box-t.nih.gov
References
- 1.Abrams CS, Wu H, Zhao W, Belmonte E, White D, Brass LF. Pleckstrin inhibits phosphoinositide hydrolysis initiated by G-protein-coupled and growth factor receptors. J Biol Chem. 1995;270:14485–14492. doi: 10.1074/jbc.270.24.14485. [DOI] [PubMed] [Google Scholar]
- 2.Allen V, Swigart P, Cheung R, Cockcroft S, Katan M. Regulation of inositol lipid-specific phospholipase Cδ by changes in Ca2+ion concentrations. Biochem J. 1998;327:545–552. doi: 10.1042/bj3270545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balla T, Baukal AJ, Guillemette G, Catt KJ. Multiple pathways of inositol polyphosphate metabolism in angiotensin-stimulated adrenal glomerulosa cells. J Biol Chem. 1988;263:4083–4091. [PubMed] [Google Scholar]
- 4.Balla T, Sim SS, Baukal AJ, Rhee SG, Catt KJ. Inositol polyphosphates are not increased by overexpression of Ins(1,4,5)P33-kinase but show cell-cycle dependent changes in growth factor-stimulated fibroblasts. Mol Biol Cell. 1994;5:17–27. doi: 10.1091/mbc.5.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carpenter CL, Cantley LC. Phosphoinositide kinases. Biochemistry. 1990;29:11147–11156. doi: 10.1021/bi00503a001. [DOI] [PubMed] [Google Scholar]
- 6.Challis, J.R.A. 1997. Mass assay of inositol 1,4,5-trisphosphate and phosphatidylinositol 4,5-bisphosphate. In Signalling by Inositides. S.B. Shears, editor. Oxford University Press, Oxford, New York, Tokyo. 151–164.
- 7.Divecha N, Banfic H, Irvine RF. Inositides and the nucleus and inositides in the nucleus. Cell. 1993;74:405–407. doi: 10.1016/0092-8674(93)80041-c. [DOI] [PubMed] [Google Scholar]
- 8.Downes CP, Michell RH. The polyphosphoinositide phosphodiesterase of erythrocyte membranes. Biochem J. 1981;198:133–140. doi: 10.1042/bj1980133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Downing GJ, Kim S, Nakanishi S, Catt KJ, Balla T. Characterization of a soluble adrenal phosphatidylinositol 4-kinase reveals wortmannin-sensitivity of Type III phosphatidylinositol 4-kinases. Biochemistry. 1996;35:3587–3594. doi: 10.1021/bi9517493. [DOI] [PubMed] [Google Scholar]
- 10.Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J. Activation of phospholipase Cγ by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO (Eur Mol Biol Organ) J. 1998;17:414–422. doi: 10.1093/emboj/17.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. Crystal structure at 2.2 Å resolution of the pleckstrin homology domain from human dynamin. Cell. 1994;79:199–209. doi: 10.1016/0092-8674(94)90190-2. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell. 1995;83:1037–1046. doi: 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 13.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt protooncogene product by PI(3,4)P2 . Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 14.Goldschmidt-Clermont PJ, Kim JW, Machesky LM, Rhee SG, Pollard TD. Regulation of phospholipase C-γ1 by profilin and tyrosine phosphorylation. Science. 1991;251:1231–1233. doi: 10.1126/science.1848725. [DOI] [PubMed] [Google Scholar]
- 15.Guillemette G, Balla T, Baukal AJ, Spät A, Catt KJ. Intracellular receptors for inositol 1,4,5-trisphosphate in angiotensin II target tissues. J Biol Chem. 1987;262:1010–1015. [PubMed] [Google Scholar]
- 16.Harlan JE, Hajduk PJ, Yoon HS, Fesik SW. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature. 1994;371:168–170. doi: 10.1038/371168a0. [DOI] [PubMed] [Google Scholar]
- 17.Hunyady L, Baukal AJ, Balla T, Catt KJ. Independence of type 1 angiotensin II receptor endocytosis from G protein coupling and signal transduction. J Biol Chem. 1994;269:24798–24804. [PubMed] [Google Scholar]
- 18.James SR, Downes CP, Gigg R, Grove SJA, Holmes AB, Alessis DR. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J. 1996;315:709–713. doi: 10.1042/bj3150709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanematsu T, Takeya H, Watanabe Y, Ozaki S, Yoshida M, Koga T, Iwanaga S, Hirata M. Putative inositol 1,4,5-trisphosphate binding proteins in rat brain cytosol. J Biol Chem. 1992;267:6518–6525. [PubMed] [Google Scholar]
- 20.King CE, Stephens LR, Hawkins PT, Guy GR, Michell RH. Multiple metabolic pools of phosphoinositides and phosphatidate in human erythrocytes incubated in a medium that permits rapid transmembrane exchange of phosphate. Biochem J. 1987;244:209–217. doi: 10.1042/bj2440209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klarlund JK, Rameh LE, Cantley LC, Buxton JM, Holik JJ, Sakelis C, Patki V, Corvera S, Czech MP. Regulation of GRP1-catalyzed ADP ribosylation factor guanine nucleotide exchange by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:1859–1862. doi: 10.1074/jbc.273.4.1859. [DOI] [PubMed] [Google Scholar]
- 22.Koreh K, Monaco ME. The relationship of hormone-sensitive and hormone-insensitive phosphatidylinositol to phosphatidylinositol 4,5-bisphosphate in the WRK-1 cell. J Biol Chem. 1986;261:88–91. [PubMed] [Google Scholar]
- 23.Lemmon MA, Falasca M, Ferguson KM, Schlessinger J. Regulatory recruitment of signalling molecules to the cell membrane by pleckstrin-homology domains. Trends Cell Biol. 1997;7:237–242. doi: 10.1016/S0962-8924(97)01065-9. [DOI] [PubMed] [Google Scholar]
- 24.Lemmon MA, Ferguson KM, O'Brien R, Sigler PB, Schlessinger J. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci USA. 1995;92:10472–10476. doi: 10.1073/pnas.92.23.10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCallum SH, Barker CJ, Hunt PA, Wong NS, Kirk CJ, Michell RH. The use of cells doubly labelled with [14C]inositol and [3H]inositol to search for a hormone-sensitive inositol lipid pool with atypically rapid metabolic turnover. J Endocrinol. 1989;122:379–389. doi: 10.1677/joe.0.1220379. [DOI] [PubMed] [Google Scholar]
- 26.Michell RH, Hawthorne JN. The site of diphosphoinositide synthesis in rat liver. Biochem Biophys Res Commun. 1965;21:333–338. doi: 10.1016/0006-291x(65)90198-1. [DOI] [PubMed] [Google Scholar]
- 27.Nakanishi S, Catt KJ, Balla T. A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc Natl Acad Sci USA. 1995;92:5317–5321. doi: 10.1073/pnas.92.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pitcher JA, Touhara K, Payne ES, Lefkowitz RJ. Pleckstrin homology domain-mediated membrane association and activation of the β-adrenergic receptor kinase requires coordinate interaction with Gbgammasubunits and lipid. J Biol Chem. 1995;270:11707–11710. doi: 10.1074/jbc.270.20.11707. [DOI] [PubMed] [Google Scholar]
- 29.Rameh LE, Arvidsson A, Carraway KL, III, Couvillon AD, Rathbun G, Crompton A, VanRentherghem B, Czech MP, Ravichandran KS, Burakoff SJ, et al. A comparative analysis of the phosphoinositide binding specificity of pleckstrin homology domains. J Biol Chem. 1997;272:22059–22066. doi: 10.1074/jbc.272.35.22059. [DOI] [PubMed] [Google Scholar]
- 30.Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem. 1997;272:15045–15048. doi: 10.1074/jbc.272.24.15045. [DOI] [PubMed] [Google Scholar]
- 31.Salim K, Bottomley MJ, Querfurth E, Zvelebil MJ, Gout I, Scaife R, Margolis RL, Gigg R, Smith CIE, Driscoll PC, et al. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton's tyrosine kinase. EMBO (Eur Mol Biol Organ) J. 1996;15:6241–6250. [PMC free article] [PubMed] [Google Scholar]
- 32.Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- 33.Stauffer TP, Meyer T. Compartmentalized IgE receptor–mediated signal transduction in living cells. J Cell Biol. 1997;139:1447–1454. doi: 10.1083/jcb.139.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiedemann C, Schäfer T, Burger MM. Chromaffin granule-associated phosphatidylinositol 4-kinase activity is required for stimulated secretion. EMBO (Eur Mol Biol Organ) J. 1996;15:2094–2101. [PMC free article] [PubMed] [Google Scholar]
- 35.Yagisawa H, Sakuma K, Paterson HF, Cheung R, Allen V, Hirata H, Watanabe Y, Hirata M, Williams RL, Katan M. Replacement of single basic amino acids in the pleckstrin homology domain of phospholipase C-δ1alter the ligand binding, phospholipase activity and interaction with the plasma membrane. J Biol Chem. 1998;273:417–424. doi: 10.1074/jbc.273.1.417. [DOI] [PubMed] [Google Scholar]