Endosome to Golgi Retrieval of the Vacuolar Protein Sorting Receptor, Vps10p, Requires the Function of the VPS29, VPS30, and VPS35 Gene Products (original) (raw)
Abstract
Mutations in the S. cerevisiae VPS29 and VPS30 genes lead to a selective protein sorting defect in which the vacuolar protein carboxypeptidase Y (CPY) is missorted and secreted from the cell, while other soluble vacuolar hydrolases like proteinase A (PrA) are delivered to the vacuole. This phenotype is similar to that seen in cells with mutations in the previously characterized VPS10 and VPS35 genes. Vps10p is a late Golgi transmembrane protein that acts as the sorting receptor for soluble vacuolar hydrolases like CPY and PrA, while Vps35p is a peripheral membrane protein which cofractionates with membranes enriched in Vps10p. The sequences of the VPS29, VPS30, and VPS35 genes do not yet give any clues to the functions of their products. Each is predicted to encode a hydrophilic protein with homologues in the human and C. elegans genomes. Interestingly, mutations in the VPS29, VPS30, or VPS35 genes change the subcellular distribution of the Vps10 protein, resulting in a shift of Vps10p from the Golgi to the vacuolar membrane. The route that Vps10p takes to reach the vacuole in a vps35 mutant does not depend upon Sec1p mediated arrival at the plasma membrane but does require the activity of the pre-vacuolar endosomal t-SNARE, Pep12p. A temperature conditional allele of the VPS35 gene was generated and has been found to cause missorting/secretion of CPY and also Vps10p to mislocalize to a vacuolar membrane fraction at the nonpermissive temperature. Vps35p continues to cofractionate with Vps10p in vps29 mutants, suggesting that Vps10p and Vps35p may directly interact. Together, the data indicate that the VPS29, VPS30, and VPS35 gene products are required for the normal recycling of Vps10p from the prevacuolar endosome back to the Golgi where it can initiate additional rounds of vacuolar hydrolase sorting.
Eukaryotic cells contain numerous distinct membrane bound organelles. To maintain the functional identities of these organelles, specific sets of proteins must be efficiently sorted and delivered to each intracellular compartment. For example, in the well-characterized lysosomal protein delivery pathway in mammalian cells (Kornfeld, 1992), soluble lysosomal hydrolases are modified in the Golgi by the attachment of a mannose 6-phosphate group on their N-linked oligosaccharide side chains. In the TGN this tag is recognized by a set of two mannose 6-phosphate receptors (MPRs)1 that mediate sorting of the hydrolases to the endosome where the hydrolases dissociate from the receptors due to the acidic environment. Ultimately, the hydrolases are delivered to the lysosome by an unknown mechanism while the receptors are recycled back to the Golgi apparatus for further rounds of sorting. Several sequence specific signals in the cytoplasmic tails of the MPRs have been identified that are important in the trafficking of the receptors to and from the endosome (Kornfeld, 1992). For instance, signals containing a casein kinase II site and a dileucine motif at the end of the cytoplasmic tail domain aid in the exit of the MPRs from the Golgi apparatus (Lobel et al., 1989; Mauxion et al., 1996). Additionally, if the receptors reach the cell surface, they can be endocytosed due to the recognition of aromatic based signals located early in the cytoplasmic tail domain (Johnson et al., 1990; Canfield et al., 1991). The cellular machinery which recognizes these signals has begun to be identified. Clathrin adaptor proteins have been shown to interact with the first two signals (Glickman et al., 1989; Mauxion et al., 1996), but the proteins responsible for sorting MPRs for retrieval from the endosome to the TGN have yet to be identified.
To define additional components of the cell's sorting apparatus, we and others have employed genetic selection schemes to isolate mutants in Saccharomyces cerevisiae that are unable to properly sort proteins to the vacuole (the yeast equivalent to the mammalian lysosome) (Jones, 1977; Bankaitis et al., 1986; Rothman et al., 1986; Robinson et al., 1988). Mutations in more than 40 complementation groups result in defective vacuolar protein sorting (vps mutants). The number of genes involved in this process reflects the numerous transport steps as well as the complexity of each stage of the vacuolar protein sorting pathway. The vps mutants have been divided into six classes (A–F) according to their vacuolar morphologies, which range from wild-type vacuoles to no recognizable vacuolar structures (Banta et al., 1988; Raymond et al., 1992). As in mammalian cells, a receptor mediated process sorts yeast vacuolar hydrolases away from secretory proteins in the late Golgi apparatus. The receptor/hydrolase complexes are transported to an endosome and then delivered to the vacuole. We have previously characterized two of the vps mutants as having a selective missorting phenotype. Mutations in VPS10 and VPS35 were shown to result in the missorting of the vacuolar hydrolase carboxypeptidase Y (CPY), while the vast majority of two other soluble hydrolases, proteinase A (PrA) and proteinase B (PrB), were properly sorted to the vacuole and matured (Paravicini et al., 1992; Marcusson et al., 1994). The VPS10 gene encodes a transmembrane sorting receptor for soluble vacuolar hydrolases (Marcusson et al., 1994; Westphal et al., 1996), and like the MPRs, cycles between the late Golgi apparatus and endosomes. Transport of Vps10p out of the Golgi is believed to be mediated by clathrin-coated vesicles (Seeger and Payne, 1992); subsequent docking and fusion of these vesicles requires an endosomal t-SNARE, Pep12p (Becherer et al., 1996). Multiple aromatic amino acid–containing signals in the cytoplasmic tail of Vps10p have been shown to be necessary for efficient maintenance of Vps10p in the Golgi and endosome compartments (Cereghino et al., 1995; Cooper and Stevens, 1996). It has been postulated that these signals may be recognized by as yet unidentified cytoplasmic proteins which participate in the recycling of Vps10p from the endosome back to the Golgi complex (Cereghino et al., 1995; Cooper and Stevens, 1996; Nothwehr et al., 1996). The VPS35 gene has previously been shown to encode a hydrophilic protein of unknown activity which is associated with the cytosolic face of a light membrane fraction.
We carried out a screen to look for other vps mutants that exhibited a selective missorting phenotype similar to vps10 and vps35 mutants. Two new mutants, vps29 and vps30, were found to belong to this group. Like vps10 and vps35 mutants, these mutants have normal vacuoles (class A morphology) and missort the vast majority of CPY while properly sorting the majority of PrA. In this paper, we report the sequence of these genes and show that the VPS29, VPS30, and VPS35 gene functions are each required to maintain Vps10p in its proper intracellular location. In strains deleted for any one of these genes or in a vps35ts mutant, Vps10p is redirected to the vacuolar membrane. However, localization of another Golgi protein, Mnn1p, is not altered in this group of mutants. We also report that Vps35p, which in wild-type cells is found in the same Golgi/endosome enriched membrane fraction as Vps10p, shifts to a vacuole enriched low speed membrane pellet in Δvps29 and Δvps30 mutants. Together, our findings suggest that the VPS29, VPS30, and VPS35 gene products direct retrieval of Vps10p as well as other late Golgi proteins (e.g, Kex2p and dipeptidylaminopeptidase A) that cycle between the Golgi and the prevacuolar endosome.
Materials and Methods
Media, Strains, and Yeast Genetic Methods
All yeast strains (Table I) were grown in yeast extract, peptone, dextrose medium (YPD), synthetic dextrose medium, or yeast nitrogen base medium (YNB) (Sherman et al., 1979). Standard media were used for bacterial strains (Miller, 1972). Standard yeast genetic techniques for sporulation, tetrad dissection, and gene disruption were employed (Sherman et al., 1979). E. coli transformations were performed using the method of Hanahan (1983). Yeast transformations were performed with the alkali cation treatment method for integrative transformations (Ito et al., 1983) or the procedure of Elble (1992) for plasmid transformations.
Table I.
Yeast Strains Used in This Study
| Name | Description | Source or Reference |
|---|---|---|
| SEY6210 | MATα leu2-3,112 ura3-52 his3Δ200 trp1-Δ901 lys2-801 suc2-Δ9 | Robinson et al., 1988 |
| SEY6211 | MATa leu2-3,112 ura3-52 his3Δ200 trp1-Δ901 ade2-101 suc2-Δ9 | Robinson et al., 1988 |
| SEY29-3 | SEY6210 vps29-3 | Robinson et al., 1988 |
| SEY30-2 | SEY6210 vps30-2 | Robinson et al., 1988 |
| BHY10 | SEY6210 leu2-3,112::pBHY11 | Horazdovsky et al., 1994 |
| BHY11 | SEY6211 leu2-3,112::BHY11 | Horazdovsky et al., 1994 |
| PSY129 | SEY6210 Δvps29::HIS3 | P. Scott, this study |
| EMY3 | SEY6210 Δvps10::HIS3 | Marcusson et al., 1994 |
| EMY18 | SEY6210 Δvps35::HIS3 | Cereghino et al., 1995 |
| EGY111-1 | sec1-1 MATa leu2-3,112 ura3-52 his3Δ200 trp1-Δ901 | E. Gaynor, unpublished |
| MSY3510 | BHY10 Δvps35::HIS3 | M. Seaman, this study |
| MSY35-4ts | SEY6210 vps35-4ts | M. Seaman, this study |
| MSY3501 | EGY111-1 Δvps35::HIS3 | M. Seaman, this study |
| MSY0435 | MBY3 Δvps35::HIS3 | M. Seaman, this study |
| MSY3512 | CBY9 Δvps35::HIS3 | M. Seaman, this study |
| MSY4012 | vps4 Δpep12::LEU2 | M. Seaman, this study |
| EMY50 | BHY11 VPS29::VPS329:TRP1 | E. Marcusson, this study |
| EMY51 | SEY 6210 vps29-3 leu2-3,112::pBHY11 | E. Marcusson, this study |
| CBY31 | SEY6210 Δpep12::HIS3 | C. Burd, unpublished |
| JCY3000 | SEY6210 Δvps30::HIS3 | J. Cereghino, this study |
| JCY3001 | BHY11 VPS30::VPS30::TRP1 | J. Cereghino, this study |
| JCY3002 | SEY 6210 vps30-2 leu2-3,112::pBHY11 | J. Cereghino, this study |
To clone the VPS29 and VPS30 genes, strains carrying the mutant alleles vps29-3 or vps30-2 were transformed with a yeast genomic library (CEN, LEU2). Library plasmids were recovered from Vps+ transformants. Re-introduction of these plasmids into the above strains resulted in a Vps+ phenotype. For VPS29, the minimal complementing fragment was mapped to a 3-kb BamH1 fragment, while for VPS30, the smallest fragment found to complement the mutant allele was mapped to a 2-kb SnaBI–HincII fragment. Integrative mapping analysis was used to confirm that the minimum complementing fragments of the VPS29 and VPS30 genes mapped to the mutant allele loci. The integrative mapping construct pEM29-50 was linearized by cutting with StyI and integrated into the strain BHY11 to create the Trp+ strain EMY50. The plasmid pBHY11 (Horazdovsky et al., 1994) was linearized by cutting with EcoRI and integrated into SEY29-3 to create the strain EMY51. EMY50 was then crossed to EMY51 and the resulting diploid strain was sporulated. Twenty tetrads were dissected and showed 2:2 segregation with respect to Vps+/ Vps−and Trp+/Trp−. All Trp+ segregants displayed a Vps+ phenotype and vice versa.
The integrative mapping construct pRS304-30 was linearized by cutting with EcoRI and integrated into the strain BHY11 to create the Trp+ strain JCY3001. The plasmid pBHY11 was linearized by cutting with EcoRI and integrated into SEY30-2 to create the strain JCY3002. JCY3001 was then crossed to JCY3002 and the resulting diploid strain was sporulated. 23 tetrads were dissected and showed 2:2 segregation with respect to Vps+/ Vps− and Trp+/Trp−. All Vps− segregants displayed a Trp− phenotype and vice versa.
Disruption of VPS29 was achieved by creating a disrupted allele in which a 1.3-kb fragment of the HIS3 gene was inserted into a ClaI site engineered into VPS29 downstream of the intron (see below). This generated a construct in which VPS29 is disrupted at the codon for amino acid 61. The wild-type copy of VPS29 in SEY6210 was replaced with the disrupted allele by homologous recombination producing the strain PSY129 which was found to be Vps−. A null allele of the VPS30 gene was created by replacing the region BstXI and XhoI sites with the HIS3 fragment resulting in deletion of ∼70% of the coding region of VPS30. The wild-type VPS30 gene in SEY6210 was replaced with the null allele by homologous recombination to create the strain JCY3000. This was found to have a Vps− phenotype similar to the vps29 mutant strain PSY129 and also to vps10 and vps35 mutant strains.
The double mutant strains were constructed as follows: MSY3501 was made by deletion of VPS35 in a sec1 mutant strain (EGY111-1) using the VPS35::HIS3 deletion construct described in Paravicini et al. (1992). MSY3512 was made by a deletion of VPS35 in the pep12ts strain CBY9 (Cowles et al., 1997). This deletion was made in a similar fashion to the VPS35::HIS3 deletion by removal of ∼90% of the VPS35 open reading frame by digestion with SnaB1. The LEU2 open reading frame was ligated into the SnaBI sites and this VPS35::LEU2 construct was used to delete VPS35. The strain MSY0435 was made by deletion of VPS35 in the strain MBY3 which was already deleted for VPS4 (Babst et al., 1997). The VPS35::HIS3 deletion construct was used for this deletion. MSY4012 was made by deletion of PEP12 in a strain carrying a vps4 mutant allele (SEY4-15). Deletion of PEP12 was achieved using a PEP12::LEU2 deletion construct similar to the one described in Becherer et al. (1996). All deletions were confirmed using PCR.
Plasmid Construction, Mutagenesis, and Sequencing
Standard molecular biology techniques for the manipulation of DNA were used throughout this study (Sambrook et al., 1989). The glass bead method for the isolation of DNA fragments was used (Volgelstein and Gillespie, 1979).
The original library plasmids with complementing activity toward vps29 and vps30 were named pVPS29-1 and pVPS30-1, respectively. The centromeric plasmid pEMY29-1 was created by subcloning the 3-kb BamHI fragment from pVPS29 into pRS414. The same fragment was subcloned into pRS304 to make the integrative mapping construct pEM29-50. To facilitate the production of a construct to disrupt VPS29, a ClaI site was engineered into VPS29 by site-directed mutagenesis (Deng and Nickoloff, 1992). The plasmid pEMYm1-29 was created by changing the codon for the sixty first amino acid from GTA to GAT, thus creating a ClaI site inside the second exon of the VPS29 gene. The BamHI fragment was cut out of the plasmid pEMYm1-29 and put into pBlueScript (Stratagene, La Jolla, CA) to make the plasmid pEM29-51. This plasmid was cut with ClaI and filled in with T4 DNA polymerase. A 1.3-kb HIS3 containing fragment was subcloned into this site to create the plasmid pPS129.
The SnaBI–HincII fragment from pVPS30-1 was subcloned into pBlueScript to form the plasmid pJC30-1. The 2-kb ApaI–BamHI fragment from pJC30-1 was subcloned into pRS414, pRS424, and pRS304, to create the plasmids pJCY30-2, pJCY30-3, pJC30-4, respectively. To produce an antisera specific for Vps30p, a TrpE-Vps30 fusion was generated. A SpeI fragment of VPS30 coding for the carboxyl terminal 292 amino acids including the stop codon was ligated into the pATH-TrpE vector which had been cut with XbaI to produce an in-frame fusion. The fusion protein was expressed and isolated as described previously (Paravicini et al., 1992). A New Zealand white rabbit was immunized with the fusion protein following a standard antibody production protocol.
The plasmid pAla-397-CPD-Y was a generous gift from Klaus Breddam and Morten Kielland-Brandt. This plasmid encodes a mutant form of CPY in which the histidine residue at position 397 has been changed to an alanine, thus reducing the CPY activity by 10,000-fold (Bech and Breddam, 1989).
Sequencing was performed by the Sequenase sequencing kit (United States Biochem. Corp., Cleveland, OH). Constructs for sequencing the VPS29 gene were made by treating pEMY29-1 with exonuclease III and mung bean nuclease as described in the Stratagene BlueScript manual. Several fragments from pVPS30-1 were subcloned into pBlueScript. The T3 and T7 primers were used to sequence one strand of DNA from each gene. Synthetic oligonucleotides were designed and made in order to sequence the second strand of each gene for confirmation purposes. Sequence analysis revealed that the VPS29 ORF was interrupted by an intron. All of the necessary sequences for splicing (GTATGT . . . TAG and TACTAAC [Langford and Gallwitz, 1983; Langford et al., 1984]) were found. The first and second exons are 49 bp and 800 bp, respectively, and are interrupted by an intron of 119 bp. Subsequent sequencing of chromosome VIII by the S. cerevisiae sequencing project confirmed the sequence obtained (designated ORF YHR012w, GenBank accession number U10400). The sequence of VPS30 obtained from sequencing the SnaBI– HincII fragment was found to be identical to sequence on chromosome XVI (designated LPH7w, GenBank accession number U43503).
The strain carrying the vps35ts allele was generated by PCR mutagenesis/gapped plasmid repair of wild-type VPS35 as outlined in Stack et al. (1995). Briefly, MSY3510 was transformed with the plasmid pGPY35 (Paravicini et al., 1992) which had been gapped by digestion with SnaBI to remove ∼90% of the coding region of VPS35. The gapped plasmid was purified by agarose gel electrophoresis. Along with gapped pGPY35, MSY3510 was transformed with PCR DNA generated under mutagenic conditions (dATP was limiting) using primers which annealed ∼150 bp either sides of the SnaBI sites. Transformants were selected for on −URA plates. After replica plating onto two sets of YP fructose plates, the transformants were screened for secretion of a CPY-Invertase fusion protein using a colorimetric plate assay (Paravicini et al., 1992). Colonies which exhibited temperature conditional secretion of the CPY-Invertase fusion were picked. After further testing, the plasmids were rescued and retransformed into MSY3510 and also EMY18. The strains carrying the plasmids were then tested for their ability to sort and mature CPY using the conventional pulse/chase assay (see below). The above strains carrying plasmids pMS35-2ts and pMS35-4ts and pMS35ts-6 were found to sort and mature ∼80–90% CPY at 26°C and secrete ∼50% of p2CPY at 38°C (with a 10-min preshift). To integrate the 35-4ts allele into the genome of wildtype cells, the plasmid pMS35-4ts was cut with NciI and XbaI to remove the vps35-4ts open reading frame. This linear fragment was gel purified and then cotransformed with pCYI50 (the plasmid carrying the CPYInvertase fusion) into SEY6210. Using the colorimetric plate assay, colonies exhibiting temperature conditional secretion of the CPY-Invertase fusion were identified. After several rounds of testing, the strain MSY354ts was transformed with pGPY35 carrying the wild-type VPS35 gene which completely complemented the temperature conditional CPY secretion phenotype.
Cell Labeling, Immunoprecipitations, Subcellular Fractionation, and Immunofluorescence
Cell labeling and immunoprecipitations were performed essentially as described (Gaynor et al., 1994; Cereghino et al., 1995). A brief description is as follows: yeast cells were grown to mid-logarithmic phase in YNB media. The cells were labeled with 20–30 μCi of Tran35S-label (ICN Radiochemicals, Irvine, CA) per OD equivalent of cells. During the chase period, unlabeled methionine and cysteine were added to concentrations of 5 and 1 mM, respectively. The chase was terminated by adding samples to an equal volume of cold 2× spheroplast stop solution (2 M sorbitol, 50 mM Tris pH 7.5, 40 mM NaN3, 40 mM NaF, 20 mM DTT). The samples were incubated on ice for 10 min, and then treated with 10 μg of zymolyase 100T (Seikagaku, Tokyo, Japan) per OD equivalent. The cell and media fractions were then separated by centrifugation for 1 min at 8,000 g. Both fractions were precipitated with 5–10% trichloroacetic acid. Immunoprecipitations were carried out as previously described (Klionsky et al., 1988; Cereghino et al., 1995). Subcellular fractionation of Vps10p experiments were carried out as described in Cereghino et al. (1995) except 10 μg of zymolyase 100T was used without glusulase. Fractionation of Vps35p was performed in an identical fashion with the exception that the homogenization buffer was as follows: 20 mM Hepes-KOH (pH 7.0), 50 mM potassium acetate, 2 mM EDTA, 0.2 M sorbitol. Immunofluorescence was performed as previously described (Cereghino et al., 1995).
Sucrose gradient fractionation of P100 membranes was performed essentially as described in Becherer et al., (1996) with the exception that the Hepes-KOH buffer described above was used in the homogenization and in the gradient buffers. Briefly, spheroplasted cells were labeled and chased as above before being lysed and homogenized as described above. The lysate was then spun at 13,000 g (for 10 min) to remove all vacuolar membranes, plasma membrane, mitochondria, and endoplasmic reticulum before being loaded (7 ODs equivalent) onto a 10–60% sucrose gradient prepared as described in Becherer et al. (1995). After centrifugation to equilibrium, the gradients were unloaded and 1-ml fractions were collected. Vps35p, Vps10p, and Kex2p were immunoprecipitated from each fraction and separated by SDS-PAGE electrophoresis. The relative amount of each protein was determined using a phosphorimager (Molecular Dynamics, Sunnyvale, CA).
Results
vps29 and vps30 Mutants Exhibit Selective Defects in Vacuolar Hydrolase Sorting
VPS10 and VPS35 are two previously characterized genes, which when mutated yield cells with morphologically normal vacuoles, but exhibit a selective vacuolar sorting defect (Banta et al., 1988; Raymond et al., 1992; Paravicini et al., 1992; Marcusson et al., 1994). These mutants missort and secrete more than 80% of their CPY, but properly localize and mature 80–90% of PrA and all other soluble vacuolar hydrolases tested. To identify other vps mutants which exhibit a similar selective sorting defect, a careful screen of the vps mutant collection was undertaken which resulted in the identification of two mutants, vps29 and vps30, with phenotypes similar to vps10 and vps35 mutants. Like vps10 and vps35 mutants, vps29 and vps30 mutants had previously been shown to contain morphologically normal vacuoles (Banta et al., 1988; Raymond et al., 1992). Pulse chase analysis revealed that these mutants also exhibited a selective vacuolar sorting defect (Fig. 1 A). For this experiment, cells were incubated in the presence of Tran35S label for 10 min at 30°C then chased in the presence of unlabeled cysteine and methionine for 30 min. Next, the cells were converted to spheroplasts, separated into intracellular (I) and extracellular (E) fractions, and CPY and PrA were recovered by immunoprecipitation. Unlike wild-type cells, which properly localize and mature both CPY and PrA (Fig. 1 A, lanes 1 and 2), cells deleted for VPS10 missort and secrete ∼90% of their CPY (lanes 3 and 4). However, these Δvps10 cells were able to properly localize and mature the vast majority of their PrA (Fig. 1 A, lanes 3 and 4). We found that vps29 mutants exhibited the same sorting phenotype as vps10 mutants for both CPY and PrA (Fig. 1 A, lanes 5 and 6). Mutations in VPS30 also led to a similar defect in CPY sorting; however, the sorting of PrA was more affected than in vps10 or vps29 mutants, as only ∼75% of PrA was properly matured (Fig. 1 A, lanes 7 and 8). The small amount of PrA which is secreted from these three strains is found as a form termed pseudo PrA and is the result of an autocatalytic cleavage of PrA in the extracellular media (Westphal et al., 1996). Unexpectedly, we found that the ability of these mutants to sort PrA was partially dependent on the media in which they were grown. The pH of the media was the most critical factor. The lower the pH, the better these mutants sorted and matured PrA (data not shown). The amount of PrA properly sorted and matured could be as low as 50% when the pH of the media was greater than 5.0, but usually was between the values shown in Table II (yeast grow rapidly in low pH media with a pH in the 3–5 range). CPY sorting was not affected by the pH of the media. At present, we do not know the mechanism of this pH-dependent PrA sorting. It may be that the Vps10p-independent recognition pathway for PrA sorting is very sensitive to changes in compartment pH induced by changes in media pH. Regardless, the selective missorting phenotype shown by vps10, vps29, vps30, and vps35 mutants distinguishes them from the rest of the characterized vps mutants, none of which show such a large difference in their ability to sort PrA vs CPY.
Figure 1.

Selective missorting of vacuolar proteins. (A) Yeast cells were pulse-labeled with Tran35S for 10 min and chased for 30 min. The cells were then spheroplasted and separated into intracellular (I) and extracellular (E) fractions. CPY or PrA was immunoprecipitated from both fractions and resolved by SDSPAGE and fluorography. The strains used were SEY6210 (WT), EMY3 (Δvps10), PSY129 (vps29), and JCY3000 (Δvps30). The position and molecular weights of both precursor and mature forms of CPY and PrA are indicated. (B) vps35ts cells (MSY354ts) were incubated at either 26°C for 0 min before labeling or 38°C for the specified time. The cells were labeled for 5 min and chased as above, and then spheroplasted and separated into intracellular (I) and extracellular (E) fractions. CPY was immunoprecipitated from both fractions and resolved by SDS-PAGE and fluorography.
Table II.
Summary of Mutant Phenotypes
| Strain | CPY sorted | PrA sorted | Vps10p in P100 | Half-life of Kex2p |
|---|---|---|---|---|
| % | % | % | ||
| Wild-type | 95–100 | 95–100 | 85–90 | >120 min |
| Δvps10 | 5–10 | 80–90 | — | >120 min |
| Δvps29 | 5–10 | 80–90 | 10–20 | ∼30 min |
| Δvps30 | 5–10 | 70–80 | 10–20 | ∼75 min |
| Δvps35 | 5–10 | 75–85 | 10–20 | ∼45 min |
To better understand the function of Vps35p in the sorting of CPY to the vacuole, a temperature conditional mutant was generated using random PCR mutagenesis. The vps35ts allele was subsequently integrated into the genome of wild-type cells as described in the Materials and Methods section. In Fig. 1 B, the CPY sorting phenotype of the vps35ts allele is shown. Cells carrying the vps35ts allele were incubated at either 26°C (lanes 1–2) or 38°C for 0, 10, 20, or 30 (lanes 3–10) min before labeling with [35S]methionine. After labeling for 5 min, the CPY was chased for 30 min before the cells were transferred to ice and then converted to spheroplasts to separate the intracellular pool (I) of CPY from the secreted extracellular pool (E). At 26°C, the vps35ts cells sort and mature 90% of the labeled CPY, with just a small proportion of p2CPY being secreted and a small intracellular pool retained in the cell. Upon shifting to the nonpermissive temperature, the cells secrete progressively more p2CPY with increasing lengths of preshift times, so that after 20 min at the nonpermissive temperature, ∼80% of the labeled CPY is secreted into the media as the late-Golgi (p2) modified form. This result is consistent with Vps35p having a direct role in sorting/transport of CPY to the vacuole and confirms that the CPY sorting defect observed in the vps35 mutant is not due to secondary effects resulting from deletion of the protein.
Cloning of VPS29 and VPS30
To characterize further the function of VPS29 and VPS30, the genes were cloned by complementation of the CPY sorting defect as previously described (Paravicini et al., 1992). The CPY sorting phenotypes of vps29 and vps30 mutants with and without the complementing fragments are shown in Fig. 2. The cells were pulse labeled with Tran35S for 10 min and chased for 30 min at 30°C. The cells were then converted to spheroplasts and separated into intracellular (I) and extracellular (E) fractions from which CPY was immunoprecipitated. The vps29 and vps30 mutant cells missorted and secreted virtually all of their CPY as the Golgi modified p2 form (Fig. 2, lanes 1, 2, 5, and 6). However, when the appropriate plasmid was introduced into each mutant, CPY was properly matured inside the cells, indicating that the vacuolar sorting defect had been corrected (Fig. 2, lanes 3, 4, 7, and 8). Sequencing of VPS29 showed that it encodes a hydrophilic protein of 282 amino acids and a pI of 4.3. The primary sequence of VPS29 does not contain any structural motifs or homologies with proteins of known function. VPS30 also encodes a hydrophilic protein containing 556 amino acids which has a predicted molecular mass of 65 kD and pI of 4.8. The central portion of Vps30p (residues 186 to 322) is predicted to form coiled-coil structures (Lupas et al., 1991).
Figure 2.

Complementation of the vps29 and vps30 mutants. Yeast cells were pulse-labeled with Tran35S for 10 min and chased for 30 min. The cells were then spheroplasted and separated into intracellular (I) and extracellular (E) fractions. CPY or PrA was immunoprecipitated from both fractions and resolved by SDSPAGE and fluorography. The strains used were PSY129 (vps29), PSY129 carrying a centromeric plasmid containing the VPS29 gene (PSY129 + p_VPS29_), JCY3000 (Δvps30), and JCY3000 carrying a centromeric plasmid containing the VPS30 gene (JCY3000 + p_VPS30_).
Vps29p, Vps30p, and Vps35p Are Highly Conserved Among Eukaryotes
BLAST searches (Altschul et al., 1990) comparing the sequences of Vps29p, Vps30p, and Vps35p to the GenBank database and a database of human expressed sequence tags (ESTs) (Boguski et al., 1994) revealed that each of these proteins is highly homologous to proteins found in the C. elegans and human genome. A mouse protein was also identified which exhibited 33% identity to Vps35p. When the sequence alignment for Vps35p and its homologues was looked at more closely, it could be seen that the proteins could be divided into three domains based on their percent identity to one another (Fig. 3 A). The degree of homology was highest at the amino and carboxyl termini. This suggests that these Vps35p-like proteins may have multiple functional domains, two of which are conserved and represented by the amino- and carboxyl-terminal regions of the proteins. Fig. 3 B shows the results of a sequence alignment through the regions of highest homology for Vps29p and Vps30p and their human and C. elegans homologues. The conservation of the Vps29, Vps30, and Vps35 proteins between organisms as widely divergent as yeast and humans indicates that these proteins may play a fundamental role in maintaining proper protein trafficking within cells.
Figure 3.
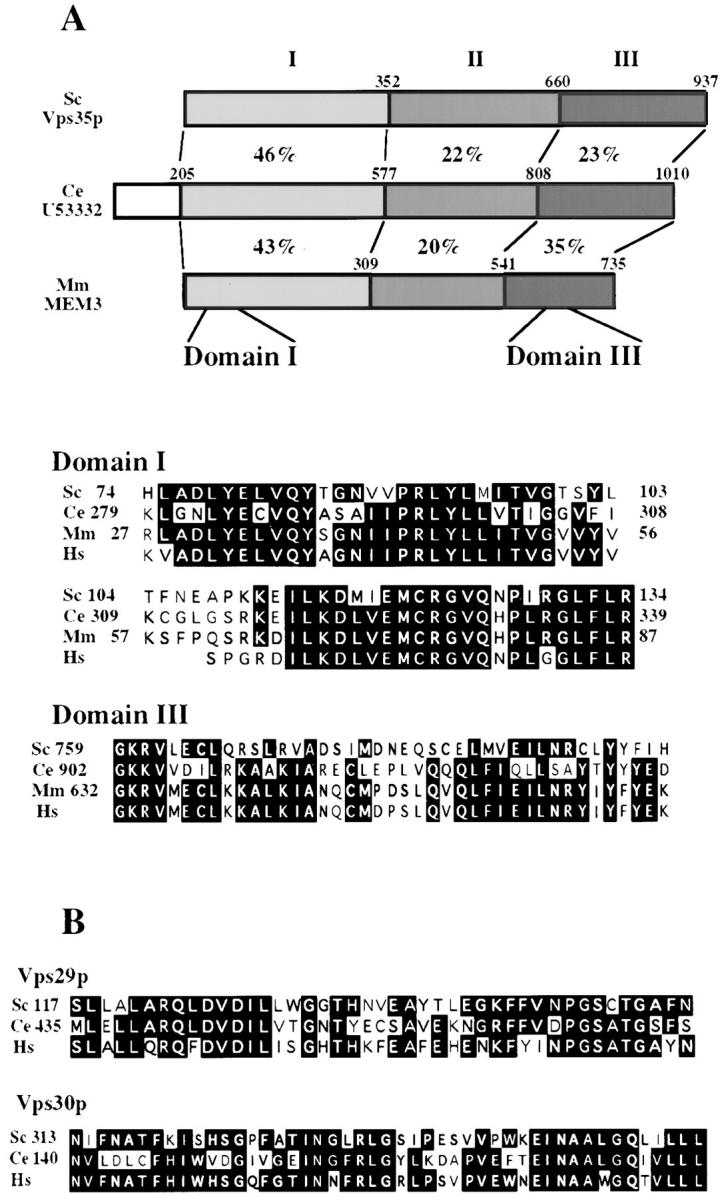
VPS35, VPS30, and VPS29 are highly conserved in eukaryotes. (A) Schematic view of homology between Vps35p and proteins found in the C. elegans and M. musculus proteomes. The numbers between the proteins are percent identities between Vps35p and the other two proteins. In the lower section, the two most highly conserved domains (designated domains I and III) are aligned to show the regions of greatest homology between the yeast, mouse, nematode, and also human homologues. The black boxes indicate completely conserved residues. (B) Alignments between Vps29p and homologues present in the proteomes of C. elegans and H. sapiens, and also alignments between Vps30p and homologues present in the proteomes of C. elegans and H. sapiens. Black shaded regions indicate areas that are completely conserved.
A Portion of Vps30p Is Peripherally Associated with a Membrane
To make an antiserum specific for Vps30p, a TrpE VPS30 fusion construct was made (see Materials and Methods). Antisera raised against the resulting fusion protein was used to further analyze the VPS30 gene product. Wildtype, Δvps30, and wild-type cells carrying a multicopy plasmid containing the VPS30 gene were pulse-labeled with Tran35S-label for 20 min at 30°C. The cells were then incubated with unlabeled cysteine and methionine for 0 or 60 min. Cell lysates were immunoprecipitated with the antisera raised against the TrpE/Vps30 fusion protein. In wildtype cells, the antisera recognized a 63-kD protein which was stable over the 60-min chase period (Fig. 4 A, lanes 2 and 3). This protein was absent from Δvps30 cells (Fig. 4 A, lane 1) and was stably expressed at a higher level in the cells carrying a multicopy plasmid containing the VPS30 gene (Fig. 4 A, lanes 4 and 5). These results show that the antisera specifically recognized the VPS30 gene product.
Figure 4.
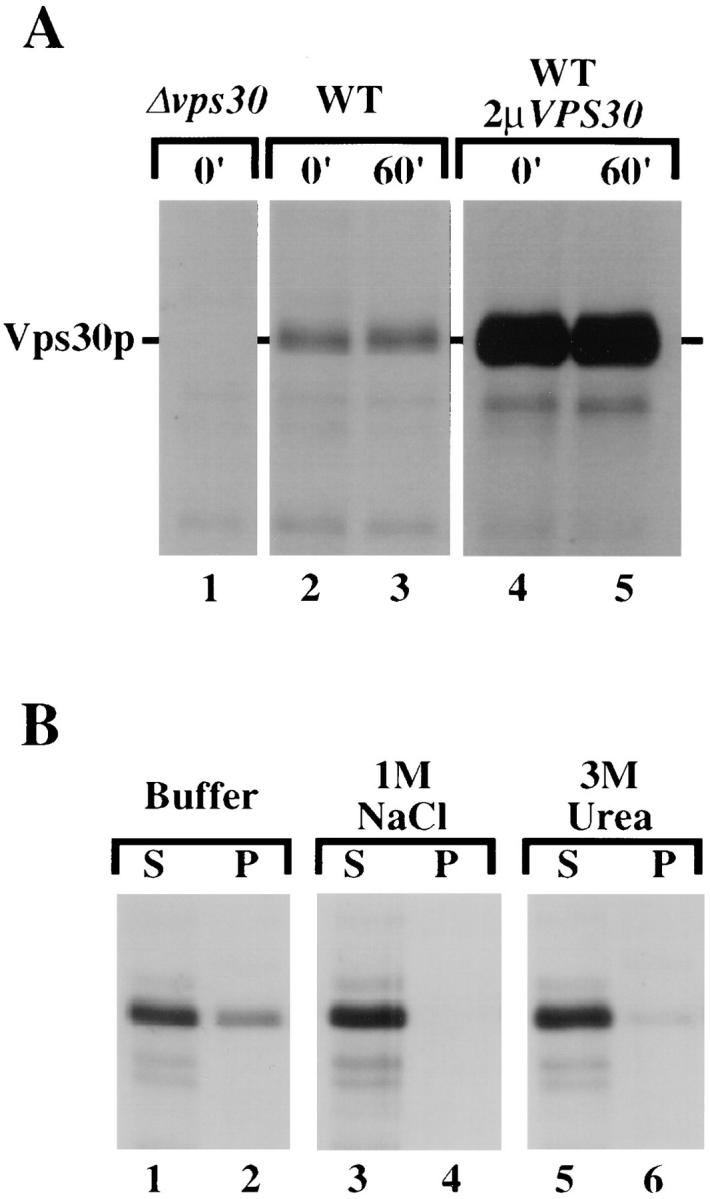
Detection of Vps30p. (A) Yeast cells were pulselabeled with Tran35S for 20 min and chased for either 0 or 60 min. The position of Vps30p is indicated. The strains used were JCY3000 (Δvps30), SEY6210 (WT), and SEY6210 carrying a 2-μ plasmid containing the VPS30 gene (WT + 2 μ VPS30). (B) Wildtype spheroplasts (SEY6210) were pulse-labeled with Tran35S for 20 min and chased for 60 min. The spheroplasts were then gently lysed by dounce homogenization in a hypotonic buffer and then washed in either 10 mM Tris/1 mM EDTA, pH 8.0 (Buffer), 1 M NaCl, or 3 M urea. The homogenates were then spun in an ultracentrifuge at 100,000 g. The pellet (P) and supernatant (S) fractions were then immunoprecipitated with antiserum specific to Vps30p and resolved by SDS-PAGE.
The intracellular location of Vps30p was investigated by subcellular fractionation experiments. Wild-type spheroplasts were pulse-labeled for 20 min with Tran35S-label and chased for 60 min in the presence of unlabeled methionine and cysteine. The labeled spheroplasts were lysed by osmotic pressure and gentle dounce homogenization, and the homogenate was separated into pellet and supernatant fractions by centrifugation at 100,000 g. Approximately 20% of the Vps30p was found in the pellet fraction (data not shown). To determine the nature of the interaction of Vps30p with the pellet fraction, the experiment was repeated, but the homogenates were washed with one of several solutions before centrifugation. When the homogenates were washed with buffer alone, the fractionation pattern was unchanged, as ∼20% of Vps30p was again found in the pellet fraction (Fig. 4 B, lanes 1 and 2). In contrast, when the homogenates were washed with either 1 M NaCl or 3 M urea, virtually all of the Vps30p could be found in the supernatant fraction (Fig. 4 B, lanes 3–6). This fractionation pattern indicates that Vps30p exists in two separate pools, a soluble or cytoplasmic pool, and a membrane-associated pool. The membrane association of Vps30p must be peripheral, as there are no apparent transmembrane domains or sites for lipid modification, and the protein is easily washed off of the membrane pellet fraction with salt or urea.
Unfortunately, to date, attempts to make fusion proteins with Vps29p for the purpose of antibody production have been unsuccessful. A variety of fusion constructs were made, but all appeared to be unstable or toxic as no product was detectable.
Deletion of VPS29, VPS30, or VPS35 Alters the Intracellular Location of Vps10p
Since vps29, vps30, and vps35 mutants show a vacuolar sorting phenotype similar to that of vps10 mutants, we reasoned that the function and/or location of Vps10p might be affected in these mutants. To investigate the subcellular location of Vps10p in these mutants, cells were converted to spheroplasts and then labeled for 15 min at 30°C with Tran35S-Label. The spheroplasts were then chased with unlabeled amino acids for 45 min to allow the labeled Vps10p to reach its steady state location in the cell. Gentle lysis of the spheroplasts was achieved by dounce homogenization in a hypotonic buffer. The lysates were then separated into pellet and supernatant fractions by sequential centrifugation at 500, 13,000, and 100,000 g. It has been previously shown that in wild-type cells ∼85–90% of the Vps10p is found in the same Golgi-enriched 100,000 g pellet (P100) fraction that contains the late Golgi protein Kex2p (Marcusson et al., 1994). The remainder of Vps10p was found in the 13,000-g pellet (P13) fraction, which has been shown to contain endoplasmic reticulum, mitochondria, plasma membrane, and vacuoles (Marcusson et al., 1994). When this same procedure was performed using Δvps29, Δvps30, or Δvps35 cells, the vast majority of Vps10p was found in the P13 fraction (Fig. 5 A, upper panels). This effect seems to be a general effect on proteins that reside late in the Golgi complex, as Kex2p is rapidly degraded in the vacuole in Δvps29 and Δvps35 mutant strains (Table II). The degradation of Kex2p occurred more slowly in the Δvps30 mutant, but it is still faster than in wild-type cells. This group of vps mutants does not seem to affect the distribution of all Golgi proteins as Mnn1p, a glycosyltransferase of the medial-Golgi, can still be found in the P100 fraction in a Δvps29 mutant (data not shown). Importantly, not all vps mutants cause Vps10p to be redistributed to the P13 fraction. The majority of Vps10p could be found in the P100 subcellular fraction from cells with mutations in PEP12 (VPS6), VPS8, VPS13, VPS18, VPS21, or VPS45 (data not shown).
Figure 5.

Subcellular fractionation. (A) Yeast spheroplasts were pulse-labeled with Tran35S for 20 min and chased for 40 min. The spheroplasts were then gently lysed by dounce homogenization in a hypotonic buffer and the lysates were fractionated by differential centrifugation. The fractions were immunoprecipitated with an antiserum specific to Vps10p and separated on an 8% SDS-polyacrylamide gel. The strains used were SEY6210 (WT), PSY129 (vps29), JCY3000 (Δvps30), and EMY18 (Δvps35). To localize Vps35p, the same procedure was followed, except antisera specific to Vps35p was used to immunoprecipitate the samples. In the wildtype strain, Vps10p is localized to the P100 fraction which contains Golgi, endosomal, and vesicular proteins; however in the vps29, Δvps30, and Δvps35 strains, Vps10p is localized to a P13 fraction which is rich in vacuolar markers. Vps35p appears to follow Vps10p to the P13 fraction in the vps29 strain and partially in the Δvps30 strain. (B) The strains SEY6210 (wild-type) and MSY35-4ts (vps35ts) were shifted to the indicated temperature for 15 min before labeling and chasing at that temperature as described above. The cells were fractionated and Vps10p was immunoprecipitated as described above. In the vps35ts strain, at the nonpermissive temperature, Vps10p is shifted to a P13 fraction (lanes 10–12). This shift of Vps10p is not observed in wild-type cells (lanes 4–6).
It has been previously shown that Vps35p is also localized to the Golgi-enriched P100 fraction, with ∼80% of the protein peripherally associated with this membrane fraction (Paravicini et al., 1992). Along with the subcellular distribution of Vps10p, the localization of Vps35p in Δvps29 and Δvps30 strains was also examined. We found that wild-type cells have a distribution of Vps35p consistent with the previous findings: ∼80% of membrane associated Vps35p is in a P100 fraction (Fig. 5 A, lower panels, lanes 1–3). However, in Δvps29 cells, there is a striking redistribution of Vps35p to the P13 fraction that mirrors the redistribution of Vps10p to the P13 fraction; in fact, 70% of the Vps35p can now be found in the P13 (Fig. 5 A, lower panels lanes 4–6). The shift of Vps35p to the P13 fraction is also observed in Δvps30 cells, although to a much lesser extent, with ∼70% of the Vps35p remaining associated with the P100 membranes (Fig. 5 A, lower panels, lanes 7–9). In Δvps35 cells, only the cross-reacting bands that migrate above and below Vps35p are visible (Fig. 5 A, lower panels, lanes 10–12). It appears, therefore, that the subcellular distribution of Vps35p is affected by the localization of Vps10p and presumably other proteins that cycle between the late Golgi and the endosome, for instance Kex2p.
The observation that Vps10p is mislocalized to the P13 fraction in a Δvps35 strain would account for the inability of the Δvps35 stain to sort p2CPY correctly at the lateGolgi for subsequent delivery to the vacuole. To determine whether the mislocalization of Vps10p to the vacuole in the Δvps35 strain was the primary defect or due to a secondary defect, perhaps caused by adaptation of the cells to the lack of Vps35 protein, the distribution of Vps10p was assessed in the vps35ts mutant. In Fig. 5 B spheroplasted cells were incubated for 15 min at the specified temperature before labeling and chasing (at either 26°C or 38°C) as described above. In the vps35ts cells, the distribution of Vps10p at the permissive temperature is similar to wildtype cells (compare Fig. 5 B, lanes 7–9, with Fig. 5 A, lanes 1–3), although marginally more Vps10p is localized to the P13 fraction. At the nonpermissive temperature, the distribution of Vps10p in the vps35ts cells is shifted to the P13 (Fig. 5 B, lanes 10–12), in a similar fashion to the Δvps35 (see Fig. 5 A, upper panels, lanes 10–12). The distribution of Vps10p in wild-type cells at 38°C was unaffected (Fig. 5 B, lanes 4–6). These data are consistent with the primary defect of Δvps35 cells being in the localization of Vps10p. These data are also consistent with the observation that the vps35ts cells have a slow onset of p2CPY secretion (see Fig. 1 B), which would be the result of a lack of available Vps10p in the late-Golgi caused by a defect in recycling of Vps10p from the prevacuolar endosomal compartment.
Vps35p Is Associated with Two Discrete Pools of Light P100 Membranes
To better characterize the compartment to which Vps35p is localized in wild-type cells, a more detailed fractionation of the light P100 membranes was carried out. Wild-type cells were spheroplasted before labeling for 15 min at 30°C and then chasing for 45 min as before. The spheroplasts were lysed as described in the Materials and Methods section, and a fraction containing the Golgi and endosomal membranes (P100) was generated by centrifugation at 13,000 g which pellets vacuolar membranes, plasma membrane, and endoplasmic reticulum to produce a supernatant that contained only P100 membranes and soluble cytosolic proteins. This was loaded onto the top of a sucrose gradient and spun to equilibrium. Twelve fractions were collected and the distribution of Vps35p, Vps10p, and Kex2p was analyzed by immunoprecipitation of the respective proteins. In Fig. 6, the amount of Vps35p in each fraction is shown by the autoradiogram, while the distribution of Vps10p and Kex2p in shown in the graph. It appears that Vps10p and Kex2p fractionate in two discrete peaks and that Vps35p is associated with both of these peaks. It is possible that these peaks correspond to Golgi membranes (fractions 3–4) and lighter endosomal membranes (fraction 6). In previous studies it has been observed that p2CPY can be separated into two discrete peaks of light membranes (Vida et al., 1993) and that a marker for endosomes, Pep12p, fractionates away from Kex2p on similar gradients (Becherer et al., 1996). Hence, it appears that the distribution of Vps35p mirrors that of the proteins that cycle between the late-Golgi and the endosome.
Figure 6.

Vps35p is associated with two discrete pools of light membranes. Wild-type cells were spheroplasted, labeled with [35S]methionine for 15 min, and then chased for 45 min. After lysis by dounce homogenization, large membranes were cleared by centrifugation at 13,000 g for 10 min. The cleared lysate was then loaded onto a 10–60% sucrose gradient and spun to equilibrium. 12 fractions were collected; Vps35p, Vps10p, and Kex2p were immunoprecipitated from the fractions and subjected to SDSPAGE and fluorography. The autoradiogram shows the distribution of Vps35p in the gradient, and the graph shows the amounts of Vps10p and Kex2p in each fraction. Vps35p appears to associate with two discrete pools of membrane.
Vps10p Is Mislocalized to the Vacuole in vps29, vps30, and vps35 Mutants
The redistribution of Vps10p to the P13 pellet in vps29, vps30, and vps35 cells is indicative of Vps10p being delivered to the vacuole. However, it was possible that Vps10p could have been in one of the other organelles which fractionates with the P13 pellet, such as the plasma membrane. To determine if the vacuole was in fact the location of Vps10p in these mutant cells, indirect immunofluorescence experiments were undertaken. Previous immunofluorescence studies using a Myc-tagged version of Vps10p encoded on a centromeric plasmid showed a dispersed punctate staining pattern in diploid wild-type cells, consistent with a late-Golgi location of Vps10p (Cereghino et al., 1995). When the same procedure was undertaken with haploid wild-type cells, once again a punctate pattern was seen (Fig. 7). In contrast, in haploid Δvps29, Δvps30, and Δvps35 cells expressing the Myc-tagged version of Vps10p from a centromeric plasmid, clear vacuolar staining was observed (Fig. 7). This indicated that the shift of Vps10p from the P100 fraction to the P13 fraction in these cells is indicative of Vps10p being mislocalized to the vacuolar membrane. As a control, the staining pattern for Vps10p in pep12 (vps6) mutant haploid cells was also investigated. In these cells, as in wild-type cells, only a dispersed punctate pattern was seen (Fig. 7). This shows that vps mutants which do not cause Vps10p to shift to the P13 pellet in subcellular fractionation experiments also do not show a vacuolar staining pattern for Vps10p.
Figure 7.
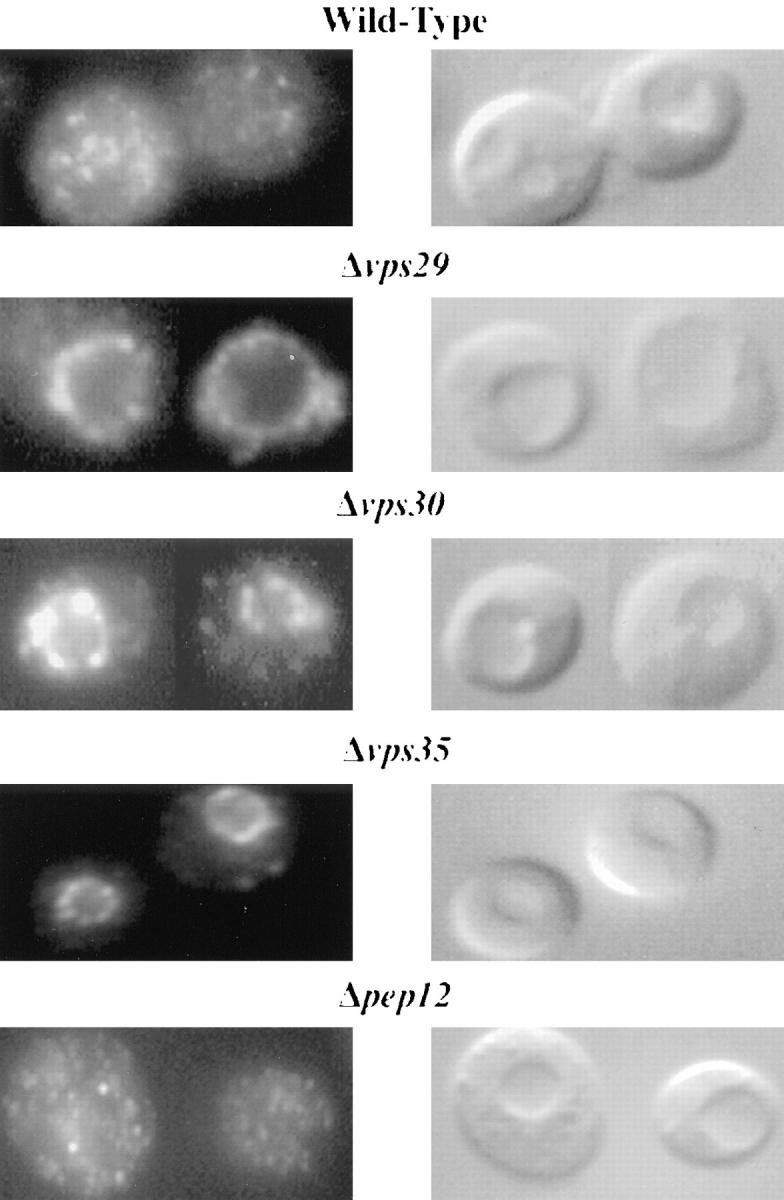
Vps10p is localized to the vacuole in vps29, vps30, and vps35 mutants. Using an epitope-tagged version of Vps10p expressed at centromeric levels, Vps10p protein was localized in wild-type cells, vps29, Δvps30 and Δvps35 cells, and also Δpep12/ vps6 cells. In wild-type cells the labeling pattern appeared punctate and characteristic of a Golgi and endosomal localization. In the vps29, Δvps30, and Δvps35 cells, Vps10p was localized to the vacuolar membrane. The redistribution of Vps10p to the vacuolar membrane is not a general phenomenon associated with disruption of the VPS pathway as in Δpep12/vps6 cells; Vps10p has a punctate distribution indistinguishable from wild-type cells.
Although the immunofluorescence experiment shows that Vps10p is localized to the vacuole in Δvps29, Δvps30, and Δvps35 cells, it is still possible that Vps10p reaches the vacuole by endocytosis from the plasma membrane. This possibility was tested using the sec1 mutant strain in which VPS35 was deleted in the same fashion used to create the strain EMY18. Sec1p is required for secretory vesicles to fuse with the plasma membrane and the allele present in the sec1 strain is temperature sensitive for function (Aalto et al., 1991). Thus, it is possible to block the arrival of proteins at the plasma membrane by elevating the temperature to 38°C which is nonpermissive for the sec1 allele. Vps10p was localized in the sec1 Δvps35 strain at both the permissive and nonpermissive temperature by subcellular fractionation. At both temperatures, Vps10p was localized to the P13 fraction (Fig. 8, lanes 1–6). If Vps10p was transported to the vacuole via the plasma membrane, it would be detected in the P100 fraction which contains small vesicles including secretory vesicles. However, as the data show, the distribution of Vps10p is unaffected by inactivation of Sec1p. Thus, it seems most likely that Vps10p is transported directly to the vacuole in Δvps35 mutants presumably through a prevacuolar compartment from which Vps10p would recycle back to the Golgi in wild-type cells.
Figure 8.

Vps10p is transported to the vacuole via the prevacuolar endosome in Δvps35 cells. The strains MSY3501 (sec1 Δvps35) and MSY3512 (pep12ts Δvps35) were converted to spheroplasts before pre-shifting to the indicated temperature for 15 min. The cells were then labeled for 15 min with [35S]methionine and chased for an additional 45 min at either 26°C or 38°C. After dounce homogenization, the lysed cells were subjected to differential centrifugation to separate small Golgi-enriched membranes (P100) from the larger vacuoles (P13). Vps10p was immunoprecipitated from the fractions. Transport of Vps10p to the P13 fraction does not require Sec1p function and therefore does not occur by arrival of Vps10p at the plasma membrane and subsequent endocytosis and delivery to the vacuole, but does require Pep12p function as Vps10p becomes trapped in a P100 membrane fraction upon inactivation of the endosomal t-SNARE Pep12p.
To test this hypothesis, Vps10p was localized in vps35 mutant cells which also contained a temperature conditional allele of the pep12 gene. Pep12p is a t-SNARE which has been localized to the prevacuolar endosome and is required for Golgi to endosome transport (Becherer et al., 1996; Cowles et al., 1997). Thus, inactivation of Pep12p would be predicted to block the arrival of Vps10p in a P13 membrane fraction, causing Vps10p to fractionate with the lighter P100 membranes. In Fig. 8 (lanes 7–12), the fractionation of Vps10p in a pep12tsΔvps35 strain is shown. At the permissive temperature (lanes 7–9), Vps10p is localized to the P13 membrane fraction that contains vacuolar membranes. However, inactivation of Pep12p by incubation at the nonpermissive temperature (lanes 10–12) results in a block in the transport of Vps10p from the lateGolgi to the vacuole, resulting in Vps10p fractionating with the lighter P100 membranes. This clearly demonstrates that transport of Vps10p to the vacuole in a vps35 mutant requires passage through a Pep12p-positive prevacuolar endosome.
Functional Endosomal t-SNARE Pep12p Is Required for Golgi to Endosome Delivery of Vps10p
In certain vps mutants, namely the class E mutants, the prevacuolar endosome becomes enlarged and acidic with active proteases contained within and is termed the “class E compartment” (Raymond et al., 1992; Reider et al., 1996; Piper et al., 1996). Vps10p traffics through this compartment in class E mutants and is proteolytically clipped with a half time of ∼30 min (Cereghino et al., 1995; Cooper and Stevens, 1996). The finding that Vps35p appears to be associated with membranes enriched in Kex2p as well as Vps10p (see Fig. 6) raised the possibility that Vps35p might also act at the late-Golgi, perhaps to affect the exit rate of Vps10p. Any effect that inactivation of Vps35p might have on the exit of Vps10p from the late-Golgi could manifest itself as an effect upon the kinetics of the clipping of Vps10p in a class E mutant. To address this question, a class E Δvps35 mutant was constructed using a null mutant of vps4 as a representative class E mutant (Babst et al., 1997). This mutant was transformed with plasmids containing either wild-type VPS35 or the vps35ts mutant gene. The clipping of Vps10p in these strains was investigated and is shown in Fig. 9 A. Cells were incubated for 15 min at 38°C to inactivate the temperature conditional allele of vps35 before labeling with [35S]methionine for 10 min, after which an excess of unlabeled methionine was added. Aliquots of cells were removed at 0 min of chase and also after 30 min and 60 min. After cell lysis, Vps10p was recovered by immunoprecipitation. As shown in Fig. 9 A, cells harboring the wild-type VPS35 allele (lanes 1–3) have ∼50% clipped Vps10p (denoted by the *) after a 30-min chase, a phenotype which is identical to class E mutant alone (Cereghino et al., 1995). Cells which contained the temperature conditional allele of vps35 (Fig. 9 A, lanes 4–6) showed no significant difference in the kinetics of the clipping of Vps10p, suggesting that inactivation of Vps35p does not affect the exit rate of Vps10p from the late-Golgi. By contrast, however, inactivation of Pep12p in a class E mutant completely prevents the clipping of Vps10p. In Fig. 9 B, cells containing a mutant allele of vps4 and also the temperature conditional allele of pep12 (Cowles et al., 1997) were pulsed for 10 min with [35S]methionine and chased at either 26°C or 38°C as described above. Vps10p was recovered from the aliquots of cells removed after the specified chase times. At permissive temperature, Vps10p is clipped with normal kinetics for a class E mutant; however, upon raising the temperature to 38°C and inactivation of Pep12p, Vps10p is stabilized and is not proteolytically clipped even after 60 min of chase. These data strongly suggest that pep12ts is epistatic to vps35 and that the vps35 mutation does not affect the exit of Vps10p from the Golgi in any way.
Figure 9.

Pep12p is epistatic to Vps35p. (A) The strain MSY 0435 (Δvps4Δvps35) harboring plasmids encoding either wild-type VPS35 (pGPY35) or the temperature conditional allele of vps35 (pMS35-4ts) was shifted to 38°C 15 min before labeling with [35S]methionine. After labeling for 10 min, excess unlabeled methionine was added and the cells were chased for the specified times. Aliquots of cells (2 OD equivalents) were removed at the specified times, the cells were lysed and Vps10p was recovered by immunoprecipitation. Vps10p was clipped (clipped Vps10p denoted by *) with a half-time of ∼30 min which is typical for a class E mutant. The clipping of Vps10p is unaffected by inactivation of Vps35p. (B) The strain MSY4012 (vps4Δpep12) harboring the plasmid encoding the temperature conditional allele of pep12 (pCBY9) was grown at 26oC, before being shifted to the specified temperature for 10 min before labeling. The cells were labeled and chased as described above at either 26oC or 38oC, and Vps10p was recovered from the lysates as described above. Vps10p was completely stabilized by inactivation of Pep12p, indicating that it never reached the class E compartment and hence that Pep12p is epistatic to Vps35p.
The observations that Vps10p was delivered to the vacuole in vps29, vps30, and vps35 mutants but remained stable was somewhat surprising, since previous studies had shown that overexpressed Vps10p arrived at the vacuole and was rapidly degraded (Cereghino et al., 1995). One possible explanation for these conflicting results is that the absence of CPY from the vacuole in these mutants makes the vacuole less competent for the degradation of Vps10p. The stability of overexpressed Vps10p was analyzed in strains producing either a catalytically inactive form (single amino acid change) of CPY (Bech and Breddam, 1989) or no CPY at all. Overexpressed Vps10p was found to be more stable in both of these strains when compared to a wildtype strain (data not shown). Since the carboxy terminus of Vps10p is found in the cytosol, it seems unlikely that vacuolar CPY can play a direct role in the degradation of Vps10p. It may be that CPY is involved in the activation of an unknown vacuolar endopeptidase which plays a role in the degradation of Vps10p. In contrast to Vps10p, Kex2p was degraded in a CPY-independent manner in the vps29, vps30, and vps35 mutants resulting in a greatly reduced half life for Kex2p (Table II).
Discussion
We previously reported the initial characterization of the S. cerevisiae protein Vps10p, the sorting receptor for the vacuolar hydrolase CPY (Marcusson et al., 1994; Cereghino et al., 1995). Mutations in VPS10 lead to a selective vacuolar missorting phenotype in which CPY is missorted and secreted from the cell while the majority of PrA is properly sorted and matured. In this paper and previously (Paravicini et al., 1992), we show that mutations in three other VPS genes (VPS29, VPS30, and VPS35) lead to the same selective CPY missorting phenotype (Fig. 1 A). A temperature conditional allele of vps35 missorts and secretes progressively more CPY with increasing incubation at the nonpermissive temperature. The relatively slow onset (∼15 min) of the CPY missorting phenotype (relative to either the vps34ts [Stack et al., 1995] or the pep12ts [Cowles et al., 1997]) suggests that either the Vps35 protein is inactivated slowly at the nonpermissive temperature, or that inactivation of Vps35p results in the gradual depletion of a factor(s) required for CPY sorting. Analysis of the other vps35ts alleles isolated at the same time indicated that they all exhibited a slow onset of CPY secretion, hence we favor the latter explanation.
Each of these mutants change the subcellular localization of the Vps10 sorting receptor. The receptor is shifted from a high speed pellet (P100) fraction which is enriched in Golgi, endosomal, and vesicular membranes, to a low speed pellet (P13) fraction which contains membranes from several organelles including the vacuole (Fig. 5 A). The shift of Vps10p from a Golgi/endosome-enriched membrane fraction to a low speed pellet fraction also occurs in the vps35ts mutant cells at the nonpermissive temperature, resulting in very little (∼20%) Vps10p remaining in the Golgi/endosome P100 fraction. Immunofluorescence experiments with a Myc-tagged version of Vps10p demonstrated that Vps10p was mislocalized to the vacuolar membrane in vps29, vps30, and vps35 mutants, but not in wildtype cells or other vps mutants that also were tested (Fig. 7). Furthermore, Vps10p appears to follow a direct route to the vacuole. It does not arrive at the vacuole via endocytosis from the plasma membrane as a double sec1 Δvps35 mutant failed to block Vps10p delivery; however, inactivation of a t-SNARE required for Golgi to endosome transport (Pep12p) did prevent the mislocalization of Vps10p to a P13 membrane fraction in a pep12ts Δvps35 double mutant (see Fig. 8). These data show that VPS29, VPS30, and VPS35 play a role in controlling the intracellular location of Vps10p and probably act at the prevacuolar endosome to direct the recycling of Vps10p to the Golgi (see Fig. 10).
Figure 10.
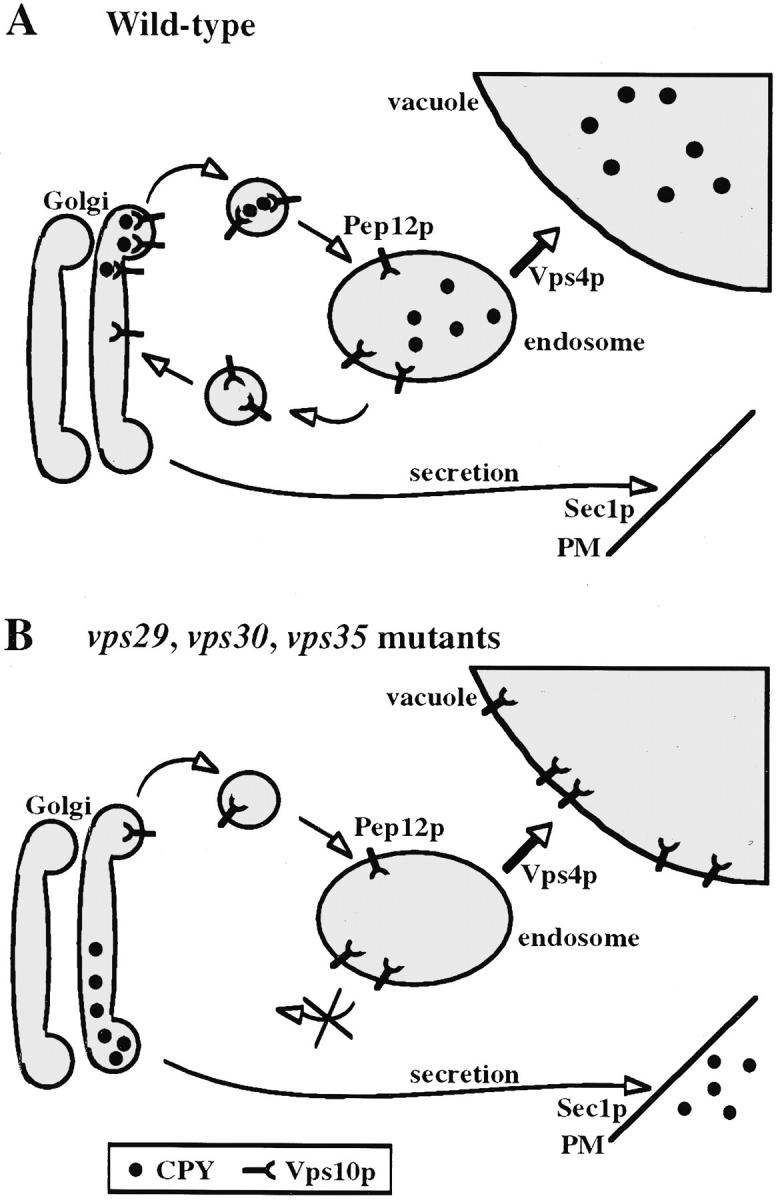
Proposed role of Vps29p, Vps30p, and Vps35p. (A) In wild-type cells, the VPS29, VPS30, and VPS35 gene products all play a role in the recycling of Vps10p and other late-Golgi proteins (e.g., Kex2p and DPAP A) from the endosome back to the TGN. Vps10p is efficiently sorted and retrieved from the prevacuolar endosomal compartment and returned to the late-Golgi. (B) In the absence of Vps29p, Vps30p, or Vps35p, Vps10p (and Kex2p and DPAP A) is transported by default to the vacuole and becomes limiting in the late-Golgi resulting in the secretion of p2CPY. Disruption of VPS35 does not block arrival of Vps10p in the class E compartment/endosome in a vps4 mutant; however, transport of Vps10p to the vacuole in a vps29, vps30, or vps35 mutant requires a functional endosomal t-SNARE, Pep12p, but does not require Sec1p-dependent arrival at the plasma membrane and subsequent endocytosis.
In mammalian cells, the MPRs sort lysosomal enzymes away from secreted proteins in the TGN. MPRs plus cargo are then concentrated in clathrin-coated vesicles and subsequently delivered to a prelysosomal endocytic compartment (Kornfeld, 1992). The pathway taken by the Vps10 receptor in the sorting of yeast vacuolar hydrolases is very similar to the MPRs. Vps10p binds vacuolar hydrolases in the late-Golgi/TGN (Graham and Emr, 1991) and transports them to the prevacuolar endosome in a process that is altered shortly after shifting a clathrin heavy chain (CHC) temperature sensitive mutant to the nonpermissive temperature, thus suggesting a role for CHC in this process (Seeger and Payne, 1992). In the endosome, the hydrolases must be released and the unoccupied Vps10 receptor is then recycled back to the Golgi for further rounds of sorting. The results of our analysis suggest that this recycling step is dependent on VPS29, VPS30, and VPS35. When any of these genes are mutated, Vps10p is not able to recycle back to the TGN and instead is mislocalized to the vacuole, presumably via default flow of membrane proteins in the yeast secretory pathway which leads to the vacuole (Roberts et al., 1992; Wilcox et al., 1992). With the recycling pathway inoperative, the Vps10 receptor becomes limiting for CPY sorting, causing CPY to be secreted from the cell by the default pathway for soluble proteins. It has been recently demonstrated that Vps10p is also responsible for sorting some PrA (Cooper and Stevens, 1996; Westphal et al., 1996); however, PrA can be delivered to the vacuole in a Vps10p-independent manner. This Vps10p-independent PrA sorting is very sensitive to media pH, (which presumably affects the pH of the lumen of intracellular organelles) as acidic media was required for efficient PrA sorting. It seems possible that PrA may be delivered to the vacuole by “piggybacking” on a membrane protein(s) destined for the vacuole. This interaction may be very sensitive to pH, and hence efficient PrA sorting requires low pH. In this way, PrA could arrive at the vacuole in a manner independent of a sorting receptor that requires efficient retrieval from the endosome, a process that would be defective in vps29, vps30, and vps35 mutants.
Our experiments suggest a role for the Vps29, Vps30, and Vps35 proteins not only for the trafficking of Vps10p, but also for other proteins that cycle between the TGN and the endosome, such as Kex2p. The subcellular location of Kex2p is also changed in vps29, vps30, and vps35 mutants resulting in a reduction in the half life of Kex2p (Table II). Consistent with this conclusion, a hybrid protein in which the cytoplasmic tail domain of Vps10p was replaced by the cytoplasmic tail of Kex2p resulted in efficient sorting of CPY to the vacuole when expressed in a Δvps10 mutant strain (Marcusson, E.G., and S.D. Emr, unpublished observations). However, this hybrid protein cannot suppress either Δvps10/Δvps29 or Δvps10/Δvps35 double mutants. This indicates that the Vps10 Kex2 hybrid protein can replace Vps10p, but its transport still requires Vps29p, Vps30p, and Vps35p function.
Vps35p is a peripheral membrane protein which probably associates with a transmembrane protein(s) via an ionic interaction as Vps35p can be removed from the membrane pellet fraction by washing with 1 M NaCl or 1% Triton X-100 (Paravicini et al., 1992). The subcellular fractionation pattern of Vps35p mimics that of Vps10p, both in wildtype cells and vps29 (and partially vps30) mutant cells. The distribution of Vps35p mimics that of Vps10p to the extent that sucrose gradient fractionation of a Golgi/endosome membrane-enriched fraction which was able to resolve Vps10p distribution into two discrete peaks indicated that Vps35p was associated with both these pools of Vps10p containing membranes. It appears therefore that the localization of Vps10p (along with proteins such as Kex2p and DPAP A) is the crucial factor in determining Vps35p localization.
What role might Vps35p, Vps30p, and Vps29p play in retrieval of Vps10p? One possibility is that these proteins act at the late-Golgi to restrain Vps10p and delay its exit in order to allow for cargo binding by Vps10p. Thus, in the absence of Vps35p, Vps30p, or Vps29p, Vps10p exits the late- Golgi before it has bound CPY and subsequently accumulates in the prevacuolar endosome resulting in saturation of the retrieval machinery. Vps10p would then become limiting in the late-Golgi and CPY would be secreted. While this possibility cannot be formally excluded, it seems unlikely because in a class E vps35ts double mutant, the kinetics of the clipping of Vps10p are unaltered by the inactivation of Vps35p (Fig. 9). Furthermore, inactivation of Pep12p in a class E pep12ts double mutant completely prevented clipping of Vps10p (Fig. 9). Thus, inactivation of Vps35p has no effect upon exit from the Golgi and arrival at the endosome/class E compartment, and PEP12 is epistatic to VPS35 which is consistent with a role for Vps35p in the recycling of Vps10p from the endosome back to the Golgi.
The data presented here are consistent with a model in which Vps35p, Vps30p, and Vps29p act in the retrieval of Vps10p from the prevacuolar endosome (see Fig. 10). The finding that the localization of Vps35p appears to be very significantly affected by the distribution of Vps10p raises the possibility that Vps35p interacts with the cytoplasmic tail of Vps10p (and other proteins such as Kex2p and dipeptidylaminopeptidase A [DPAP A]). One possibility is that Vps35p is required for cargo selection at the endosome during formation of recycling vesicles destined for the Golgi. Vps29p might act in a similar fashion, possibly in a complex with Vps35p and other proteins. As Vps30p is predominantly a soluble protein, of which only a small fraction is membrane associated, it seems likely that Vps30p acts at a slightly different step. It is interesting to note that in vps30 cells Kex2p has a longer half-life compared to the half-life of Kex2p in vps29 and vps35 cells (Table II), suggesting that it is not mislocalized to the vacuole as quickly as it is in vps29 and vps35 cells. Thus, significant amounts of Kex2p and possibly other proteins such as DPAP A will remain in the late-Golgi and endosomes. This could account for the reduced shift of Vps35p to the vacuolar fraction in vps30 mutants. Consistent with this, a vps35 mutant was recently identified by a completely different genetic approach designed to identify mutants that were unable to properly localize another late-Golgi protein, DPAP A (Nothwehr et al., 1996). DPAP A is a type II transmembrane protein which, like Vps10p and Kex2p, contains an aromatic motif which has been shown to be important for maintaining the protein in the Golgi/endosome pathway (Wilcox et al., 1992; Nothwehr and Stevens, 1994; Cereghino et al., 1995; Cooper and Stevens, 1996; Bryant and Stevens, 1996). This observation, combined with the data presented in this paper, indicates that Vps35p may play a general role in recycling transmembrane proteins (both type I and type II) back to the late Golgi apparatus, possibly by interaction with the aromatic residue-containing sorting signals present in these proteins. Consistent with this hypothesis is the finding that the localization of the type II resident Golgi membrane protein, Mnn1p, is not affected in vps29 mutants. Mnn1p does not have an aromatic residue-containing sorting signal in its cytoplasmic tail domain (Graham et al., 1994).
While the precise molecular mechanisms involved in Vps35p, Vps30p, and Vps29p function remain to be elucidated, it seems likely that these proteins act together to retrieve not only Vps10p, but also Kex2p and DPAP A from a prevacuolar endosome to the late-Golgi. Conservation between yeast and higher eukaryotes of the proteins involved in membrane trafficking has recently been shown to occur at virtually every membrane traffic step in the secretory and endocytic pathways (reviewed in Rothman, 1994; Schekman and Orci, 1996; Pfeffer, 1996). The fact that Vps35p, Vps30p, and Vps29p are also conserved in higher eukaryotes underscores their important roles in membrane trafficking, and it is likely that these proteins will have similar functions in all eukaryotes.
Acknowledgments
We would like to thank the members of the Emr lab for many helpful discussions. We are indebted to Peggy Mustol for her expert technical assistance. We would like to thank Klaus Breddam and Morten KiellandBrandt for supplying the plasmid, pAla-397-CPD-Y. We are also grateful to Steven Nothwehr, Tom Stevens, and Robert Fuller for the communication of unpublished results and for many useful discussions.
Abbreviations used in this paper
CPY
carboxypeptidase Y
DPAP
dipeptidylaminopeptidase A
MPR
mannose 6-phosphate receptor
PrA
proteinase A
Footnotes
This work was partially supported by a grant from the National Institutes of Health (NIH) to S.D. Emr (GM32703) and by an NIH Postdoctoral Fellowship to J.L. Cereghino (GM15638). M.N.J. Seaman is the recipient of a long term postdoctoral fellowship from the European Molecular Biology Organisation. S.D. Emr is supported as an investigator of the Howard Hughes Medical Institute.
Please address all correspondence to S.D. Emr, Division of Cellular and Molecular Medicine, Howard Hughes Medical Institute, University of California, San Diego, School of Medicine, La Jolla, CA 92093-0668. Fax: (619) 534-6414.
The current address of E.G. Marcusson is Isis Pharmaceuticals, Carlsbad Research Center, 2292 Faraday Avenue, Carlsbad, CA 92008.
References
- Aalto MK, Ruohonen L, Hosano K, Keranen S. Cloning and sequencing of the yeast Saccharomyces cerevisiae SEC1gene localized on chromosome IV. Yeast. 1991;7:643–650. doi: 10.1002/yea.320070613. [DOI] [PubMed] [Google Scholar]
- Altschul S, Gish W, Miller W, Lipman D. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Babst, M., T.K. Sato, L.M. Banta, and S.D. Emr. 1997 Endosomal transport in yeast requires a novel AAA-type ATPase, Vps4p. EMBO (Eur. Mol. Biol. Organ.) J. In press. [DOI] [PMC free article] [PubMed]
- Bankaitis VA, Johnson LM, Emr SD. Isolation of yeast mutants defective in protein targeting to the vacuole. Proc Natl Acad Sci USA. 1986;83:9075–9079. doi: 10.1073/pnas.83.23.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banta LM, Robinson JS, Klionsky DJ, Emr SD. Organelle assembly in yeast: characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. J Cell Biol. 1988;107:1369–1383. doi: 10.1083/jcb.107.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bech LM, Breddam K. Inactivation of carboxypeptidase Y by mutational removal of the putative essential histidyl residue. Carlsberg Res Commun. 1989;54:165–171. doi: 10.1007/BF02904470. [DOI] [PubMed] [Google Scholar]
- Becherer KA, Rieder SE, Emr SD, Jones EW. Novel syntaxin homologue Pep12p, required for the sorting of lumenal hydrolases to the lysosome-like vacuole in yeast. Mol Biol Cell. 1996;7:579–594. doi: 10.1091/mbc.7.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguski MS, Tolstoshev CM, Bassett D., Jr Gene discovery in dbEST [letter] Science (Wash DC) 1994;265:1993–1994. doi: 10.1126/science.8091218. [DOI] [PubMed] [Google Scholar]
- Bryant, N.J., and T.H. Stevens. 1996. Two separate signals act independently to localize a yeast late Golgi membrane protein through a combination of retrieval and retention. J. Cell Biol. In press. [DOI] [PMC free article] [PubMed]
- Canfield WM, Johnson KF, Ye RD, Gregory W, Kornfeld S. Localization of the signal for rapid internalization of the bovine cation-independent mannose 6-phosphate/insulin-like growth factor-II receptor to amino acids 24-29 of the cytoplasmic tail. J Biol Chem. 1991;266:5682–5688. [PubMed] [Google Scholar]
- Cereghino JL, Marcusson EG, Emr SD. The cytoplasmic tail domain of the vacuolar sorting receptor Vps10p and a subset of VPSgene products regulate receptor stability, function, and localization. Mol Biol Cell. 1995;6:1089–1102. doi: 10.1091/mbc.6.9.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Stevens TH. Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J Cell Biol. 1996;133:529–542. doi: 10.1083/jcb.133.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles, C.R., W.B. Snyder, C.G. Burd, and S.D. Emr. 1997. Novel Golgi to vacuole delivery pathway in yeast: identification of a sorting determinant and required transport component. EMBO (Eur. Mol. Biol. Organ.) J. In press. [DOI] [PMC free article] [PubMed]
- Deng WP, Nickoloff JA. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal Biochem. 1992;200:81–88. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- Elble R. A simple and efficient procedure for transformation of yeasts. Biotechniques. 1992;13:18–20. [PubMed] [Google Scholar]
- Gaynor EC, te Heesen S, Graham TR, Aebi M, Emr SD. Signalmediated retrieval of a membrane protein from the Golgi to the ER in yeast. J Cell Biol. 1994;127:653–665. doi: 10.1083/jcb.127.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman JN, Conibear E, Pearse BMF. Specificity of binding of clathrin adaptors to signals on the mannose-6-phosphate/insulin-like growth factor II receptor. EMBO (Eur Mol Biol Organ) J. 1989;8:1041–1047. doi: 10.1002/j.1460-2075.1989.tb03471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TR, Emr SD. Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined in a yeast sec18(NSF) mutant. J Cell Biol. 1991;114:207–218. doi: 10.1083/jcb.114.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TR, Seeger M, Payne GS, MacKay VL, Emr SD. Clathrin-dependent localization of α1,3 mannosyltransferase to the Golgi complex of Saccharomyces cerevisiae. . J Cell Biol. 1994;127:667–678. doi: 10.1083/jcb.127.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coliwith plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Horazdovsky BF, Busch GR, Emr SD. VPS21encodes a rab5like GTP binding protein that is required for the sorting of yeast vacuolar proteins. EMBO (Eur Mol Biol Organ) J. 1994;13:1297–1309. doi: 10.1002/j.1460-2075.1994.tb06382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukada Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KF, Chan W, Kornfeld S. Cation-dependent mannose 6-phosphate receptor contains two internalization signals in its cytoplasmic domain. Proc Natl Acad Sci USA. 1990;87:10010–10014. doi: 10.1073/pnas.87.24.10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EW. Proteinase mutants of Saccharomyces cerevisiae. . Genetics. 1977;85:23–33. doi: 10.1093/genetics/85.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Banta LM, Emr SD. Intracellular sorting and processing of a yeast vacuolar hydrolase: proteinase A propeptide contains vacuolar targeting information. Mol Cell Biol. 1988;8:2105–2116. doi: 10.1128/mcb.8.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu Rev Biochem. 1992;61:307–330. doi: 10.1146/annurev.bi.61.070192.001515. [DOI] [PubMed] [Google Scholar]
- Langford CJ, Gallwitz D. Evidence for an intron-contained sequence required for the splicing of yeast RNA polymerase II transcripts. Cell. 1983;33:519–527. doi: 10.1016/0092-8674(83)90433-6. [DOI] [PubMed] [Google Scholar]
- Langford CJ, Klinz FJ, Donath C, Gallwitz D. Point mutations identify the conserved, intron-contained TACTAAC box as an essential splicing signal sequence in yeast. Cell. 1984;36:645–653. doi: 10.1016/0092-8674(84)90344-1. [DOI] [PubMed] [Google Scholar]
- Lobel P, Fujimoto K, Ye RD, Griffiths G, Kornfeld S. Mutations in the cytoplasmic domain of the 275 kD mannose 6-phosphate receptor differentially alter lysosomal enzyme sorting and endocytosis. Cell. 1989;57:787–796. doi: 10.1016/0092-8674(89)90793-9. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science (Wash DC) 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Marcusson EG, Horazdovsky BF, Cereghino JL, Gharakhanian E, Emr SD. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10gene. Cell. 1994;77:579–586. doi: 10.1016/0092-8674(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Mauxion F, Le Borgne R, Munier-Lehmann H, Hoflack B. A casein kinase II phosphorylation site in the cytoplasmic domain of the cation-dependent mannose 6-phosphate receptor determines the high affinity interaction of the AP-1 Golgi assembly proteins with membranes. J Biol Chem. 1996;271:2171–2178. doi: 10.1074/jbc.271.4.2171. [DOI] [PubMed] [Google Scholar]
- Miller, J. 1972. Experiments in Molecular Genetics. In Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 431–435.
- Nothwehr SF, Bryant NJ, Stevens TH. The newly identified yeast GRDgenes are required for retention of late–Golgi membrane proteins. Mol Cell Biol. 1996;16:2700–2707. doi: 10.1128/mcb.16.6.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothwehr SF, Stevens TH. Sorting of membrane proteins in the yeast secretory pathway. J Biol Chem. 1994;269:10185–10188. [PubMed] [Google Scholar]
- Paravicini G, Horazdovsky BF, Emr SD. Alternative pathways for the sorting of soluble vacuolar proteins in yeast: a vps35null mutant missorts and secretes only a subset of vacuolar hydrolases. Mol Biol Cell. 1992;3:415–427. doi: 10.1091/mbc.3.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR. Transport vesicle docking: SNAREs and associates. Annu Rev Cell Dev Biol. 1996;12:441–461. doi: 10.1146/annurev.cellbio.12.1.441. [DOI] [PubMed] [Google Scholar]
- Piper RC, Cooper AA, Yang H, Stevens TH. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae. . J Cell Biol. 1996;131:603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CK, Howald-Stevenson I, Vater CA, Stevens TH. Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vpsmutants. Mol Biol Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reider SE, Banta LM, Kohrer K, McCaffery JM, Emr SD. Multilamellar endosome-like compartment accumulates in the yeast vps28vacuolar protein sorting mutant. Mol Biol Cell. 1996;7:985–999. doi: 10.1091/mbc.7.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CJ, Nothwehr SF, Stevens TH. Membrane protein sorting in the yeast secretory pathway: evidence that the vacuole may be the default compartment. J Cell Biol. 1992;119:69–83. doi: 10.1083/jcb.119.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta LM, Emr SD. Protein sorting in Saccharomyces cerevisiae:isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature (Lond) 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Rothman JH, Hunter CP, Valls LA, Stevens TH. Overproduction-induced mislocalization of a yeast vacuolar protein allows isolation of its structural gene. Proc Natl Acad Sci USA. 1986;83:3248–3252. doi: 10.1073/pnas.83.10.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. 2nd Edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 545 pp.
- Schekman R, Orci L. Coat proteins and vesicle budding. Science (Wash DC) 1996;271:1526–1532. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Seeger M, Payne GS. A role for clathrin in the sorting of vacuolar proteins in the Golgi complex of yeast. EMBO (Eur Mol Biol Organ) J. 1992;11:2811–2818. doi: 10.1002/j.1460-2075.1992.tb05348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and L.W. Lawrence. 1979. Methods in Yeast Genetics: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 1–98.
- Stack JH, DeWald DB, Takegawa K, Emr SD. Vesicle-mediated protein transport: regulatory interactions between Vps15 protein kinase and the Vps34 PtdIns 3-kinase essential for protein sorting to the vacuole in yeast. J Cell Biol. 1995;129:321–334. doi: 10.1083/jcb.129.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida TA, Huyer G, Emr SD. Yeast vacuolar proenzymes are sorted in the late Golgi complex and transported to the vacuole via a prevacuolar late endosome-like compartment. J Cell Biol. 1993;121:1245–1256. doi: 10.1083/jcb.121.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgelstein D, Gillespie D. Preparative and analytical purification of DNA from agarose. Proc Natl Acad Sci USA. 1979;76:615–619. doi: 10.1073/pnas.76.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal V, Marcusson EG, Winther JR, Emr SD, van den Hazel HB. Multiple pathways for vacuolar sorting of yeast proteinase A. J Biol Chem. 1996;271:11865–11870. doi: 10.1074/jbc.271.20.11865. [DOI] [PubMed] [Google Scholar]
- Wilcox CA, Redding K, Wright R, Fuller RS. Mutation of a tyrosine localization signal in the cytosolic tail of yeast Kex2 protease disrupts Golgi retention and results in default transport to the vacuole. Mol Biol Cell. 1992;3:1353–1371. doi: 10.1091/mbc.3.12.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]