Sli15 Associates with the Ipl1 Protein Kinase to Promote Proper Chromosome Segregation in Saccharomyces cerevisiae (original) (raw)
Abstract
The conserved Ipl1 protein kinase is essential for proper chromosome segregation and thus cell viability in the budding yeast Saccharomyces cerevisiae. Its human homologue has been implicated in the tumorigenesis of diverse forms of cancer. We show here that sister chromatids that have separated from each other are not properly segregated to opposite poles of ipl1-2 cells. Failures in chromosome segregation are often associated with abnormal distribution of the spindle pole–associated Nuf2-GFP protein, thus suggesting a link between potential spindle pole defects and chromosome missegregation in ipl1 mutant cells. A small fraction of ipl1-2 cells also appears to be defective in nuclear migration or bipolar spindle formation. Ipl1 associates, probably directly, with the novel and essential Sli15 protein in vivo, and both proteins are localized to the mitotic spindle. Conditional sli15 mutant cells have cytological phenotypes very similar to those of ipl1 cells, and the ipl1-2 mutation exhibits synthetic lethal genetic interaction with sli15 mutations. sli15 mutant phenotype, like ipl1 mutant phenotype, is partially suppressed by perturbations that reduce protein phosphatase 1 function. These genetic and biochemical studies indicate that Sli15 associates with Ipl1 to promote its function in chromosome segregation.
Keywords: chromosome segregation, Ipl1, Sli15, protein kinase, Nuf2
Chromosome segregation is a complicated process that is critical to the proliferation of all cell types. In this process, the functions of many proteins must be activated and inactivated coordinately in a strict temporal and spatial order, thereby leading to an orchestrated series of events that culminate in the segregation of sister chromatids to opposite poles of a mitotic spindle. These events include chromosome condensation, sister chromatid cohesion, bipolar mitotic spindle assembly and elongation, microtubule capturing by kinetochores, sister chromatid separation, and kinetochore microtubule-mediated poleward movement of separated sister chromatids (for reviews see references 23, 27, and 43). Protein phosphorylation plays important roles in the control of chromosome segregation. In animal cells, many components of the mitotic spindle apparatus are known to be specifically phosphorylated during M phase (19, 75). In the budding yeast Saccharomyces cerevisiae, phosphorylation is known to regulate the function of a number of proteins that play critical roles in chromosome segregation, including those required for kinetochore function and sister chromatid separation (6, 7, 32, 36).
Using a chromosome-gain genetic assay, we have previously identified the Ipl1 protein kinase as being essential for proper chromosome segregation and cell viability in the budding yeast S. cerevisiae (8, 13). At the restrictive temperature, most conditional temperature-sensitive (Ts−)1 ipl1-2 mutant cells retain the ability to assemble mitotic spindles that can undergo elongation. However, chromosomes become severely missegregated in the process, resulting in cell death within a single cell cycle. Interestingly, severe chromosome missegregation is not associated with cell cycle arrest, as spindle disassembly and cell division occur in ipl1-2 cells that are destined to be inviable. These results suggest that ipl1-2 mutant cells are defective in the execution of a mitotic process(es) that is not monitored by mitotic spindle checkpoint control (for review see reference 56), or that ipl1 mutant cells are defective in both the mechanics of chromosome segregation and mitotic spindle checkpoint control. However, it is clear that ipl1-2 cells are not totally defective in mitotic spindle checkpoint control, since such cells do become arrested in G2/M phase when exposed to the microtubule-destabilizing drug nocodazole (13). The molecular basis of the chromosome segregation defect in ipl1 mutant cells has not been well characterized, but genetic analysis has shown that ipl1 mutant phenotypes can be partially suppressed by perturbations that reduce protein phosphatase 1 (PP1) function, thus suggesting that PP1 acts in opposition to the Ipl1 protein kinase in vivo (13, 74).
Since our initial report of the Ipl1 protein kinase, several structural homologues of Ipl1 have been described from diverse organisms, including Drosophila (15), Xenopus (49), Caenorhabditis elegans (60, 61), mouse (17, 18, 44, 63, 73, 80), rat (72), and humans (4, 33, 62, 63, 73). Like Ipl1, many of these Ipl1-related protein kinases are known or predicted to play important roles in chromosome segregation. Drosophila cells with mutational alteration of the Ipl1-related Aurora kinase are defective in centrosome separation and form monopolar spindles (15). C. elegans embryonic cells lacking the AIR-1 kinase, which normally localizes to the centrosome, are defective in chromosome segregation (60). Eg2, a Xenopus homologue of Ipl1, can bind microtubules directly and is associated with the centrosome and mitotic spindle in cultured cells. In an in vitro mitotic spindle assembly assay, Eg2 is required for bipolar spindle assembly (49). Furthermore, Eg2 also plays a role in the progesterone-activated signaling pathway that triggers oocyte maturation (1). While the biological function of other Ipl1-related kinases is less understood, several are known to be associated with the centrosome and/or mitotic spindle (4, 17, 33, 60, 72). Furthermore, a chimera containing the nonkinase domain of Ipl1 and the kinase domain of human Aurora2 can partially complement the Ts− phenotype of ipl1 mutant cells (4), whereas expression of the murine IAK1 kinase causes lethality in ipl1 mutant but not wild-type yeast cells, possibly because IAK1 may interfere with Ipl1 function by associating nonproductively with normal binding partners or substrates of Ipl1 (17). These observations suggest that at least some members of the Ipl1 family of protein kinases may perform related functions in vivo.
Aneuploidy is known to be associated with many forms of human cancer (25). However, relatively few proteins that are involved in chromosome segregation have been found to be mutationally altered in cancer cells. Recently, the gene encoding the Ipl1-related Aurora2 kinase was found to be amplified and/or overexpressed in a variety of human tumors, including a significant fraction of colorectal and breast tumors (4, 62). Furthermore, ectopic expression of Aurora2 in rodent fibroblasts leads to chromosome missegregation, centrosome amplification, and cellular transformation, and the transformed cells are tumorigenic in nude mice (4, 81). Thus, the unregulated expression of a human homologue of Ipl1 may be a common contributor to tumorigenesis.
We report here further characterization of the chromosome segregation defect observed in ipl1 mutant cells. In addition, we describe the identification and characterization of Sli15, a regulatory binding partner of Ipl1.
Materials and Methods
Strains, Media, and Genetic Techniques
The yeast strains used in this study are listed in Table I. The diploid strain CBY1830-53 was constructed by a one-step gene disruption procedure (53), replacing one of the two SLI15 genes in DBY1830 with the sli15-Δ2:: HIS3 allele present on the ∼4.6-kb PvuII-ScaI fragment of pCC923. The sli15-3 strain CCY482-13D-1-1 was constructed by a recombination-mediated two-step gene replacement procedure (58), first by transforming CCY482-13D to Ura+ with the integrating _URA3_-plasmid pCC1147 that had been linearized at the unique SnaBI site, and then by screening for Ura− Ts− segregants of the Ura+ Ts+ transformant. This resulted in the replacement of the chromosomal copy of SLI15 gene in CCY482-13D with the sli15-3 mutant allele present on pCC1147. These gene replacements were confirmed by DNA hybridization. CCY598-49C and CCY598-52B, which contain a LEU2 marker integrated adjacent to the ipl1-2 locus, were constructed by transforming CCY405-10B with the integrating _LEU2_-plasmid pCC512 that had been linearized at the unique NdeI site. The resulting Leu+ transformant was backcrossed to yield CCY598-49C and CCY598-52B. CCY941-2C, which contains a URA3 marker integrated adjacent to the ipl1-2 locus, was similarly constructed, using the integrating _URA3_-plasmid pCC821 that was linearized at the ClaI sites to transform CCY915-2B. The resulting Ura+ transformant was backcrossed to yield CCY941-2C. CCY405-10B-2 and CCY1060-1D-4, which contain a LEU2 marker integrated adjacent to the SLI15 and sli15-3 locus, respectively, were constructed by transforming CCY405-10B and CCY1060-1D, respectively, to Leu+ with the integrating _LEU2_-plasmid pCC912 that had been linearized at the unique NheI site. The Escherichia coli strain DB1142 (leu pro thr hsdR hsdM recA) was used routinely as a host for plasmids, except in experiments involving recombinant protein expression, where the E. coli strains TOP10 (F− _mcrA Δ_[_mrr-hsdRMS-mcrBC_] _φ80lacZΔM15 ΔlacX74 deoR recA1 araD139 Δ_[_ara-leu_]7697 galU galK rpsL [_StrR_] endA1 nupG), BL21 (F− ompT hsdS [rB − mB −] gal dcm), and RR1 (proA2 leuB6 galK2 xyl-5 mtl-1 ara-14 rpsL20 supE44 hsdS λ −) were used.
Table I.
Yeast Strains Used in This Study
| Strain | Genotype | |
|---|---|---|
| DBY1830 | a/α | ade2/+ lys2-801/+ his3-Δ200/his3-Δ200 ura3-52/ura3-52 leu2-3,112/leu2-3,112 trp1-1/trp1-1 |
| CBY1830-53 | a/α | ade2/+ lys2-801/+ his3-Δ200/his3-Δ200 ura3-52/ura3-52 leu2-3,112/leu2-3,112 trp1-1/trp1-1 sli15-Δ2::HIS3/+ |
| CCY108-15C-1 | a | ade2 his3-Δ200 ura3-52 lys2-Δ101::HIS3::lys2-Δ102 ipl1-2 |
| CCY396-8D | a | ade2 ade3-130 lys2-801 ura3-52 leu2-3,112 his3-Δ200 ipl1-2 [IPL1, URA3, ADE3] (i.e., with the 2μ-plasmid pCC476) |
| CCY405-10B | α | ade2 ade3-130 lys2-801 his3-Δ200 ura3-52 leu2-3,112 ipl1-2 |
| CCY405-10B-2 | α | _ade2 ade3-130 lys2-801 his3-Δ200 ura3-52 leu2-3,112 ipl1-2 LEU2 (_at SLI15) |
| CCY482-13D | a | ade2 his3-Δ200 ura3-52 leu2-3,112 lys2-Δ101::HIS3::lys2-Δ102 |
| CCY482-13D-1-1 | a | ade2 his3-Δ200 ura3-52 leu2-3,112 lys2-Δ101::HIS3::lys2-Δ102 sli15-3 |
| CCY598-49C | α | ade2 ade3-130 ura3-52 leu2-3,112 his3-Δ200 ipl1-2 LEU2 (at ipl1-2) |
| CCY598-52B | a | ade2 ade3-130 ura3-52 leu2-3,112 his3-Δ200 ipl1-2 LEU2 (at ipl1-2) |
| CCY757-1D | a | ade2 ade3-130 lys2-801 ura3-52 leu2-3,112 his3-Δ200 ipl1-2 sli15-13 [IPL1, URA3, ADE3] (i.e., with the 2μ-plasmid pCC476) |
| CCY766-9D-1 | a | lys2-801 his3-Δ200 ura3-52 leu2-3,112 trp1-1 NUF2::URA3-NUF2-SGFP |
| CCY822-6B | α | ade2 ade3-130 ura3-52 leu2-3,112 his3-Δ200 ipl1-2 sli15-1 [IPL1, URA3, ADE3] (i.e., with the 2μ-plasmid pCC476) |
| CCY822-7B | a | ade2 ade3-130 ura3-52 leu2-3,112 his3-Δ200 ipl1-2 sli15-1 [IPL1, URA3, ADE3] (i.e., with the 2μ-plasmid pCC476) |
| CCY915-2B | α | lys2-801 ura3-52 his3-Δ200 trp1-1 ipl1-2 |
| CCY915-13C-5 | a | lys2-801 his3-Δ200 ura3-52 leu2-3,112 trp1-1 ipl1-2 NUF2::URA3-NUF2-SGFP |
| CCY941-2C | α | lys2-801 ura3-52 leu2-3,112 his3-Δ200 ipl1-2 URA3 (at ipl1-2) |
| CCY1022-10C | a | lys2-801 his3-Δ200 ura3-52 leu2-3,112 trp1-1 sli15-Δ2::HIS3 [URA3, SLI15] (i.e., with the CEN-plasmid pCC977) |
| CCY1060-1D | a | ade2 ura3-52 leu2-3,112 his3-Δ200 lys2-Δ101::HIS3::lys2-Δ102 sli15-3 |
| CCY1060-1D-4 | a | ade2 ura3-52 leu2-3,112 his3-Δ200 lys2-Δ101::HIS3::lys2-Δ102 sli15-3 LEU2 (at sli15-3) |
| CCY1076-28B | α | lys2 his3 ura3 leu2 trp1-1 ura3::URA3-tetO112 leu2::LEU2-tetR-GFP |
| CCY1077-6C | α | lys2 his3 ura3 leu2 ipl1-2 ura3::URA3-tetO112 leu2::LEU2-tetR-GFP |
| CCY1078-37B | α | lys2 his3 ura3 leu2 sli15-3 ura3::URA3-tetO112 leu2::LEU2-tetR-GFP |
| CCY1083-8B | a | lys2-801 his3-Δ200 ura3-52 leu2-3,112 sli15-3 NUF2::URA3-NUF2-SGFP |
| TD4 | a | ura3-52 leu2-3,112 trp1-289 his4-519 |
Yeast genetic manipulation as well as the preparation of rich medium (YEPD), synthetic complete medium (SC) lacking some amino acids, synthetic minimal medium (SD), and SD with necessary supplements were performed as described (52). 5-fluoroorotic acid (5-FOA; US Biological) was used at 1 mg/ml for plates to be incubated at 26°C and at 0.5 mg/ml for plates to be incubated at 37°C. Yeast cells were grown at 26°C unless otherwise specified.
Identification of sli15-1 and sli15-13 Mutants
sli mutations that confer a lethal or very slow-growth phenotype only when combined with the ipl1-2 mutation were identified by a colony sectoring assay (2, 35). The details of this genetic screen will be described elsewhere. In brief, an ade2 ade3 ura3 leu2 ipl1-2 haploid strain that contains IPL1, URA3, and ADE3 on a 2μ-plasmid (CCY396-8D) can grow well at 26°C even after spontaneous loss of the plasmid. Thus, it forms sectoring (red and white) colonies on medium containing adenine and uracil, and it is resistant to 5-FOA. This strain was mutagenized by treatment with ethyl methanesulfonate, and mutagenized colonies that were nonsectoring and 5-FOA–sensitive (i.e., could not grow well upon loss of the _IPL1-URA3-ADE3_–plasmid) at 26°C were identified as potentially containing sli mutations. Standard backcrossing with ade2 ade3 ura3 leu2 ipl1-2 strains that were marked at the ipl1-2 locus with LEU2 (CCY598-49C and CCY598-52B) led to the identification of 23 sli mutant strains, including two that are relevant to this study. As expected, three ipl1 mutant strains were also identified. The cloned SLI15 gene complemented the mutant phenotypes of two of the sli mutants (sli15-1 and sli15-13) (CCY757-1D and CCY822-6B), thus suggesting that these two sli mutants have mutations in the same gene. This was confirmed by linkage analysis.
Molecular Cloning of SLI15 and Construction of Temperature-sensitive sli15 Mutant Alleles
The SLI15 gene was cloned by complementation of the nonsectoring and 5-FOA–sensitive phenotypes of ade2 ade3 ura3 leu2 ipl1-2 sli15-1 cells that contained an _IPL1-URA3-ADE3_–plasmid. Strain CCY822-6B was transformed with plasmid DNA from a yeast genomic library constructed in the _LEU2_-CEN–plasmid p366 (gift of P. Hieter, University of British Columbia, Vancouver, Canada). Leu+ transformants were selected on SC medium lacking leucine. Sectoring transformant colonies were identified and tested for their ability to grow on supplemented SD medium containing 5-FOA. Plasmids were recovered into E. coli from sectoring colonies that were 5-FOA–resistant. These plasmids were retested for their ability to complement the nonsectoring and 5-FOA–sensitive phenotypes of CCY822-6B. From ∼120,000 Leu+ transformants screened, we obtained 20 plasmids that contained IPL1 and one plasmid (pCC879) that contained a different sequence. To confirm that pCC879 contained SLI15, the integrating _LEU2_-plasmid pCC912, which contained an ∼4.8-kb SacI-XhoI yeast genomic DNA fragment derived from the insert present within pCC879, was linearized at the unique NheI site present within this fragment and used to transform the ade2 ade3 leu2 ura3 ipl1-2 strain CCY405-10B. The resulting Leu+ transformant (CCY405-10B-2) was mated with an ade2 ade3 leu2 ura3 ipl1-2 sli15-1 strain that contained an _IPL1-URA3-ADE3_–plasmid (CCY822-7B). Sporulation and tetrad analysis of the resulting diploid revealed absolute linkage between the Leu+ and 5-FOA– sensitive phenotypes. Thus, the insert DNA from pCC879 was derived from the SLI15 locus.
Mutagenesis of SLI15 was carried out by in vitro error-prone PCR and in vivo gapped-repair as described (42). In brief, T3 and T7 primers (Promega) were used in a PCR reaction with Taq DNA polymerase (Promega) to amplify the SLI15 gene present on the low copy number _LEU2_-plasmid pCC982. Approximately 0.5 μg of the ∼3.6-kb PCR product and ∼0.5 μg of the ∼7.5-kb NruI-SnaBI fragment of unmutagenized pCC982 were used to transform the yeast strain CCY1022-10C, which contained SLI15 on a _URA3_-CEN–plasmid as the only source of SLI15. Leu+ transformants were selected at 26°C on SC medium lacking leucine and were tested for their ability to grow at 26 and 37°C on supplemented SD medium lacking leucine but containing uracil and 5-FOA. Transformant colonies that could grow on 5-FOA plates at 26 but not 37°C were chosen. Such transformants were taken from the 5-FOA plates that had been incubated at 26°C and retested for their ability to grow on YEPD plates at 26 but not 37°C. The _LEU2_-plasmids were recovered from such Ts− transformants into E. coli and retested for their ability to support growth of CCY1022-10C on 5-FOA plates at 26 but not 37°C. From ∼10,500 Leu+ transformants screened by this method, 10 plasmids containing temperature-sensitive sli15 alleles (sli15-3 to sli15-12) were isolated. One such plasmid, pCC1137, contains the sli15-3 mutant allele.
Plasmid Constructions
Subcloning experiments were routinely carried out with the high copy number plasmids pSM217 and pSM218 (gift of P. Hieter), the low copy number plasmids pRS315 and pRS316 (64), and the integrating plasmids pRS305, pRS306, and YIp5 (58, 64). Plasmids encoding epitope-tagged versions of Ipl1 (pCC1128) or Sli15 (pCC1173 and pCC1193) were constructed by inserting a DNA fragment encoding three tandem copies of the HA- or Myc-epitope (from pMPY-3xHA or pMPY-3xMYC [59]) into the coding sequence of IPL1 (after the initiation codon) or SLI15 (before the stop codon). The HA-Ipl1 and Sli15-Myc fusion proteins are functional since they can complement the Ts− phenotype of ipl1-2 and sli15-3 cells, respectively (data not shown). Plasmids encoding the GST-Sli15 fusion protein (pCC1061 and pCC1062) were constructed in pEG(KT) (41) (for expression in yeast) and pGEX-2T (65) (for expression in E. coli). The GST-Sli15 fusion protein is functional since it can complement the inviability phenotype of a sli15-Δ2::HIS3 mutant (data not shown). Plasmids encoding chimeric proteins containing the green fluorescent protein (GFP) (24) fused to Ipl1 (pCC959) or Sli15 (pCC1060) were constructed with pRB2138 (11). The GFP-Ipl1 and GFP-Sli15 fusion proteins are functional since they can complement the Ts− phenotype of ipl1-2 and the inviability phenotype of sli15-Δ2::HIS3 cells, respectively (data not shown). The plasmid encoding His6-Ipl1 (pCC1167), which has six tandem histidine residues fused to the NH2 terminus of Ipl1, was constructed with pTrcHis A (Invitrogen Corp.). The plasmid encoding the sli15-Δ2::HIS3 mutant allele (pCC923) was constructed by replacing the DNA sequence between the AvrII and NruI sites present in the low copy number _URA3_-plasmid pCC883 with the ∼1.8-kb XbaI-SmaI fragment (containing HIS3) of pJJ217 (30). The plasmid encoding the TrpE-Ipl1 fusion protein (pCC134-16) was constructed by inserting the ∼1.5-kb EcoRI DNA fragment of pCC100 (13) into the EcoRI site of pATH10 (34), resulting in an in-frame fusion between trpE and codon 45 of IPL1.
Antibody Production
The TrpE-Ipl1 fusion protein was partially purified from E. coli cells (RR1) harboring pCC134-16 by a previously described method (78) and used as antigen in injections of guinea pigs. Anti-Ipl1 antibodies were affinity-purified with TrpE-Ipl1 that was immobilized on a nitrocellulose membrane (78).
Immunoprecipitation and GST Pulldown Assays
To prepare extracts from yeast cells that coexpressed HA-Ipl1 and GST or HA-Ipl1 and GST-Sli15, a saturated culture of TD4 that contained the plasmids pCC1128 (encoding HA-Ipl1) and pEG(KT) (encoding GST) or the plasmids pCC1128 and pCC1061 (encoding GST-Sli15) was diluted 20-fold into 50 ml of SC medium lacking uracil and leucine and with 2% raffinose (US Biological) instead of glucose as carbon source. After 6 h at 30°C, galactose (US Biological) was added to a final concentration of 4%, and the culture was incubated at 30°C for another 4 h. Cells were harvested and rinsed once with 10 ml of lysis buffer, which consists of 50 mM Hepes-KOH (pH 7.4), 200 mM KCl, 10% glycerol (vol/vol), 1% NP-40 (vol/vol), 1 mM EDTA, 1 mM dithiothreitol, 25 mM NaF, 1 mM NaVO4, and the following protease inhibitors (Sigma Chemical Co.): 2 μg/ml each of antipain, leupeptin, pepstatin A, chymostatin, and aprotinin; 10 μg/ml of phenanthroline; 16 μg/ml of benzamidine-HCl; and 1 mM PMSF. Cells were resuspended in 0.45 ml of lysis buffer and aliquoted into three 1.5-ml microcentrifuge tubes. Acid-washed glass beads (425–600-μm diam; Sigma Chemical Co.) were added to each tube to give a final volume of ∼0.2 ml. After chilling on ice, the tubes were mixed by being vortexed for 1 min. This cycle of chilling and vortexing was repeated six more times. The lysates from the three tubes were combined, followed by a 10-min centrifugation at 20,800 g to remove cell debris. 140-μl aliquots of the resulting supernatant were distributed into three 1.5-ml microcentrifuge tubes, each containing 50 μl of a 50% slurry of glutathione-agarose beads (Sigma Chemical Co.) in equilibration buffer (50 mM Hepes-KOH, pH 7.4, 200 mM KCl, 10% glycerol [vol/vol], 1% NP-40 [vol/vol], 1 mM EDTA). The tubes were incubated at 4°C with constant agitation for 2 h. The glutathione-agarose beads were harvested by a 2-min centrifugation at 960 g, followed by one, three, or five 5-min washes with 250 μl of lysis buffer. The proteins bound on the glutathione-agarose beads in each tube were eluted by the addition of 60 μl of sample buffer (50 mM Tris-HCl, pH 6.8, 100 mM dithiothreitol, 2% sodium dodecyl sulfate [wt/vol], 0.1% bromophenol blue [wt/vol], 10% glycerol [vol/vol]).
To prepare extracts from yeast cells that coexpressed HA-Ipl1 and Sli15 or HA-Ipl1 and Sli15-Myc, a saturated culture of TD4 that contained the plasmids pCC1128 (encoding HA-Ipl1) and pCC1192 (encoding Sli15) or the plasmids pCC1128 and pCC1193 (encoding Sli15-Myc) was diluted into 50 ml of SC medium lacking leucine and uracil to give a cell density of ∼2 × 106/ml. After 5 h at 30°C (when cultures had reached a cell density of ∼1.5 × 107/ml), cells were harvested and cell extracts were prepared as described above for GST pulldown experiments, except that lysis buffer with protease inhibitors was replaced by buffer B (50 mM MOPS, pH 7.0, 200 mM KCl, 5 mM dithiothreitol, 10% glycerol [vol/vol], 1% NP-40 [vol/ vol], 25 mM NaF, 0.5 mM NaVO4) with protease inhibitors. After clearing of cell debris, 140 μl of cell lysate was added to a 1.5-ml microcentrifuge tube that contained 50 μl of a 50% slurry of BSA-coated protein A–agarose beads (Pharmacia) in buffer A, which consists of 50 mM MOPS, pH 7.0, 200 mM KCl, 3 mM Na azide, 5% glycerol (vol/vol). The tube was incubated at 4°C with constant agitation for 30 min. After a 2-min centrifugation at 960 g, the supernatant was transferred to a 1.5-ml microcentrifuge tube that contained 1 μl of mouse anti-Myc ascites fluid (gift of B. Haarer, University of Texas at Austin, Austin, TX). After a 1-h incubation at 4°C with constant agitation, 50 μl of a 50% slurry of BSA-coated protein A–agarose beads (in buffer A) was added, followed by a 30-min incubation at 4°C with constant agitation. The protein A–agarose beads were harvested by a 2-min centrifugation at 960 g, followed by two 5-min washes with 250 μl of buffer B. The proteins bound on the protein A–agarose beads in each tube were eluted by the addition of 50 μl of sample buffer.
To prepare extracts from E. coli that expressed GST, GST-Sli15, His6-Ipl1, or His6-NonO-C protein, a saturated culture of BL21 that contained the plasmid pGEX-2T (encoding GST) or pCC1062 (encoding GST-Sli15) or a saturated culture of TOP10 that contained the plasmid pCC1167 (encoding His6-Ipl1) or pRBD2-nonO (encoding His6-NonO-C; gift of C. Huang and P. Tucker, University of Texas at Austin, Austin, TX) was diluted 100-fold into 50 ml of Luria broth with ampicillin. After 3 h at 37°C, IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a final concentration of 1 mM, followed by a 4-h incubation at 26°C. Cells were harvested, rinsed once in 10 ml of ice cold lysis buffer (see above), and resuspended in 2.5 ml of the same buffer. Cells were lysed in a French press at 16,000 psi, and cell debris was removed by a 10-min centrifugation at 20,800 g, thus generating the supernatant fraction. 0.4 ml of supernatant containing GST or GST-Sli15 was added to a 1.5-ml microcentrifuge tube that contained 0.1 ml of a 50% slurry of BSA-coated glutathione-agarose beads in equilibration buffer (see above). After a 0.5-h incubation at 4°C with constant agitation, the supernatant was removed and replaced by 0.8 ml of supernatant that contained His6-Ipl1 or His6-NonO-C. After a 1.5-h incubation at 4°C with constant agitation, the supernatant was removed and the glutathione-agarose beads were rinsed twice with 1 ml of lysis buffer. The proteins bound on the glutathione-agarose beads were eluted by the addition of 60 μl of sample buffer.
Protein samples were electrophoretically separated in SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with 2,000-fold diluted rabbit anti-GST antibodies (Molecular Probes, Inc.), 1,000-fold diluted mouse anti-HA ascites fluid (BABCO), 1,000-fold diluted mouse anti-Myc ascites fluid (gift of B. Haarer, University of Texas at Austin), 200-fold diluted affinity-purified guinea pig anti-Ipl1 antibodies, or 2,500-fold diluted rabbit anti-NonO antibodies (gift of C. Huang and P. Tucker, University of Texas at Austin). Proteins recognized by primary antibodies were visualized by the ECL chemiluminescent system (Amersham Corp.).
Cytological Techniques
Immunofluorescence staining of yeast cells was carried out as described (47). In experiments that involved immunostaining of microtubules in yeast cells that expressed GFP fusion proteins, cells were fixed in 3.7% formaldehyde at room temperature for 30 min, followed by standard procedures for immunofluorescence staining (47), except that the methanol- and acetone-fixation steps were omitted. In experiments that did not involve immunostaining, GFP fusion proteins were observed either in live yeast cells or in cells that had been fixed for 10 min at room temperature in 3.7% formaldehyde. In experiments in which visualization of DNA as well as GFP fusion proteins was desired, 4′,6-diamidino-2-phenylindole (DAPI; Accurate Chemical Co.) was added to the growth medium to a final concentration of 2.5 μg/ml 15 min before preparation of cells for observation.
Results
Separated Sister Chromatids Are Not Properly Segregated in ipl1 Mutant Cells
We have shown previously that ipl1-2 mutant cells do not have a uniform arrest phenotype at the restrictive temperature of 37°C (13). Instead, they go through the cell cycle, missegregate chromosomes severely, undergo cytokinesis, and become inviable. In a temperature-shift experiment, over 20% of the large-budded cells in an asynchronous culture of ipl1-2 cells that had been incubated at 37°C for 3–4 h had clearly failed to segregate chromosomal DNA evenly to the opposite poles of apparently normal looking mitotic spindles (Fig. 1) (13). The percentage of ipl1-2 cells that had missegregated chromosomes was probably much higher (see below and reference 8), since we were conservative in our scoring of uneven chromosomal DNA masses. To find out whether this failure in chromosome segregation was caused by a failure in sister chromatid separation, we examined the distribution of chromosome V that was marked by the binding of Tet repressor-green fluorescent protein (TetR-GFP) to Tet operator sites located adjacent to the centromere of chromosome V (40). At both 26°C and after a 2-h incubation at 37°C, ≥90% of large-budded wild-type haploid cells that were in early anaphase had clearly separated sister chromatids, as indicated by the presence of two dots of TetR-GFP signal (Fig. 2, a and b, and Fig. 3). As these cells reached late anaphase or early G1, essentially all cells had separated and properly segregated sister chromatids, as indicated by the presence of a single dot of TetR-GFP signal within each of the two evenly segregated chromosomal DNA masses (Fig. 2, c and d, and Fig. 3).
Figure 1.

Cytological phenotype of ipl1-2 and sli15-3 mutants. ipl1-2 (CCY108-15C-1) and sli15-3 (CCY1060-1D) cells were incubated at 37°C for 4 h and processed for immunofluorescence microscopy. Phase-contrast, DAPI-stained, and anti-tubulin– stained images are shown. Top panels: uneven chromosome segregation; middle panels: nuclear migration defect; bottom panels: monopolar spindle.
Figure 2.
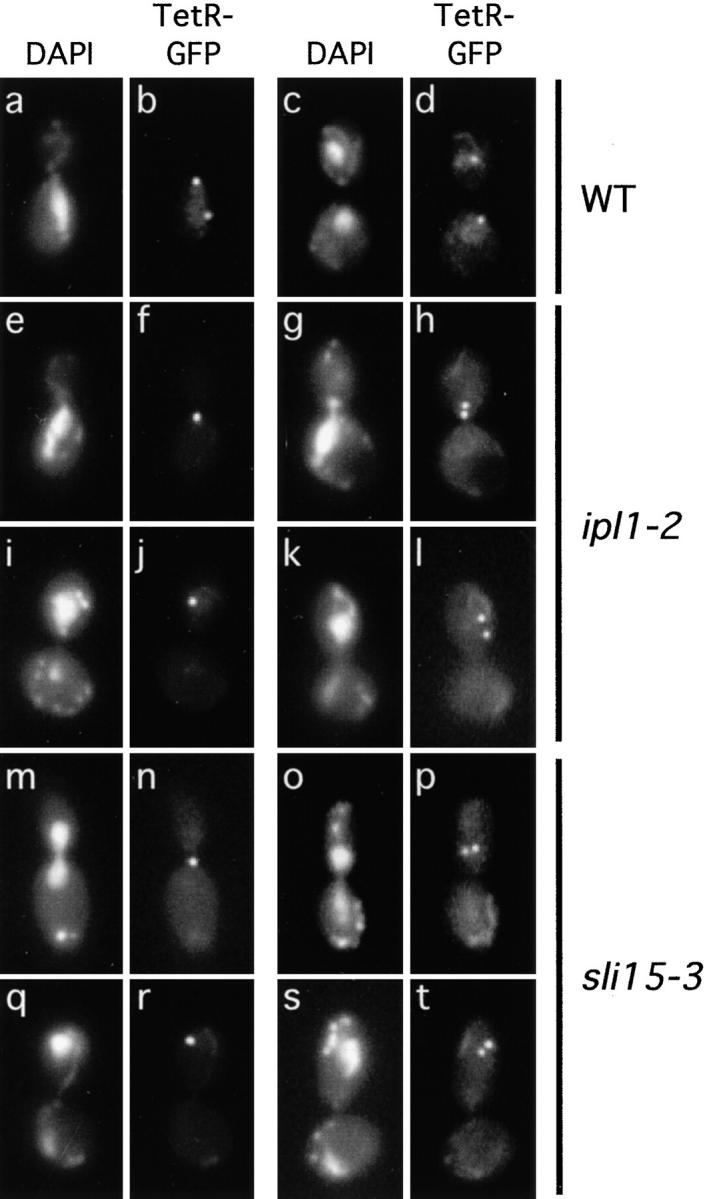
Defective sister chromatid segregation in ipl1-2 and sli15-3 cells. Wild-type (WT) (CCY1076-28B), ipl1-2 (CCY1077-6C), and sli15-3 (CCY1078-37B) cells that had the centromere region of chromosome V marked by the binding of TetR-GFP to Tet operator sites were incubated at 37°C for 2 h and briefly fixed. Pairs of DAPI-stained and TetR-GFP images taken from large-budded cells are shown.
Figure 3.

Summary of sister chromatid separation/segregation defects in ipl1-2 and sli15-3 cells. The localization of TetR-GFP (small dots) and chromosomal DNA masses (shaded) were examined in wild-type (WT) (CCY1076-28B), ipl1-2 (CCY1077-6C), and sli15-3 (CCY1078-37B) cells that were incubated at 26°C (0 h) or for 2 h at 37°C. Uneven segregation of chromosomal DNA masses was indicated by shaded areas of different sizes. For each sample, 100 large-budded cells that were in early anaphase and 100 large-budded cells that were in late anaphase or early G1 were scored. No distinction was made between the mother and bud of these large-budded cells. Whether the centromere region of chromosome V (marked by TetR-GFP) present on sister chromatids appeared separated (sep.) or unseparated (unsep.) was indicated.
In contrast, ipl1-2 cells had properly separated and segregated sister chromatids at 26 but not 37°C. After a 2-h incubation at 37°C, among ipl1-2 cells that were in early anaphase, ∼60% had only a single dot of TetR-GFP signal (Fig. 2, e and f, and Fig. 3), and ∼22% had two dots that were located unusually close to each other (Fig. 2, g and h, and Fig. 3). As ipl1-2 cells reached late anaphase or early G1, only ∼30% of them appeared to have properly separated and segregated the sister chromatids of chromosome V. Approximately 43% had a single dot of TetR-GFP signal (Fig. 2, i and j, and Fig. 3), and ∼27% had two dots that were located within only one of the two (often unevenly segregated) chromosomal DNA masses (Fig. 2, k and l, and Fig. 3). The presence of a single dot of TetR-GFP signal in ipl1-2 cells suggested that sister chromatids had failed to separate, or that sister chromatids had separated but had failed to be properly segregated to opposite poles, thus resulting in the juxtaposition of two dots of TetR-GFP signal that were scored mistakenly as a single dot (70). While we cannot distinguish between these two possibilities, the presence of a large fraction of ipl1-2 cells with two dots of TetR-GFP signal within only one of the two chromosomal DNA masses indicated that sister chromatids had separated in these cells, but these separated sister chromatids had failed to segregate away from each other towards the two opposite poles. Thus, if a defect in sister chromatid separation exists in ipl1-2 cells, it cannot be the only cause of chromosome missegregation in these cells, as sister chromatids that have separated also fail to be properly segregated.
Mislocalization of Nuf2-GFP in ipl1-2 Cells
In addition to the most prominent phenotype of uneven chromosome segregation described in the last section, a small fraction of ipl1-2 cells exhibits some other cytological defects. After 3–4 h at 37°C, ∼10% of the large-budded cells in an asynchronous culture of ipl1-2 cells exhibited defects in nuclear migration and/or mitotic spindle orientation (Fig. 1). A smaller fraction of ipl1-2 cells appeared to have monopolar spindles (Fig. 1), which are suggestive of defects in spindle pole body duplication or separation. This latter phenotype is more noticeable in ipl1-2 cells that had been presynchronized at G1 before being released into the cell cycle at 37°C (data not shown). To find out whether ipl1-2 cells have normal spindle poles, we examined the localization of Nuf2-GFP in ipl1-2 cells. Previous immunofluorescence and biochemical studies have shown that Nuf2 is associated with the intranuclear region of the spindle pole body (45, 77), and Nuf2-GFP is a marker commonly used for observing spindle pole dynamics (31). Consistent with previous reports, our immunofluorescence study with wild-type cells showed that at 26°C and after a 2–3 h incubation at 37°C, Nuf2-GFP was found concentrated at the spindle poles. In cells that had a mitotic spindle, the intensities of the Nuf2-GFP signal were even at the two poles (Fig. 4, a–f, and Fig. 5).
Figure 4.

Abnormal distribution of Nuf2-GFP in ipl1-2 and sli15-3 cells. DNA (DAPI), microtubules (anti-tubulin), and Nuf2-GFP were localized in wild-type (CCY766-9D-1) (a–f), ipl1-2 (CCY915-13C-5) (g–o), and sli15-3 (CCY1083-8B) (p–x) cells that had been incubated at 37°C for 2 h and briefly fixed.
Figure 5.

Summary of Nuf2-GFP distribution. The localization of Nuf2-GFP (dots) and chromosomal DNA masses (shaded) were examined in wild-type (WT) (CCY766-9D-1), ipl1-2 (CCY915-13C-5), and sli15-3 (CCY1083-8B) cells that were incubated at 26°C (0 h) or for 3 h at 37°C. Uneven distribution of Nuf2-GFP was indicated by dots of different sizes within the same cell. Uneven segregation of chromosomal DNA masses was indicated by shaded areas of different sizes. For each sample, 100 large-budded cells that were in preanaphase and 100 large-budded cells that were in late anaphase or early G1 were scored. No distinction was made between the mother and bud of these large-budded cells. Whether the distribution of Nuf2-GFP appeared abnormal is indicated.
In contrast, the localization of Nuf2-GFP appeared abnormal in ipl1-2 cells, especially after a 2–3-h incubation at 37°C. First, Nuf2-GFP was no longer restricted to the spindle poles. Instead, it was also found in many ipl1-2 cells as extra dots that colocalized with mitotic spindles that were short to medium in length (Fig. 4, g–i, and Fig. 5). This pattern of Nuf2-GFP was found at much lower frequencies in wild-type cells. A similarly abnormal localization of Nuf2 has been reported in the nuf2-61 mutant (45), thus suggesting that Nuf2 function may be compromised in ipl1-2 cells. However, we have so far not detected genetic interaction between IPL1 and NUF2. The Ts− phenotype of ipl1-2 or nuf2-61 cells is not complemented by a high copy number plasmid carrying NUF2 or IPL1, respectively, and ipl1-2 nuf2-61 double mutant cells are no more defective in growth at elevated temperatures than ipl1-2 and nuf2-61 single mutant cells. Furthermore, the abundance and electrophoretic mobility of Nuf2 are not affected in ipl1-2 cells (data not shown).
Second, for a large fraction of ipl1-2 cells with mitotic spindles, the intensities of Nuf2-GFP signal were no longer even at the two poles (Fig. 4, j–l, and Fig. 5). In extreme cases, the Nuf2-GFP signal was barely or not detectable from one pole (Fig. 4, m–o, and Fig. 5). There was a general correlation between the pattern of uneven Nuf2-GFP distribution and the pattern of uneven chromosome segregation. Examination of ipl1-2 cells that were in late anaphase or early G1 revealed that the pole with the greater intensity of Nuf2-GFP signal was most often associated with a greater-than-normal amount of chromosomal DNA, and in no case was stronger Nuf2-GFP signal associated with a smaller-than-normal amount of chromosomal DNA (Fig. 5). The presence of ipl1-2 cells with uneven distribution of Nuf2-GFP but apparently even distribution of chromosomal DNA masses, and cells with uneven distribution of chromosomal DNA masses but apparently even distribution of Nuf2-GFP, may simply reflect the intrinsic difficulty in scoring (smaller) differences in the intensity of Nuf2-GFP signal and chromosomal DNA masses. Furthermore, since yeast chromosomes vary greatly in size, chromosomal DNA mass may not be strictly correlated with chromosome number.
To find out whether other spindle pole–associated components may also be mislocalized in ipl1-2 cells, we examined the localization of the spindle pole body central plaque component Spc42 (55). At 26°C, a functional Spc42-GFP fusion protein (gift of J. Kilmartin, Medical Research Council, Cambridge, England) was evenly distributed at opposite spindle poles in 100% of wild-type and ipl1-2 cells. After a 3-h incubation at 37°C, the Spc42-GFP signal at opposite spindle poles was slightly uneven in <3% of wild-type and ipl1-2 cells. Furthermore, Spc42-GFP was never detected along the mitotic spindle in either cell type (data not shown). Thus, Nuf2-GFP, but not Spc42-GFP, becomes distributed abnormally in ipl1-2 cells. This abnormality suggests that potential defects in the spindle poles may contribute to chromosome missegregation in ipl1-2 cells. This subject will be taken up further in the Discussion.
Localization of GFP-Ipl1 to the Mitotic Spindle
To determine the subcellular localization of Ipl1, we generated a fusion gene encoding the GFP and full-length Ipl1. The GFP-IPL1 fusion gene, which was under the control of the ACT1 promoter, was functional (see Materials and Methods). In wild-type yeast cells that carried pCC959, a low level of GFP-Ipl1 signal could be detected in the cytoplasm (Fig. 6). The intensity of this cytoplasmic signal varied somewhat in different cells, perhaps due to the small variations in the copy number of pCC959. GFP-Ipl1 was also found enriched in the nucleus, with special concentration on the mitotic spindle apparatus. In unbudded and small-budded cells, GFP-Ipl1 was sometimes found in a dot-like structure that typically colocalized with the edge of the nucleus (Fig. 6). Immunofluorescent staining of microtubules in these cells indicated that this dot-like structure represented the spindle pole body (data not shown). In large-budded cells that had completed chromosome segregation, GFP-Ipl1 was often found concentrated on elongated or disassembling spindles (Fig. 6 and data not shown). At a lower frequency, GFP-Ipl1 was also found concentrated on mitotic spindles that were short to medium in length. We do not know whether the apparent differences in our ability to detect GFP-Ipl1 on spindles of different lengths were due to cell cycle-specific changes in the localization pattern of GFP-Ipl1, or the possibility that GFP-Ipl1 signal on short/medium-length spindles might be more readily obscured by the overall nuclear signal. We have so far not detected GFP-Ipl1 signal on cytoplasmic microtubules. However, we cannot rule out the possibility that GFP-Ipl1 might be present at low levels on cytoplasmic microtubules.
Figure 6.

Subcellular localization of GFP-Ipl1 and GFP-Sli15. The DIC, DAPI-stained, and GFP-fusion protein images were obtained from unfixed wild-type diploid (DBY1830) cells that carried the low copy number plasmid pCC959 (encoding GFP-Ipl1) or pCC1060 (encoding GFP-Sli15).
Mutations in SLI15 Cause ipl1-2 Cells to Be Inviable at 26°C
To identify other proteins that may play a role in the Ipl1-mediated chromosome segregation process, we reasoned that nonlethal mutations that lower or abolish the function of proteins that play a positive role in this process (e.g., as substrates or positive regulators of Ipl1) may be tolerated in wild-type cells but not in mutant cells with reduced Ipl1 function (21). ipl1-2 mutant cells have a normal growth rate at 26°C but they do not have normal Ipl1 protein kinase function, since ipl1-2 cells exhibit an ∼10-fold increase in the frequency of chromosome gain at this temperature (8). Thus, we carried out a genetic screen at 26°C for nonlethal sli mutations that confer a lethal or very slow-growth phenotype only when combined with the ipl1-2 mutation (see Materials and Methods). Among 23 sli mutants isolated, two contain recessive mutations in the SLI15 gene.
Sli15 Is an Essential Protein That Is Localized to the Mitotic Spindle
The wild-type SLI15 gene was cloned by complementation of the mutant phenotypes of sli15-1 ipl1-2 cells (see Materials and Methods). Subcloning and partial sequencing revealed that SLI15 is identical to YBR156C, which potentially encodes a protein of 698 residues, with a predicted molecular mass of ∼79 kD and a predicted pI of ∼10. The predicted Sli15 protein sequence is not highly similar to that of any protein listed in the sequence databases at the National Center for Biotechnology Information. A putative nuclear localization signal is present at residues 530– 546 (48), and the region (residues 517–565) surrounding this putative localization signal is predicted to have a high probability of adopting a coiled-coil conformation (38).
It has been reported previously that disruption of YBR156C (SLI15) leads to loss of cell viability (51). We have confirmed this result by creating a diploid yeast strain (CBY1830-53) that is heterozygous for the sli15-Δ2:: HIS3 null mutation. Tetrad analysis of this diploid strain revealed that sli15-Δ2::HIS3 haploid cells are inviable. Thus, Sli15, like Ipl1, is essential for yeast cell viability.
To determine the subcellular localization of Sli15, we generated a fusion gene encoding GFP and full-length Sli15. The GFP-SLI15 fusion gene, which was under the control of the ACT1 promoter, was functional (see Materials and Methods). In wild-type yeast cells, GFP-Sli15 was found to be present at a low level throughout the nucleoplasm (Fig. 6). Furthermore, GFP-Sli15 was highly concentrated on mitotic spindles. In unbudded cells, GFP-Sli15 was present in a dot-like structure that represented the spindle pole body, as indicated by its colocalization with the vertex of microtubule staining (Fig. 6 and data not shown). In a small fraction of unbudded cells, GFP-Sli15 was also found on what appeared to be intranuclear microtubules. In budded cells, GFP-Sli15 was concentrated on mitotic spindles of all different lengths, including those that were in the process of disassembly (Fig. 6 and data not shown). We did not detect GFP-Sli15 signal on cytoplasmic microtubules, but we cannot exclude the possibility that GFP-Sli15 might be present at low levels on cytoplasmic microtubules.
sli15-3 Mutant Cells Have Phenotypes Similar to Those of ipl1-2 Cells
The sli15-1 and sli15-13 mutants that we identified originally in the synthetic lethal screen do not have major growth phenotypes at 13–37°C. Thus, we carried out in vitro mutagenesis of SLI15 and screened for sli15 mutant alleles that confer a Ts− growth phenotype at 37°C (see Materials and Methods). One such allele, sli15-3, was used to replace the chromosomal copy of the wild-type SLI15 gene in a haploid IPL1 strain (see Materials and Methods). The resulting sli15-3 strain is Ts− for growth at ≥33°C (see Fig. 8). The sli15-3 mutation, like the sli15-1 and sli15-13 mutations, causes a synthetic lethal phenotype at 26°C when combined with the ipl1-2 mutation. Tetrad analysis of a diploid heterozygous for ipl1-2 and sli15-3 (CCY941-2C × CCY1060-1D-4) revealed that 0 of 7 ipl1-2 sli15-3 haploid meiotic products were viable at 26°C, whereas 12 of 13 ipl1-2 SLI15 haploids and 13 of 13 IPL1 sli15-3 haploids were viable at this temperature.
Figure 8.

Suppression of sli15-3 by increased dosage of IPL1 and GLC8. Suspensions of sli15-3 mutant cells (CCY1060-1D) carrying different plasmids were spotted on YEPD and allowed to grow at the indicated temperatures for 1.75 d. The plasmids used were high copy number control plasmid pSM217, low copy number _SLI15_-plasmid pCC977, low copy number _IPL1_-plasmid pCC100, high copy number _glc7-Δ186–312_–plasmid pCC418, and high copy number _GLC8_-plasmid pCC638.
To examine the phenotype of sli15-3 cells, we have carried out temperature-shift experiments with asynchronous cultures of sli15-3 cells. After a 4-h shift to 37°C, only 15– 20% of sli15-3 cells remained viable, and they did not arrest with a uniform cell morphology. Immunofluorescence microscopy showed that >20% of large-budded sli15-3 cells had clearly failed to segregate chromosomal DNA evenly to the opposite poles of apparently normal looking mitotic spindles (Fig. 1); ∼10% of large-budded cells seemed to be defective in nuclear migration and/or mitotic spindle orientation (Fig. 1); and a smaller fraction of large-budded cells appeared to have monopolar spindles (Fig. 1). These phenotypes of sli15-3 cells are very similar to those of ipl1-2 cells.
We have also examined the separation and segregation of sister chromatids in sli15-3 cells, using the TetR-GFP assay described above. After a 2-h shift to 37°C, among sli15-3 cells that were in early anaphase, ∼53% had only a single dot of TetR-GFP signal (Fig. 2, m and n, and Fig. 3), and ∼25% had two dots that were located unusually close to each other (Fig. 2, o and p, and Fig. 3). As sli15-3 cells reached late anaphase or early G1, only ∼39% of them appeared to have separated properly and segregated the sister chromatids of chromosome V. Approximately 32% had a single dot of TetR-GFP signal (Fig. 2, q and r, and Fig. 3), and ∼29% had two dots that were located within only one of the two (often unevenly segregated) chromosomal DNA masses (Fig. 2, s and t, and Fig. 3). Thus sli15-3 cells, like ipl1-2 cells, are defective in sister chromatid segregation (and possibly sister chromatid separation).
Finally, we also examined the distribution of Nuf2-GFP in sli15-3 cells. After 2–3 h at 37°C, Nuf2-GFP was no longer restricted to the spindle poles. Instead, it was also found in some sli15-3 cells as extra dots that colocalized with mitotic spindles that were short to medium in length (Fig. 4, p–r, and Fig. 5). Furthermore, the intensities of Nuf2-GFP signal were no longer even at the two poles of a large fraction of mitotic spindles (Fig. 4, s–x, and Fig. 5). Examination of sli15-3 cells that were in late anaphase or early G1 revealed that the pole with greater intensity of Nuf2-GFP signal was most often associated with a greater-than-normal amount of chromosomal DNA, and in no case was stronger Nuf2-GFP signal associated with a smaller-than-normal amount of chromosomal DNA (Fig. 5). These abnormal patterns of Nuf2-GFP distribution are very similar to those observed in ipl1-2 cells. Furthermore, this abnormality is limited to Nuf2-GFP, as a functional Spc42-GFP fusion protein is evenly distributed to the spindle poles in sli15-3 cells (data not shown).
Sli15 Binds Ipl1 In Vivo
The genetic interaction between ipl1-2 and sli15 mutations, the similar phenotypes of ipl1-2 and sli15-3 cells, and the similar subcellular localization of GFP-Ipl1 and GFP-Sli15 suggest that these two proteins may associate with each other. To examine this possibility, we used a high copy number plasmid to express in yeast a functional version of Ipl1 that was fused to the HA-epitope (HA-Ipl1). We coexpressed in the same cells GST or a functional version of Sli15 that was fused to GST (GST-Sli15). The latter proteins were expressed under the control of the GAL1/10 promoter and were thus present in great excess. As shown in Fig. 7 A, affinity purification of GST-Sli15, but not the more abundant GST, led to the copurification of HA-Ipl1. The association between HA-Ipl1 and GST-Sli15 was moderately stable, as repeated rounds of washing led to only partial disruption of this association. Comparison of the level and electrophoretic mobility of HA-Ipl1 in cells that coexpressed GST or GST-Sli15 revealed two interesting features. First, the level of HA-Ipl1 was much higher in cells that coexpressed GST-Sli15, thus suggesting that Sli15 may serve to stabilize Ipl1. Second, in cells that coexpressed GST, HA-Ipl1 could be detected as two forms that differed in electrophoretic mobility, with the slower-migrating form being much less abundant (Fig. 7 A). In cells that coexpressed GST-Sli15, the relative abundance of the slower-migrating form was increased and this form of HA-Ipl1 appeared to be more readily copurified with GST-Sli15. The slower-migrating form could be converted to the faster-migrating form by endogenous phosphatases that were present in the yeast extract, as the slower-migrating form was detected only when the yeast extract was prepared in the presence of phosphatase inhibitors (Fig. 7 B). These results indicated that the slower-migrating form of HA-Ipl1 is phosphorylated and that GST-Sli15 (and probably Sli15) may promote the phosphorylation of HA-Ipl1.
Figure 7.
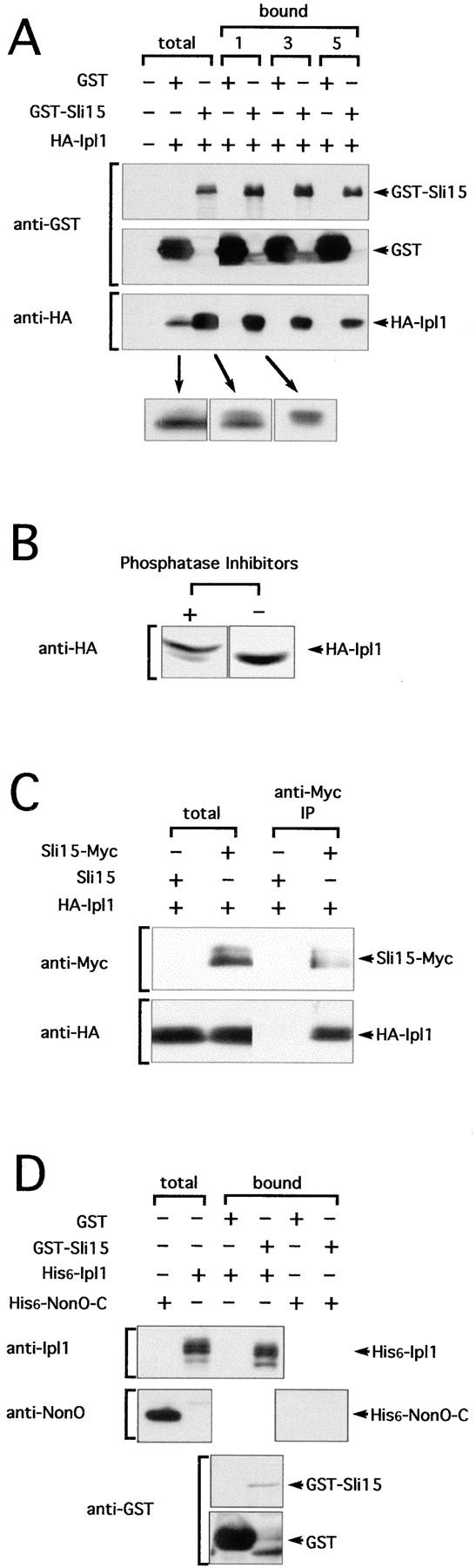
In vivo association of Sli15 with Ipl1. (A) Copurification of HA-Ipl1 with GST-Sli15. GST and GST-Sli15 were affinity-purified in the presence of phosphatase inhibitors (NaF and NaVO4) with glutathione-agarose beads from a yeast strain (TD4) that carried the plasmids pEG(KT) (encoding GST) and pCC1128 (encoding HA-Ipl1) or the plasmids pCC1061 (encoding GST-Sli15) and pCC1128. Aliquots of proteins from total protein extracts (total) or from the affinity-purified (bound) fractions (after 1, 3, or 5 rounds of washing with a buffer containing 200 mM KCl) were analyzed by immunoblotting, using antibodies against GST or the HA-epitope. The enlarged images shown at the bottom are from films that had been exposed for different times. (B) HA-Ipl1 is phosphorylated in vivo. Crude yeast extracts prepared in the presence or absence of the phosphatase inhibitors NaF and NaVO4 from a yeast strain (TD4) that carried the GST-Sli15 plasmid pCC1061 and the HA-Ipl1 plasmid pCC1128 were analyzed by immunoblotting with antibodies against the HA-epitope. (C) Coimmunoprecipitation of HA-Ipl1 with Sli15-Myc. Sli15-Myc was immunoprecipitated with antibodies against the Myc-epitope from a yeast strain (TD4) that carried the plasmids pCC1128 (encoding HA-Ipl1) and pCC1192 (encoding Sli15) or the plasmids pCC1128 and pCC1193 (encoding Sli15-Myc). Aliquots of proteins from total protein extracts (total) or from the immunoprecipitated fractions (anti-Myc IP) were analyzed by immunoblotting, using antibodies against the Myc-epitope or the HA-epitope. (D) Direct binding of His6-Ipl1 to GST-Sli15. GST and GST-Sli15 were affinity-purified with glutathione-agarose beads from an E. coli strain (BL21) that carried pGEX-2T (encoding GST) or pCC1062 (encoding GST-Sli15). Proteins in a crude extract (total) from an E. coli strain (TOP10) that carried pCC1167 (encoding His6-Ipl1) or pRBD2-nonO (encoding His6-NonO-C) were allowed to bind to the immobilized GST or GST-Sli15. Aliquots of proteins from the crude extract (total) or from the fractions that bound to GST or GST-Sli15 were analyzed by immunoblotting, using antibodies against GST, Ipl1, or NonO.
We have also used high copy number plasmids to coexpress in yeast HA-Ipl1 and Sli15 or a functional version of Sli15 that was fused to the Myc-epitope (Sli15-Myc). As shown in Fig. 7 C, immunoprecipitation of Sli15-Myc from a crude yeast lysate with anti-Myc antibodies led to the coprecipitation of HA-Ipl1. Interestingly, Sli15-Myc appeared as multiple electrophoretic forms, thus suggesting that Sli15 may also be a phosphoprotein.
To find out whether Sli15 binds Ipl1 directly, we expressed GST-Sli15 and His6-Ipl1 in E. coli. As shown in Fig. 7 D, His6-Ipl1 that was present in an E. coli crude extract associated with GST-Sli15, but not GST, that was immobilized on glutathione-agarose beads. The association between GST-Sli15 and His6-Ipl1 was specific, as the control protein His6-NonO-C failed to associate with GST-Sli15. These results together indicate that Sli15 is a binding partner of Ipl1 in vivo and that these two proteins most probably bind to each other directly.
Suppression of sli15-3 by Increased Dosage of IPL1 or GLC8
The results described so far strongly suggest that Ipl1 and Sli15 function in a complex to promote proper chromosome segregation. Mutations that affect the function of one component of a protein complex can sometimes be suppressed by overproduction of other components of the complex (22). Thus, we examined whether overproduction of Ipl1 or Sli15 can suppress the sli15-3 or ipl1-2 mutation, respectively. Our results showed that a high copy number _SLI15_-plasmid had no effect on the Ts− growth phenotype of ipl1-2 cells (data not shown). In contrast, a low copy number _IPL1_-plasmid could suppress partially (Fig. 8) and a high copy number _IPL1_-plasmid could suppress almost completely (data not shown) the Ts− phenotype of sli15-3 cells at 37°C. Suppression requires residual Sli15 function, as a high copy number _IPL1_-plasmid could not suppress the inviability of sli15-Δ2::HIS3 cells (data not shown). One possible interpretation of these results is that the sli15-3 mutation compromises the ability of mutant Sli15 to associate with Ipl1, whereas the ipl1-2 mutation compromises the catalytic activity of the Ipl1 protein kinase. In fact, the ipl1-2 mutation is known to alter a residue located in the COOH-terminal catalytic domain of Ipl1 (13).
We have shown previously that the Ts− phenotype of some ipl1 mutants can be suppressed partially by overproduction of a truncated and dominant negative form (Glc7-Δ186-312) of PP1 or by overproduction of Glc8, an inhibitor of PP1, thus suggesting that PP1 acts in opposition to the Ipl1 protein kinase in regulating chromosome segregation (13, 74). If the sli15-3 mutation indeed leads to a reduction in Ipl1 protein kinase function, we might expect the perturbations described above also to result in suppression of sli15-3 mutant phenotype. We found this to be true, as high copy number plasmids containing either the glc7-Δ186-312 dominant negative allele or the wild-type GLC8 gene could suppress the Ts− phenotype of sli15-3 cells at 33°C (Fig. 8). Thus, SLI15 behaves genetically as a positive regulator of IPL1.
Discussion
Roles of Ipl1 in Chromosome Segregation
The most prominent cytological phenotype of ipl1-2 mutant cells is the uneven segregation of chromosomes to opposite poles of apparently normal looking mitotic spindles that are capable of undergoing elongation and disassembly (13). At the restrictive temperature, sister chromatids of chromosome V often appear unseparated in ipl1-2 cells (Figs. 2 and 3), suggesting that sister chromatid separation has failed, or that sister chromatids that have separated are not properly segregated away from each other to opposite poles of the mitotic spindle. In a separate study, it has been shown that the sister chromatid cohesion protein Mcd1/Scc1 (20, 40) dissociates with wild-type kinetics from the chromosomes of ipl1 cells, thus suggesting that sister chromatids probably are separated normally in ipl1 cells (3). Consistent with the idea that ipl1-2 cells are defective in sister chromatid segregation but not separation, sister chromatids of chromosome V are clearly separated in the majority of ipl1-2 cells, although such separated sister chromatids very often are not segregated to opposite poles (Figs. 2 and 3). However, sister chromatid segregation clearly does not fail in all ipl1-2 cells. This observation is consistent with our previous finding that at least some sister chromatids segregate away from each other in ipl1-2 cells, since the mitotic spindle-mediated poleward forces (acting on sister kinetochores) that cause the breakage of topologically intertwined sister chromatids in top2-4 mutant cells (26, 67) also cause chromosome breakage in top2-4 ipl1-2 cells (12).
Defects in the spindle pole bodies, the kinetochore microtubules, or the kinetochores themselves can all lead to a failure in sister chromatid segregation. Nuf2 is a spindle pole–associated protein that can be copurified with yeast spindle poles (77), and immunofluorescence microscopy has shown that Nuf2 colocalizes with Ndc80 to the intranuclear region of spindle poles (45). Interestingly, the phenotype of ndc80 mutant cells is similar, although not identical, to that of ipl1-2 mutant cells. During mitosis, most of the chromosomal DNA remains at one pole of the elongated mitotic spindle in ndc80-1 cells (77). Immunoelectron microscopy has shown that Ndc80 is associated with spindle microtubules, particularly at regions close to the spindle pole body (54). It was proposed that Ndc80 may be associated specifically with kinetochore microtubules (54, 77). If Nuf2 is localized similarly to the kinetochore microtubules, the observation that uneven amounts of Nuf2-GFP are often found at the spindle poles of ipl1-2 cells (Figs. 4 and 5) would suggest quantitative or qualitative differences between the kinetochore microtubules that emanate from opposite spindle poles, possibly as consequences of defects in spindle pole body. For example, the number of kinetochore microtubules emanating from opposite spindle poles may differ greatly in ipl1-2 cells. We do not favor this idea because kinetochore microtubules make up most of the spindle microtubules (79), and immunofluorescent staining of microtubules has not revealed major differences in the intensity of half spindles. Alternatively, the properties of the kinetochore microtubules that emanate from the opposite poles may differ, with those from one pole being less proficient in bringing about sister chromatid segregation to that pole. Such properties may include the ability of kinetochore microtubules to attach to kinetochores and undergo polymerization or depolymerization.
In addition to ndc80 mutants, some ndc10 mutants also have phenotypes that are similar, but not identical, to those of ipl1-2 cells. During mitosis, essentially all chromosomes remain at one pole of the elongated mitotic spindle in ndc10-1 and ndc10-2 cells, and chromosome missegregation is not associated with cell cycle arrest (16, 66). NDC10 encodes an essential component of yeast kinetochores (10, 29). We have examined possible genetic interaction between ipl1 and ndc10 mutations and found that some, but not all, ipl1-2 ndc10-2 double mutants have a restrictive growth temperature lower than that of either single mutant (our unpublished results). Immunofluorescence microscopy has shown that Ndc10 localizes to the spindle pole body region of nearly all cells and also along some short mitotic spindles (16). This observation raises the question whether Nuf2 may actually be associated with yeast kinetochores. We do not favor this idea since Nuf2, but not Ndc10 (or other kinetochore components), is known to be copurified with spindle poles (54, 77). Furthermore, our preliminary results suggest that whereas Nuf2-GFP is concentrated in a single dot-like structure at the spindle poles of wild-type cells, Ndc10-GFP (gift of J. Kahana, Harvard University Medical School, Boston, MA) is more often found in multiple dots that cluster around the spindle poles and also less frequently along mitotic spindles, including those that are elongated. If Nuf2 is associated with kinetochores, the uneven amounts of Nuf2-GFP found at opposite spindle poles in some ipl1-2 mutant cells may simply reflect unequal numbers of chromosomes (and their kinetochores) that are segregated to the two poles. Furthermore, the presence of Nuf2-GFP in dot-like structures along some mitotic spindles in ipl1-2 cells may reflect a failure of kinetochores to move to opposite spindle poles (Figs. 4 and 5), and it would also suggest that the kinetochores are attached to the kinetochore microtubules in ipl1-2 cells. In a separate study, it has been shown that Ipl1 can phosphorylate Ndc10 in vitro, and that the kinetochores assembled in extracts from ipl1 mutants show altered binding to microtubules, thus suggesting that Ipl1 may affect kinetochore functions (3). A better understanding of the actual site of Nuf2 localization and the quantity and quality of kinetochore microtubules present in ipl1 cells will help us understand to what degree the chromosome missegregation observed in ipl1 cells is due to defects in spindle pole or kinetochore function.
A functional GFP-Ipl1 fusion protein is localized to the entire mitotic spindle (Fig. 6), thus suggesting that Ipl1 may play important roles not only at spindle poles, kinetochore microtubules, or kinetochores. Furthermore, instead of the most prominent phenotype of uneven chromosome segregation, a small fraction of ipl1-2 cells exhibits one of two other phenotypes. First, some ipl1-2 cells exhibit a nuclear migration defect (Fig. 1), which has also been reported for the pac15-1/ipl1 mutant (14). Nuclear migration in yeast is dependent on cytoplasmic microtubules (46, 71) and microtubule-based motor proteins (for review see reference 69). We have not detected GFP-Ipl1 on cytoplasmic microtubules. However, it is possible that spindle pole–associated Ipl1 acts on (motor proteins present at) the minus ends of cytoplasmic microtubules to influence their functions. Second, some ipl1-2 cells appear to have monopolar spindles (Fig. 1), which are suggestive of defects in spindle pole body duplication or separation. This phenotype of ipl1 cells is reminiscent of that of Drosophila aurora mutants, which are defective in centrosome separation and form monopolar spindles due to mutations in the gene encoding a homologue of Ipl1 (15). In budding yeast, spindle pole body separation requires the function of the kinesin-related Cin8 and Kip1 motor proteins (28, 50, 57). Interestingly, the nonessential Cin8 motor protein becomes indispensable in ipl1 mutant cells (14 and our unpublished results), thus suggesting that Ipl1 may act on kinesin-related motor proteins. In this regard, it is interesting to note that microinjection of antibodies against HsEg5, a human homologue of Cin8 and Kip1, leads to the abnormal distribution of some centrosome-associated proteins in HeLa cells (76). Furthermore, C. elegans embryonic cells lacking the centrosome-associated Ipl1-homologue AIR-1 kinase are also defective in the localization of the centrosome-associated protein PIE-1 (60).
Sli15 as a Binding Partner of Ipl1
Several lines of evidence indicate that there is a very close functional relationship between Ipl1 and Sli15. First, nonlethal mutations in SLI15 exacerbate the Ts− growth phenotype of ipl1-2 cells, leading to cell inviability at 26°C. Second, sli15-3 and ipl1-2 cells have very similar mutant phenotypes, including failure of separated sister chromatids to be properly segregated (Figs. 2 and 3), abnormal distribution of Nuf2-GFP (Figs. 4 and 5), and minor defects in nuclear migration and bipolar spindle formation (Fig. 1). Third, GFP-Ipl1 and GFP-Sli15 are both localized on the mitotic spindle (Fig. 6). Fourth, Ipl1 and Sli15 associate with each other in vivo, most probably through direct binding (Fig. 7). These results suggest that Sli15 may function as a major physiological substrate and/or as a positive regulatory binding partner of Ipl1.
The following observations support the idea that Sli15 functions as a positive regulatory binding partner of Ipl1, although they by no means preclude the possibility that Sli15 may also function as a major physiological substrate of Ipl1. First, the Ts− phenotype of sli15-3 cells can be suppressed by a small increase in the gene dosage of IPL1 or perturbations that lower the in vivo function of PP1 (Fig. 8). Such perturbations also suppress the Ts− phenotype of ipl1 mutant cells (13, 74). Second, the sli15-3 mutation exhibits synthetic lethal genetic interaction with the same spectrum of mutations that are synthetic lethal with ipl1-2 (our unpublished results). Third, the abundance of HA-Ipl1 is increased in cells that overexpress GST-Sli15, and the phosphorylation state of HA-Ipl1 is also altered in such cells (Fig. 7). Furthermore, the phosphorylated form of HA-Ipl1 appears to be copurified preferentially with GST-Sli15, thus suggesting that GST-Sli15 may promote the phosphorylation of HA-Ipl1.
As a binding partner of Ipl1, Sli15 potentially may stimulate the protein kinase activity of Ipl1 or it may stabilize Ipl1 and target it to its sites of action. We are currently testing the ability of Sli15 to stimulate the in vitro kinase activity of Ipl1. We do not know whether the mitotic spindle association of Ipl1 is dependent on Sli15 function, since we have not been able to localize Ipl1 when it is expressed from the chromosomal copy of IPL1, and a small increase in the dosage of IPL1 suppresses the Ts− phenotype of sli15-3 cells (Fig. 8). It is known that the transcript level of IPL1 varies through the cell cycle, peaking in late G1 (9, 68), presumably due to the presence of a putative MluI cell cycle box (39) in the promoter of IPL1. Interestingly, the transcript level of SLI15 fluctuates less so through the cell cycle (9). This raises the possibility of the existence of two populations of Ipl1, one of which is in association with Sli15. Consistent with this possibility, GFP-Sli15 appears to be mostly restricted to the mitotic spindle whereas GFP-Ipl1 appears to be more broadly distributed (Fig. 6).
The cells from a majority of human colorectal tumors are aneuploid (5), probably due to increased rates of chromosome missegregation in such cells (37). The gene encoding the Ipl1-related Aurora2 kinase is amplified and/or overexpressed in a variety of human tumors, including a significant fraction of colorectal and breast tumors (4, 62, 81). Ectopic expression of Aurora2 in cultured fibroblasts leads to chromosome missegregation, centrosome amplification, and cellular transformation. Since overexpression of GST-Sli15 leads to an increase in the level of HA-Ipl1, overexpression of a Sli15-related binding partner of human Aurora2 may also lead to an increase in the level of Aurora2. A human homologue of Sli15 has not yet been identified. If such a homologue exists, it will be important to find out whether amplification and/or overexpression of the gene encoding this Sli15-homologue may also be correlated with diverse forms of human cancer.
Acknowledgments
We thank David Botstein, Bob Deschenes, Gerry Fink, Bruce Futcher, Phil Hieter, Connie Holm, Brian Haarer, John Kilmartin, Ching-Jung Huang, and Phil Tucker for the supply of strains, plasmids, and antibodies; Peter Reddien for the construction of some of the plasmids used in this study; and Sue Biggins and Andrew Murray for the communication of results before publication.
This work was supported by National Institutes of Health grant GM45185.
Abbreviations used in this paper
GFP
green fluorescent protein
PP1
protein phosphatase 1
TetR-GFP
Tet repressor-green fluorescent protein
Ts−
temperature-sensitive
References
- 1.Andrésson T, Ruderman JV. The kinase Eg2 is a component of the Xenopusoocyte progesterone-activated signaling pathway. EMBO (Eur Mol Biol Org) J. 1998;17:5627–5637. doi: 10.1093/emboj/17.19.5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bender A, Pringle J. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:1295–1305. doi: 10.1128/mcb.11.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biggins S, Severin FF, Bhalla N, Sassoon I, Hyman AA, Murray AW. The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev. 1999;13:532–544. doi: 10.1101/gad.13.5.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et al. A homologue of Drosophila aurorakinase is oncogenic and amplified in human colorectal cancers. EMBO (Eur Mol Biol Org) J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bocker T, Schlegel J, Kullmann F, Stumm G, Zirngibl H, Epplen JT, Ruschoff J. Genomic instability in colorectal carcinomas: comparison of different evaluation methods and their biological significance. J Pathol. 1996;179:15–19. doi: 10.1002/(SICI)1096-9896(199605)179:1<15::AID-PATH553>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 6.Cardenas ME, Dang Q, Glover CVC, Gasser SM. Casein kinase II phosphorylates the eukaryote-specific C-terminal domain of topoisomerase II in vivo. . EMBO (Eur Mol Biol Org) J. 1992;11:1785–1796. doi: 10.1002/j.1460-2075.1992.tb05230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cardenas ME, Walter R, Hanna D, Gasser SM. Casein kinase II copurifies with yeast DNA topoisomerase II and re-activates the dephosphorylated enzyme. J Cell Sci. 1993;104:533–543. doi: 10.1242/jcs.104.2.533. [DOI] [PubMed] [Google Scholar]
- 8.Chan CSM, Botstein D. Isolation and characterization of chromosome-gain and increase-in-ploidy mutants in yeast. Genetics. 1993;135:677–691. doi: 10.1093/genetics/135.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, Davis RW. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 10.Doheny KF, Sorger PK, Hyman AA, Tugendreich S, Spencer F, Hieter P. Identification of essential components of the S. cerevisiaekinetochore. Cell. 1993;73:761–774. doi: 10.1016/0092-8674(93)90255-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyle T, Botstein D. Movement of yeast cortical actin cytoskeleton visualized in vivo. . Proc Natl Acad Sci USA. 1996;93:3886–3891. doi: 10.1073/pnas.93.9.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francisco L, Chan CSM. Regulation of yeast chromosome segregation by Ipl1 protein kinase and type 1 protein phosphatase. Cell Mol Biol Res. 1994;40:207–213. [PubMed] [Google Scholar]
- 13.Francisco L, Wang W, Chan CSM. Type-1 protein phosphatase acts in opposition to the Ipl1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol. 1994;14:4731–4740. doi: 10.1128/mcb.14.7.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiser JR, Schott EJ, Kingsbury TJ, Cole NB, Totis LJ, Bhattacharyya G, He L, Hoyt MA. Saccharomyces cerevisiae genes required in the absence of the CIN8-encoded spindle motor act in functionally diverse mitotic pathways. Mol Biol Cell. 1997;8:1035–1050. doi: 10.1091/mbc.8.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in auroraprevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 16.Goh P-Y, Kilmartin JV. NDC10: a gene involved in chromosome segregation in Saccharomyces cerevisiae. . J Cell Biol. 1993;121:503–512. doi: 10.1083/jcb.121.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gopalan G, Chan CSM, Donovan PJ. A novel mammalian, mitotic spindle-associated kinase is related to yeast and fly chromosome segregation regulators. J Cell Biol. 1997;138:643–656. doi: 10.1083/jcb.138.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gopalan G, Centanni J, Gilbert DJ, Copeland NG, Jenkins NA, Donovan PJ. Novel mammalian kinase related to yeast and fly chromosome segregation regulators is exclusively expressed in the germline. Mol Reprod Dev. 1999;52:18–28. [Google Scholar]
- 19.Gorbsky GJ, Ricketts WA. Differential expression of a phosphoepitope at the kinetochores of moving chromosomes. J Cell Biol. 1993;122:1311–1321. doi: 10.1083/jcb.122.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation through the analysis of MCD1 in S. cerevisiae. . Cell. 1997;91:47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guarente L. Synthetic enhancement in gene interaction: a genetic tool come of age. Trends Genet. 1993;9:362–366. doi: 10.1016/0168-9525(93)90042-g. [DOI] [PubMed] [Google Scholar]
- 22.Hadwiger JA, Wittenberg C, Richardson HE, de Barros M, Lopes, Reed SI. A family of cyclin homologs that control the G1phase in yeast. Proc Natl Acad Sci USA. 1989;86:6255–6259. doi: 10.1073/pnas.86.16.6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heck MMS. Condensins, cohesins, and chromosome architecture: how to make and break a mitotic chromosome. Cell. 1997;91:5–8. doi: 10.1016/s0092-8674(01)80002-7. [DOI] [PubMed] [Google Scholar]
- 24.Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- 25.Heim, S., and F. Mitelman. 1995. Cancer Cytogenetics. 2nd ed. Wiley-Liss, Inc., New York. 530 pp.
- 26.Holm C, Stearns T, Botstein D. DNA topoisomerase II must act at mitosis to prevent nondisjunction and chromosome breakage. Mol Cell Biol. 1989;9:159–168. doi: 10.1128/mcb.9.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoyt MA, Geiser JR. Genetic analysis of the mitotic spindle. Annu Rev Genet. 1996;30:7–33. doi: 10.1146/annurev.genet.30.1.7. [DOI] [PubMed] [Google Scholar]
- 28.Hoyt MA, He L, Loo KK, Saunders WS. Two Saccharomyces cerevisiaekinesin-related gene products required for mitotic spindle assembly. J Cell Biol. 1992;118:109–120. doi: 10.1083/jcb.118.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang W, Lechner J, Carbon J. Isolation and characterization of a gene (CBF2)specifying a protein component of the budding yeast kinetochore. J Cell Biol. 1993;121:513–519. doi: 10.1083/jcb.121.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones JS, Prakash L. Yeast Saccharomyces cerevisiaeselectable markers in pUC18 polylinkers. Yeast. 1990;6:363–366. doi: 10.1002/yea.320060502. [DOI] [PubMed] [Google Scholar]
- 31.Kahana JA, Schnapp BJ, Silver PA. Kinetics of spindle pole body separation in budding yeast. Proc Natl Acad Sci USA. 1995;92:9707–9711. doi: 10.1073/pnas.92.21.9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell. 1997;91:491–500. doi: 10.1016/s0092-8674(00)80435-3. [DOI] [PubMed] [Google Scholar]
- 33.Kimura M, Kotani S, Hattori T, Sumi N, Yoshioka T, Todokoro K, Okano Y. Cell cycle-dependent expression and spindle pole localization of a novel human protein kinase, Aik, related to Aurora of Drosophilaand yeast Ipl1. J Biol Chem. 1997;272:13766–13771. doi: 10.1074/jbc.272.21.13766. [DOI] [PubMed] [Google Scholar]
- 34.Koerner TJ, Hill JE, Myers AM, Tzagoloff A. High-expression vectors with multiple cloning sites for construction of trpE fusion genes: pATH vectors. Methods Enzymol. 1991;194:477–490. doi: 10.1016/0076-6879(91)94036-c. [DOI] [PubMed] [Google Scholar]
- 35.Kranz JE, Holm C. Cloning by function: an alternative approach for identifying yeast homologs of genes from other organisms. Proc Natl Acad Sci USA. 1990;87:6629–6633. doi: 10.1073/pnas.87.17.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lechner J, Carbon J. A 240 kd multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–725. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- 37.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 38.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–525. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 39.McIntosh EM, Atkinson T, Storms RK, Smith M. Characterization of a short, cis-acting DNA sequence which conveys cell cycle stage-dependent transcription in Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:329–337. doi: 10.1128/mcb.11.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- 41.Mitchell DA, Marshall TK, Deschenes RJ. Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast. 1993;9:715–723. doi: 10.1002/yea.320090705. [DOI] [PubMed] [Google Scholar]
- 42.Muhlrad D, Hunter R, Parker R. A rapid method for localized mutagenesis of yeast genes. Yeast. 1992;8:79–82. doi: 10.1002/yea.320080202. [DOI] [PubMed] [Google Scholar]
- 43.Nicklas RB. How cells get the right chromosomes. Science. 1997;275:632–637. doi: 10.1126/science.275.5300.632. [DOI] [PubMed] [Google Scholar]
- 44.Niwa H, Abe K, Kunisada T, Yamamura K. Cell-cycle-dependent expression of the STK-1gene encoding a novel murine putative protein kinase. Gene. 1996;169:197–201. doi: 10.1016/0378-1119(95)00809-8. [DOI] [PubMed] [Google Scholar]
- 45.Osborne MA, Schlenstedt G, Jinks T, Silver PA. Nuf2, a spindle pole body-associated protein required for nuclear division in yeast. J Cell Biol. 1994;125:853–866. doi: 10.1083/jcb.125.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmer RE, Sullivan DS, Huffaker T, Koshland D. Role of astral microtubules and actin in spindle orientation and migration in the budding yeast Saccharomyces cerevisiae. . J Cell Biol. 1992;119:583–593. doi: 10.1083/jcb.119.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pringle JR, Preston RA, Adams A, Stearns T, Drubin D, Haarer BK, Jones E. Fluorescence microscopy methods for yeast. Methods Cell Biol. 1989;31:357–435. doi: 10.1016/s0091-679x(08)61620-9. [DOI] [PubMed] [Google Scholar]
- 48.Robbins J, Dilworth SM, Laskey RA, Dingwall C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell. 1991;64:615–623. doi: 10.1016/0092-8674(91)90245-t. [DOI] [PubMed] [Google Scholar]
- 49.Roghi C, Giet R, Uzbekov R, Morin N, Chartrain I, Le Guellec R, Courturier A, Dorée M, Philippe M, Prigent C. The Xenopusprotein kinase pEg2 associates with the centrosome in a cell cycle-dependent manner, binds to the spindle microtubules and is involved in bipolar mitotic spindle assembly. J Cell Sci. 1998;111:557–572. doi: 10.1242/jcs.111.5.557. [DOI] [PubMed] [Google Scholar]
- 50.Roof DM, Meluh PB, Rose MD. Kinesin-related proteins required for assembly of the mitotic spindle. J Cell Biol. 1992;118:95–108. doi: 10.1083/jcb.118.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose M, Kiesau P, Proft M, Entian K-D. Sequence and functional analysis of a 7.2 kb DNA fragment containing four open reading frames located between RPB5 and CDC28on the right arm of chromosome II. Yeast. 1995;11:865–871. doi: 10.1002/yea.320110908. [DOI] [PubMed] [Google Scholar]
- 52.Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 198 pp.
- 53.Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 54.Rout MP, Kilmartin JV. Components of the yeast spindle and spindle pole body. J Cell Biol. 1990;111:1913–1927. doi: 10.1083/jcb.111.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rout MP, Kilmartin JV. Yeast spindle pole body components. Cold Spring Harbor Symp Quant Biol. 1991;56:687–691. doi: 10.1101/sqb.1991.056.01.077. [DOI] [PubMed] [Google Scholar]
- 56.Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- 57.Saunders WS, Hoyt MA. Kinesin-related proteins required for structural integrity of the mitotic spindle. Cell. 1992;70:451–458. doi: 10.1016/0092-8674(92)90169-d. [DOI] [PubMed] [Google Scholar]
- 58.Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. . Proc Natl Acad Sci USA. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schneider BL, Seufert W, Steiner B, Yang QH, Futcher AB. Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. . Yeast. 1995;11:1265–1274. doi: 10.1002/yea.320111306. [DOI] [PubMed] [Google Scholar]
- 60.Schumacher JM, Ashcroft N, Donovan PJ, Golden A. A highly conserved centrosomal kinase, AIR-1, is required for accurate cell cycle progression and segregation of developmental factors in Caenorhabditis elegansembryos. Development. 1998;125:4391–4402. doi: 10.1242/dev.125.22.4391. [DOI] [PubMed] [Google Scholar]
- 61.Schumacher JM, Golden A, Donovan PJ. AIR-2: an Aurora/ Ipl1-related protein kinase associated with chromosomes and midbody microtubules is required for polar body extrusion and cytokinesis in Caenorhabditis elegansembryos. J Cell Biol. 1998;143:1635–1646. doi: 10.1083/jcb.143.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAKon chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–2200. doi: 10.1038/sj.onc.1201065. [DOI] [PubMed] [Google Scholar]
- 63.Shindo M, Nakano H, Kuroyanagi H, Shirasawa T, Mihara M, Gilbert DJ, Jenkins NA, Copeland NG, Yagita H, Okumura K. cDNA cloning, expression, subcellular localization, and chromosomal assignment of mammalian aurora homologues, aurora-related kinase (ARK) 1 and 2. Biochem Biophys Res Commun. 1998;244:285–292. doi: 10.1006/bbrc.1998.8250. [DOI] [PubMed] [Google Scholar]
- 64.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia colias fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 66.Sorger PK, Doheny KF, Hieter P, Kopski KM, Huffaker TC, Hyman AA. Two genes required for the binding of an essential Saccharomyces cerevisiaekinetochore complex to DNA. Proc Natl Acad Sci USA. 1995;92:12026–12030. doi: 10.1073/pnas.92.26.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spell RM, Holm C. Nature and distribution of chromosomal intertwining in Saccharomyces cerevisiae. . Mol Cell Biol. 1994;14:1465–1476. doi: 10.1128/mcb.14.2.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiaeby microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stearns T. Motoring to the finish: kinesin and dynein work together to orient the yeast mitotic spindle. J Cell Biol. 1997;138:957–960. doi: 10.1083/jcb.138.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Straight AF, Marshall WF, Sedat JW, Murray AW. Mitosis in living budding yeast: anaphase A but no metaphase plate. Science. 1997;277:574–578. doi: 10.1126/science.277.5325.574. [DOI] [PubMed] [Google Scholar]
- 71.Sullivan DS, Huffaker TC. Astral microtubules are not required for anaphase B in Saccharomyces cerevisiae. . J Cell Biol. 1992;119:379–388. doi: 10.1083/jcb.119.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO (Eur Mol Biol Org) J. 1998;17:667–676. doi: 10.1093/emboj/17.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tseng T-C, Chen S-H, Hsu Y-P P, Tang TK. Protein kinase profile of sperm and eggs: cloning and characterization of two novel testis-specific protein kinases (AIE1, AIE2) related to yeast and fly chromosome segregation regulators. DNA Cell Biol. 1998;17:823–833. doi: 10.1089/dna.1998.17.823. [DOI] [PubMed] [Google Scholar]
- 74.Tung HYL, Wang W, Chan CSM. Regulation of chromosome segregation by Glc8p, a structural homolog of mammalian inhibitor 2 that functions as both an activator and an inhibitor of yeast protein phosphatase 1. Mol Cell Biol. 1995;15:6064–6074. doi: 10.1128/mcb.15.11.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vandré DD, Davis FM, Rao PN, Borisy GG. Phosphoproteins are components of mitotic microtubule organizing centers. Proc Natl Acad Sci USA. 1984;81:4439–4443. doi: 10.1073/pnas.81.14.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whitehead CM, Rattner JB. Expanding the role of HsEg5 within the mitotic and post-mitotic phases of the cell cycle. J Cell Sci. 1998;111:2551–2561. doi: 10.1242/jcs.111.17.2551. [DOI] [PubMed] [Google Scholar]
- 77.Wigge PA, Jensen ON, Holmes S, Souès S, Mann M, Kilmartin JV. Analysis of the Saccharomycesspindle pole by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. J Cell Biol. 1998;141:967–977. doi: 10.1083/jcb.141.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Winey M, Hoyt MA, Chan C, Goetsch L, Botstein D, Byers B. NDC1: a nuclear periphery component required for yeast spindle pole body duplication. J Cell Biol. 1993;122:743–751. doi: 10.1083/jcb.122.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Winey M, Mamay CL, O'Toole ET, Mastronarde DN, Giddings TH, McDonald KL, McIntosh JR. Three-dimensional ultrastructural analysis of the Saccharomyces cerevisiaemitotic spindle. J Cell Biol. 1995;129:1601–1615. doi: 10.1083/jcb.129.6.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yanai A, Arama E, Kilfin G, Motro B. ayk1, a novel mammalian gene related to Drosophilaaurora centrosome separation kinase, is specifically expressed during meiosis. Oncogene. 1997;14:2943–2950. doi: 10.1038/sj.onc.1201144. [DOI] [PubMed] [Google Scholar]
- 81.Zhou H, Kuang J, Zhong L, Kuo W-l, Gray JW, Sahin A, Brinkley BR, Sen S. Tumour amplified kinase STK15/BTAKinduces centrosome amplification, aneuploidy and transformation. Nature Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]