Integration of Endothelial Cells in Multicellular Spheroids Prevents Apoptosis and Induces Differentiation (original) (raw)
Abstract
Single endothelial cells (EC) seeded in suspension culture rapidly undergo apoptosis. Addition of survival factors, such as VEGF and FGF-2, does not prevent apoptosis of suspended EC. However, when cells are allowed to establish cell–cell contacts, they become responsive to the activities of survival factors. These observations have led to the development of a three-dimensional spheroid model of EC differentiation. EC spheroids remodel over time to establish a differentiated surface layer of EC and a center of unorganized EC that subsequently undergo apoptosis. Surface EC become quiescent, establish firm cell–cell contacts, and can be induced to express differentiation antigens (e.g., induction of CD34 expression by VEGF). In contrast, the unorganized center spheroid cells undergo apoptosis if they are not rescued by survival factors. The responsiveness to the survival factor activities of VEGF and FGF-2 was not dependent on cell shape changes since it was retained after cytochalasin D treatment. Taken together, these findings characterize survival factor requirements of unorganized EC and indicate that polarized surface EC differentiate to become independent of exogenous survival factors. Furthermore, they demonstrate that spheroid cell culture systems are useful not just for the study of tumor cells and embryonic stem cells but also for the analysis of differentiated functions of nontransformed cells.
Keywords: endothelium, differentiation, apoptosis, spheroid, VEGF, FGF-2
Endothelial cells (EC)1 are mesenchyme-derived cells that line the inside of all blood vessels as an epithelioid monocellular layer. Correspondingly, endothelial cells can be grown on tissue culture surfaces to form a monocellular layer of cells that mimics many of the phenotypic and functional properties of endothelial cells in vivo (37). Originally isolated from large blood vessels such as the bovine aorta (45) and the human umbilical vein (21, 26), EC can now essentially be isolated from any capillary bed (23, 18) and serially propagated in culture as nontransformed cells with a finite life span. Bovine aortic EC (BAEC) have been cultured for >50 passages (>100 population doublings; reference 5) and human umbilical vein endothelial cells (HUVEC) can be grown up to 15–20 passages (30–40 population doublings; reference 32).
EC maintained in standard two-dimensional cell culture tend to lose many of their differentiated phenotypic properties that limits the study of differentiated cell functions in vitro. The distinct organ-specific morphological characteristics of EC (continuous nonfenestrated [brain, lung]; discontinuous with intracellular fenestrae [renal glomeruli, endocrine glands]; discontinuous with interendothelial gaps [sinusoids of liver, spleen, and bone marrow]; references 6, 41) are gradually lost when EC are propagated in culture. Cultured brain EC lose their tight junction-dependent blood–brain barrier characteristics (macromolecular impermeability and high electrical resistance; 54). Likewise, EC differentiation antigens such as CD34 are downregulated upon transfer of EC in culture (16). Cultured EC grow to form a confluent monolayer. Nevertheless, established two-dimensional EC culture models are not capable to generate a quiescent, resting EC phenotype in vitro that corresponds to the very low turnover of EC in vivo (for review see reference 15).
With the increasing appreciation of the functional and phenotypic diversity of EC (20), we reasoned that the study of differentiated endothelial cell functions would require the development of novel experimental cell culture systems that reflect the in vivo microenvironment of EC more closely than standard monolayer culture on plastic substrata or the use of nonphysiological matrices (37). Our search for novel endothelial cell culture and differentiation models was furthermore inspired by recent in vivo observations that have shed light into the complexity of differentiated endothelial cell functions to demonstrate the requirement of immature EC to be supported by the activities of survival factors (1, 11).
The search for cell culture systems that facilitate the study of cell differentiation and the analysis of the activities of survival factors in a defined microenvironment has led us to the development of a spheroid model of endothelial cell differentiation. Spheroid models of tumor cells and embryonic stem cells have been widely used to study cellular differentiation, cell–cell interactions, hypoxia responses, and even in therapeutically oriented studies (7, 29, 31, 36, 49, 52). Few investigators have applied spheroid cell culture models for the study of differentiated functions of nontransformed cells. However, as shown in this study a spheroid cell culture model of nontransformed EC proved to be a powerful tool to study the differentiation of EC as well as the functions of endothelial survival factors in a defined experimental system. The data presented in this study characterize the target cell population of endothelial survival factors and illustrate a differentiation potential of spheroid EC that goes beyond the degree of differentiation that can be achieved in standard two-dimensional culture systems. The experiments suggest that spheroid culture models may be useful not just for the study of differentiated endothelial cell functions but also for the analysis of the organotypic differentiation of other nontransformed cell populations.
Materials and Methods
Antibodies, Growth Factors, and Reagents
FGF-2, TNF-α, and IL-1 (all human recombinant) were obtained from Promega (Mannheim, Germany). Human recombinant VEGF was from PAN-Systems (Nürnberg, Germany). Neutralizing (type I) and nonneutralizing (type II) monoclonal mouse anti–bovine FGF-2 antibody were purchased from Upstate Biotechnology (Biomol, Hamburg, Germany) and the neutralizing monoclonal mouse anti-human VEGF antibody was obtained from R&D Systems GmbH (Wiesbaden, Germany) just as the polyclonal goat anti–human ICAM-1 and goat anti–human VCAM-1 antibodies. The monoclonal mouse anti-CD34 antibody (clone QBEnd/10) was purchased from Novocastra Laboratories (Loxo GmbH, Dossenheim, Germany). The monoclonal mouse anti-CD31 antibody and the monoclonal mouse anti-BrdU antibody were from Dako (Glostrup, Denmark). Cytochalasin D and carboxymethylcellulose were obtained from Sigma (Deisenhofen, Germany). RGD-containing peptides (GRGDSP) as well as control RAD-peptides were from Biomol (Hamburg, Germany).
Cell Culture
Endothelial cell growth medium (ECGM) and endothelial cell growth supplement (human umbilical vein endothelial cell culture) were purchased from Promocell (Heidelberg, Germany). DME and other cell culture media were from Life Technologies (GIBCO BRL, Eggenstein, Germany). FCS was obtained from Biochrom (Berlin, Germany). Bovine aortic EC (BAEC) were isolated from thoracic aortas of healthy cattle by collagenase digestion following standard protocols. Cells were cultured at 37°C in 75-cm2 tissue culture dishes in DME containing 10% heat-inactivated FCS and frozen in liquid nitrogen at passage 2 or 3. Cells were routinely split at a 1:5 ratio and cultured <50 passages. Only BAE cells cultured from passage 15–30 were used for experiments. HUVEC were freshly isolated from human umbilical veins of newborn babies by collagenase digestion. Cells were cultured at 37°C in 75-cm2 tissue culture dishes in ECGM containing 10% heat-inactivated FCS and frozen in liquid nitrogen at passage 2 or 3. Only HUVEC cultured from passage 4 to 8 were used for experiments. Nontransformed bovine esophageal epithelial cells (KOP) were obtained from Dr. R. Riebe (BFAF, Insel Riems, Germany) and C6 glioma cells were provided by Dr. G. Breier (MPI, Bad Nauheim, Germany).
Generation of Endothelial Spheroids
Confluent monolayers of HUVEC or BAEC were trypsinized. Cells were suspended in corresponding culture medium containing 20% methocel, seeded into nonadhesive 75-cm2 bacteriological dishes (Greiner, Frickenhausen, Germany), and cultured at 37°C (5% CO2, 100% humidity). Under these conditions suspended EC aggregate spontaneously within 4 h to form cellular aggregates of varying size and cell number. The methocel used for these experiments was diluted from a stock solution that was generated by dissolving 6 g of carboxymethylcellulose in 500 ml of medium (DME or ECGM basal medium). After centrifugation the clear, gel-like supernatant was used for experiments. Methocel prevents adhesion of cells and acts as an inert viscosity modulating substance. Variation of the methocel concentration during spheroid formation was, thus, used to control the average size of the spheroids. These multicellular spheroids were designated as random spheroids and used for all experiments that employed larger populations of cells. To generate endothelial cell spheroids of defined size and cell number, a specific number of cells (varying between 500 and 3,000 cells per spheroid, depending on the experiment) was suspended in culture medium and seeded in nonadherent round-bottom 96-well plates (Greiner, Frickenhausen, Germany). Under these conditions all suspended cell contribute to the formation of a single endothelial cell spheroid. These spheroids, designated as standard spheroids, were harvested within 24 h and used for the corresponding experiments.
BrdU Labeling
Proliferating cells were labeled by adding BrdU (concentration in medium: 100 μM; Sigma, Deisenhofen, Germany) for 20 h into the culture medium without changing the medium. Subconfluent cells were labeled 2 d after seeding. To assess BrdU incorporation into confluent monolayers, cells were split at a low ratio (1:3), grown to confluence for 2 d to 3 d and cultured for an additional day after which BrdU was added. BrdU incorporation into spheroids was assessed at early time points (1 d) as well as later time points (BAEC, 7 d; HUVEC, 5 d). Monolayer cells and spheroids were processed for paraffin embedding as outlined below.
Morphological and Immunohistochemical Analysis
Random spheroids or standard spheroids were harvested and centrifuged for 3 min at 200 g. Cultured monolayer cells were harvested by trypsinization and collected by centrifugation. Spheroids and pelleted monolayer cells were fixed in HBSS containing 4% formaldehyde and processed for paraffin embedding: after dehydration (graded series of ethanol and isopropanol, 1 h each), the specimen were first immersed with paraffin I (melting temperature 42°C) for 12 h at 60°C. Spheroids and monolayer cells were again collected by centrifugation and immersed with paraffin II (melting temperature 56°C) for 12 h at 70°C. Finally, the resulting paraffin block was cooled to room temperature and trimmed for sectioning. For histochemical analyses, paraffin sections (3 μm) were cut, deparaffinized, and rehydrated. Sections were then incubated with 3% H2O2 in H2O to inhibit endogenous peroxidase. Antigen retrieval was performed by boiling the sections in 500 ml of 0.1 M citrate buffer (pH 6.0) in a microwave oven for 30 min (ICAM-1, VCAM-1) or incubating them in 0.5% Triton X-100 in 2 N HCl (BrdU). After washings in PBS, the sections were incubated for 30 min with blocking solution (10% normal goat serum [adhesion molecules]; 0.5% Tween-20 and 0.5% BSA in PBS [BrdU]) followed by incubation with the corresponding primary antibody in a humid chamber at 4°C overnight. They were then incubated with secondary antibody (biotinylated goat anti–rabbit immunoglobulin or biotinylated goat anti– mouse immunoglobulin antibody; Zymed, San Francisco, CA), exposed to streptavidin peroxidase, developed with diaminobenzidine as substrate, and weakly counterstained with methylgreen.
Ultrastructural Analysis
Spheroids were fixed in Karnovsky's fixative, postfixed in 1.0% osmium tetroxide, dehydrated in a graded series of ethanol, and embedded in epon. 0.5-μm sections were cut and stained with azure 11 methylene blue for light microscopic evaluation. Ultrathin sections (50–80 nm) were cut, collected on copper grids, and automatically stained with uranyl acetate and lead citrate for observation with a Zeiss EM 10 electron microscope.
Detection of Apoptotic Cells in Spheroids
Native Spheroids.
Apoptotic and living cells in native spheroids were stained with two discriminating fluorescence dyes (Live/Dead-Viability/ Cytotoxicity Kit; Molecular Probes, MoBiTec, Göttingen, Germany). 10 standard spheroids were harvested and incubated for 30 min with calcein AM and ethidiumbromide-homodimer following the manufacturer's instructions. After centrifugation for 1 min at 500 g, the supernatant was aspirated and the spheroids were transferred on a glass slide and analyzed by confocal laser scanning microscopy (LSM410; Zeiss, Jena, Germany).
Fixed Spheroids.
Apoptotic cells were visualized by histochemical detection of nucleosomal fragmentation products (TUNEL) applying the in situ Cell Death Detection Kit (Boehringer Mannheim, Germany) following the manufacturer's instructions. In brief, nucleosomal fragmentation products in sections of paraffin embedded spheroids were detected after deparaffination and proteinase K digestion by 3′ end labeling with fluorescein-dUTP using terminal deoxynucleotidyl transferase. Labeling was visualized either directly by fluorescence microscopy or indirectly after incubating the sections with peroxidase labeled anti-fluorescein antibody and developing with diaminobenzidine as substrate.
DNA Fragmentation Assays
DNA Laddering.
To assess DNA fragmentation in nonadherent, suspended EC, cells were harvested and pelleted by centrifugation at 13,000 g (4°C) for 20 min. The pellet was diluted in 500 μl 10 mM Tris containing 10 mM EDTA (pH 8.0) and incubated with 20 μg/ml RNase A (Sigma, Deisenhofen, Germany) for 25 min on ice. Subsequently, 500 μl of 10 mM Tris containing 10 mM EDTA and 2% SDS (pH 8.0) was added and the lysate was incubated with 200 μg/ml Proteinase K (Sigma, Deisenhofen, Germany) at 37°C for 3 h. DNA was extracted by double chloroform/phenol-extraction-method and precipitated overnight in 0.5 M NaCl containing 50% isopropanol (−70°C). After centrifugation at 13,000 g for 15 min at 4°C, the DNA was dissolved in 10 mM Tris containing 10 mM EDTA (pH 8.0) and analyzed on a 1.6% agarose gel.
DNA Fragmentation ELISA.
Quantitation of fragmented DNA was performed by ELISA (Cell Death Detection Elisa Kit; Boehringer Mannheim, Germany). Fragmented DNA of 10 standard spheroids was extracted by lysis for 60 min at room temperature with vigorous shaking. The extracts were centrifuged for 10 min at 13,000 g and 300 μl of the supernatant was incubated with peroxidase-labeled anti-DNA antibody and biotinylated anti-histone antibody in streptavidin-coated microtiter plates following the manufacturer's instructions. After washing, binding of mono- and oligonucleosomal DNA was visualized by developing with the peroxidase substrate ABTS (2,2′-Azino-di[3-ethylbenzthiazolin-sulfonat]). Plates were analyzed at 405 nm using an automated microtiter plate reader (EAR 400AT; SLT Lab Instruments, Salzburg, Austria).
Results
Endothelial Cell Spheroids Form Spontaneously and Differentiate Over Time
To establish procedures for the generation of stable endothelial cell spheroids, we employed similar techniques that have been developed for the generation of tumor cell spheroids. Seeding of suspended EC in nonadhesive tissue culture dishes led to the formation of multicellular aggregates within 4 h. Depending on the methocel concentration in the medium, the average size of the resulting endothelial cell spheroids varied from very small aggregates (<50 cells) to larger aggregates of several thousand cells with several hundred micrometer in diameter. Routinely, we used 20% methocel which resulted in the formation of spheroids with an average diameter between 100 and 300 μm. As an alternative spheroid generation technique, we seeded a defined number of suspended EC in nonadhesive 96-well round bottom plates. Applying this technique, essentially all cells seeded in one well contributed to the formation of a single standardized spheroid (Fig. 1).
Figure 1.

Formation of a standard spheroid of BAE cells. A defined number of cells (3,000) was seeded in nonadhesive 96-well round-bottom plates. As cells sediment over time, they aggregate within 2–4 h after which they remodel to form a compact rounded spheroid within 18 h. Essentially all suspended cells contribute to the formation of a single spheroid. Similar results are obtained with HUVEC. Bar, 200 μm.
Both, random as well as standard spheroids differentiated spontaneously over time if they were maintained in suspension culture. Within 24 h of spheroid formation, the cells in the spheroids organized to establish a surface layer of elongated cells that formed a monolayer and a center of unorganized cells (Fig. 2 A). Spheroids of EC could be maintained in suspension culture for several weeks. After 7 d, the unorganized center cells had disappeared and the endothelial cell spheroids consisted of an almost acellular core and a surface layer of elongated cells (Fig. 2 B).
Figure 2.
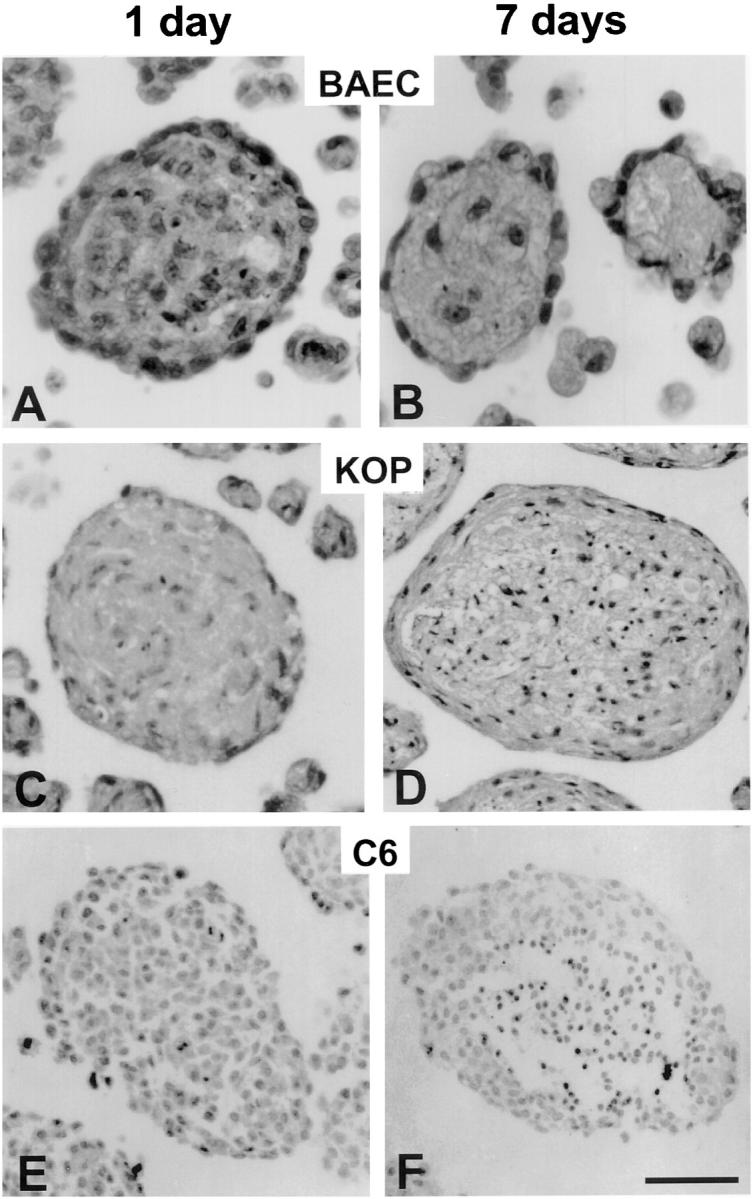
Differentiation of endothelial cell spheroids (A and B) compared with spheroids generated from esophageal epithelial cells (C and D) and C6 glioma cells (E and F). (A) 1-d BAEC spheroid with surface layer of elongated cells and a core of unorganized cells and apoptotic bodies. (B) 7-d BAEC spheroids with surface monolayer and an almost acellular core. (C) 1-d KOP cell spheroid. (D) 7-d KOP cell spheroids with multilayered elongated surface cells and a core of unorganized cells with numerous apoptotic bodies. (E) 1-d C6 glioma spheroid. (F) 7-d C6 glioma spheroids with a necrotic center and a peripheral ring of viable, but undifferentiated surface cells. Bar, 50 μm.
To examine if other nontransformed cells differentiate spontaneously in three-dimensional cell culture systems, we formed spheroids of esophageal epithelial (KOP) cells. These cells grow in two-dimensional cell culture to form a growth arrested monolayer. Like EC, spheroids of KOP cells form multicellular aggregates in suspension culture with a core of unorganized cells and a surface layer of elongated cells (Fig. 2 C). After 7 d, KOP spheroids consisted of a core of dead cells with numerous apoptotic bodies and several layers of elongated viable surface cells indicative of an attempt to form a multilayered epithelium (Fig. 2 D). As control tumor cells that have been widely used in spheroid experiments (for example see reference 46), we used C6 glioma cells. As expected, these cells readily form multicellular aggregates within hours. They do, however, fail to organize and form a uniform mass of cells (Fig. 2 E). After prolonged periods of culture, C6 glioma spheroids develop a necrotic center surrounded by several layers of viable but unorganized surface cells (Fig. 2 F).
Integration of EC in the Organized Spheroid Surface Monolayer Induces Polar Endothelial Cell Differentiation and Cellular Quiescence
The initial series of experiments had indicated that EC maintained in suspension in three-dimensional spheroids organize spontaneously to develop a two compartment system consisting of a surface layer of differentiated cells and a core of unorganized cells that disappear over time. Differentiation into a two compartment system could be confirmed by ultrastructural analyses of cultured endothelial cell spheroids. The surface layer of cells was found to consist of a continuous monolayer of elongated cells (Fig. 3 A). Well-differentiated cell–cell contacts with parallel electron dense strands were indicative of the formation of tight junctions (Fig. 3 B) that indicated a high degree of cellular differentiation (30). Numerous apoptotic bodies were present in the center of the spheroids (Fig. 3 C) as well as in surface EC that had failed to integrate into the surface monolayer (Fig. 3 A, arrows).
Figure 3.

Ultrastructural analysis of a 5-d BAEC spheroid. (A) BAEC spheroids consist of a fully differentiated and polarized surface layer of cells and a core of unorganized cells. The surface cells form a continuous monolayer that may develop electron dense strands indicative of tight junctional cell–cell contacts (magnification in B). The center cells undergo apoptosis as evidenced by the presence of numerous apoptotic bodies (magnification in C). Likewise, cells on the spheroid surface that did not integrate into the monolayer undergo apoptosis (A, arrows). Bars: (A and C) 10 μm; (B) 0.5 μm.
To assess the degree of cellular quiescence of spheroid EC compared with monolayer EC, we determined the fraction of proliferating cells using BrdU labeling. EC in the spheroid center showed no detectable incorporation of BrdU. In contrast, EC in the surface monolayer of 1 d spheroids had a very low labeling index (BAEC: 0.97 ± 1.43%; HUVEC: 0.08 ± 0.35%) that declined to undetectable levels within 5 d. In comparison, cultured monolayer EC had much higher BrdU labeling indices even when analyzing cell populations that were cultured for an additional day after acquiring confluence (BAEC [subconfluent]: 72.6 ± 8.8%; BAEC [confluent]: 9.0 ± 1.6%; HUVEC [subconfluent]: 48.7 ± 7.9%; HUVEC [confluent]: 8.6 ± 3.3%).
Organized Surface Spheroid EC Can Be Induced to Express Differentiation and Activation Antigens
We next studied the capacity of spheroid EC to express differentiation and activation antigens. For these experiments, 1-d spheroids were incubated with various cytokines and the expression of CD31 (PECAM-1), ICAM-1, VCAM-1, and CD34 was analyzed cytochemically. Most of the spheroid EC expressed the panendothelial cell surface molecule CD31 (Fig. 4 A). Expression was most intense in the cells closest to the spheroid surface and gradually declined towards the spheroid center. The center EC expressed CD31 uniformly on the cell surface. In contrast, the surface layer of EC expressed CD31 selectively at lateral and basal cell–cell contacts and only minimally on the luminal cell surface. Cytokine stimulation experiments with FGF-2, VEGF, IL-1, and TNF-α did not affect endothelial CD31 expression in multicellular spheroids (Fig. 5).
Figure 4.
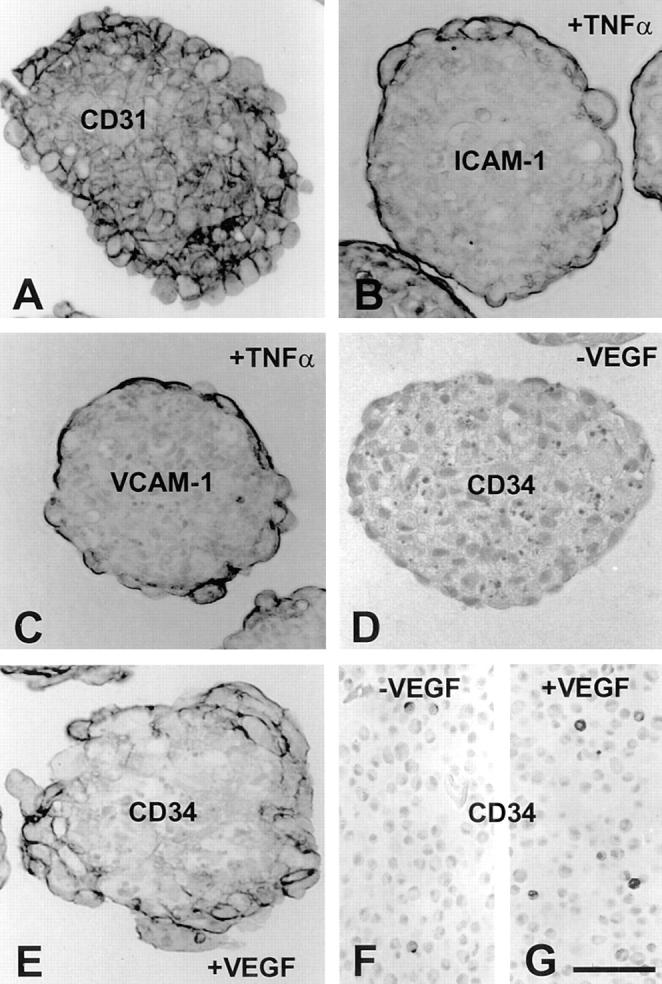
Expression of endothelial cell surface adhesion molecules by HUVEC spheroids. (A) Most spheroid EC express the panendothelial cell marker CD31. (B) Expression of ICAM-1 in HUVEC spheroids after stimulation with TNF-α for 24 h. (C) Expression of VCAM-1 in HUVEC spheroids after stimulation with TNF-α for 24 h. (D) Untreated HUVEC spheroids express barely detectable levels of CD34. (E) Expression of CD34 in HUVEC spheroids after stimulation with VEGF for 24 h. (F and G) Expression of CD34 in cross sections of harvested HUVEC monolayer embedded in paraffin as single cells. CD34 expression is downregulated in two-dimensional culture (F). Stimulation of monolayer cultures with VEGF only minimally increases the number of CD34-positive cells (G). Bar, 50 μm.
Figure 5.

Summary of expression pattern of endothelial cell adhesion molecules in monolayer culture and spheroids. EC constitutively express CD31. Intensity of expression does not change after cytokine stimulation. Stimulation of EC with IL-1 or TNF-α induces expression of ICAM-1 and VCAM-1, which is in the spheroids limited to the luminal aspect of the surface monolayer. CD34 is only expressed after VEGF stimulation. The induction of CD34 by VEGF is dependent on the differentiation status of the cells and their microenvironment as it is only inducible in spheroid EC and only minimally in monolayer cells.
IL-1 and TNF-α stimulation of endothelial cell spheroids was capable to induce surface expression of inducible endothelial cell adhesion molecules such as ICAM-1 (CD54) and VCAM-1 (CD106). As shown in Fig. 4, B (ICAM-1) and C (VCAM-1) expression of these molecules was limited to the differentiated surface layer of the spheroids. These cells expressed ICAM-1 and VCAM-1 selectively on the luminal cell surface.
Based on these experiments, we next asked if other differentiation antigens of EC could be induced in spheroid culture. CD34 is a surface molecule that is expressed by most EC in vivo (9). However, upon transfer of cells in culture endothelial cell CD34 expression is rapidly downregulated (16). Endothelial cell CD34 expression can be maintained to some extend by restraining cell proliferation and promoting cell contact (16). However, no cytokines or specific microenvironmental requirements have been characterized that are capable to upregulate endothelial CD34 expression. Spheroid EC that were generated from EC previously maintained in two-dimensional monolayer culture expressed barely detectable levels of CD34 (Fig. 4 D). Exposure of spheroids to different cytokines revealed that VEGF is capable to prominently induce endothelial cell CD34 expression (Fig. 4 E). Expression is limited to the surface layer of EC. However, in contrast to the polar expression of CD31, ICAM-1, and VCAM-1 by the surface cells CD34 is uniformly expressed on the cell surface of the responsive cells. The induction of endothelial CD34 expression was found to be a VEGF specific effect as other cytokines failed to induce CD34 expression (Fig. 5). Exposure of VEGF-treated spheroids to TNF-α led to a complete downregulation of CD34 expression in spheroid EC (data not shown). Finally, the responsiveness to VEGF in the induction of CD34 was found to be critically microenvironment dependent since it could be observed in spheroid culture (Fig. 4 E) and only to a lesser degree in monolayer culture (Fig. 4, F vs. G). The microenvironment dependency of VEGF-mediated CD34 expression was also indicated by the fact that 2-d spheroids were more responsive to VEGF stimulation than 1-d spheroids (data not shown).
Spheroid Organization Is Dependent on Integrin-mediated Cell–Matrix Contacts That Are Crucial for the Survival of the Organized Surface Spheroid EC
To identify adhesive interactions of the surface monolayer cells that might be responsible for preventing these cells from undergoing apoptosis, endothelial cell spheroids were treated with RGD peptides or control RAD peptides (30 μM) to inhibit integrin-dependent adhesive interactions. Quantitative analysis of apoptosis using the DNA fragmentation ELISA indicated a significant increase in total EC apoptosis in RGD-treated spheroids (P < 0.01; RGD-treated vs. untreated control: 216.1 ± 40.1%; RAD-treated vs. untreated control: 113.8 ± 15.3%). To qualitatively trace apoptotic cells, apoptosis was monitored using TUNEL staining (Fig. 6 A) or staining with ethidium bromide (Fig. 6 B). As indicated in Fig. 6 D, treatment of spheroids with RGD peptides led to apoptosis of the surface monolayer EC as well as to an increased level of apoptosis in the center of the spheroids. If maintained in suspension culture, RGD-treated endothelial cell spheroids gradually lost all signs of differentiation and began to fall apart (Fig. 6 F). In contrast, control RAD revealed the specificity of the RGD mediated antiadhesive effects demonstrating intact spheroids (Fig. 6 E) without apoptosis of the surface cells (Fig. 6 C).
Figure 6.
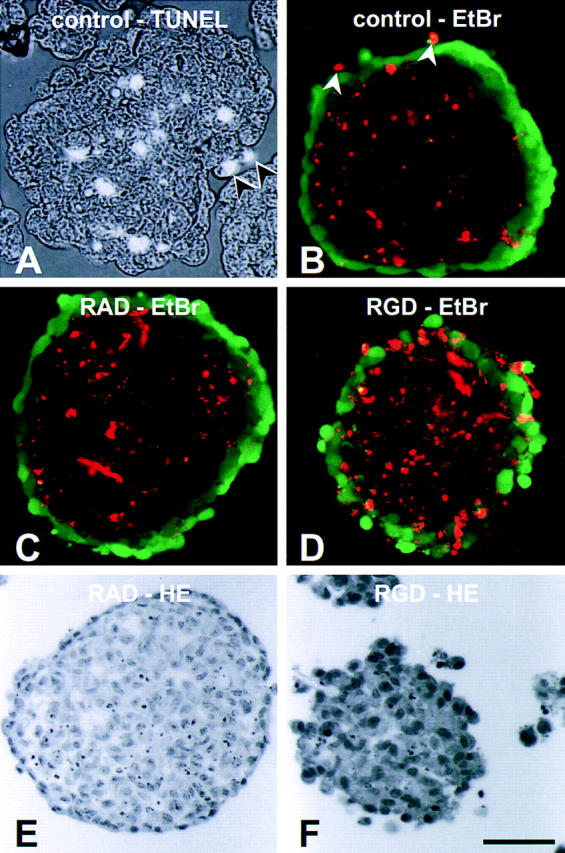
Analysis of endothelial cell apoptosis in 1-d BAEC spheroids. Apoptosis of EC was detected either by TUNEL staining (A, random spheroids) or by ethidium bromide staining (B, standard spheroid; calcein AM used as surface stain). Both techniques led to similar results. Apoptosis of BAE cells is limited to the cells in the center of the spheroids (A and B) as well as to surface BAE cells that have not integrated into the surface monolayer (A and B, arrows). (C) Treatment of BAEC spheroids with RAD peptides (30 μM) does not affect endothelial cell apoptosis ([_E_] hematoxylin-stained sectioned RAD-treated spheroid). (D) Treatment of BAEC spheroids with RGD peptides (30 μM) for 24 h leads to a disruption of the integrity of the surface monolayer and induces surface cell apoptosis ([_F_] hematoxylin-stained sectioned RGD-treated disintegrating BAEC spheroid). Bar, 50 μm.
Center Spheroid EC Do Not Differentiate and Are Dependent on Survival Factors to Prevent Apoptosis
The experiments performed so far had indicated that apoptosis of the center EC will eventually lead to the formation of a spheroid with an acellular core. We, thus reasoned that the unorganized center spheroid cell population might be a suitable target cell population to study the effects of endothelial survival factors. VEGF and FGF-2 regulate numerous critical endothelial cell functions and both of these cytokines have been proposed to act as endothelial survival factors (1, 11, 27, 43, 47, 53).
Treatment of HUVEC spheroids with VEGF and FGF-2 significantly inhibited apoptosis of center spheroid EC (Fig. 7, A, C, and D). VEGF (100 ng/ml) and FGF-2 (30 ng/ml) exerted additive effects reducing spheroid endothelial cell apoptosis by >50% (Fig. 7 A). Treatment with a neutralizing monoclonal α-FGF-2 antibody increased center endothelial cell apoptosis above the already high baseline levels (Fig. 7 A). Exogenous VEGF rescued from neutralizing α-FGF-2 antibody mediated increased apoptosis to a similar degree as would be expected from the observed additive effects of VEGF and FGF-2 (Fig. 7 A). In contrast, addition of a neutralizing α-VEGF antibody had no effect on baseline apoptosis, suggesting that FGF-2 and not VEGF is capable of controlling survival of immature EC in an autocrine manner.
Figure 7.


Quantitative (A and B) and qualitative (C–F) analysis of endothelial cell apoptosis (A, C, and D, HUVE cells; B, E, and F, BAE cells) after treatment with different cytokines. (A and B) A defined number of spheroids was stimulated with different cytokines for 24 h and the presence of nucleosomal fragmentation products was quantitated by ELISA (see Materials and Methods). Data are expressed as percentage deviation of the level of apoptosis from the untreated control spheroid population (shown in C [HUVEC] and E [BAEC]). The figure shows the mean ± SEM of three different experiments performed in duplicate (*P < 0.05; **P < 0.01). (C) control HUVEC spheroid; (D) VEGF + FGF-2 stimulated HUVEC spheroid. (E) control BAEC spheroid treated with isotype matched nonneutralizing monoclonal antibody to FGF-2 (4 μg/ml). (F) BAEC spheroid treated with neutralizing monoclonal antibody to FGF-2 [4 μg/ml]). Bar, 50 μm.
Similar, albeit qualitatively different results were obtained in the analysis of VEGF and FGF-2 mediated survival effects on BAE cells. Compared with HUVEC, BAE cells had a much lower baseline rate of apoptosis (Fig. 7, E vs. C). BAE cell behavior in culture is known to be strongly influenced by the endogenous expression of FGF-2 and BAEC have, thus, been widely studied to assess FGF-2 mediated autocrine endothelial cell growth control (35, 51, 56). Corresponding to these findings, strongest effects on the survival of BAEC in spheroids were observed when BAEC spheroids were treated with a neutralizing α-FGF-2 antibody (4 μg/ml) that increased endothelial cell apoptosis by 60% (Fig. 7, B and E vs. F). Treatment with a neutralizing α-FGF-2 antibody affected the center cells and not the differentiated cells of the surface monolayer (Fig. 7 F). Corresponding to the induction of endothelial cell apoptosis by neutralizing α-FGF-2 antibody, addition of exogenous FGF-2 further reduced baseline endothelial cell apoptosis by >30%. Addition of exogenous VEGF or neutralizing α-VEGF antibody had no effect on BAEC survival in spheroids (Fig. 7 B), despite the fact that BAEC in spheroids expressed VEGF-R1 (Flt-1) and VEGF-R2 (Flk-1/KDR; data not shown). Surprisingly and much in contrast to the additive effect of VEGF and FGF-2 in HUVEC, VEGF attenuated the exogenous FGF-2–mediated reduction of baseline BAE cell apoptosis (Fig. 7 B). This effect was reproducibly observed in all experiments. Several studies have indicated synergistic activities of VEGF and FGF-2 on endothelial cell function (3, 22, 38) and some reports even suggest that VEGF and FGF-2 mediated activities are functionally dependent on each other (33, 48). In light of these recent findings, the molecular basis of the observed differences in the responsiveness of the two cell populations to VEGF is focus of ongoing experiments that may well shed further light into the mechanistic functional interaction of VEGF and FGF-2.
The survival mediating effect of VEGF and FGF-2 was analyzed in 24 h short-term experiments. To verify if exogenous survival factors could rescue the center spheroid cells for prolonged periods of time, spheroids of HUVEC and BAE cells were either incubated with VEGF and FGF-2 or with neutralizing antibodies to these cytokines for 4 d. Reflecting their low baseline autocrine activity, HUVEC spheroids cultured for 4 d without exogenous survival factors consisted of an almost acellular core and a differentiated surface monolayer (Fig. 8 A). In contrast, HUVEC spheroids continuously maintained in 50 ng/ml of VEGF and 30 ng/ml of FGF-2 had a core of viable EC (Fig. 8 C). BAEC spheroids had a higher rate of autocrine activity as indicated by a lower baseline rate of center spheroid cell apoptosis. After 4 d in culture without exogenous survival factors, BAEC spheroids had a differentiated surface monolayer and a center of still viable unorganized cells along with numerous dying cells with condensed apoptotic bodies (Fig. 8 B). Addition of a neutralizing α-FGF-2 antibody led to an increase in the rate of apoptosis of the center spheroid BAE cells, but did not affect the survival of the monolayer BAE cells (Fig. 8 D). This observation provided additional evidence for the notion that the cells of the surface monolayer matured to a degree that rendered them independent of the activities of exogenous as well as endogenous survival factors.
Figure 8.

Effect of long-term treatment (4 d) of EC spheroids with the survival factors VEGF and FGF-2 and a neutralizing monoclonal antibody to FGF-2. Cytokines and antibodies were added at the beginning of the experiment and again after 2 d. Spheroids were analyzed after 4 d. (A) control HUVEC spheroid. (B) Control BAEC spheroid. (C) HUVEC spheroid treated with VEGF (50 ng/ml) and FGF-2 (30 ng/ml). (D) BAEC spheroid treated with a neutralizing monoclonal antibody to FGF-2 (4 μg/ml). Bars: (A and C) 30 μm; (B and D) 50 μm.
Single Suspended EC Die by Apoptosis and Can Not Be Rescued by Exogenous Survival Factors
The experiments described above had indicated that the center spheroid EC were the target cell population of the activities of endothelial survival factors. Consequently, we next studied the responsiveness of single suspended EC to exogenously administered survival factors. Inhibition of anchorage-dependent cell spreading of single cells has previously been demonstrated to trigger apoptosis of cultured EC (40). Corresponding to these findings, single suspended EC (nonadhesive tissue culture dishes, high concentrations of methocel) were not capable to survive and died within hours (HUVEC) and days (BAEC), respectively. Viability of EC in suspension culture was assessed by quantitating their adhesiveness after transfer to adhesive tissue culture dishes (Fig. 9 A). The reduction in endothelial cell readhesiveness after transfer to adhesive tissue culture dishes was paralleled by massive nucleosomal fragmentation as evidenced by DNA laddering that suggested that the nonadhesive cells died by apoptosis (Fig. 9 B). Based on these observations, we exposed single suspended EC to the survival factors that had proved to be effective in spheroid culture. As shown in Fig. 9 C and corresponding to recent studies with FGF-2 (27), none of the treatments (VEGF and FGF-2 alone or in combination) affected the rate of apoptosis of single suspended EC suggesting that these cells are not responsive to the survival factor activities of VEGF and FGF-2.
Figure 9.

Analysis of the fate of single nonadherent EC and of their responsiveness to exogenously administered survival factors. The experiments shown were performed with HUVEC, similar results were obtained with BAE cells. (A) HUVEC were grown at low seeding density in nonadhesive tissue culture dishes in the presence of high concentrations of methocel to prevent cellular aggregation (see Materials and Methods). The cells were transferred to adhesive tissue culture dishes after the indicated periods of time and the number of adherent cells per microscopic field of view (MFV) was quantitated after 2 h. Adherence was quantitated by counting the cells that had adhered as single cells (gray bars) and the cells that had adhered in clustered groups that was indicative of cellular aggregation in the methocel medium (black bar). The data shown represent the mean ± SD of three independent experiments performed in triplicate. (B) The reduction of single cell readhesiveness as shown in A is paralleled by a pronounced increase of single cell apoptosis as evidenced by intense DNA laddering. (C) Quantitation of nucleosomal fragmentation by ELISA (see Materials and Methods) to analyze the responsiveness of single suspended HUVEC to exogenously administered survival factors.
Cell–Cell Contacts of Unorganized EC are a Prerequisite for Survival Factor Responsiveness
To identify parameters that mediate the responsiveness of EC to survival factors, we assessed the responsiveness of single and aggregated EC to survival factors after treatment of the cells with cytochalasin D. The rationale of these experiments was based on the idea that cytochalasin D–treated cells are still able to establish cell–cell contacts. Disruption of the cytoskeleton does, however, inhibit three-dimensional spheroidal organization. The experiment was consequently aimed at answering if cell–cell contacts are sufficient to mediate endothelial survival factor responsiveness or if the complex cellular interactions including shape changes as they occur in the center of the spheroids would be required to mediate survival factor responsiveness.
As shown in Fig. 10 A, cytochalasin D treatment during HUVEC spheroid formation did not affect the rate of apoptosis. As expected, however, cytochalasin D treatment prevented spheroid formation leaving an aggregate of loosely adherent cells that failed to organize three-dimensionally (Fig. 10, C vs. B). Cytochalasin D–treated cellular aggregates were responsive to the survival factor activities of exogenously administered VEGF and FGF-2 (Fig. 10 A) reducing HUVE cell apoptosis by ∼50% (P < 0.05). In contrast, deprivation of Ca++-dependent cellular adhesion by EGTA treatment inhibited cellular aggregation and, thus, left the cells unresponsive to the activities of survival factors (Fig. 10 A). Taken together, the data indicate that cell–cell contacts are a requisite for rendering EC survival factor responsive.
Figure 10.
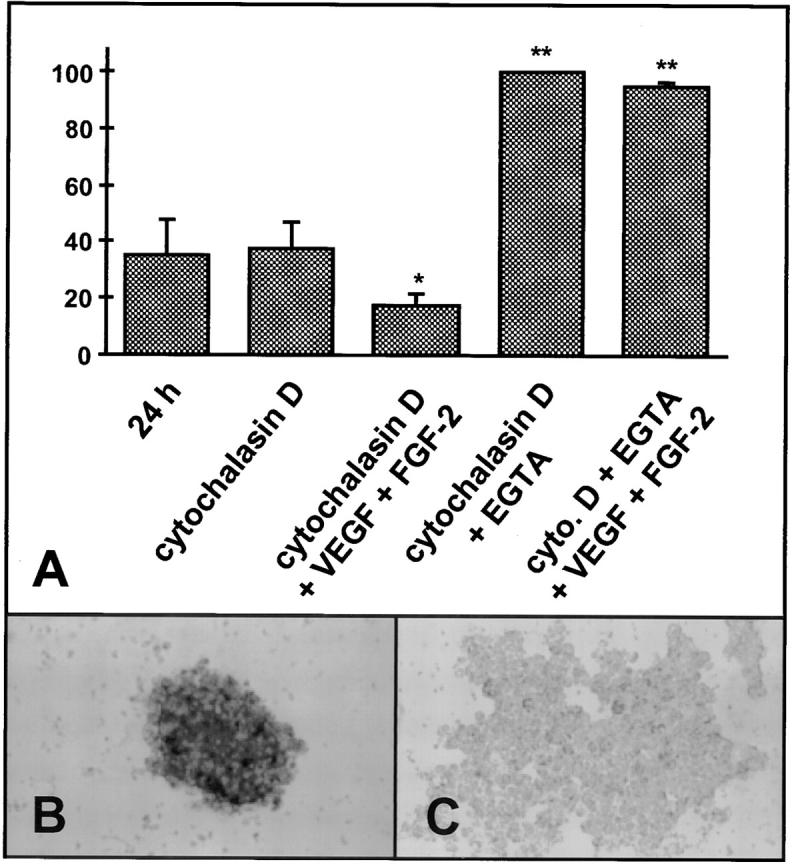
Analysis of the effect of cytochalasin D treatment on VEGF and FGF-2 mediated survival factor activities in HUVEC spheroids (quantitation by ELISA; see Materials and Methods; results [mean ± SD of three independent experiments performed in duplicate] are expressed in% of the maximum). (A) Cytochalasin D treatment (5 μg/ml) does not affect apoptosis of aggregated EC. (B) Control HUVEC spheroid. (C) HUVEC aggregation after cytochalasin D treatment. Cytochalasin D–treated HUVEC are responsive to the survival factor activities of VEGF and FGF-2. Treatment of cells with EGTA (5 mM, 24 h) disrupts calcium-dependent cell–cell contacts resulting in highest levels of apoptosis. In contrast to cytochalasin–treated aggregated cells with cell– cell contacts, EGTA-treated single cells are not responsive to VEGF and FGF-2.
Discussion
Three-dimensional tissue culture systems have been used widely in the context of tumor cell biology (7, 31, 36, 49). Likewise, the differentiation of individual cell types in three-dimensional cultures of embryonic stem cells has contributed to the understanding of cell lineages and differentiation phenomena (10, 29, 52). Spheroid cultures of embryonic stem cells can be used for the organotypic differentiation of EC in embryoid bodies that are lined by a peripheral layer of angioblastic cells and a core of hematopoietic cells (10, 52). We, thus, hypothesized that spheroid cultures of nontransformed EC may provide a suitable differentiation model to study differentiated endothelial cell functions.
EC maintained in suspension cultures under conditions that allow cellular aggregation readily form three-dimensional spheroids. However, in contrast to tumor cell spheroids endothelial cell spheroids organize to establish a two compartment system consisting of a surface monolayer of differentiated cells and a center of unorganized cells that will die by apoptosis if they are not rescued by survival factors. This organoid differentiation is not restricted to EC. Instead, we observed that nontransformed epithelial cells that grow as a monolayer in two-dimensional culture form a multilayered epithelium in three-dimensional spheroid culture suggesting that spheroid culture techniques may be used to study differentiation phenomena of a variety of different nontransformed cell types.
We used the endothelial cell spheroid differentiation model to trace the fate of the unorganized center EC and their responsiveness to survival factors as well as the differentiation capacity of the surface monolayer. The data support a model of endothelial cell organization and differentiation in spheroid culture that is summarized in Fig. 11. Single suspended EC are not capable to survive and rapidly die by apoptosis. They are not responsive to the activities of survival factors. Spheroidal aggregation of EC mediates cell–cell contacts and renders the cells survival factor responsive. Survival factor responsiveness is mediated by cell–cell contacts and does not require cell shape changes. Upon spheroid organization, the surface cells establish a polarized elongated continuous monolayer. The differentiation characteristics of this surface monolayer goes beyond the degree of differentiation that can be obtained in two dimensional culture. This conclusion is supported by (a) the observed polarized expression of endothelial cell surface molecules (basal and lateral expression of CD31; luminal expression of ICAM-1 and VCAM-1), (b) the ultrastructural characteristics of the surface monolayer with well developed cell–cell contacts, (c) the prominent induction of surface molecules that can only minimally be induced in two-dimensional culture systems (VEGF mediated induction of CD34), and (d) the lower turnover rates of spheroid surface EC compared with confluent monolayer EC reflecting a higher degree of cellular quiescence. Finally, if EC spheroids are not exposed to survival factors, the center cells completely undergo apoptosis leaving an EC spheroid consisting of an acellular core and a surface monolayer that has become independent of the activities of survival factors.
Figure 11.

Model of endothelial cell organization and differentiation in three-dimensional spheroids. Single suspended EC are neither capable to survive nor responsive to the activities of survival factors and die by apoptosis (anoikis; apoptosis as a consequence of a loss of matrix anchorage). EC that aggregate to form three-dimensional spheroids establish cell–cell contacts that renders the cell survival factor responsive. Subsequently, nontransformed EC in spheroids organize to establish a two compartment system consisting of a peripheral surface monolayer of differentiated, polarized, survival factor–independent cells, and a core of survival factor–dependent unorganized cells. If not continuously exposed to survival factors, the center EC will die by apoptosis to produce a three-dimensional spheroid that consists of an acellular core and a differentiated surface monolayer of EC.
One of the most surprising findings of this study was the observed regulation of endothelial cell CD34 expression by VEGF. CD34 is a differentiation antigen expressed by most EC in vivo that is downregulated upon transfer of cells into standard in vitro cell culture systems (9, 16, 17, 20). Endothelial cell CD34 expression has been reported to be reciprocally regulated to the expression of inflammation associated endothelial cell adhesion molecules (16). Likewise, expression of CD34 has been shown to be upregulated by EC during angiogenesis (25, 57). Both findings are compatible with the observed regulation of CD34 by VEGF. More intriguingly, however, CD34 has been characterized as a marker of hemangioblastic stem cells, which is downregulated in the hematopoietic lineage and maintained in the angioblastic lineage (4, 25, 57). This expression pattern parallels the expression of VEGF-R2 (KDR/Flk-1), which is similarly expressed by hemangioblastic stem cells being downregulated in the hematopoietic lineage and not in the angioblastic lineage (55). CD34 has functionally been characterized as a ligand for L-selectin (8, 39), but the function of its endothelial cell expression has not been well defined. Likewise, analysis of the phenotype CD34 deficient mice has not been very instructive in defining the function of endothelial cell CD34 expression (14).
In addition to studying the differentiation of the surface endothelial monolayer, the endothelial cell spheroid model proved to be useful to analyze the functions and to characterize the phenotypic properties of the target cell population of endothelial survival factors. A number of elegant in vivo studies have demonstrated the dependence of EC in immature capillaries on exogenous survival factors (1, 11). VEGF appears to play a central role as endothelial survival factor as was originally demonstrated in the immature retinal vasculature of newborn rats (1). Likewise, the sudden downregulation of VEGF in conditionally VEGF overexpressing tumors leads to a collapse of the vasculature with detachment and subsequent apoptosis of immature capillary EC (11). Several in vitro studies have provided additional evidence for the central role of VEGF in acting as an endothelial survival factor (43, 47, 53) and VEGF has also been proposed to act as survival factor for hematopoietic stem cells (28).
This study employed the endothelial cell spheroid model to analyze the survival mediating activities of VEGF and FGF-2. The data indicate that both VEGF and FGF-2 are capable to act as endothelial survival factors with endogenous expression of FGF-2 playing a critical role as an autocrine growth factor in regulating endothelial cell survival. More importantly, this study could characterize the phenotype of EC that is responsive and dependent on survival factors. Single suspended EC can only survive for short periods of times and are not responsive to survival factors. Circulating hematopoietic cells as well as the recently characterized circulating angioblastic stem cell (4) can survive without cell–cell contacts or anchorage dependence and are apparently not dependent on survival factors. Correspondingly, techniques have been described to differentiate EC from circulating angioblastic cells using VEGF and adhesion to fibronectin as selection and differentiation pressure (4). In light of the findings presented in this study, it will be interesting to see at which stage of differentiation angioblastic cells become survival factor dependent.
In contrast to the survival factor dependence of the unorganized cells in the spheroid center, the cells of the surface monolayer differentiated to a degree that rendered them survival factor independent. A number of studies have demonstrated that cellular functions including cell survival depend on the tensional integrity (tensegrity) of the cells that is directly affected by the shape of the cells (13, 42). Likewise, the endothelial cell extracellular matrix does not only act as a morphogenetic regulator (24), but is also known to mediate survival signals (34) that involves integrin mediated signaling (19, 42, 44, 50). Integrin-mediated signal transduction is known to be at least partially controlled by cell shape as evidenced by the inability of RGD-coated beads to inhibit apoptosis of rounded nonadherent EC (40). Corresponding to these findings, the unorganized center spheroid cells are surrounded by extracellular matrix components and fail to organize into a polarized elongated monolayer. Cell–cell contacts may render these cells survival factor responsive, but not survival factor independent. In contrast, the surface cells of the spheroids grow on an extracellular matrix and differentiate to form a polarized monolayer of elongated cells that has become survival factor independent. The nature of the survival mediating extracellular matrix in vivo and in vitro remains to be elucidated but it appears that endothelial cell spheroids offer a useful approach to this end. Recent experiments have suggested that pericyte contact renders EC survival factor independent as long as the cell are exposed to a provisional matrix (12). According to these findings, synthesis of a mature extracellular matrix is a critical step in vessel wall assembly and endothelial cell maturation.
Taken together this study has developed a spheroid model for the analysis of specific differentiation phenomena of EC as well as the functions of endothelial survival factors. It is concluded that spheroid models of nontransformed cells are not just applicable to the analysis of differentiated endothelial cell functions, but that they may prove to be powerful tools to study the differentiation of numerous other nontransformed cell types. Likewise, the endothelial cell spheroid model may prove to be useful for a number of other aspects of endothelial cell differentiation such as three-dimensional angiogenic maturation (Korff, T., and H.G. Augustin, manuscript in preparation), regulation of endothelial cell surface receptor expression, and the analysis of cellular interactions of EC with mural cells. Finally, the described techniques to generate endothelial cell spheroids of defined size may prove to be useful for therapeutic strategies aimed at reintroducing genetically manipulated EC into specific capillary beds after similar experimental approaches as have been described for the microsphere mediated delivery of angiogenic cytokines to sites of cardiac ischemia (2).
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB500, C3).
Abbreviations used in this paper
BAEC
bovine aortic endothelial cells
EC
endothelial cells
HUVEC
human umbilical vein endothelial cells
Footnotes
The authors would like to acknowledge the excellent technical assistance of Mrs. Renate Dietrich. We thank Dr. Franz-J. Kaup (DPZ, Göttingen, Germany) for assistance with the electron microscopic analyses as well as Dr. Irina Majoul and Dr. Reinhard Jahn (MPI, Göttingen, Germany) for providing access to their laser scanning microscope. We thank Dr. Georg Breier (MPI, Bad Nauheim, Germany) for providing C6 glioma cells and Dr. R. Riebe (BFAF, Insel Riems, Germany) for providing KOP cells.
Address all correspondence to Dr. Hellmut G. Augustin, Cell Biology Laboratory, Department of Gynecology and Obstetrics, University of Göttingen Medical School, Robert-Koch-Str. 40, 37075 Göttingen, Germany. Tel.: 49 551 396573. Fax: 49 551 396711. E-mail: haugust@med.uni-goettingen.de
References
- 1.Alon T, Hemo I, Itin A, Pe'er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- 2.Arras M, Mollnau H, Strasser R, Wenz R, Ito WD, Schaper J, Schaper W. The delivery of angiogenic factors to the heart by microsphere therapy. Nat Biotechnol. 1998;16:159–162. doi: 10.1038/nbt0298-159. [DOI] [PubMed] [Google Scholar]
- 3.Asahara T, Bauters C, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, Isner JM. Synergistic effect of vascular endothelial growth factor and basic fibroblast growth factor on angiogenesis in vivo. Circulation. 1995;92:II365–II371. doi: 10.1161/01.cir.92.9.365. [DOI] [PubMed] [Google Scholar]
- 4.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:965–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 5.Augustin HG, Voss AK, Pauli BU. Senescence of aortic endothelial cells in culture: effects of basic fibroblast growth factor expression on cell phenotype, migration, and proliferation. J Cell Physiol. 1993;157:279–288. doi: 10.1002/jcp.1041570210. [DOI] [PubMed] [Google Scholar]
- 6.Augustin HG, Kozian DH, Johnson RC. Differentiation of endothelial cells: analysis of the constitutive and activated endothelial cell phenotypes. Bioessays. 1994;16:901–906. doi: 10.1002/bies.950161208. [DOI] [PubMed] [Google Scholar]
- 7.Bates RC, Buret A, van Helden DF, Horton MA, Burns GF. Apoptosis induced by inhibition of intercellular contact. J Cell Biol. 1994;125:403–415. doi: 10.1083/jcb.125.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baumhueter S, Singer MS, Henzel W, Hemmerich S, Renz M, Rosen SD, Lasky LA. Binding of L-selectin to the vascular sialomucin CD34. Science. 1993;262:436–438. doi: 10.1126/science.7692600. [DOI] [PubMed] [Google Scholar]
- 9.Baumhueter S, Dybdal N, Kyle C, Lasky LA. Global vascular expression of murine CD34, a sialomucin-like endothelial ligand for L-selectin. Blood. 1994;84:2554–2565. [PubMed] [Google Scholar]
- 10.Bautch VL, Stanford WL, Rapoport R, Russell S, Byrum RS, Futch TA. Blood island formation in attached cultures of murine embryonic stem cells. Dev Dyn. 1996;205:1–12. doi: 10.1002/(SICI)1097-0177(199601)205:1<1::AID-AJA1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 11.Benjamin LE, Keshet E. Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: induction of endothelial cell shedding and regression of hemangioblastoma-like vessels by VEGF withdrawal. Proc Natl Acad Sci USA. 1997;94:8761–8766. doi: 10.1073/pnas.94.16.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benjamin L, Hemo I, Keshet E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development. 1998;125:1591–1598. doi: 10.1242/dev.125.9.1591. [DOI] [PubMed] [Google Scholar]
- 13.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 14.Cheng J, Baumhueter S, Cacalano G, Carver-Moore K, Thibodeaux H, Thomas R, Broxmeyer HE, Cooper S, Hague N, Moore M, Lasky LA. Hematopoietic defects in mice lacking the sialomucin CD34. Blood. 1996;87:479–490. [PubMed] [Google Scholar]
- 15.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- 16.Delia D, Lampugnani MG, Resnati M, Dejana E, Aiello A, Fontanella E, Soligo D, Pierotti MA, Greaves MF. CD34 expression is regulated reciprocally with adhesion molecules in vascular endothelial cells in vitro. Blood. 1993;81:1001–1008. [PubMed] [Google Scholar]
- 17.Fina J, Molgard MV, Robertson D, Bradley NJ, Managhan PD, Sutherland DR, Baker MA, Graeves MF. Expression of the CD34 gene in vascular endothelial cells. Blood. 1990;75:2417–2426. [PubMed] [Google Scholar]
- 18.Folkman J, Haudenschild CC, Zetter BR. Long-term culture of capillary endothelial cells. Proc Natl Acad Sci USA. 1979;76:5217–5221. doi: 10.1073/pnas.76.10.5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- 20.Garlanda C, Dejana E. Heterogeneity of endothelial cells. Specific markers. Arterioscler Thromb Vasc Biol. 1997;17:1193–1202. doi: 10.1161/01.atv.17.7.1193. [DOI] [PubMed] [Google Scholar]
- 21.Gimbrone MA, Cotran RS, Folkman J. Human vascular endothelial cells in culture. Growth and DNA synthesis. J Cell Biol. 1974;60:673–684. doi: 10.1083/jcb.60.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goto F, Goto K, Weindel K, Folkman J. Synergistic effects of vascular endothelial growth factor and basic fibroblast growth factor on the proliferation and cord formation of bovine capillary endothelial cells within collagen gels. Lab Invest. 1993;69:508–517. [PubMed] [Google Scholar]
- 23.Gumkowski F, Kaminska G, Kaminski M, Morrissey LW, Auerbach R. Heterogeneity of mouse vascular endothelium. In vitro studies of lymphatic, large blood vessel and microvascular endothelial cells. Blood Vessels. 1987;24:11–23. [PubMed] [Google Scholar]
- 24.Ingber DE, Folkman J. How does extracellular-matrix control capillary morphogenesis. Cell. 1989;58:803–805. doi: 10.1016/0092-8674(89)90928-8. [DOI] [PubMed] [Google Scholar]
- 25.Ito A, Nomura S, Hirota S, Suda T, Kitamura Y. Enhanced expression of CD34 messenger RNA by developing endothelial cells of mice. Lab Invest. 1995;72:532–538. [PubMed] [Google Scholar]
- 26.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karsan A, Yee E, Poirier GG, Zhou P, Craig R, Harlan JM. Fibroblast growth factor-2 inhibits endothelial cell apoptosis by Bcl-2-dependent and independent mechanisms. Am J Pathol. 1997;151:1775–1784. [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh O, Tauchi H, Kawaishi K, Kimura A, Satow Y. Expression of the vascular endothelial growth factor (VEGF) receptor gene KDR, in hematopoietic cells and inhibitory effect of VEGF on apoptotic cell death caused by ionizing radiation. Cancer Res. 1995;55:5687–5692. [PubMed] [Google Scholar]
- 29.Keller GM. In vitro differentiation of embryonic stem cells. Curr Opin Cell Biol. 1995;7:862–869. doi: 10.1016/0955-0674(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 30.Lampugnani MG, Dejana E. Interendothelial junctions: structure, signalling and functional roles. Curr Opin Cell Biol. 1997;9:674–682. doi: 10.1016/s0955-0674(97)80121-4. [DOI] [PubMed] [Google Scholar]
- 31.Lincz LF, Buret A, Burns GF. Formation of spheroid structures in a human colon carcinoma cell line involves a complex series of intercellular rearrangements. Differentiation. 1997;61:261–274. doi: 10.1046/j.1432-0436.1997.6140261.x. [DOI] [PubMed] [Google Scholar]
- 32.Maciag T, Hoover GA, Stemerman MB, Weinstein R. Serial propagation of human endothelial cells in vitro. J Cell Biol. 1981;91:420–426. doi: 10.1083/jcb.91.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandriota SJ, Pepper MS. Vascular endothelial growth factor-induced in vitro angiogenesis and plasminogen activator expression are dependent on endogenous basic fibroblast growth factor. J Cell Sci. 1997;110:2293–2302. doi: 10.1242/jcs.110.18.2293. [DOI] [PubMed] [Google Scholar]
- 34.Meredith JE, Jr, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mignatti P, Morimoto T, Rifkin DB. Basic fibroblast growth factor released by single, isolated cells stimulates their migration in an autocrine manner. Proc Natl Acad Sci USA. 1991;88:11007–11011. doi: 10.1073/pnas.88.24.11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller-Klieser W. Three-dimensional cell cultures: from molecular mechanisms to clinical applications. Am J Physiol. 1997;273:C1109–C1123. doi: 10.1152/ajpcell.1997.273.4.C1109. [DOI] [PubMed] [Google Scholar]
- 37.Pauly RR, Passaniti A, Crow M, Kinsella JL, Papadopoulos N, Monticone R, Lakatta EG, Martin GR. Experimental models that mimic the differentiation and dedifferentiation of vascular cells. Circulation. 1992;86:III68–III73. [PubMed] [Google Scholar]
- 38.Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun. 1992;189:824–831. doi: 10.1016/0006-291x(92)92277-5. [DOI] [PubMed] [Google Scholar]
- 39.Puri KD, Finger EB, Gaudernack G, Springer TA. Sialomucin CD34 is the major L-selectin ligand in human tonsil high endothelial venules. J Cell Biol. 1995;131:261–270. doi: 10.1083/jcb.131.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Re F, Zanetti A, Sironi M, Polentarutti N, Lanfrancone L, Dejana E, Colotta F. Inhibition of anchorage-dependent cell spreading triggers apoptosis in cultured human endothelial cells. J Cell Biol. 1994;127:537–546. doi: 10.1083/jcb.127.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Risau W. Differentiation of endothelium. FASEB J. 1995;9:923–933. [PubMed] [Google Scholar]
- 42.Ruoslahti E, Reed JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 43.Satake S, Kuzuya M, Ramos MA, Kanda S, Iguchi A. Angiogenic stimuli are essential for survival of vascular endothelial cells in three-dimensional collagen lattice. Biochem Biophys Res Commun. 1998;244:642–646. doi: 10.1006/bbrc.1998.8313. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz MA. Integrins, oncogenes, and anchorage independence. J Cell Biol. 1997;139:575–578. doi: 10.1083/jcb.139.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwartz SM. Selection and characterization of bovine aortic endothelial cells. In Vitro. 1978;14:966–980. doi: 10.1007/BF02616210. [DOI] [PubMed] [Google Scholar]
- 46.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 47.Spyridopoulos I, Brogi E, Kearney M, Sullivan AB, Cetrulo C, Isner JM, Losordo DW. Vascular endothelial growth factor inhibits endothelial cell apoptosis induced by tumor necrosis factor-α: balance between growth and death signals. J Mol Cell Cardiol. 1997;29:1321–1330. doi: 10.1006/jmcc.1996.0365. [DOI] [PubMed] [Google Scholar]
- 48.Stavri GT, Zachary IC, Baskerville PA, Martin JF, Erusalimsky JD. Basic fibroblast growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells: synergistic interaction with hypoxia. Circulation. 1995;92:11–14. doi: 10.1161/01.cir.92.1.11. [DOI] [PubMed] [Google Scholar]
- 49.Sutherland RM. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 1988;240:177–184. doi: 10.1126/science.2451290. [DOI] [PubMed] [Google Scholar]
- 50.Varner JA, Brooks PC, Cheresh DA. Review: the integrin αVβ3: angiogenesis and apoptosis. Cell Adhes Commun. 1995;3:367–374. doi: 10.3109/15419069509081020. [DOI] [PubMed] [Google Scholar]
- 51.Villaschi S, Nicosia RF. Angiogenic role of endogenous basic fibroblast growth factor released by rat aorta after injury. Am J Pathol. 1993;143:181–190. [PMC free article] [PubMed] [Google Scholar]
- 52.Vittet D, Prandini MH, Berthier R, Schweitzer A, Martin-Sisteron H, Uzan G, Dejana E. Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood. 1996;88:3424–3431. [PubMed] [Google Scholar]
- 53.Watanabe Y, Dvorak HF. Vascular permeability factor/vascular endothelial growth factor inhibits anchorage-disruption-induced apoptosis in microvessel endothelial cells by inducing scaffold formation. Exp Cell Res. 1997;233:340–349. doi: 10.1006/excr.1997.3583. [DOI] [PubMed] [Google Scholar]
- 54.Wolburg H, Neuhaus J, Kniesel U, Krauss B, Schmid EM, Ocalan M, Farrell C, Risau W. Modulation of tight junction structure in blood-brain barrier endothelial cells. Effects of tissue culture, second messengers and cocultured astrocytes. J Cell Sci. 1994;107:1347–1357. doi: 10.1242/jcs.107.5.1347. [DOI] [PubMed] [Google Scholar]
- 55.Yamaguchi TP, Dumont DJ, Conlon RA, Breitman ML, Rossant J. flk-1, and flt-related receptor tyrosine kinase is an early marker for endothelial cell precursors. Development. 1993;118:489–498. doi: 10.1242/dev.118.2.489. [DOI] [PubMed] [Google Scholar]
- 56.Yayon A, Klagsbrun M. Autocrine regulation of cell growth and transformation by basic fibroblast growth factor. Cancer Metastasis Rev. 1990;9:191–202. doi: 10.1007/BF00046360. [DOI] [PubMed] [Google Scholar]
- 57.Young PE, Baumhueter S, Lasky LA. The sialomucin CD34 is expressed on hematopoietic cells and blood vessels during murine development. Blood. 1995;85:96–105. [PubMed] [Google Scholar]