Domains of Axin Involved in Protein–Protein Interactions, Wnt Pathway Inhibition, and Intracellular Localization (original) (raw)
Abstract
Axin was identified as a regulator of embryonic axis induction in vertebrates that inhibits the Wnt signal transduction pathway. Epistasis experiments in frog embryos indicated that Axin functioned downstream of glycogen synthase kinase 3β (GSK3β) and upstream of β-catenin, and subsequent studies showed that Axin is part of a complex including these two proteins and adenomatous polyposis coli (APC). Here, we examine the role of different Axin domains in the effects on axis formation and β-catenin levels. We find that the regulators of G-protein signaling domain (major APC-binding site) and GSK3β-binding site are required, whereas the COOH-terminal sequences, including a protein phosphatase 2A binding site and the DIX domain, are not essential. Some forms of Axin lacking the β-catenin binding site can still interact indirectly with β-catenin and regulate β-catenin levels and axis formation. Thus in normal embryonic cells, interaction with APC and GSK3β is critical for the ability of Axin to regulate signaling via β-catenin. Myc-tagged Axin is localized in a characteristic pattern of intracellular spots as well as at the plasma membrane. NH2-terminal sequences were required for targeting to either of these sites, whereas COOH-terminal sequences increased localization at the spots. Coexpression of hemagglutinin-tagged Dishevelled (Dsh) revealed strong colocalization with Axin, suggesting that Dsh can interact with the Axin/APC/GSK3/β-catenin complex, and may thus modulate its activity.
Keywords: β-catenin, glycogen synthase kinase 3β (GSK3β), adenomatous polyposis coli (APC), Dishevelled (Dsh), dorsal axis formation
Axin is the product of the murine genetic locus originally called Fused, in which mutations cause a variety of developmental defects (Gluecksohn-Schoenheimer, 1949; Theiler and Gluecksohn-Waelsch, 1956; Jacobs-Cohen et al., 1984). The Axin gene was cloned with the aid of an insertional mutation and found to potentially encode a protein of up to 992 amino acids (aa)1 (Perry et al., 1995; Zeng et al., 1997). Axin contains two conserved domains, a regulators of G-protein signaling domain (RGS; Dohlman and Thorner, 1997) near its NH2 terminus and a COOH-terminal DIX domain (also found in Dishevelled [Dsh]; Cadigan and Nusse, 1997) that suggested a role in signal transduction. More specific insight into the function of Axin came from studies of its effects on vertebrate embryogenesis. The occurrence of axial duplications in loss-of-function Axin mutants in the mouse suggested that the gene might play a negative regulatory role in an early step in axis formation. This hypothesis was tested and confirmed by the ability of overexpressed Axin to block dorsal axis formation in Xenopus embryos. Further studies showed that the effect of Axin is due to its specific ability to inhibit signal transduction through components of the Wnt pathway and suggested that Axin functioned downstream of glycogen synthase kinase 3β (GSK3β) and upstream of β-catenin (Zeng et al., 1997).
β-Catenin is thought to serve as a key mediator of Wnt signal transduction that is regulated through the following mechanism (for review see Gumbiner, 1995; Peifer, 1995; Miller and Moon, 1996; Cadigan and Nusse, 1997). In the absence of a Wnt signal, β-catenin is confined to the plasma membrane, where it stably is associated with cadherin adhesion molecules. Cytosolic β-catenin levels are very low because free β-catenin is a target for GSK3β-dependent phosphorylation and is degraded rapidly via the ubiquitin pathway. In the presence of a Wnt signal, GSK3β phosphorylation of β-catenin is inhibited, free β-catenin is stabilized, accumulates in the cytoplasm, and is imported into the nucleus. β-Catenin can interact with HMG-box transcription factors of the TCF/Lef-1 family, leading to activation of specific target genes.
Despite the apparent simplicity of this signaling cascade, the mechanisms involved in the regulation of β-catenin are still rather obscure. For instance, it is still not known whether upstream components (Wnt, Frizzled, Dsh) affect GSK3β activity or the accessibility of β-catenin to GSK3β. The tumor suppressor gene product adenomatous polyposis coli (APC), which also binds directly to β-catenin, appears to be required to maintain low levels of β-catenin in mammalian cell lines (Munemitsu et al., 1995). However, experiments in embryonic systems are inconsistent with APC being only a negative regulator of β-catenin and suggest that it might, on the contrary, be an activator of the pathway (Rocheleau et al., 1997; Vleminckx et al., 1997). Finally, there is still no definitive evidence that regulation of the β-catenin level is the only important parameter in β-catenin signaling, or whether phosphorylation could affect directly its signaling activity. The apparent involvement of Axin in Wnt signal transduction, at a level close to GSK3β and β-catenin, indicated that Axin might be at the heart of the process of β-catenin phosphorylation/degradation.
To investigate the mechanism by which Axin participates in the regulation of Wnt signal transduction, we undertook a study of the interaction of Axin with various components of the Wnt pathway. We also carried out a functional dissection of the role of different regions of Axin in its ability to ventralize frog embryos (an in vivo assay for inhibition of β-catenin signaling) or to influence the levels of β-catenin in the frog embryo. During the course of this work, several publications have described the direct interaction of Axin with APC, GSK3β, and β-catenin (Hart et al., 1998; Ikeda et al., 1998; Itoh et al., 1998; Kishida et al., 1998; Sakanaka et al., 1998). These reports suggested that the primary function of Axin was to bind GSK3β and β-catenin simultaneously, thus promoting the phosphorylation of β-catenin and its subsequent degradation. Similar results have been reported for an Axin homologue called Axil or Conductin (Behrens et al., 1998; Yamamoto et al., 1998). Both Axin and Axil/Conductin were found to bind to APC through their RGS domains (Behrens et al., 1998; Hart et al., 1998) and to GSK3β and β-catenin through two distinct central domains (Behrens et al., 1998; Ikeda et al., 1998), thus mediating the formation of a multiprotein complex. However, the RGS (APC-binding) domain was not required for Axin to stimulate the phosphorylation of β-catenin in vitro or its turnover in SW480 colon cancer cells (Behrens et al., 1998; Hart et al., 1998; Ikeda et al., 1998), raising questions about the role of APC in this complex. In addition to these proteins, Axin has been found to have a binding site for the serine/threonine protein phosphatase 2A (PP2A), which might modulate phosphorylation of proteins in the complex, and another region that mediates multimerization with other Axin molecules. The latter region includes the DIX domain, which is highly conserved between Axin and Dsh, suggesting that Axin and Dsh might also interact through this domain (Hsu et al., 1999).
In the studies reported here, we have examined the regions of Axin required for its role in the regulation of axis formation and β-catenin levels in the frog embryo. We have found that in this system both the RGS domain and GSK3β binding sites, but not necessarily the β-catenin site, are essential for activity. We have shown that epitope-tagged Axin expressed in frog embryos is localized to the plasma membrane and to a characteristic type of intracellular spots and have delimited the sequences in Axin that mediate this pattern of localization. Finally, we have found that epitope-tagged forms of Axin and Dsh show strongly overlapping patterns of localization when coexpressed, suggesting that Dsh can also form part of the Axin complex.
Materials and Methods
Plasmid Construction
To express Myc-tagged forms of Axin, the coding region of mouse Axin (form 1) sequence was inserted downstream of the SP6 promoter in the vector pCS2-MT (Rupp et al., 1994). DNA inserts amplified by PCR with Pfu DNA polymerase (Stratagene) were used for the construction of plasmids containing small fragments, such as Ax455-552, Ax497-672, etc. Full-length (FL) Xenopus APC cDNA (Vleminckx et al., 1997) was inserted downstream of the CMV/SP6 promoter in the pCS2 vector, with a 5′ 5× VSV-G (vesicular stomatitis virus glycoprotein) tag (YTDIEMNRLGK). Human Myc epitope-tagged APC constructs used for direct binding assays were described previously (Vleminckx et al., 1997). pET32 vector (Novagen, Inc.) was used to produce the His·S tagged Axin fusion proteins. The reading frame of all constructs was confirmed by sequencing and detection of expected sized bands in Western blot or Coomassie brilliant blue R250 stained SDS–polyacrylamide gels. Plasmids for transfection and in vitro transcription/translation were isolated using the midi-prep kit (QIAGEN Inc.). Other constructs used included: Myc-tagged Xenopus β-catenin (Fagotto et al., 1996), hemagglutinin (HA)-tagged Xenopus β-catenin (Funayama et al., 1995), HA-tagged Xenopus Dsh (gift of Dr. U. Rothbächer, University of Marseille, France), HA-tagged dominant negative human GSK3β (gift of Dr. X. He, Harvard Medical School, Boston, MA).
Antibodies
Antibodies were purchased from the indicated sources: anti–β-catenin mouse mAb, clone 14, and anti-GSK3β mAb, clone 7, Transduction Laboratories; anti–VSV-G mAb P5D4, Boehringer Mannheim; anti-Myc 9E10.2 mAb, Calbiochem-Novabiochem; anti–HA-tag rabbit polyclonal antibodies (pAb), Santa Cruz Biotechnology; and anti–β-galactosidase rabbit pAb, Cappel Laboratories and Organon Teknika Corp. Anti-Myc rabbit pAb was raised against the c–Myc-epitope tag (EQKLISEEDL) and affinity-purified. Anti-HA mAb 12CA5 was a gift from Dr. P. McCrea (M.D. Anderson Cancer Center, Houston, TX).
Tissue Culture and Transient Transfection
293 cells, obtained from the American Type Culture Collection, were cultured in DME/F12 medium (Mediatech) supplemented with 10% FBS (HyClone Laboratories Inc.) in humidified 6% CO2. Cells were transfected using a calcium phosphate mammalian cell transfection kit (5 Prime → 3 Prime, Inc.). The next day cells were collected and lysed and analyzed for transient expression of transfected DNAs.
Immunoprecipitation (IP) and Western Blot
For IP and Western blot analysis, 293 cells were washed with PBS, pH 7.2, and lysed in lysis buffer (150 mM NaCl, 5 mM EDTA, 50 mM NaF, 50 mM KH2PO4, 10 mM sodium molybdate, 20 mM Tris-HCl, pH 7.4, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 0.6 mM DTT, 2 mM sodium orthovanadate, 0.2 mM PMSF, 1% Triton X-100). After 20 min at 4°C with constant rotation, the lysate was centrifuged at 14,000 g for 15 min and the supernatant was saved. Protein concentration was measured by the Lowry method (Lowry et al., 1951). For coimmunoprecipitation (coIP) of Axin/APC/ β-catenin/GSK3β complex, 150–300 μg (total protein) of cell lysate was incubated with 1–2 μg of an appropriate antibody in lysis buffer for 2 h at 4°C with constant rotation. 30 μl of protein A/G plus–agarose (Santa Cruz Biotechnology) was added and the incubation continued for an additional 1.5 h. Immunoprecipitates were pelleted and washed three times with lysis buffer. Immunoprecipitates were analyzed by SDS-PAGE (5% acrylamide for APC detection, 10% for detection of other proteins) and Western blot, using HRP-conjugated donkey anti–rabbit and sheep anti– mouse secondary antibodies (Amersham Life Science) and the chemiluminescence system (RENAISSANCE™; NEN Life Science Products). Approximately 10–20 μg of protein was used to detect expression of the various constructs.
Direct Binding Assay
Myc-APC constructs were produced with TNT coupled wheat germ extract system (Promega). These [35S]Met-labeled proteins were incubated with 2 μg of bacterially expressed S-tagged Axin fusion proteins in 500 μl of buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 5 mM DTT, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 0.1 mM PMSF, 0.5% NP-40 at 37°C for 10 min and at 4°C for 10 min. The protein complexes were precipitated with 30 μl S-protein agarose (Novagen), washed three times with the same incubation buffer, and analyzed by SDS-PAGE (7.5% gel) and autoradiography.
Mobility Shift Assay
Lysates from cells transiently expressing VSV-G–tagged APC (VSV– APC) and Axin constructs were used for IP with anti–VSV-G P5D4 mAb. Immunoprecipitates were incubated with 1,000 U of λ-protein phosphatase (New England Biolabs Inc.) in 50 μl reaction buffer (50 mM Tris-HCl, pH 7.8, 5 mM DTT, 2 mM MnCl2, and 100 μg/ml BSA) at 30°C for 30 min. λ-Protein phosphatase-treated samples were separated by SDS-PAGE (5% gels) and VSV–APC was detected by immunoblot using the P5D4 mAb.
Embryo Injection and Scoring
mRNA was synthesized using SP6 polymerase (Promega) and dissolved in diethyl pyrocarbonate–treated water. For ventralization assays, 15 nl mRNA was injected in the subequatorial region of the two dorsal blastomeres of a four cell stage embryo. For axis duplication assays, 15 nl mRNA was injected in one ventral blastomere. Ventralization was scored at tailbud/tadpole stages according to the dorsoanterior index (Kao and Elinson, 1988). Axis duplication was scored at neurula/tailbud stage, in four categories (Fagotto et al., 1997): (1) complete secondary axis (including cement gland), (2) partial axis (no cement gland), (3) vestigial axis (small remnants, blebs), and (4) normal. Only complete and partial axes were considered as bonafide secondary axes.
Expression Levels in Embryos
Expression levels of Myc-tagged Axin constructs were determined from NP-40 extracts of late blastula–early gastrula embryos (stage 9–10 1/2) by SDS-PAGE and immunoblot, using the anti–Myc-tag 9E10.2 mAb. NP-40 buffer had the following: 1% NP-40, 100 mM NaCl, 10 mM Hepes, pH 7.4, 1 mM EDTA, with a cocktail of protease inhibitors (1 mM PMSF, 1 μg/ml pepstatin A, 2 μg/ml leupeptin, 4 μg/ml aprotinin, 10 μg/ml antipain, 50 μg/ml benzamidine, 10 μg/ml soybean trypsin inhibitor, 100 μg/ml iodoacetamide).
Cell Fractionation
Early cleaving embryos were coinjected with 1 ng Myc-tagged Axin (Myc– Axin) and 3 ng β-galactosidase mRNA. At stage 9–10, 10 embryos were homogenized in 500 μl 250 mM sucrose, 110 mM potassium acetate, 10 mM Hepes, pH 7.4, 2 mM magnesium acetate, 2 mM DTT, 1 mM EDTA supplemented with protease inhibitors. The homogenate was centrifuged for 5 min at 1,500 g, and the low speed pellet was extracted in NP-40 buffer. The low speed supernatant was fractionated further by centrifugation for 30 min at 100,000 g in a tabletop ultracentrifuge (TL-100; Beckman Instruments Inc.) into a high speed pellet and supernatant. The fractions were analyzed for Myc–Axin and β-galactosidase by SDS-PAGE and immunoblot.
Con A Precipitation
FL Axin or various mutant constructs mRNA were injected into 4–8-cell stage embryos. At stage 9–10, pools of six embryos were extracted in 500 μl NP-40 buffer and each extract was incubated with 50 μl of Con A–agarose beads (75% slurry; Sigma Chemical Co.) for 1–2 h at 4°C. The beads were spun down, the supernatant was collected (unbound fraction), the beads washed three times with 1 ml NP-40 buffer, and extracted by boiling in SDS-PAGE sample buffer (bound fraction). Levels of Myc–Axin constructs in bound and unbound fractions were analyzed by SDS-PAGE and immunoblot using the anti-Myc 9E10 mAb.
β-Catenin Stability
HA-tagged β-catenin mRNA (75 pg) was coinjected with β-galactosidase mRNA (control) or various Axin mutant mRNAs. Amounts of mRNA injected were the following: β-galactosidase, 1 ng; FL Axin (Ax12-956), 1 ng; Ax12-531, 0.5 ng; Ax194-530, 0.5 ng; Ax194-672, 0.25 ng; AxΔ251-351, 1 ng; Ax331-956, 0.5 ng; and Ax531-956, 0.5 ng. Total amounts of injected mRNA were adjusted to 1.075 ng by addition of β-galactosidase mRNA. In some experiments (see Fig. 5 C), higher levels of β-catenin were tested using 0.75–1.5 ng mRNA. Embryos were extracted in NP-40 buffer at stage 9–10 and either directly analyzed by SDS-PAGE and immunoblot, or cleared from cadherin-bound β-catenin as follows: six embryos were extracted in 200 μl NP-40 buffer. 50 μl of Con A–agarose beads (75% slurry) were added, and the samples were incubated with constant mixing for 1–2 h. The beads were spun down and discarded and the supernatant was analyzed for β-catenin levels using an anti–HA-tag rabbit antibody (Santa Cruz Biotech.), as well as for Axin mutant levels using the 9E10.2 mAb.
Figure 5.

Expression of Axin and Axin mutants affects β-catenin stability. (A) Diagram of Axin molecule. (B) Dorsal injection of FL Axin and several mutant constructs downregulate free β-catenin levels. HA-tagged β-catenin mRNA (75 pg) was coinjected with β-galactosidase mRNA (control), or the indicated Myc-tagged forms of Axin, in the dorsal side of early cleaving embryos. mRNA amounts (see Materials and Methods) were selected to produce equivalent levels of expression of Axin constructs, and β-galactosidase mRNA was added to equalize amounts of injected mRNA. Extracts from late blastula embryos were depleted of cadherin-bound β-catenin using Con A beads and the levels of free HA–β-catenin were analyzed by immunoblot. Duplicate samples correspond to two independent pools of seven embryos each. FL and several mutant forms (Ax12-531, Ax194-531, and Ax194-672) strongly reduced β-catenin levels. On the other hand, AxΔ251-351 and Ax331-956 caused an increase in β-catenin levels. Ax531-956 had no effect. (C) Stabilization of total β-catenin by ventral injection of AxΔ231-351. HA–β-catenin mRNA was coinjected with β-galactosidase, AxΔ251-351, FL Axin, or HA-dnGSK3β mRNAs in the ventral side of cleaving embryos. Total levels of HA–β-catenin were compared directly by immunoblot. AxΔ251-351, but not FL Axin, induced an increase in β-catenin comparable to the increase obtained with dnGSK3β (Yost et al., 1996). The lower panels show expression of HA-dnGSK and of the Myc-tagged Axin constructs. (D) Axin downregulates FL β-catenin, but not NH2-terminally truncated β-catenin. HA-tagged β-catenin or ΔNβ-catenin mRNA (750 pg) were coinjected dorsally with β-galactosidase mRNA (control, 2 ng) or FL Axin mRNA (2 ng). Total levels of HA–β-catenin were compared.
Immunofluorescence (IF)
Stage 9–11 embryos were fixed in 4% paraformaldehyde, 100 mM Hepes, pH 7.4, 100 mM NaCl for 1 h at room temperature, then in Dent's fixative (20% DMSO, 80% methanol) overnight at -20°C. They were rinsed in 100 mM Tris-HCl, 100 mM NaCl, and embedded in 15%, then 25% fish gelatin, and 10-μm cryosections were prepared as described (Fagotto and Gumbiner, 1994; Fagotto, 1999). Sections were labeled with 9E10.2 mAb and anti–mouse Oregon green488 or Alexa488 secondary antibodies (Molecular Probes Inc.), the yolk counterstained with Eriochrome back, and nuclei with 4′,6-diamidino-2-phenylindole (DAPI) as described (Fagotto, 1999). For double staining, sections from embryos coinjected with Myc– Axin and HA-tagged Dsh (HA–Dsh) mRNAs (1 ng each) were stained simultaneously with anti-Myc rabbit pAb and anti-HA mAb 12CA5, followed by Alexa488 goat anti–rabbit and Cy3 donkey anti–mouse (Dianova) secondary antibodies.
For localization of Myc–Axin in cultured cells, HeLa cells cultured in DMEM were transfected with pCS2-Myc–Axin using LipofectAmine (GIBCO BRL). 36-48 h after transfection, cells were fixed in 4% paraformaldehyde/PBS, permeabilized with 0.05% Triton X-100, and labeled with anti-Myc 9E10.2 mAb and Cy3 goat anti–mouse (Dianova) secondary antibodies. Nuclei were counterstained with DAPI. Samples were observed with an Axioplan epifluorescence microscope (Zeiss) using standard fluorescein and Cy3 filters, and digital images were collected using a camera (768x576 3CCD color video; Sony).
Electron Microscopy and Immunogold Labeling
Preembedding labeling was performed as described (Kurth, 1997; Fagotto, 1999). In brief, embryos expressing FL Myc–Axin or Myc-AxΔ531-810 were fixed at stage 10 with 4% paraformaldehyde, 0.02% glutaraldehyde, and 100 mM Hepes-NaOH. Labeling was performed by incubating 100-μm vibrotome sections with the 9E10.2 mAb and a Nanogold-coupled anti– mouse secondary antibody, followed by silver enhancement. The reaction produced electron dense aggregates with a diameter of ∼20–60 nm (see Fig. 8, C–E). The sections were embedded in Spurr resin and ultrathin sections were prepared.
Figure 8.



Cell fractionation of Myc–Axin in Xenopus embryos: sedimentability and Con A binding. (A) Diagram of Axin molecule. (B) Myc–Axin is associated with a sedimentable fraction. Homogenates of late blastula embryos were fractionated into a low speed sedimentable fraction (P, pellet), a high speed sedimentable fraction (M, membranes), and a high speed supernatant (S, soluble fraction) as described in Materials and Methods. β-Galactosidase was coexpressed and used as a control for soluble cytosolic proteins. Unlike β-galactosidase, Myc–Axin was sedimentable under these conditions. (C) Myc–Axin is fully extractable in NP-40. Embryos expressing Myc–Axin were extracted in NP-40–containing buffer (sol). The insoluble pellet was reextracted in the presence of SDS (insol, NP-40– insoluble). (D) A pool of Axin is associated with a membrane glycoprotein, and this association requires the NH2-terminal domain. Myc-tagged FL Axin and various Axin mutant constructs were expressed in embryos, and NP-40 extracts were fractionated using Con A beads. Bound fractions (B) were four times concentrated relative to unbound fractions (U). FL Axin showed significant association with Con A beads that indicates a stable interaction with a membrane glycoprotein. Stronger binding was observed for the NH2-terminal fragments Ax12-531. On the other hand, constructs lacking the NH2-terminal domain showed no (Ax-531-956 and Ax194-353) or very weak binding (Ax194-956).
Postembedding labeling was performed on small blebs obtained from the wounds of injected embryos. These blebs contained a large number of Axin-expressing cells, and their ultrastructure was better preserved than in whole embryos (Kurth, 1997). Paraformaldehyde/glutaraldehyde-fixed samples were processed for Lowycryl embedding, ultrathin sectioning, and immunogold labeling (9E10.2 mAb and 15 nm gold-coupled protein G) according to standard procedures.
Results
Binding of Full-length and Mutant Forms of Axin to APC, GSK3β, and β-Catenin
When epitope-tagged full-length Axin (amino acids 12– 956) was expressed in 293 cells, endogenous GSK3β and β-catenin, as well as VSV-epitope tagged APC (VSV– APC), could be coimmunoprecipitated (coIP) with Axin. A variety of mutant forms of Axin were used next for coIP and direct binding assays to further delimit the regions of Axin required for these interactions (Figs. 1 and 2 and data not shown) and to compare binding abilities with activity in functional assays (see below). The results are summarized in Fig. 3, which includes a schematic diagram of Axin, indicating the locations of the major binding sites for these proteins as well as PP2A binding and Axin self-binding (Hsu et al., 1999).
Figure 1.


In vivo interaction of Myc-tagged Axin and mutant Axin constructs with endogenous β-catenin (A) or GSK3β (B). Myc-tagged Axin constructs were expressed in 293 cells. Cell lysates were immunoprecipitated with anti-Myc mAb and immunoprecipitates were analyzed for endogenous β-catenin or GSK3β by Western blot (upper panels in A and B). Expression of the transfected Myc– Axin constructs (asterisks) is shown in the lower panels.
Figure 2.

Axin binds to APC and induces APC phosphorylation in vivo. (A) Diagram showing the regions of human APC included in three APC constructs. (B) Direct in vitro binding of APC25 to Axin fragments containing the RGS domain. Sulfur-35–labeled APC25 protein was incubated with S-tagged fusion proteins containing the indicated regions of Axin. After S-protein agarose IP, bound APC25 was detected by SDS-PAGE and autoradiography. Coomassie blue staining of the Axin fusion proteins is shown at the bottom. (C) In vitro binding with NH2-terminal Axin fragments reveals a second APC-binding site upstream of the RGS domain. Binding of APC2 and APC25 required the RGS domain (Ax96-353 and Ax96-480). However, FL hAPC also interacted with Ax96-253, and hAPC2 bound Ax96-253 very strongly. Interaction of Sulfur-35–labeled FL APC, APC2, APC21 and APC25 with S-tagged fusion proteins was determined as in B. (D) CoIP of VSV–APC with FL Axin and several mutant forms of Axin. 293 cells were cotransfected with VSV-G–tagged Xenopus APC (VSV–APC) and the indicated Axin constructs. The cell lysates were immunoprecipitated with anti-Myc and probed with anti–VSV-G antibody. Failure to detect interaction with Ax12-355 was due to the very low levels of VSV–APC observed when coexpressed with this particular construct (E). (E and F) Axin-dependent phosphorylation of APC. (E) Axin-induced mobility shift of VSV–APC. Total lysates from cotransfected cells were analyzed by Western blot using an anti-VSV mAb. FL Axin and mutants Ax12-810, 12-600, and 12-531 induced a mobility shift, whereas AxΔ251-351, Ax12-355, and Ax497-622 did not. (F) Phosphatase treatment eliminated the mobility shift. Cotransfected cell lysates were IP with anti-VSV mAb, and the products were analyzed by SDS-PAGE and Western blot before (left) or after (right) incubation with λ-protein phosphatase.
Figure 3.

Summary of properties of FL and mutant forms of Myc-tagged Axin. The diagram at the top indicates homology domains and binding sites for other proteins based on data reported here and previously (see text for references). Numbers at left indicate the NH2- and COOH-termini, and any internal deletion (Δ). Dotted lines indicate deletions. Vent., extent of ventralization (+++ or ++) or dorsalization (D, strong or d, weaker, and only at high concentration) upon injection into Xenopus embryos. β-cat level indicates the effect on the level of expression of coinjected HA-tagged β-catenin: up arrow, increase; side arrow, no change; and down arrow, decrease. coIP indicates coimmunoprecipitation with the indicated protein (VSV–APC, endogenous GSK3β, or endogenous β-catenin) after transient transfection into 293 cells. +/− indicates that coIP was weak and sporadic in multiple assays. The question mark indicates that the coIP of APC could not be determined because expression of these two Axin mutants greatly reduced the total level of cellular APC (E and data not shown). Localiz. indicates a pattern of intracellular localization after injection into Xenopus embryos. Sp and sp, strong or weak localization to spots, respectively; M and m, strong or weak plasma membrane staining, respectively; df, diffuse cytoplasmic staining; asterisk, pattern was slightly particulate, in contrast to other mutants labeled df. A blank space indicates not determined.
The region of rAxin corresponding to aa 561–630 of mAxin has been shown to contain a β-catenin binding site (Ikeda et al., 1998) and our results confirmed that all Axin mutants containing this region could coIP with endogenous β-catenin. However, several mutants that lacked this region but included the RGS domain (APC binding site) were also able to coIP with β-catenin (Ax12-531, Ax194-531, and AxΔ531-810) (Fig. 1 A and data not shown). This may be due to indirect interaction with β-catenin via APC, because deletion of the RGS domain from this mutant Axin (Ax12-531Δ251-351), which eliminated APC binding (see below), also eliminated coIP with β-catenin (Fig. 1 A). The 85-aa region of rAxin corresponding to mAxin 477–561 was reported to bind to GSK3β (Ikeda et al., 1998). We found that Ax497-600, Ax403-552, and Ax455-552 all showed strong coIP with GSK3β (Fig. 1 B), indicating that the binding site is located in the 55-aa segment between aa 497 and 552. Mutants that terminate at aa 531 either coIP very weakly (Ax12-531 and Ax194-531) or fail to coIP with GSK3β (Ax12-531Δ251-351), suggesting that truncation at this site partially disrupts the GSK3β binding site (Figs. 1 B and 3).
The RGS domain of Axin has been identified as a site for direct interaction with the 20-aa repeat region of APC (Hart et al., 1998; Kishida et al., 1998). We confirmed that only Axin fragments including the RGS domain could bind to the 20-aa repeat region of APC (APC2 or APC25) (Fig. 2, A–C). However, we also found that a second region of Axin, between aa 96–253, could bind directly to the NH2-terminal region of APC (APC21), which contains the Armadillo repeats and 15 aa repeats (Fig. 2 C). coIPs of the Axin mutants with VSV–APC generally were dependent on the RGS domain: all mutants containing this domain were able to coIP with VSV–APC, whereas all but one of those lacking it failed to coIP with VSV–APC (Figs. 2 D and 3). However, Ax331-956 also was able to coIP with APC, probably via β-catenin (Fig. 2 D). The second APC binding region (aa 96–253) was not sufficient for coIP with VSV–APC (Fig. 2 D, AxΔ251-351). Whether Ax12-355 or Ax12-167 could coIP with VSV–APC could not be determined because expression of these mutant Axins resulted in a strong reduction in the level of VSV– APC (Fig. 2 E and data not shown).
Axin Overexpression Induces Phosphorylation of APC In Vivo
When FL Axin and VSV–APC were cotransfected into 293 cells, the electrophoretic mobility of VSV–APC was reduced compared with control cells cotransfected with VSV–APC plus pCS2 vector. Ax12-810 and Ax12-531 caused a similar mobility shift, whereas AxΔ231-351, Ax12-355, and Ax497-672, which lack either the GSK3β or APC binding site, did not (Fig. 2 E). It has been shown that phosphorylation of APC by GSK3β (Rubinfeld et al., 1996) can be stimulated by Axin in vitro (Hart et al., 1998). To test whether this Axin-induced mobility shift was due to phosphorylation, the immunoprecipitated proteins were treated with λ-protein phosphatase before immunoblot analysis with anti-VSV. This treatment eliminated the mobility shift, indicating that it was due to phosphorylation (Fig. 2 F). This suggests that binding of APC and GSK3β to Axin promotes the phosphorylation of APC in vivo, presumably by GSK3β (Rubinfeld et al., 1996).
Axin Sequences Necessary for Ventralization of Xenopus Embryos
We previously have shown that the ability of Axin to inhibit dorsal axis formation, when expressed in early Xenopus embryos, is due to its inhibitory effect on the Wnt signaling pathway (Zeng et al., 1997). Therefore, we used this assay to delimit the sequences in Axin required for its negative effects on signaling through the Wnt pathway. 22 mutant forms of Axin were expressed by mRNA injection on the dorsal side of 4-cell stage embryos that were cultured to the tadpole stage and examined for the extent of dorsal axis formation (fraction of embryos ventralized and dorso-anterior index). The amount of injected mRNAs was systematically titrated to obtain comparable levels of expression for the various mutants. The results are summarized in Fig. 3, the data are listed in Table I and examples are shown in Fig. 4.
Table I.
Effect of Axin Mutants on Axis Development: Dorsal Injections
| Mutant | Amount mRNA injected | Total injected embryos | Percent ventralized§ | Average DAI‖ | Ventralization | No. exp ¶ |
|---|---|---|---|---|---|---|
| ng‡ | ||||||
| FL (12-956) | 2 × 0.13 | 28 | 46 | 3.6 | +/− | 2 |
| FL (12-956) | 2 × 0.25 | 57 | 70 | 2.5 | + | 2 |
| FL (12-956) | 2 × 2 | 103 | 94 | 1.0 | +++ | 6 |
| Δ251-351 | 2 × 1–2 | 60 | 4 | ? | dorsalized | 5 |
| Δ352-631 | 2 × 1 | 61 | 26 | 4.3 | − | 4 |
| Δ531-810 | 2 × 0.5–2 | 63 | 89 | 1.6 | ++ | 5 |
| 194-956 | 2 × 0.5–1 | 107 | 91 | 1.6 | ++ | 5 |
| 331-956 | 2 × 0.25 | 40 | 10 | 4.9 | − | 3 |
| 331-956 | 2 × 0.5–1 | 63 | 0 | ? | dorsalized | 3 |
| 531-956 | 2 × 0.5–1.5 | 107 | 11 | 4.6 | − | 6 |
| 810-956 | 2 × 0.5–1 | 50 | 4 | 4.8 | − | 4 |
| 194-810 | 2 × 0.5–1 | 98 | 84 | 2.1 | ++ (*) | 5 |
| 194-672 | 2 × 0.125–0.5 | 168 | 94 | 0.6 | +++ (*) | 7 |
| 194-531 | 2 × 0.125–1 | 204 | 12 | ? | dorsalized | 10 |
| 194-353 | 2 × 1–2 | 47 | 11 | 4.6 | − | 3 |
| 12-810 | 2 × 0.5–2 | 104 | 86 | 1.8 | ++ | 5 |
| 12-672 | 2 × 1 | 63 | 87 | 1.6 | ++ | 3 |
| 12-531 | 2 × 0.5–2 | 149 | 85 | 1.9 | ++ | 7 |
| 12-355 | 2 × 0.5–1 | 105 | 11 | 4.6 | − | 6 |
| 12-167 | 2 × 0.5–2 | 63 | 16 | 4.5 | − | 4 |
| 531-810 | 2 × 0.75–2 | 42 | 12 | 4.4 | − | 3 |
| 12-810Δ251-351 | 2 × 1 | 34 | 0 | ? | dorsalized | 3 |
| 12-762Δ251-351 | 2 × 1–2 | 54 | 2 | 4.8 | − | 3 |
| 12-531Δ251-351 | 2 × 0.25–1 | 27 | 0 | 5.0 | − | 2 |
| 497-672 | 2 × 0.5–1 | 78 | 9 | 4.8 | − (d) | 4 |
| 403-600 | 2 × 0.25–1 | 119 | 8 | 4.9 | − (d) | 7 |
Figure 4.


Ventralization and dorsalization by expression of Axin mutants in Xenopus embryos. (A) Diagram of Axin molecule. The main binding domains are indicated as well as the aa numbers corresponding to most restriction sites used in the generation of mutant Axin constructs. (B) Examples of ventralized embryos (tailbud stage) obtained by dorsal injection of Ax194-672 mRNA. Two embryos have greatly reduced axes (arrowheads) and no head. The other four embryos are completely ventralized (i.e., they lack dorsal structures and have no body axis). (C) Embryos expressing Ax531-956 develop normally. (D) Examples of hyperdorsalized embryos obtained by dorsal expression of Ax331-956. These embryos have enlarged heads and cement glands (large arrows) or double cement glands (small arrows). (E) Partially dorsalized embryos resulting from dorsal expression of Ax194-531. These embryos typically form very large semicircular cement glands (arrows) and their heads are generally strongly disorganized (arrowheads). (F) Axis duplication by ventral expression of Ax331-956. Most of the embryos form complete twin embryos, including two cement glands (arrows). (G) Expression levels of FL and mutant Axin constructs in embryos. (Left) An example of a series of constructs expressed dorsally. (Right) Expression levels of a series of constructs with internal deletion of the RGS domain, expressed ventrally. Note that 1–2 ng Ax12-956Δ251-351 mRNA induced complete duplicated axes (see Table II), whereas the other constructs expressed at similar or even higher levels induced mostly partial or no secondary axis.
As we previously reported (Zeng et al., 1997), an internal deletion of the RGS domain (AxΔ251-351) eliminated the ability to ventralize and instead caused dorsalization. Deletion of the GSK3β and β-catenin binding sites (AxΔ352-631) also abolished ventralizing activity. A small fragment containing the GSK3β and β-catenin binding sites (Ax497-672) was insufficient to ventralize the embryo, although similar Axin fragments were able to promote phosphorylation of β-catenin in vitro (Ikeda et al., 1998). A fragment containing only the RGS domain (Ax194-353) was also ineffective.
Successive truncation from the NH2 terminus of Axin confirmed the importance of the RGS domain for ventralization. Whereas removal of the first 193 aa had no significant effect, further truncation to aa 331 eliminated ventralizing activity and resulted in dorsalizing activity, similar to the internal RGS deletion (Fig. 4, D and F). Truncation at aa 531, removing both the RGS domain and GSK3β binding region, eliminated all activity (Fig. 4 C) as did truncation to aa 810 (Ax810-956).
Mutant Axins with NH2-termini at aa 194 were subjected to COOH-terminal truncation to examine the importance of the DIX, PP2A binding, β-catenin binding, and GSK3β binding domains. Removal of the DIX domain (Ax194-810) had little if any effect, whereas removal of the DIX and PP2A binding domains (Ax194-672) caused an increase in ventralizing activity (Fig. 4 B). This observation is consistent with the hypothesis that the binding of PP2A to the Axin complex may negatively regulate the phosphorylation of β-catenin by GSK3β (Hsu et al., 1999). Further truncation to aa 531, removing the β-catenin binding site, abolished ventralizing activity, and instead resulted in some dorsalizing activity (i.e., shorter axis, larger head, and circular or double cement gland; Fig. 4 E). When injected at high concentrations, the other Axin mutants lacking the NH2-terminal and the COOH-terminal regions (Ax194-672 and Ax194-810) also showed dorsalizing activity, as discussed below.
When the NH2 terminus of Axin was left intact, truncation of the COOH terminus to remove the DIX domain, or both the DIX and PP2A domains, caused only a slight reduction, if any, in the ability to ventralize. Surprisingly, there was no further reduction in activity when the region including the β-catenin binding site was truncated (Ax12-531) or removed by an internal deletion (AxΔ531-810). Unlike the mutants with NH2-termini truncated at aa 194, no dominant negative effect (dorsalization) was seen when high concentrations were injected (Table II). Further truncation, removing the GSK3β site, eliminated all activity (Ax12-355). Internal deletion of only the RGS domain, in the context of COOH-terminal truncations (mutant Ax12-810Δ251-351, Ax12-672Δ251-351, and Ax12-531Δ251-351), also eliminated ventralizing activity and instead cause weak dorsalizing activity.
Table II.
Effect of Axin Mutants on Axis Development: Ventral Injections
| Mutant | Amount mRNA injected | Total injected embryos | Percent DA | Dorsalization | cDA | pDA | vDA | N | No. exp |
|---|---|---|---|---|---|---|---|---|---|
| ng | |||||||||
| FL (12-956) | 1–4 | 58 | 0 | − | 0 | 0 | 0 | 58 | 4 |
| Δ251-351 | 1–2* | 148 | 80 | +++ | 54 | 65 | 12 | 17 | 9 |
| Δ352-631 | 1 | 39 | 0 | − | 0 | 0 | 0 | 39 | 3 |
| 331-956 | 0.25 | 59 | 19 | + | 0 | 11 | 0 | 48 | 3 |
| 331-956 | 1* | 129 | 87 | +++ | 83 | 29 | 3 | 14 | 4 |
| 531-956 | 0.5–1.5 | 62 | 0 | − | 0 | 0 | 0 | 62 | 3 |
| 194-531 | 0.5 | 97 | 24 | + | 9 | 14 | 36 | 38 | 4 |
| 194-353 | 0.5–1 | 34 | 0 | − | 0 | 0 | 4 | 30 | 2 |
| 12-672 | 1 | 49 | 0 | − | 0 | 0 | 0 | 49 | 2 |
| 531-810 | 0.75–1 | 34 | 0 | − | 0 | 0 | 0 | 34 | 2 |
| 12-810Δ251-351 | 0.25* | 23 | 0 | − | 0 | 0 | 2 | 21 | 2 |
| 12-810Δ251-351 | 0.5–2‡ | 149 | 56 | ++ | 15 | 69 | 6 | 59 | 9 |
| 12-762Δ251-351 | 0.25–2 | 168 | 1 | − | 0 | 1 | 10 | 157 | 9 |
| 12-531Δ251-351 | 0.25* | 28 | 0 | − | 0 | 0 | 0 | 28 | 2 |
| 12-531Δ251-351 | 0.5–2‡ | 53 | 30 | + | 4 | 12 | 6 | 31 | 4 |
| 497-672 | 0.5 | 45 | 0 | − | 0 | 0 | 0 | 45 | 2 |
| 403-600 | 0.5–1 | 91 | 3 | − | 0 | 3 | 0 | 88 | 4 |
Dorsalization
In contrast to the ability of Axin and other inhibitors of Wnt signaling (e.g., GSK3β) to ventralize when injected dorsally, factors that stimulate this pathway (e.g., certain Wnts, Dsh, dnGSK3β, or β-catenin) have dorsalizing activity; when injected dorsally, they can hyperdorsalize (i.e., they induce formation of a larger head, large or multiple cement glands, shorter axis, and double anterior axis). However, their activity is best seen in ventral injections, where they can induce a secondary axis (Miller and Moon, 1996; Fagotto et al., 1997; Fagotto, 1999). We previously showed that a mutant Axin lacking the RGS domain (AxΔ251-351) behaved as such a dorsalizing factor. This activity could be competed by coexpression of FL Axin, supporting the conclusion that it was due to a dominant negative effect (Zeng et al., 1997). To identify the domains of Axin required for this activity, several additional mutant forms of Axin were also injected into the ventral side of the embryo to assay their ability to induce axis duplication.
Sequences upstream from the RGS domain were not required, as an NH2-terminal truncation at aa 331 (Ax331-956) induced axis duplication as efficiently as the RGS deletion (Figs. 3 and 4 F, Table II). However, the GSK3β binding site was required as truncation at aa 531 abolished the effect (Ax531-956). The COOH-terminal sequences were also important: in the presence of the RGS deletion, COOH-terminal truncation at aa 810, 672, or 530 strongly reduced the dorsalizing activity so that axis duplication was observed only when high concentrations of RNA were injected. When the amount of injected RNA was titrated down to yield expression levels at which AxΔ251-351 showed optimal activity, only Ax331-956 was active (Table II).
Effects of Axin and Axin Mutants on β-Catenin levels
Modulation of the Wnt pathway has a striking effect on β-catenin levels; in the absence of a Wnt signal, constitutively active GSK3β phosphorylates β-catenin and causes its rapid turnover, whereas Wnt signaling induces stabilization of β-catenin. Therefore, we tested the effect of Axin on β-catenin levels in Xenopus embryos by coexpressing HA-tagged β-catenin with FL or mutant forms of Axin, or with β-galactosidase as a control. Low amounts of HA-tagged β-catenin mRNA were used to mimic the behavior of endogenous β-catenin. At the late blastula stage, when endogenous β-catenin signaling peaks (Lemaire et al., 1995; Schneider et al., 1996), the levels of HA-tagged β-catenin in embryo extracts were analyzed. The membrane (cadherin-bound) pool of β-catenin is known to be very stable (Kofron et al., 1997) and changes in β-catenin levels by the Wnt signaling pathway affect mostly the unbound, soluble pool (Riggleman et al., 1990; Peifer et al., 1994; Pai et al., 1997). Removing the cadherin-bound pool of β-catenin by Con A precipitation made it possible to obtain samples enriched in soluble β-catenin, allowing the effect of Axin on β-catenin levels to be analyzed more accurately. As shown in Fig. 5 B, FL Axin caused a dramatic decrease in exogenous β-catenin. Several mutant Axin constructs were coexpressed similarly; mutants with ventralizing activity (Ax12-531 and Ax194-672) also proved to be effective in reducing β-catenin levels. A mutant lacking ventralizing activity (Ax531-956) had no effect on β-catenin levels. On the other hand, mutants with strong dominant negative activity (AxΔ251-351 and Ax331-956), induced a clear increase in β-catenin levels. Unexpectedly, mutant Ax194-531, which failed to ventralize over a wide range of concentrations, but instead showed some dorsalizing activity, caused a strong decrease in β-catenin levels.
The activity of the dominant negative mutant AxΔ251-351 was also tested on the ventral side, where the β-catenin degradation machinery is maximally active. Under these conditions, stabilization of β-catenin by AxΔ251- 351 could be observed even in total extracts (Fig. 5 C). NH2 terminally deleted β-catenin (hemagglutinin epitope– tagged Δ-NH2 terminus β-catenin; Funayama et al., 1995), which lacks the GSK3β-dependent phosphorylation site (Munemitsu et al., 1996; Yost et al., 1996), was found to be insensitive to Axin overexpression (Fig. 5 D), suggesting that Axin-induced destabilization of β-catenin requires phosphorylation by GSK3β.
Intracellular Distribution of Axin
As the subcellular localization of endogenous Axin is so far unknown, we examined the distribution of the ectopically expressed Myc-tagged Axin in Xenopus embryos. As shown in Fig. 6, FL Myc–Axin exhibited a striking and unusual pattern. The signal was mostly concentrated in very bright spots, which were found singly or in clusters of variable size, mainly, but not exclusively, at the cell periphery (Fig. 6, A and A′, arrows). The rest of the cytoplasm was devoid completely of the signal. In addition, some plasma membrane staining was also observed. However, the membrane staining was quite variable: absent in many cells, weak in others (Fig. 6, A and A′′, arrowhead), and very strong in a few rare cells (not shown). The punctate pattern and the absence of diffuse cytoplasmic staining were observed at all mRNA concentrations used from 0.15 ng, the limit of detection by IF, to 2 ng. A similar pattern was observed in Axin-transfected cultured HeLa (Fig. 6, B and B′) and A6 cells (not shown).
Figure 6.
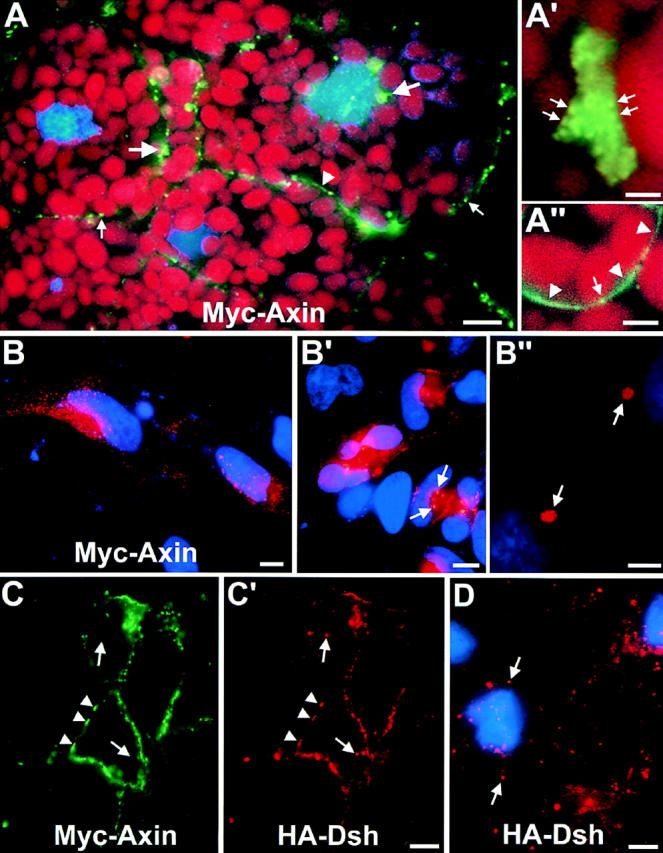
Myc-tagged Axin localizes in discrete spots and colocalizes with HA–Dsh. (A) Cellular distribution of FL Myc–Axin (green, Alexa488 staining) in Xenopus blastulae, detected by indirect IF on frozen sections. Myc–Axin localized mainly in spots (small arrows) or clusters of spots (large arrows), found generally but not exclusively at the cell periphery. Some plasma membrane staining was also observed (arrowhead). Red shows yolk platelets counterstained with Eriochrome Black; blue shows the nuclei (DAPI staining). (A′) Large cluster of Myc–Axin-positive spots (arrows) at higher magnification. (A′′) Detail of membrane localization of Myc–Axin (arrowheads). Spots also are found at or near the cell surface (arrow). (B and B′) Localization of FL Myc–Axin in transfected HeLa cells. Myc–Axin (red, Cy3 staining) is found in spots, mostly at the cell periphery. In high expressing cells, bright cytoplasmic clusters were observed (B′, arrows). (B′′) Detail of two clusters at high magnification (arrows). Note that images in A and A′′ and B and B′′ were collected under very different conditions (exposure, filters), since the signal was one order of magnitude stronger in transfected HeLa cells compared with mRNA-injected embryos. Blue shows the nuclei (DAPI staining). (C and C′) Colocalization of FL Myc–Axin and HA–Dsh. Coexpressed Myc–Axin (green, Alexa488 staining) and HA–Dsh (red, Cy3 staining) were detected on sections from Xenopus blastulae by double IF. All Myc–Axin colocalized perfectly with HA–Dsh (arrowheads), but some HA–Dsh spots did not stain for Myc–Axin (arrows). Note that most Axin-Dsh spots are distributed along the periphery of the cells. (D) Localization of HA–Dsh expressed alone: Dsh (red, Cy3 staining) localizes in spots, distributed throughout the cytoplasm. Blue shows nuclei (DAPI). Bars: (A) 10 μm; (A′) 2 μm; (A′′) 5 μm; (B and B′) 10 μm; (B′′) 5 μm; (C and D) 20 μm.
Myc–Axin localization in Xenopus embryos was further studied at the EM level by two different techniques: on-section staining of Lowycryl sections (Fig. 7, A–C) and preembedding labeling using Nanogold and silver enhancement (Fig. 7, D–F). Notwithstanding differences in ultrastructure preservation and labeling sensitivity (see legends), both methods gave similar results. Consistent with IF data, Myc–Axin was found to be concentrated highly in discrete areas of the cell. These areas were characterized by clusters of vesicles (asterisks) surrounded by gold-decorated electron dense material (arrows). Labeled clusters varied largely in size and density, apparently as a function of expression levels. Part of a loose cluster is shown in Fig. 7 C. Small groups of gold particles associated with a few vesicles and electron dense material could be resolved, probably corresponding to the individual spots detected by IF (Figs. 6 A′ and 7 A, arrows). On the other hand, Fig. 7, D and E, shows very large dense Myc-positive areas, where vesicles were tightly packed and consequently the dense cytoplasm appeared less prominent. Fig. 7 B shows a cluster of intermediate size and vesicle density. Plasma membrane localization of FL Axin could not be detected unambiguously by EM, probably because it was generally too weak (Fig. 6 A, IF). However, strong plasma membrane staining could be observed for the mutant Axin AxΔ531-810 that is consistent with IF results (Fig. 3).
Figure 7.
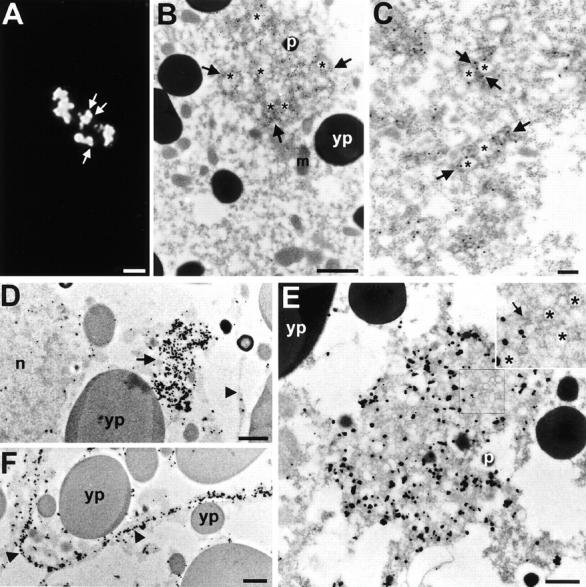
Electron microscopic localization of Myc– Axin. (A–C) EM localization of FL Myc–Axin by on-section staining on Lowycryl sections. (A) Low magnification view of an Axin-positive cluster detected by indirect IF on a thin section. Axin expression in this cell is relatively low and individual spots can be resolved (arrows). (B) Low magnification EM image of a cluster of gold particles (15 nm) labeling an area of dense cytoplasm (arrows) containing numerous vesicles (asterisks). Note that outside the cluster the surrounding cytoplasm is devoid of gold particles. (C) High magnification view of portion of a less dense cluster. The cluster is composed of small groups of gold particles decorating electron dense cytoplasm (arrows) associated with a few vesicles (asterisks). Each small group probably corresponds to a single “spot” observed by IF (A and Fig. 6, A and B). Note that the membranes surrounding the vesicular structures appear much less contrasted in B and C compared with E. This is due to the difference in the methods used (low contrast Lowycryl sections in B and high contrast conventional Spurr sections in D). (D and E) EM localization of Myc–Axin by preembedding Nanogold labeling and silver enhancement. (D) Low magnification view of a Myc–Axin positive area (arrow) in a high expressing cell. Single gold aggregates found both in the cytoplasm and in the nucleus (n) represent background that is higher with this preembedding technique. The arrowhead points to the plasma membrane that shows no significant staining in this cell. (E) High magnification view of a similar area, packed with vesicles of variable size (∼50-200 nm) embedded in electron dense cytoplasm. The irregular shape of the gold/silver particles is due to the silver enhancement method. Insert shows enlarged view (2×) of the outlined area, with tightly-packed vesicles (asterisks) and electron dense cytoplasm (arrow). Note that gold labeling tends to be somewhat excluded from the areas particularly packed with vesicles. This could be due to limited diffusion of Nanogold in these preparations. (F) Nanogold localization of Myc–Axin at the plasma membrane. Because FL Axin localization at the membrane is weak (D, and Fig. 7 A), a mutant Axin, AxΔ531-810 was used in this experiment. In this case, the plasma membrane is heavily decorated with gold/silver particles (arrowheads). m, mitochondrion; p, pigment granules; and yp, yolk platelets. Bars: (A) 2 μm; (B) 1 mm; (C) 0.2 μm; (D) 1 μm; (E) 0.5 μm; (F) 1 μm.
Consistent with the IF data, Axin was found to be largely particulate/sedimentable in differential centrifugation experiments (Fig. 8 B). On the other hand, it was completely solubilized in the presence of a mild nonionic detergent, NP-40 (Fig. 8 C). Thus, the sedimentation properties of Axin are not due to interaction with detergent-insoluble cytoskeletal elements. In the presence of NP-40, Axin could be partially precipitated using Con A beads (Fig. 8 C), indicating that a pool of Axin is associated with a membrane glycoprotein. We believe that this Axin– membrane association involves the plasma membrane pool of Axin, but not that in the intracellular spots, because binding to Con A of all Axin deletion mutants tested (Fig. 8 D) strictly correlated with plasma membrane localization (as detected by IF, see below).
Colocalization of Dsh with Axin
The punctate distribution of Axin strongly was reminiscent of the localization pattern of ectopically expressed Dsh (Yang-Snyder et al., 1996; Axelrod et al., 1998) (the distribution of endogenous Dsh in Xenopus is not known). Thus, we compared the localization of coexpressed Myc-tagged Axin and HA–Dsh by double IF. We observed a very good colocalization of these two proteins (Fig. 6, C and C′): HA–Dsh was detected at all sites positive for Myc–Axin (arrowheads), although some other spots were positive for Dsh but negative for Axin (arrows). The Myc– Axin pattern in these embryos was indistinguishable from the pattern observed in the absence of exogenous Dsh, suggesting that Dsh does not influence Axin localization. In contrast, Myc–Axin overexpression clearly affected HA–Dsh distribution: when HA–Dsh was expressed alone, it localized exclusively in single cytoplasmic spots, or small clusters of spots, distributed throughout the cell (Fig. 6 D). No membrane staining was observed. However, when coexpressed HA–Dsh and Myc–Axin colocalized in a pattern typical for overexpressed Axin (enrichment of spots at the cell periphery, presence of large clusters, and plasma membrane staining). These results suggest that Dsh may bind, directly or indirectly, to the Axin complex.
Sequences Required for Axin Localization
To examine the sequences in Axin that target it to its specific locations, and the functional significance of this localization, we examined the intracellular distribution of the same mutant forms used above. Internal deletion of the RGS domain had little or no effect on localization; the mutant protein localizing primarily in the spots and less at the plasma membrane (Fig. 3 and Fig. 9, B and C). Deletion of the GSK3β and β-catenin binding sites (AxΔ352-631) also had no effect on localization to the spots, but eliminated the membrane staining (Fig. 9, H and I). Deletion of the COOH-terminal 146-aa resulted in localization mainly at the membrane, with little or no labeling of the spots, e.g., Ax12-810 (not shown) Ax12-672 (Fig. 9 F), Ax12-531 (Fig. 9 G), and Ax12-355 (Fig. 9 J).
Figure 9.
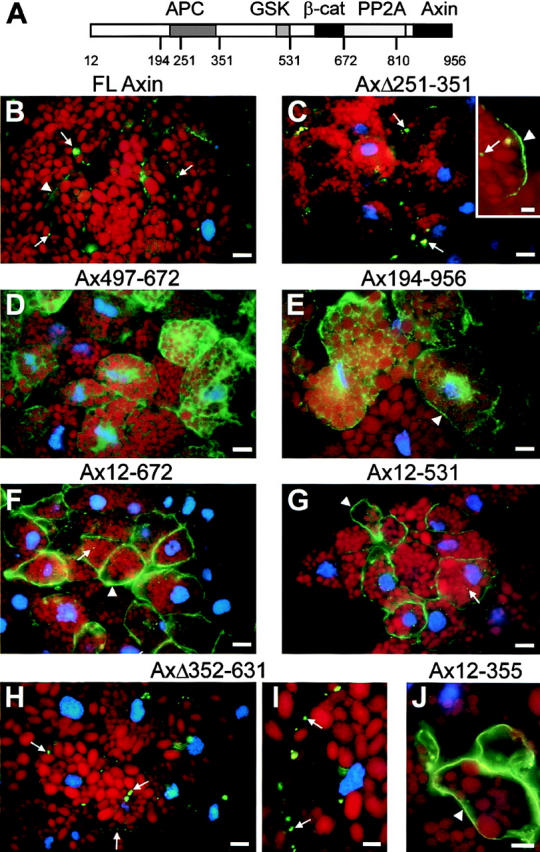
Sequence specific patterns of cellular distribution of Axin mutants. FL and various mutant forms of Myc-tagged Axin were expressed in Xenopus embryos and localized by IF (green) on frozen sections. Diagram of Axin molecule. (B and C) FL and AxΔ251-351 localized preferentially to spots or clusters of spots (arrows). Variable membrane staining was detected in some cells (B and insert C, arrowheads). This pattern was found for all constructs containing both NH2- and COOH-termini. (D) Diffuse cytoplasmic distribution of Ax497-672 found in most constructs lacking the NH2 terminus. (E) Example of a mutant (Ax194-956) showing a cytoplasmic, but somewhat particulate, distribution with some weak enrichment at the cell periphery (arrowhead). Partial cell surface enrichment is characteristic of mutants lacking the NH2 terminus but with intact APC and GSK3β-binding sites. (F and G) Constructs containing the NH2 terminus but lacking the COOH terminus, such as Ax12-672 and Ax12-531, are mainly localized at the plasma membrane (arrowheads). Some intracellular punctate staining is also observed (arrows). (H) Localization of AxΔ352-631 mimics the punctate staining of FL Axin (arrows). (I and J) High magnification views of punctate staining (I, AxΔ352-631) and membrane staining (J, Ax12-355). Bars: (A–G) 20 μm; (H, I, and insert B) 5 μm.
Forms of Axin lacking the NH2-terminal half displayed a mostly diffuse cytoplasmic localization, e.g., Ax497-672 (Fig. 9 D). When the APC and GSK3β-binding domains were left intact, there seemed to be some enrichment at the cell periphery (Fig. 9 E, Ax194-956), although it was difficult to assess the extent of membrane enrichment, because of the high cytoplasmic signal.
Thus, the NH2 terminus of Axin appears to be required for the characteristic pattern of localization, both in the cytoplasmic spots and at the membrane. The presence of the normal COOH terminus tends to cause localization to the spots, although it is not absolutely required for this. The COOH terminus, which includes a dimerization domain, might bind to endogenous Axin or to other cellular components. The APC and GSK3β binding sites appear to have a weaker effect on localization at the membrane. The membrane localization of mutants containing the NH2 terminus correlates very well with Con A binding (Fig. 8 C). For instance, Ax12-531 binds very efficiently to Con A, whereas Ax531-956 does not bind at all. However, localization to the spots appears to depend on a different mechanism, only a small fraction of FL Axin and an even smaller fraction of AxΔ351-630 (found mostly in spots) bind to Con A (Fig. 8 C and data not shown).
Discussion
Axin has been shown to negatively regulate signaling through components of the Wnt pathway. Coinjection experiments in Xenopus embryos previously suggested that it acts downstream of GSK3β and upstream of β-catenin. Subsequent studies have shown that Axin is part of a complex including these two proteins as well as APC and that it promotes the phosphorylation of β-catenin by GSK3β and its subsequent degradation (Hart et al., 1998; Ikeda et al., 1998; Itoh et al., 1998; Kishida et al., 1998; Sakanaka et al., 1998). The aims of the experiments reported here were to understand the relationship between Axin's ability to bind to these and other proteins and its capacity to function in the regulation of this pathway. To this end, we have examined a series of Axin mutants for their ability to (1) bind to APC, GSK3β, and β-catenin; (2) ventralize or dorsalize Xenopus embryos, an established assay for effects on β-catenin signaling; and (3) alter the stability of β-catenin expressed from coinjected mRNA. In addition, we have examined the intracellular localization of FL Axin and a series of Axin mutants.
Interaction of Axin with Other Components of the Wnt Signaling Pathway
Through direct binding in vitro and coIP from mammalian cell extracts, we have confirmed that Axin forms a complex with APC, GSK3β, and β-catenin, and we have further delimited some of the binding sites for these proteins. Based on coIP, the region of mAxin required for interaction with GSK3β lies between aa 497 and 531. The COOH-terminal boundary of the minimal binding region appears to lie between aa 526 and 531. Whereas Itoh et al. (1998) did not detect coIP of GSK3β with Axin 12-526, we detected weak interaction of GSK3β with some Axin mutants terminating at aa 531. We confirmed that the RGS domain (aa 220–340) includes a major binding site for APC and interacts with the 20-aa repeat region of APC. Furthermore, we identified a second region of Axin, between aa 96–253, that can bind directly to the NH2-terminal region of APC containing the Armadillo and 15-aa repeats. This region of Axin was neither necessary nor sufficient for coIP with VSV-G–tagged APC, suggesting that it plays a secondary role to the RGS domain in vivo.
We observed good agreement between the presence of the direct binding site for GSK3β (Ikeda et al., 1998) and the ability of Axin mutants to coIP with GSK3β (Fig. 4). Axin mutants lacking this region, some of which were able to coIP with APC and/or β-catenin, all failed to coIP with GSK3β, suggesting that GSK3β must bind directly to Axin to join the complex. However, several forms of Axin that lacked the direct binding site for either APC or β-catenin were able to coIP with both of these proteins. APC was found to coIP not only with all forms of Axin containing the RGS domain, but also with Ax331-956, which lacks any direct binding site for APC. Similarly, β-catenin could coIP not only with all Axin mutants containing aa 600–622, but also with Ax12-531 and Ax194-531. Both of these discrepancies likely are due to the ability of APC and β-catenin to bind to each other (Rubinfeld et al., 1993; Su et al., 1993). Since each of these three components can interact directly with the other two, we suggest that they form in vivo a triangular complex (Fig. 10).
Figure 10.

Axin domains, molecular interactions, and their functions. Binding domains are represented by boxes and direct molecular interactions are indicated by thick lines. Axin, APC, and β-catenin can bind to each other and form a triangular complex. A secondary Axin–APC interaction is shown by a dashed line. GSK3β binding to Axin is required for phosphorylation (arrows and P) of Axin itself, as well as APC, and β-catenin. Phosphorylation of APC regulates APC-β-catenin binding and phosphorylation of β-catenin is required for its degradation. The function of Axin phosphorylation is not known. The catalytic subunit of PP2A binds to a site adjacent to the β-catenin binding site and may counteract the activity of GSK3β (dotted lines with arrows). The COOH terminus contains a dimerization domain, whose function is unclear. Binding to Dsh (dotted line) is suggested by the presence of a DIX domain in both molecules and by colocalization with Axin. Localization of Axin depends on its NH2-terminal and COOH-terminal sequences. The NH2 terminus is required for both plasma membrane and cytoplasmic spot localization, whereas the COOH terminus enhances localization at spots. The RGS domain and GSK3β binding site are required for ventralizing activity. A segment of Axin including the RGS, GSK3β and β-catenin binding domains is sufficient for activity. However, the NH2 terminus can substitute for the β-catenin binding domain, possibly by stabilizing an Axin•APC•β-catenin complex in the absence of direct Axin–β-catenin binding.
The GSK3β binding domain was dispensable for direct binding to APC and β-catenin, but required for indirect binding to β-catenin (presumably via APC). Indeed, a small fragment of Axin containing only the RGS domain (Ax194-353), while able to coIP with APC, did not coIP with β-catenin. Conversely, Ax531-956 was found to coIP with β-catenin but not with APC. We also found that Axin induced a mobility shift in APC that appeared to be due to phosphorylation. This activity required the GSK3β-binding domain as well as the APC-binding region, suggesting that GSK3β is responsible for this modification. It previously has been shown that GSK3β can phosphorylate APC in vitro (Rubinfeld et al., 1996), that Axin promotes this event (Hart et al., 1998), and that this phosphorylation enhances the ability of APC to bind to β-catenin (Rubinfeld et al., 1996). Our observations argue that Axin performs a similar function in vivo.
Axin Sequences Required to Influence Axis Formation and Regulate β-Catenin Levels in Frog Embryos
In general, we found a good correlation between the ability of Axin mutants to ventralize frog embryos and to lower the levels of coinjected HA-tagged β-catenin, presumably by promoting its degradation. The RGS domain and GSK3β binding site were both required, although not sufficient, for Axin activity. In addition, either the β-catenin binding site or the NH2-terminal region upstream of the RGS domain (but not necessarily both) was required. The activity of mutant forms of Axin lacking the β-catenin binding site (e.g., Ax12-531 and AxΔ531-810) is consistent with the observation that such forms can coIP with β-catenin in 293 cells, apparently via an indirect interaction. Why the NH2-terminal sequence can substitute for the β-catenin binding site is not clear, but this could be related to its ability to bind to APC or its influence on the intracellular localization of Axin.
Fragments of Axin containing the GSK3β and β-catenin binding sites, but lacking the RGS domain, can promote the phosphorylation of β-catenin by GSK3β in vitro (Ikeda et al., 1998) as well as the degradation of endogenous β-catenin in cultured SW480 cells (Hart et al., 1998). However, in our experiments, all forms of Axin lacking the RGS domain either had no effect on axis formation and β-catenin levels or they dorsalized rather than ventralized the frog embryo and raised β-catenin levels. This indicates that direct binding to APC plays a critical role in the ability of Axin to promote the degradation of β-catenin.
These discrepancies may be due to the peculiar properties of SW480 cells that lack FL APC and have high levels of soluble β-catenin. In contrast, we used very low levels of exogenous β-catenin that can still be effectively regulated by the Wnt pathway (Yost et al., 1996). In other experiments (Fig. 5 C), we found that FL Axin was able to downregulate even very large amounts of coinjected β-catenin. However, under these conditions several Axin mutants gave results that were inconsistent with their effects at more physiological β-catenin levels and with their activity on axis induction. In particular, both dominant negative mutants AxΔ251-351 and Ax331-956 failed to stabilize β-catenin expressed at high levels, but rather caused some destabilization (not shown). Thus, Axin is capable of stimulating β-catenin degradation independently of the RGS domain, but only provided high levels of free β-catenin and/or absence of APC. Consistent with this observation, the Axin-like protein Conductin/Axil/Axin-2 appears to behave similarly. Indeed, a ΔRGS Conductin construct could downregulate β-catenin levels in SW480 cells, yet acted as a dominant negative (i.e., increased β-catenin levels) in Neuro2A cells, which have low levels of endogenous β-catenin and FL APC (Behrens et al., 1998). It is likely that high levels of β-catenin are flooding the system, bypassing normal regulatory mechanisms. Nevertheless, it is also possible that two different mechanisms exist, one APC-dependent, and one APC-independent, active at low and at high β-catenin concentrations, respectively (see below).
Surprisingly, in SW480 cells, truncation of the NH2 terminus, including the RGS domain, increased the activity of Axin that led to the proposal that the RGS domain may repress Axin activity in the absence of APC (Hart et al., 1998). However, this mutant construct could not discriminate between a role of the RGS domain itself and an effect of upstream sequences. In contrast, ΔRGS Conductin, a mutant with an internally deleted RGS domain and an intact NH2 terminus, showed weaker activity than FL Conductin in SW480 cells, arguing that the RGS domain is not responsible for the apparent repression reported by Hart et al. (1998). In fact, SW480 cells are not null for APC, but still contain an NH2-terminal fragment, which can bind β-catenin (Polakis, 1995), and can also interact with the NH2 terminus of Axin upstream of the RGS domain (our data). Thus, it is conceivable that the truncated APC may interfere with Axin activity.
In embryos, deletion of the RGS domain caused a strong dominant negative effect. The self-binding region (including the DIX domain) appeared to play a role in this activity, because deletion of this region from AxΔ251-351 substantially reduced activity. As the COOH-terminal 100 aa of Axin can mediate multimerization, the strong dominant negative forms of Axin may act by binding to endogenous Axin. However, forms of Axin lacking the DIX domain also showed weak or moderate dorsalizing activity (Ax12-810Δ251-351 and Ax12-531Δ251-351), suggesting that dominant negative activity may be generated in more than one way. One possibility is that these forms of Axin bind to GSK3β but not to APC, thus, interfering with the formation of the complete complex. Also, two mutants lacking both the NH2 and COOH termini (Ax194-672 and Ax194-810) efficiently ventralized at low concentrations, but dorsalized at high concentrations. This dual activity might be related to their subcellular localization (see below).
The NH2- and COOH-terminal portions of Axin were not required for its ventralizing activity in our assays, as also observed by Itoh et al. (1998), although they may modulate its function. Deletion of the DIX domain had little or no effect on activity. Deletion of the PP2A binding region caused some increase in activity of the forms of Axin that initiated at aa 194, although this difference was not apparent when the NH2 terminus of Axin was left intact. We have hypothesized that the binding of PP2A to the Axin complex might counteract the phosphorylation of β-catenin by GSK3β (Hsu et al., 1999) that could account for the increased ventralization activity in the absence of this domain.
The Intracellular Localization of FL and Mutant Forms of Axin
FL Myc–Axin expressed in Xenopus embryos was found primarily in characteristic spots, singly or in clusters of variable size. Ultrastructural analyses indicated that the spotty distribution is not merely due to aggregation of an overexpressed protein, but corresponds to particular (though as yet ill-defined) subcellular structures, consisting of clustered vesicles associated with dense cytoplasm. Several arguments suggest that endogenous Axin has a similar distribution including: (1) identical spots were observed over a wide range of Axin expression levels. (2) HA–Dsh (Fig. 6) accumulated in similar spots in the absence of overexpressed Axin. (3) Myc–Axin colocalized with HA–Dsh (Fig. 6). However, the formation of large clusters of spots very likely is due to Axin overexpression, as it was not observed for HA–Dsh in the absence of Axin expression. However, it may reveal the ability of Axin to act as a scaffold through multiple interactions with other cytoplasmic proteins. A small, variable fraction of Myc– Axin was also associated with the plasma membrane, and Con A-binding showed that Axin interacts with a cell surface glycoprotein. The NH2 terminus is sufficient for membrane targeting, although some other internal sequences may also confer weaker binding. Interestingly, the sequences of Axin crucial for its subcellular localization (i.e., the NH2 and COOH termini) do not bind any of the core components related to its activity (APC, β-catenin, and GSK3β), and are apparently dispensable, at least under conditions of overexpression (Fig. 10). Clearly, additional molecular interactions must take place at both ends of the molecules.
The occurrence of two well-defined locations for Myc– Axin may reflect the existence of two functionally distinct pools, possibly an active and an inactive one. Ectopically expressed Dsh shows a similar dual localization at spots/ membrane, which can be manipulated by overexpression of Wnt/Frizzled, and that may correspond to different functional states (Yanagawa et al., 1995; Axelrod et al., 1996; Steitz et al., 1996; Yang-Snyder et al., 1996). However, which would be the active and inactive sites remains unclear. The comparison of various Axin mutants did not reveal any simple correlation between their activity in functional assays and their intracellular distribution, although all active mutants can to some extent localize at the plasma membrane. However, it is quite possible that localization per se is not required for activity, but that regulation is achieved by sequestering various components of the signaling pathway in different compartments of the cell. It is then easy to conceive that overexpression may bypass such regulation and allow Axin/Axin mutants to interact with other components of the complex independently of upstream signals.
Our observations provide some hints for a role of Axin localization. For instance, the concentration-dependent dual activity of Ax194-672 and Ax194-810 may be related to their diffuse distribution; at high concentrations, they could act as dominant-negatives by affecting the balance between various endogenous components otherwise strictly compartmentalized. This dual activity was also found for the Axin-like protein Axil/Conductin/Axin-2 that has a similar diffuse distribution (Zhang, T., F. Fagotto, and F. Costantini, manuscript in preparation), but was never observed with Axin constructs showing a well-defined localization (spots and/or plasma membrane).
Conclusions and Models
Our functional data emphasize the essential role of the RGS domain for Axin activity (Fig. 10). They demonstrate that binding to GSK3β and to β-catenin, which was reported to stimulate β-catenin phosphorylation in vitro (Hart et al., 1998; Ikeda et al., 1998), is not sufficient in vivo either for β-catenin degradation or for inhibition of its signaling. Although the RGS domain may interact with other yet uncharacterized molecules, its importance most likely resides in its ability to bind APC, thus inducing the formation of a trimeric complex Axin•β-catenin•APC (Fig. 10). Note that the RGS domain of Axin is significantly diverged from the sequences of bona fide RGS proteins (Tesmer et al., 1997; Zeng et al., 1997), and does not appear to bind to G-proteins (our unpublished data) or to have RGS activity (Mao et al., 1998). Apparently, it has diverged toward other interactions and functions.
The apparent discrepancy with other data that suggests, under certain conditions, Axin can function without the RGS domain (Hart et al., 1998), may be best reconciled by postulating two mechanisms, active at different levels of free β-catenin: When β-catenin levels are low, β-catenin degradation would depend primarily of an Axin• β-catenin• APC complex, whereas when β-catenin levels are high, β-catenin• Axin complexes may form and function in the absence of APC. Both mechanisms may be physiologically important: in the absence of Wnt signal, very low levels of free β-catenin might be maintained by the combined action of Axin and APC. However, after a burst of Wnt signal, excess β-catenin would be first downregulated by an APC-independent coarse mechanism, before fine tuning by APC could eventually restore normal levels.
The occurrence of an Axin-based complex has further potential implications on the regulation of the pathway by Wnt. It had been assumed that Wnt caused β-catenin stabilization by inhibiting GSK3β activity (for review see Miller and Moon, 1996). However, GSK3β inhibition could hardly account for the specificity of the pathway, considering the many other substrates and pleiotropic functions of GSK3β. We now know that binding of GSK3β to Axin is required for phosphorylation of Axin, APC, and β-catenin, and ultimately for activity of the complex (Hart et al., 1998; Ikeda et al., 1998; this paper; Jho, E.-H., manuscript submitted for publication). Therefore, inhibition of Axin-GSK3β binding, or in fact any other interactions within the complex, could be a far more specific way to regulate this pathway. This might be precisely the function of Dsh. In this context, the distinct cellular pools of Axin may reflect the existence of different, active and inactive, complexes. The challenge will be to characterize the nature of these complexes and their regulation by upstream components of the pathway.
Acknowledgments
The authors thank Anne Schohl for technical assistance.
This work was supported by a National Institutes of Health grant GM56934 to F. Costantini.
Abbreviations used in this paper
aa
amino acid
APC
adenomatous polyposis coli
coIP
coimmunoprecipitate/coimmunoprecipitation
DAPI
4′,6-diamidino-2-phenylindole
Dsh
Dishevelled
FL
full-length
GSK3β
glycogen synthase kinase 3β
HA
hemagglutinin
HA–Dsh
hemagglutinin-tagged Dishevelled
IF
immunofluorescence
IP
immunoprecipitation
Myc–Axin
Myc-tagged Axin
pAb
polyclonal antibody
PP2A
protein phosphatase 2A
RGS
regulators of G-protein signaling
VSV-G vesicular stomatitis virus glycoprotein
VSV–APC, VSV-G–epitope-tagged APC
Footnotes
F. Fagotto and E.-H. Jho contributed equally to this work.
The current address of Li Zeng is Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Avenue, Boston, MA 02115.
The current address of Thomas Joos is Department of Biochemistry, Naturwissenschaftliches und Medizinishces Institut, Markwiesenstrasse 55, 72770 Reutlingen, Germany.
References
- Axelrod JD, Matsuno K, Artavanis-Tsakonas S, Perrimon N. Interaction between Wingless and Notch signaling pathways mediated by dishevelled. Science. 1996;271:1826–1832. doi: 10.1126/science.271.5257.1826. [DOI] [PubMed] [Google Scholar]
- Axelrod JD, Miller JR, Shulman JM, Moon RT, Perrimon N. Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and wingless signaling pathway. Genes Dev. 1998;12:2610–2622. doi: 10.1101/gad.12.16.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Thorner J. RGS proteins and signaling by heterotrimeric G proteins. J Biol Chem. 1997;272:3871–3874. doi: 10.1074/jbc.272.7.3871. [DOI] [PubMed] [Google Scholar]
- Fagotto, F. 1999. Wnt signaling in Xenopus embryos. In Signaling through cell adhesion molecules. J.-L. Guan, editor. CRC Press. In press.
- Fagotto F, Gumbiner BM. Beta-catenin localization during Xenopusembryogenesis: accumulation at tissue and somite boundaries. Development. 1994;120:3667–3679. doi: 10.1242/dev.120.12.3667. [DOI] [PubMed] [Google Scholar]
- Fagotto F, Funayama N, Gluck U, Gumbiner BM. Binding to cadherins antagonizes the signaling activity of β-catenin during axis formation in Xenopus. . J Cell Biol. 1996;132:1105–1114. doi: 10.1083/jcb.132.6.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto F, Guger K, Gumbiner BM. Induction of the primary dorsalizing center in Xenopusby the Wnt/GSK/β-catenin signaling pathway, but not by Vg1, Activin, or Noggin. Development. 1997;124:453–460. doi: 10.1242/dev.124.2.453. [DOI] [PubMed] [Google Scholar]
- Funayama N, Fagotto F, McCrea P, Gumbiner BM. Embryonic axis induction by the armadillo repeat domain of β-catenin: evidence for intracellular signaling. J Cell Biol. 1995;128:959–968. doi: 10.1083/jcb.128.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluecksohn-Schoenheimer S. The effects of a lethal mutation responsible for duplications and twinning in mouse embryos. J Exp Zool. 1949;110:47–76. doi: 10.1002/jez.1401100105. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Signal transduction of beta-catenin. Curr Opin Cell Biol. 1995;7:634–640. doi: 10.1016/0955-0674(95)80104-9. [DOI] [PubMed] [Google Scholar]
- Hart, M.J., R. de los Santos, I.N. Albert, B. Rubinfeld, and P. Polakis. 1998. Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr. Biol. 8:573–581. [DOI] [PubMed]
- Hsu W, Zeng L, Costantini F. Identification of a domain of Axin that binds to the serine/threonine protein phosphatase 2A (PP2A) and a self-binding domain. J Biol Chem. 1999;274:3439–3445. doi: 10.1074/jbc.274.6.3439. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO (Eur Mol Biol Organ) J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Krupnik VE, Sokol SY. Axis determination in Xenopusinvolves biochemical interactions of axin, glycogen synthase kinase 3 and beta-catenin. Curr Biol. 1998;8:591–594. doi: 10.1016/s0960-9822(98)70229-5. [DOI] [PubMed] [Google Scholar]
- Jacobs-Cohen RJ, Spiegelman M, Cookingham JC, Bennett D. Knobbly, a new dominant mutation in the mouse that affects embryonic ectoderm organization. Genet Res. 1984;43:43–50. doi: 10.1017/s0016672300025702. [DOI] [PubMed] [Google Scholar]
- Kao KR, Elinson RP. The entire mesodermal mantle behaves as Spemann's organizer in dorsoanterior enhanced Xenopus laevisembryos. Dev Biol. 1988;127:64–77. doi: 10.1016/0012-1606(88)90189-3. [DOI] [PubMed] [Google Scholar]
- Kishida S, Yamamoto H, Ikeda S, Kishida M, Sakamoto I, Koyama S, Kikuchi A. Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of beta-catenin. J Biol Chem. 1998;273:10823–10826. doi: 10.1074/jbc.273.18.10823. [DOI] [PubMed] [Google Scholar]
- Kofron M, Spagnuolo A, Klymkowsky M, Wylie C, Heasman J. The roles of maternal alpha-catenin and plakoglobin in the early Xenopusembryo. Development. 1997;124:1553–1560. doi: 10.1242/dev.124.8.1553. [DOI] [PubMed] [Google Scholar]
- Kurth, T. 1997. Licht- und electronenmikrosopische Untersuchungen zur Lokalisation von Zelladhäsionsmolekülen und deren Wechselwirkungen in adulten und embryonalen Geweben von Xenopus laevis (Daudin, 1802). PhD thesis. University of Tübingen, Germany. 156 pp.
- Lemaire P, Garrett N, Gurdon JB. Expression cloning of Siamois, a Xenopushomeobox gene expressed in dorsal-vegetal cells of blastulae and able to induce a complete secondary axis. Cell. 1995;81:85–94. doi: 10.1016/0092-8674(95)90373-9. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mao J, Yuan H, Xie W, Simon MI, Wu D. Specific involvement of G proteins in regulation of serum response factor-mediated gene transcription by different receptors. J Biol Chem. 1998;273:27118–27123. doi: 10.1074/jbc.273.42.27118. [DOI] [PubMed] [Google Scholar]
- Miller JR, Moon RT. Signal transduction through beta-catenin and specification of cell fate during embryogenesis. Genes Dev. 1996;10:2527–2539. doi: 10.1101/gad.10.20.2527. [DOI] [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Rubinfeld B, Polakis P. Deletion of an amino-terminal sequence beta-catenin in vivo and promotes hyperphosporylation of the adenomatous polyposis coli tumor suppressor protein. Mol Cell Biol. 1996;16:4088–4094. doi: 10.1128/mcb.16.8.4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai LM, Orsulic S, Bejsovec A, Peifer M. Negative regulation of Armadillo, a Wingless effector in Drosophila. . Development. 1997;124:2255–2266. doi: 10.1242/dev.124.11.2255. [DOI] [PubMed] [Google Scholar]
- Peifer M. Cell adhesion and signal transduction: the Armadillo connection. Trends Cell Biol. 1995;5:224–229. doi: 10.1016/s0962-8924(00)89015-7. [DOI] [PubMed] [Google Scholar]
- Peifer M, Sweeton D, Casey M, Wieschaus E. wingless signal and Zeste-white 3 kinase trigger opposing changes in the intracellular distribution of Armadillo. Development. 1994;120:369–380. doi: 10.1242/dev.120.2.369. [DOI] [PubMed] [Google Scholar]
- Perry WL, III, Vasicek TJ, Lee JJ, Rossi JM, Zeng L, Zhang T, Tilghman SM, Costantini F. Phenotypic and molecular analysis of a transgenic insertional allele of the mouse Fusedlocus. Genetics. 1995;141:321–332. doi: 10.1093/genetics/141.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. Mutations in the APC gene and their implications for protein structure and function. Curr Opin Genet Dev. 1995;5:66–71. doi: 10.1016/s0959-437x(95)90055-1. [DOI] [PubMed] [Google Scholar]
- Riggleman B, Schedl P, Wieschaus E. Spatial expression of the Drosophilasegment polarity gene armadillo is posttranscriptionally regulated by wingless. Cell. 1990;63:549–560. doi: 10.1016/0092-8674(90)90451-j. [DOI] [PubMed] [Google Scholar]
- Rocheleau CE, Downs WD, Lin R, Wittmann C, Bei Y, Cha YH, Ali M, Priess JR, Mello CC. Wnt signaling and an APC-related gene specify endoderm in early C. elegansembryos. Cell. 1997;90:707–716. doi: 10.1016/s0092-8674(00)80531-0. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with beta-catenin. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Rupp RA, Snider L, Weintraub H. Xenopusembryos regulate the nuclear localization of XMyoD. Genes Dev. 1994;8:1311–1323. doi: 10.1101/gad.8.11.1311. [DOI] [PubMed] [Google Scholar]
- Sakanaka C, Weiss JB, Williams LT. Bridging of beta-catenin and glycogen synthase kinase-3beta by Axin and inhibition of beta-catenin-mediated transcription. Proc Natl Acad Sci USA. 1998;95:3020–3023. doi: 10.1073/pnas.95.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Steinbeisser H, Warga RM, Hausen P. Beta-catenin translocation into nuclei demarcates the dorsalizing centers in frog and fish embryos. Mech Dev. 1996;57:191–198. doi: 10.1016/0925-4773(96)00546-1. [DOI] [PubMed] [Google Scholar]
- Steitz SA, Tsang M, Sussman DJ. Wnt-mediated relocalization of dishevelled proteins. In Vitro Cell Dev Biol Anim. 1996;32:441–445. doi: 10.1007/BF02723007. [DOI] [PubMed] [Google Scholar]
- Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4–activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Theiler K, Gluecksohn-Waelsch S. The morphological effects and the development of the Fused mutation in the mouse. Anat Rec. 1956;125:83–104. doi: 10.1002/ar.1091250107. [DOI] [PubMed] [Google Scholar]
- Vleminckx K, Wong E, Guger K, Rubinfeld B, Polakis P, Gumbiner BM. Adenomatous polyposis coli tumor suppressor protein has signaling activity in Xenopus laevisembryos resulting in the induction of an ectopic dorsoanterior axis. J Cell Biol. 1997;136:411–420. doi: 10.1083/jcb.136.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kishida S, Uochi T, Ikeda S, Koyama S, Asashima M, Kikuchi A. Axil, a member of the Axin family, interacts with both glycogen synthase kinase 3beta and beta-catenin and inhibits axis formation of Xenopusembryos. Mol Cell Biol. 1998;18:2867–2875. doi: 10.1128/mcb.18.5.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa S, van Leeuwen F, Wodarz A, Klingensmith J, Nusse R. The dishevelled protein is modified by wingless signaling in Drosophila. . Genes Dev. 1995;9:1087–1097. doi: 10.1101/gad.9.9.1087. [DOI] [PubMed] [Google Scholar]
- Yang-Snyder J, Miller JR, Brown JD, Lai C-J, Moon RT. A frizzled homolog functions in a vertebrate Wntsignaling pathway. Curr Biol. 1996;6:1302–1306. doi: 10.1016/s0960-9822(02)70716-1. [DOI] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopusembryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL, III, Lee JJ, Tilghman SM, Gumbiner BM, Costantini F. The mouse Fusedlocus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell. 1997;90:181–192. doi: 10.1016/s0092-8674(00)80324-4. [DOI] [PubMed] [Google Scholar]