ARF6 Targets Recycling Vesicles to the Plasma Membrane: Insights from an Ultrastructural Investigation (original) (raw)
Abstract
We have shown previously that the ADP- ribosylation factor (ARF)-6 GTPase localizes to the plasma membrane and intracellular endosomal compartments. Expression of ARF6 mutants perturbs endosomal trafficking and the morphology of the peripheral membrane system. However, another study on the distribution of ARF6 in subcellular fractions of Chinese hamster ovary (CHO) cells suggested that ARF6 did not localize to endosomes labeled after 10 min of horseradish peroxidase (HRP) uptake, but instead was uniquely localized to the plasma membrane, and that its reported endosomal localization may have been a result of overexpression. Here we demonstrate that at the lowest detectable levels of protein expression by cryoimmunogold electron microscopy, ARF6 localized predominantly to an intracellular compartment at the pericentriolar region of the cell. The ARF6-labeled vesicles were partially accessible to HRP only on prolonged exposure to the endocytic tracer but did not localize to early endocytic structures that labeled with HRP shortly after uptake. Furthermore, we have shown that the ARF6-containing intracellular compartment partially colocalized with transferrin receptors and cellubrevin and morphologically resembled the recycling endocytic compartment previously described in CHO cells. HRP labeling in cells expressing ARF6(Q67L), a GTP-bound mutant of ARF6, was restricted to small peripheral vesicles, whereas the mutant protein was enriched on plasma membrane invaginations. On the other hand, expression of ARF6(T27N), a mutant of ARF6 defective in GDP binding, resulted in an accumulation of perinuclear ARF6-positive vesicles that partially colocalized with HRP on prolonged exposure to the tracer. Taken together, our findings suggest that ARF activation is required for the targeted delivery of ARF6-positive, recycling endosomal vesicles to the plasma membrane.
Vesicular transport along the endocytic and biosynthetic pathways is essential for the biogenesis and maintenance of subcellular organelle integrity and the trafficking of proteins and lipids within the cell and between the cell and its extracellular environment. Along the endocytic pathway, a variety of macromolecules are internalized via clathrin-dependent or -independent mechanisms into early endocytic vesicles (reviewed in Sandvig and Van Deurs, 1994; Gruenberg and Maxfield, 1995; Lamaze and Schmid, 1996; Mellman, 1996), from which they are delivered to tubulovesicular sorting endosomes (Geuze et al., 1983, 1987; Griffiths et al., 1989; Gruenberg et al., 1989). At this junction, lysosomal proteins are sorted from those proteins that recycle via an iterative process in which lysosome-targeted ligands accumulate in the sorting compartment and are then delivered to lysosomes (Stoorvogel et al., 1991; Dunn et al., 1989; van Deurs, 1993). On the other hand, recycling markers are delivered to a pericentriolar recycling compartment before they are transported back to the plasma membrane (Hopkins and Trowbridge, 1983; Yashimoro et al., 1984; Hopkins et al., 1994; Marsh et al., 1995). Although considerable efforts have been made in delineating the general features of the endocytic pathway, the mechanisms that regulate these transport pathways are still incompletely understood and have been the subject of active research.
The vectorial transfer of membrane between intracellular membrane-bound endocytic organelles involves a series of tightly regulated membrane budding and fusion events. The regulatory machinery includes several cytosolic and membrane-bound GTP-binding proteins (or GTPases) that function as molecular switches cycling between their GTP- and GDP-bound states. Members of the Rab GTPase family of the Ras superfamily of low molecular mass GTPases, have been implicated in the control of various steps along the endocytic pathway. Rab4 appears to play a role in recycling of ligands from the sorting endosome, bypassing the recycling endosome (van der Sluijs et al., 1992; Daro et al., 1996), Rab5 has been localized to, and promotes fusion of, early endosomes (Bucci et al., 1992; Barbieri et al., 1994), whereas Rab11 has been thought to regulate transport between sorting and recycling endosomes (Ulrich et al., 1996).
In addition to the Rab GTPases, it is well documented that the ADP-ribosylation factor (ARF)1 GTPases are required for maintaining the integrity of organelle structure and intracellular transport. As with the GTPases of the Rab family, it is likely that the different ARF proteins may control membrane trafficking from the organelles to which they are localized. The ARF proteins were originally identified as cofactors required for cholera toxin catalyzed ADP ribosylation of Gs (Kahn and Gilman, 1986). The ARF family currently includes five structurally homologous proteins (ARFs 1, 3, 4, 5, and 6) whose structure is well conserved across the species (Tsuchiya et al., 1991). The best-characterized ARF protein, ARF1, is localized to the Golgi complex (Peters et al., 1995). It is required for the binding of coat proteins, COP I and the AP1–clathrin complex, to Golgi membranes, a process critical for maintaining the integrity of Golgi structure and for vesicle transport along the biosynthetic pathway (reviewed in Rothman, 1994; Bowman and Kahn, 1995; Donaldson and Klausner, 1994; Moss and Vaughan, 1995). The ARFs have been shown to activate phospholipase D (Brown et al., 1993; Cockroft et al., 1994; Massenburg et al., 1994). It has been speculated that ARF-mediated effects on lipid metabolism may relate to its function in maintaining organelle structure and transport, and although some studies have implied that a connection between these functions may indeed exist (Kahn et al., 1995; Liscovitch and Cantley, 1995; Ktistakis, 1996), the exact mechanism involved remains unclear.
Our previous studies have shown that ARF6, the least conserved ARF protein, is localized to the cell periphery and cycles between the plasma membrane and an intracellular endosomal compartment depending on its nucleotide status (D'Souza-Schorey et al., 1995; Peters et al., 1995). Unlike ARF1, the cellular distribution of ARF6 appears to be insensitive to brefeldin A (Peters et al., 1995). Expression of mutant forms of ARF6 defective in GTP binding and hydrolysis in CHO cells has a pronounced effect on receptor-mediated endocytic trafficking. A mutant form of ARF6, defective in GTP hydrolysis and hence locked in its GTP-bound conformation, ARF6(Q67L), is localized almost exclusively to the plasma membrane and its expression decreases the rate of uptake of transferrin (D'Souza-Schorey et al., 1995). Conversely, the dominant negative and GDP-bound mutant of ARF6, ARF6(T27N), is localized to intracellular compartments (D'Souza-Schorey et al., 1995; Peters et al., 1995), and its expression results in an intracellular accumulation of transferrin receptors and an inhibition of recycling of ligands to the cell surface. These alterations in endocytic trafficking are accompanied by gross alterations in the morphology of the peripheral membrane system (Peters et al., 1995). Expression of wild-type ARF6 and its GTPase-defective mutant, ARF6(Q67L), results in the elaboration of the plasma membrane, characterized by the formation of extensive membrane invaginations. On the other hand, expression of the ARF6(T27N) mutant results in the massive accumulation of intracellular vesicles that exhibit a novel coat structure on the cytoplasmic surface. In addition to eliciting a redistribution of peripheral membrane, the expression of ARF6(Q67L) has also been shown to induce a remodeling of the actin cytoskeleton (Radhakrishna et al., 1996; D'Souza-Schorey et al., 1997). Whether these two cellular activities of ARF6 are codependent processes or are controlled by divergent signaling pathways remains to be established. In a separate study, Cavenagh et al. (1996) investigated the distribution of ARF6 in CHO cells by analysis of subcellular fractions generated by free flow electrophoresis, wherein the early endosome fraction was defined by its accessibility to HRP. They reported that ARF6 localized exclusively to the plasma membrane and that the previously reported intracellular localization of ARF6 may have been a consequence of high levels of protein expression (Cavenagh et al., 1996). In a more recent study, endogenous ARF6 has been shown to cofractionate with chromaffin granules in adrenal chromaffin cells (Galas et al., 1997). Triggering of exocytosis resulted in the dissociation of ARF6 from the granule fraction onto an unidentified membrane compartment. Based on the above, an understanding of the precise compartments to which ARF6 is localized will be pivotal to elucidate its biological role.
In this study, by use of the high resolution technique of cryoimmunogold electron microscopy, we have undertaken a further characterization of the membrane alterations induced by expression of wild-type ARF6 and its GTP- and GDP-bound mutants in CHO cells. These studies have revealed that at low levels of protein expression, wild-type ARF6 localizes predominantly to intracellular compartments around the pericentriolar region of the cell juxtaposed to the TGN. This compartment colocalized with transferrin receptors and with cellubrevin, a member of the v-SNARE family of proteins that has been shown to play a role in membrane recycling (Galli et al., 1994). Furthermore, we have analyzed the distribution of ARF6 and its mutants relative to the endocytic tracer, HRP. We show that a small fraction of wild-type ARF6 and its mutant defective in GTP-binding, ARF6(T27N), colocalize with HRP only after prolonged exposure to the tracer. ARF6(Q67L), the GTP-bound mutant, localizes to the plasma membrane and HRP labeling in cells expressing this mutant was restricted to small peripheral vesicles. Our findings indicate that ARF6 is localized to a recycling compartment and may serve to regulate the outward flow of recycling membrane.
Materials and Methods
Cells, Reagents, and Antibodies
TRVb-1 cells, a CHO cell line that overexpresses transferrin receptors (McGraw et al., 1987), were grown and maintained in Hams F12 medium (GIBCO BRL, Gaithersburg, MD), supplemented with 5% fetal bovine serum, penicillin/streptomycin, and 100 μg/ml G418. HRP-FITC was obtained from Sigma Chemical Co. (St. Louis, MO) and rhodamine-transferrin was kindly provided by Richard Roberts (Washington University, St. Louis, MO). For the studies described, the following antibodies were used: affinity-purified polyclonal antisera directed against a COOH-terminal ARF6 peptide (D'Souza-Schorey et al., 1995); anti-HRP mouse monoclonal antibody (Sigma Chemical Co.); antibiotin mouse monoclonal antibody (Rockland, Inc., Gilbertsville, PA); anti–lysosome–associated membrane protein (LAMP)-3 (CD63) mouse monoclonal antibody (CLB, Amsterdam, The Netherlands); antitransferrin receptor H68.4 (generous gift from Ian Trowbridge, The Salk Institute, San Diego, CA; White et al., 1992); anti γ-adaptin mouse monoclonal antibody, 100/3 (Sigma Chemical Co.); anti–LAMP-1 (kindly provided by Minori Fukuda, La Jolla Cancer Research Institute, La Jolla, CA); anti–mannose-6-phosphate receptor rabbit polyclonal antisera (kindly provided by Kurt Von Figura, University of Göttingen, Göttingen, Germany); rabbit antisera against β-COP (generous gift from Jennifer Lippincott-Schwartz, National Institutes of Health, Bethesda, MD); and affinity-purified anti-cellubrevin rabbit antisera, MC16 (kindly provided by Thierry Galli, Curie Institute, Paris, France; Galli et al., 1994).
Expression of ARF6 Proteins
ARF6 and its mutants were expressed in TRVb-1 cells using the Sindbis virus as an expression vector. Recombinant virus encoding ARF6 and its mutant derivatives were generated as previously described (D'Souza-Schorey et al., 1995). A virus titer of 50 plaque-forming units/cell was used for cell infection. Adsorption was conducted at room temperature for 1 h in 250 μl PBS containing 1% FBS. The infection mixtures were replaced by 2 ml Hams F12 medium containing 3% FBS and cells were maintained at 37°C. Experiments were carried out 4–5 h after infection. The above procedure allowed us to examine ARF6 localization at varying levels of protein expression. Six batches of infected cells were analyzed for localization of each ARF protein.
Cell Surface Biotinylation
Cell monolayers in 35-mm dishes were cooled to 4°C and rinsed with 2 ml of ice-cold PBS containing 5 mM glucose. The cells were then incubated with 1 mg/ml sulpho-NHS-biotin (Pierce Chemical Co., Rockford, IL) for 30 min at 4°C. After biotinylation, the cells were rinsed with PBS and fixed with 2% glutaraldehyde in phosphate buffer and processed for cryosectioning and immunogold electron microscopy. Cells were labeled with antibodies directed against ARF6 and biotin. For controls, cells infected with vector virus were incubated with HRP (4 mg/ml) for 30 min in serum-free media, followed by washes with PBS containing 0.2% BSA and 5 mM glucose. Cells were chilled, biotinylated as described above, and then processed for cryoimmunogold electron microscopy and labeling with antibodies against HRP and biotin.
Expression of ARF6(T27N) Using Vaccinia Virus
The ARF6(T27N)-pTM3 plasmid was generated by PCR amplification of hemagglutinin (HA)-tagged ARF6(T27N) cDNA (Peters et al., 1995) with sequences encoding NcoI and BglII restriction sites at the NH2- and COOH-terminal ends, respectively, that was then cloned into the vaccinia expression vector pTM3 (Fuerst et al., 1988). The resulting plasmid was stably integrated into vaccinia virus by homologous recombination as previously described (Anusbel et al., 1987). HEK 293 cells were then coinfected with recombinant viruses that encode for ARF6(T27N) and T7 RNA polymerase (virus 7.3) for 9 h as previously described (Fuerst et al., 1988). Cells were fixed and processed for cryosectioning and immunogold labeling as described below.
Analysis of HRP Distribution
TRVb-1 cells were infected with either vector virus alone or with recombinant virus encoding wild-type ARF6, ARF6(Q67L), or ARF6(T27N). At 4.5 h after infection, cells were incubated with 5 mg/ml HRP for 5 or 60 min in Hams F12 serum-free media containing 0.2% BSA. The cells were quickly rinsed with media followed by fixation with 2% glutaraldehyde in 0.1 M phosphate buffer. Cells were scraped gently from the dish and collected in 0.1 M phosphate buffer containing 0.2% glutaraldehyde and then processed for embedding and cryosectioning (see below). Ultrathin cryosections were double labeled with antibodies directed against ARF6 and HRP as described below.
Cryoimmunogold Electron Microscopy Procedures
Cell monolayers on petri dishes were fixed at room temperature in 2% paraformaldehyde in 0.1 M phosphate buffer for 12 h, or with 2% glutaraldehyde for 1 h. Cells were scraped and collected into 0.1 M phosphate buffer containing 0.2% of paraformaldehyde and then processed for ultrathin cryosectioning as previously described (Peters et al., 1991). 45-nm cryosections were cut at −125°C using diamond knives (Drukker Cuijk, The Netherlands) in an Utracryomicrotome (Leica Aktiengesellschaft, Vienna, Austria) and transferred with a mixture of sucrose and cellulose onto formvar-coated copper grids (Liou et al., 1996). The grids were placed on 35-mm petri dishes containing 2% gelatin, and immunolabeling of sections was done as previously described (Raposo et al., 1997). In brief, sections were incubated with primary antibody for 45 min, washed, and then incubated with protein A–gold (EM Lab., Utrecht University) for 30 min. For double labeling experiments labeling with a second antibody followed by protein A–gold was carried out consecutively after labeling with the first antibody. For colocalization experiments to rule out the possibility that the absence of apparent colocalization could be due to the first antibody preventing the binding of the second antibody, we compared the density of labeling of these antibodies in single immunolabeling procedure first. No significant blocking effect was observed. Labeled sections were viewed with a JOEL 1010 electron microscope at 80 kV.
Quantitation of Immunogold Labeling
Quantitative evaluations were carried out on ultrathin cryosections of the ARF6 wt, mutants, and controls. Cross sections of 25 cells that were labeled for anti-ARF6–10-nm gold (Table I) or with ARF6–10 nm gold and HRP–5 nm gold (Table II) were randomly chosen and photographed and the negatives were printed at a final magnification of 25,000×. Gold particles located at no more than 20 nm from a visible membrane structure were assigned to the structure listed. ARF6 label was assigned to the plasma membrane/invaginations and to pericentriolar vesicles and tubules (Table I). For the assessment of size the 200-nm bar on the electron micrograph was used as an arbitrary line. The criteria for colocalization was as follows: (a) at least one small and one large gold particle should be present within the vesicle and (b) colabeling artifacts of small and large particles that were <10 nm from each other were seldom seen and excluded from quantitation.
Table I.
Quantitation of the Intracellular Distribution of Wild-type and Mutant ARF6 Proteins
| Cells transfected with | Plasma membrane/ invaginations | Pericentriolar vesicles and tubules |
|---|---|---|
| ARF6 (low expression) | 4 | 38 |
| ARF6 (high expression) | 125 | 66 |
| ARF6(Q67L) | 159 | 34 |
| ARF6(T27N) | 18 | 163 |
Table II.
Quantitation of HRP Label in Pericentriolar ARF6-positive Vesicles Relative to Other Small Vesicles (<200-nm Diameter)
| Cells transfected with | Small vesicles (<200 nm) | ARF6-positive vesicles | ||
|---|---|---|---|---|
| Minutes post uptake | ||||
| 5 | 60 | 5 | 60 | |
| ARF6 | 34 | 173 | 0 | 12 |
| ARF6(Q67L) | 21 | 112 | 0 | 0 |
| ARF6(T27N) | 59 | 189 | 0 | 28 |
| Vector | 89 | 342 | − | − |
Results
Localization of ARF6 in CHO Cells
In a previous study on the subcellular distribution of epitope-tagged ARF6 at the ultrastructural level in HEK 293 fibroblasts, ARF6 was shown to be present on the plasma membrane and on intracellular compartments (Peters et al., 1995). However, high levels of protein were expressed using the calcium phosphate method of transfection, followed by analysis of expressed proteins 48 h after transfection. In this study, we have investigated the distribution of wild-type ARF6 in CHO cells using the Sindbis virus expression system, which allows for protein detection at relatively shorter intervals after virus infection. Therefore, we could analyze the subcellular distribution of untagged ARF6 at varying levels of protein expression, by the technique of cryoimmunogold electron microscopy using antibodies directed against ARF6. At relatively high levels of ARF6 expression, implicit by higher amounts of gold labeling, the plasma membrane exhibited tortuous invaginations resembling membrane alterations seen in cells expressing the GTPase-defective mutant, ARF6(Q67L). 50–60% of gold label was seen along these invaginations (Fig. 1 A, Table I). Gold label was also seen on intracellular vesicles, some of which were peripherally distributed while most were localized around the pericentriolar region of the cell (see below). About 20% of the ARF6 label was found scattered throughout the cytosol. Interestingly, in cells expressing ARF6 at the lowest detectable level, gold label was seen predominantly on vesicles around the centriole, juxtaposed to the TGN (Fig. 1 B, Table I), whereas <15% of the label was detected on the plasma membrane. Also, the plasma membrane of these cells was relatively unperturbed compared with the surface of cells expressing relatively higher levels of ARF6. Thus, ARF6 localization in CHO cells is predominantly intracellular; with increased expression, the cell phenotype resembles that observed in cells expressing the GTPase-defective mutant, most likely due to sequestration of factors required for GTP hydrolysis by overexpressed protein. The ARF6-labeled intracellular vesicles were small and ranged in diameter from 60 to 200 nm and resembled the recycling endosomal compartment previously described in CHO cells (Yashimoro et al., 1984; Johnson et al., 1996). This compartment is distinct from the TGN, late endosomes, or lysosomes. These findings were unexpected since a previous study that used subcellular fractionation procedures, had found endogenous ARF6 to be localized exclusively to the plasma membrane of CHO cells (Cavenagh et al., 1996). This point is addressed further below.
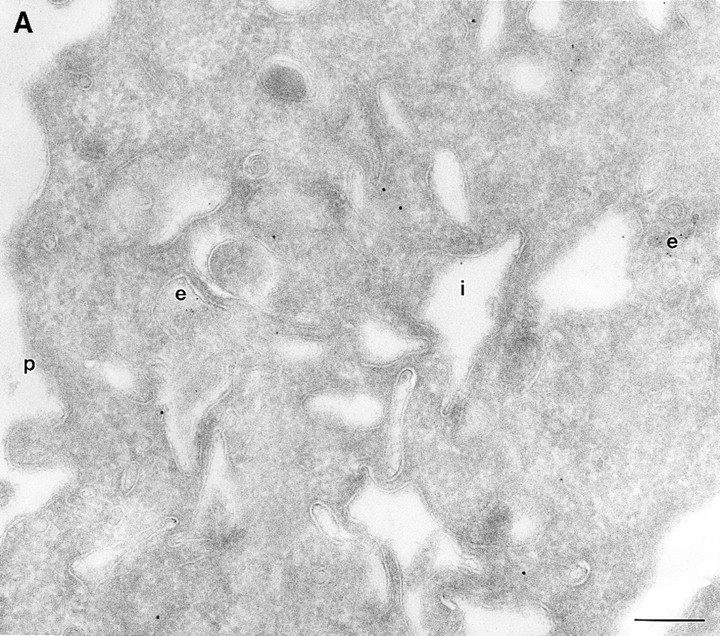
Figure 1.
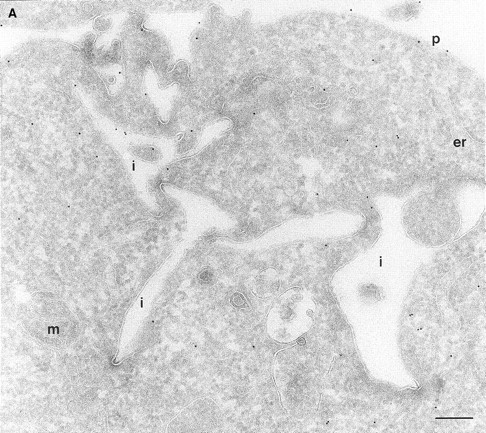
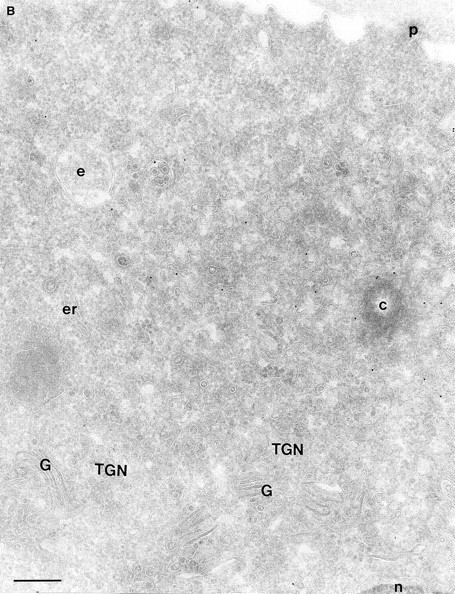
(A and B) Localization of wild-type ARF6. CHO cells expressing wild-type ARF6 were fixed and processed for ultrathin cryosections. Sections were labeled with anti-ARF6 polyclonal antibody followed by protein A–gold (10 nm). (A) ARF6 localizes to the plasma membrane and intracellular vesicles. (B) At relatively lower levels of expression, ARF6 localizes predominantly to intracellular vesicles that are situated near the TGN. i, invaginations; p, plasma membrane; er, endoplasmic reticulum; m, mitochondria; G, Golgi complex; TGN, _trans_-Golgi network; e, endosome; c, centriole; n, nucleus. Bar, 200 nm.
Ultrastructural Characterization of Membrane Rearrangements Induced by Expression of ARF6:GTP and ARF6:GDP Mutants
On analysis of the distribution of the ARF6(Q67L), the GTP-bound mutant, consistent with previous findings in HEK 293 cells, we observed that the mutant protein localized almost exclusively to discrete sites on the plasma membrane of CHO cells, in regions that exhibited numerous membrane folds that lacked clathrin-coated pits and to sac-like structures (see Table I). To determine whether these sac-like structures were cross-sections through plasma membrane folds or were internalized vesicular compartments, we tested for their accessibility to cell surface biotin. For these experiments, cells expressing ARF6(Q67L) were treated with sulpho-NHS-biotin, a biotinylating agent, at 4°C, fixed, and then double labeled with antibodies against ARF6 and biotin. Biotin couples to the entire accessible surface of the fibroblasts. In control cells, labeling for biotin was observed on the plasma membrane and in some invaginations along the cell periphery (Fig. 2 A). In cells expressing ARF6(Q67L), the membrane folds and sac-like structures that labeled positively for ARF6, also labeled with antibiotin antibodies (Fig. 2 B). This accessibility to surface biotinylation suggests that these structures were indeed sections through plasma membrane folds/invaginations and hence continuous with the extracellular environment.
Figure 2.
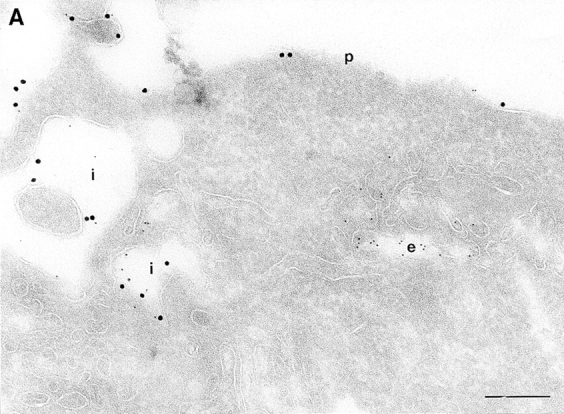
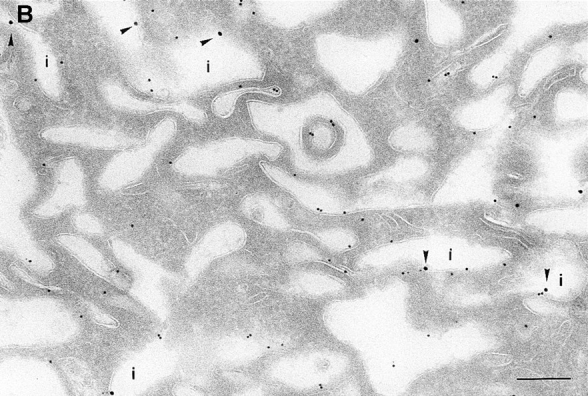
(A and B) Biotinylation of the surface of cells expressing ARF6(Q67L). Cells were either infected with vector virus and incubated with HRP for 10 min (A) or infected with recombinant virus encoding ARF6(Q67L) (B), and then incubated with sulfo-NHS-biotin and processed for immunolabeling. Sections were labeled first with an antibiotin antibody followed by protein A–gold (15 nm), and then with an antibody against HRP followed by protein A–gold (5 nm, A), or with antibody against ARF6 followed by protein A–gold (10 nm, B). (A) Only the plasma membrane and cross-sections across membrane invaginations were labeled with biotin. Early endocytic structures labeled with HRP were inaccessible to biotin. (B) In ARF6(Q67L)- expressing cells, plasma membrane invaginations were accessible to biotin (arrowheads) and thus are in continuity with the extracellular milieu. i, invaginations; p, plasma membrane; e, endosomes. Bar, 200 nm.
In cells expressing the GTP binding–defective mutant, ARF6(T27N), ARF6-positive vesicles were observed around the pericentriolar region of the cell whereas the plasma membrane exhibited little gold label (Fig. 3, Table I). Notably, these vesicles did not exhibit the prominent coat structure along their cytoplasmic face, as previously observed by Peters et al. (1995) in HEK 293 cells (see below). Further, the ARF6(T27N) vesicles in CHO cells were less densely packed and were present as much smaller clusters as compared with the grape-like clusters seen in HEK 293 cells. These differences may reflect differences in cell types. The ARF6(T27N) compartment was inaccessible to surface biotinylation clearly indicating that it was intracellular. Morphologically, it resembled an amplified perinuclear endocytic compartment observed in cells expressing wild-type protein.
Figure 3.
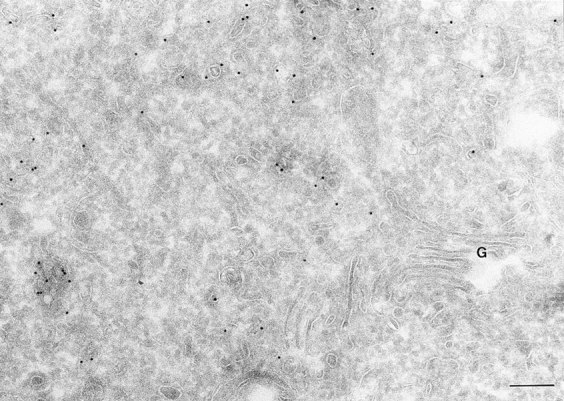
Localization of ARF6(T27N). Cells expressing ARF6 (T27N) were processed and labeled as described for Fig. 1. The ARF6 mutant localizes predominantly to clusters of vesicles juxtaposed to the TGN at the perinuclear region of the cell. G, Golgi. Bar, 200 nm.
Characterization of the ARF6 Intracellular Compartment
Double immunogold labeling of cells expressing wild-type ARF6, with antibodies directed against ARF6 and the transferrin receptor (Tfn-R), revealed that much of the ARF6 and Tfn-R label was either colocalized or found on vesicles that were either docked or in close proximity of each other at the pericentriolar region of the cell (Fig. 4 A). This compartment appeared as a compact fenestrated network of vesicles and tubules that ranged from 60 to 200 nm in diameter. The intracellular ARF6 compartment was amplified in cells expressing the ARF6(T27N) mutant and as seen in wild-type expressing cells, colocalization of ARF6 and Tfn-Rs, was also seen in cells expressing the ARF6 mutant (Fig. 4 A, inset). Approximately 90% of the Tfn-Rs were intracellular in cells expressing ARF6(T27N). When analyzed in HEK 293 cells, most of the Tfn-Rs were found in ARF6-containing vesicles, although in these cells, the ARF6/Tfn-R–containing vesicles exhibited an electron-dense coat structure along their cytoplasmic face (Fig. 4 B). Interestingly, Tfn-R–positive compartments that were negative for ARF6 and vice versa were also observed in cells expressing the wild-type and mutant ARF6 proteins, suggesting that ARF6 may not be present on Tfn-R–containing compartments throughout the Tfn-R cycle. Also, the existence of ARF6-positive vesicles that do not label with the Tfn-R implies that ARF6 may have a broader subcellular distribution than the Tfn-R–labeled recycling compartment. A statistical analysis of the number of vesicles that labeled with both proteins, as opposed to those that labeled with each of them, was not feasible because of the low numbers of gold particles per vesicle.
Figure 4.
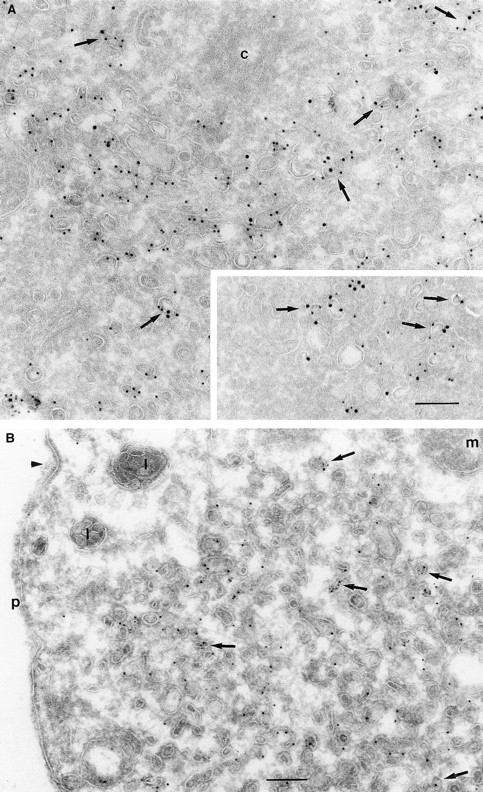
(A and B) Transferrin receptors localize to ARF6 and ARF6(T27N) vesicles. CHO cells (A) or HEK 293 cells (B) expressing ARF6 (A) and ARF6(T27N) (A, inset and B) were fixed and processed for cryoimmunogold labeling. Sections were labeled first with the anti-ARF6 antibody followed by protein A–gold (A, 15 nm; B, 10 nm) and then with the anti–Tfn-R antibody followed by protein A–gold (A, 10 nm; B, 5 nm). ARF6 and Tfn-R labeling is seen on the same vesicle (arrows) or on vesicles in near proximity of each other. Arrowhead indicates a clathrin-coated pit. c, centriole; p, plasma membrane. Bar, 200 nm.
Since we had previously shown that expression of ARF6(T27N) decreases the efficiency of Tfn recycling, we were prompted to examine whether proteins implicated in Tfn recycling colocalized with the ARF6 compartment. Cellubrevin, a member of the synaptobrevin/VAMP family of SNARES, is present on Tfn-R–containing vesicles in fibroblasts (McMohan et al., 1993) and tetanus toxin- mediated cleavage of cellubrevin impairs the recycling of Tfn-R–containing vesicles in CHO cells (Galli et al., 1994). Fig. 5 A shows the distribution of Tfn-Rs and cellubrevin in the pericentriolar endosomal compartment. We compared the distribution of ARF6 with that of cellubrevin. For these studies, cells expressing either wild-type ARF6 or ARF6(T27N) were double labeled with antibodies directed against ARF6 and cellubrevin. We found that cellubrevin-labeled, ARF6-positive vesicles in cells expressing wild-type ARF6 (Fig. 5 B) or ARF6(T27N; data not shown) and most of the colocalization was concentrated at the pericentriolar region of the cell. Double labeling for cation-dependent mannose-6-phosphate receptor (MPR; Fig 5 B) a late endosome marker, and LAMP-1 (not shown) and LAMP-3 (Fig. 5 D), markers for lysosomes, revealed little or no labeling of any of these markers in ARF6-positive vesicles. Furthermore, the ARF6-positive vesicles did not colocalize with γ-adaptin and other Golgi and endoplasmic reticulum markers (data not shown). Taken together, these findings indicate that intracellular ARF6 localizes to the perinuclear endosomal recycling compartment in CHO cells.
Figure 5.
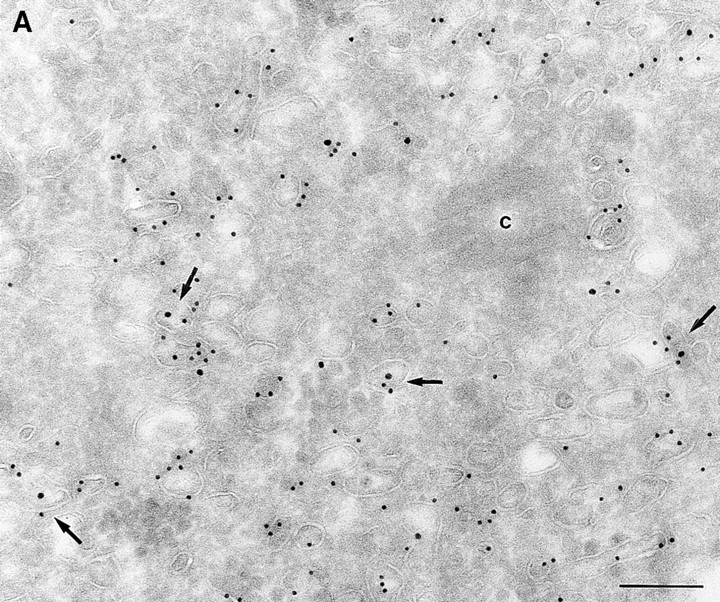
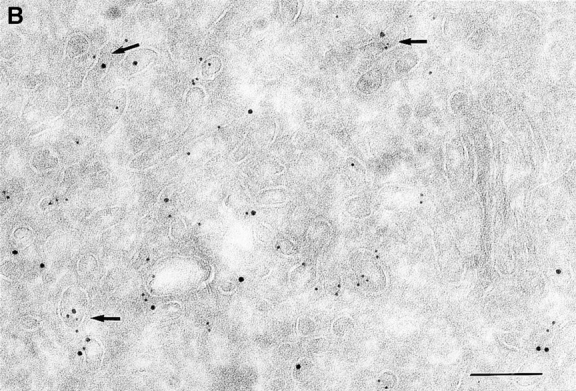
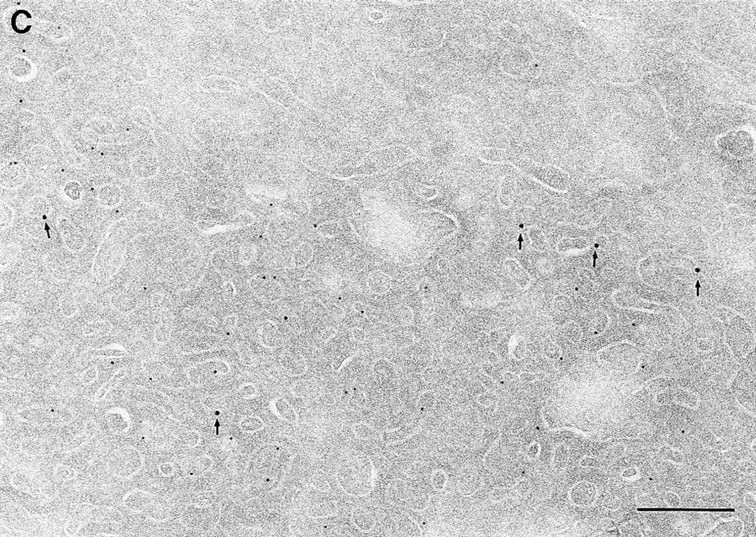
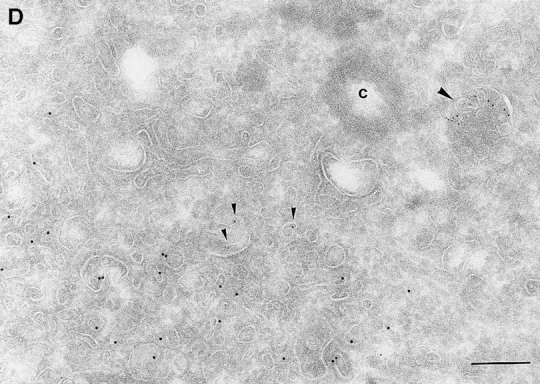
ARF6 colocalizes with cellubrevin but not with MPR or Lamp. CHO cells expressing wild-type ARF6 were doubled labeled with an anticellubrevin antibody (15 nm gold), followed by anti–Tfn-R antibody (A, 10 nm), or with an anti-ARF6 antibody (5 nm gold) followed by an anticellubrevin antibody (B, 10 nm gold). Arrows show clear examples of labeling for cellubrevin on ARF6-containing vesicles. HEK-293 cells expressing ARF6 (T27N) were double labeled with antibodies against ARF6 (5 nm gold) followed by labeling with antisera against MPR (C, 10 nm gold), or with anti-ARF6 antibody (10 nm gold) followed by anti–Lamp-3 antibody (D, 5 nm gold). MPR and Lamp-3 labeling was done in 293 cells due to low cross-reactivity in CHO cells. c, centriole. Bar, 200 nm.
Comparative Intracellular Distribution of ARF6 and Endocytosed HRP
We have compared the distribution of ARF6 and its mutants, relative to that of the endocytic tracer, HRP. HRP has been extensively used as a morphological and biochemical marker in endocytosis assays. Swanson et al. (1987) have shown that HRP is taken up by clathrin- dependent endocytosis in activated peritoneal macrophages. In BHK fibroblasts, most of the internalized HRP was shown to be delivered to lysosomes, while a small proportion recycled back to the cell surface (Griffiths et al., 1989). To analyze the distribution of endocytosed HRP, CHO cells were incubated with serum-free media containing HRP, quickly rinsed, fixed, and then processed for immunogold labeling with antibodies against HRP. With 5 min of HRP uptake, numerous early endocytic structures along the cell periphery that varied from tubular elements to large vesicles and that ranged from 50 to 400 nm in diameter were loaded with HRP (data not shown). HRP labeling was also detected in clathrin-coated vesicles, indicating that in CHO cells HRP was being internalized via clathrin-dependent in addition to clathrin-independent pathways. With 60 min of continuous HRP uptake, gold labeling became apparent in large vacuoles (<1 μM in diameter), some of which contained internal membrane and resembled multivesicular late endosomal and lysosomal compartments (Fig. 6). In addition, smaller vesicles in the perinuclear region that were ∼50–60 nm in diameter were also labeled for HRP (Fig. 6). These latter vesicles may be recycling endocytic vesicles that regurgitate HRP back to the extracellular environment. Analysis by confocal immunofluorescence microscopy of the distribution of Tfn-Rs relative to fluorescein-conjugated HRP revealed that only a subpopulation of these makers (⩽15%) was colocalized at the perinuclear compartment (data not shown).
Figure 6.
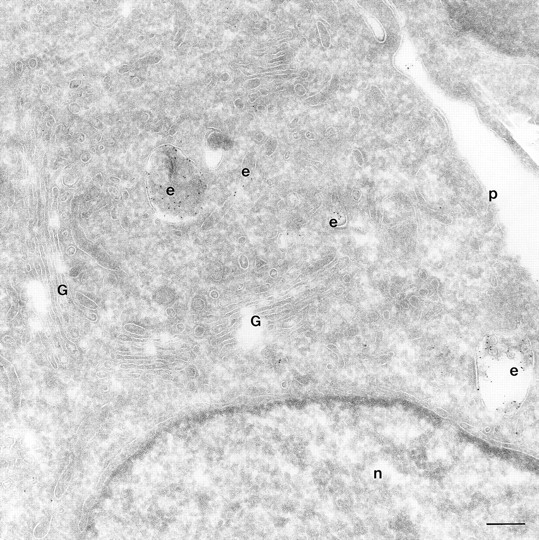
HRP distribution in CHO cells. Cells mock infected with the vector virus, were incubated with HRP for 60 min, fixed and processed for ultrathin cryosections. Sections were labeled with an anti-HRP antibody followed by protein A conjugated to gold particles (5 nm). The figure shows HRP labeling in the _trans_-Golgi region of the cell on small and multivesicular endocytic structures (e). Bar, 200 nm.
To determine the subcellular distribution of ARF6 relative to HRP under conditions of both short and more prolonged exposure to the tracer, cells expressing either wild-type ARF6 or its mutants defective in either GTP binding or hydrolysis were incubated with HRP for 5 or 60 min, fixed, processed for cryoimmunoelectron microscopy, and then labeled with antibodies directed against ARF6 and HRP. In cells expressing wild-type ARF6, no colocalization between these markers was observed with 5 min of HRP uptake (Table II), and ARF6-positive vesicles were clearly distinct from the numerous early endocytic structures that labeled with HRP. This strongly suggests that ARF6-positive vesicles were not early endosomes. On more prolonged exposure to HRP however, <10% of ARF6-positive vesicles were also labeled with HRP (Fig. 7). In cells expressing ARF6(Q67L), the GTPase-defective mutant, HRP label appeared to be significantly lower and exhibited <50% of gold labeling compared with control cells, thus indicating that the amount of HRP internalized into these cells was significantly reduced. This is similar to what was observed previously for the internalization of transferrin in CHO cells on expression of ARF6(Q67L; D'Souza-Schorey et al., 1995). The reduced efficiency of endocytosis in these cells may most likely be due to the dramatic alterations at the plasma membrane induced on expression of the GTPase-defective mutant. Labeling for HRP was restricted to small 80–100-nm vesicles at the periphery (Fig. 8 A), and on occasion, some label was also seen “entrapped” in membrane invaginations, but in 95% of these cells the ARF6-positive plasma membrane structures were devoid of HRP labeling. In cells expressing the GTP binding–defective mutant, ARF6(T27N), similar to what was observed with wild-type ARF6, no colocalization between the ARF6-positive and HRP-positive vesicles was observed with a short 5-min exposure to HRP (see Table II). However, after 60 min of HRP uptake, ∼10–15% of ARF6-positive vesicles that accumulate at the pericentriolar region of the cell also labeled for HRP (Fig. 8 B). Vesicles that label for ARF6(T27N), but not HRP, may be preformed vesicles or vesicles that are not accessible to HRP. These findings indicate that at some step after internalization, most likely at the recycling endosome, a small proportion of HRP has access to ARF6-positive vesicles. Based on the these observations and the findings described above on the colocalization of ARF6 with Tfn-Rs and cellubrevin, we propose that ARF6 localizes to and regulates the transport of recycling vesicles at the pericentriolar region of CHO cells to the plasma membrane.
Figure 7.
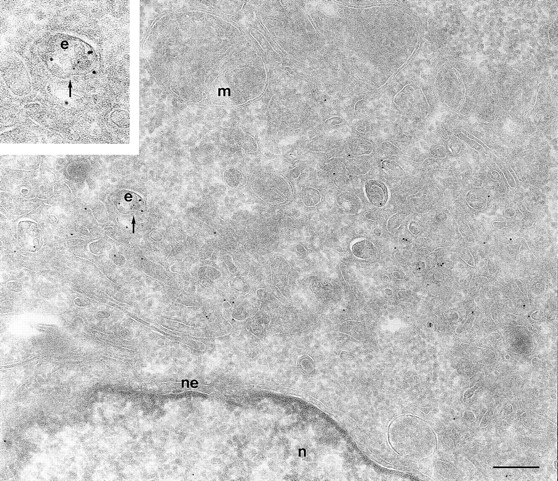
ARF6 colocalize with HRP only on prolonged exposure to the tracer. Cells expressing wild-type ARF6 were incubated for 60 min with HRP, fixed, and then processed for cryoimmunogold labeling. Samples were labeled with an anti-ARF6 polyclonal antibody followed by protein A–gold particles (10 nm) and then with an anti-HRP antibody followed by protein A–gold particles (5 nm). With prolonged exposure to HRP some colocalization (⩽10%) with ARF6-positive vesicles was observed (arrow, inset at higher magnification). e, endosomes; m, mitochondria; n, nucleus; ne, nuclear envelope; p, plasma membrane. Bar, 200 nm.
Figure 8.
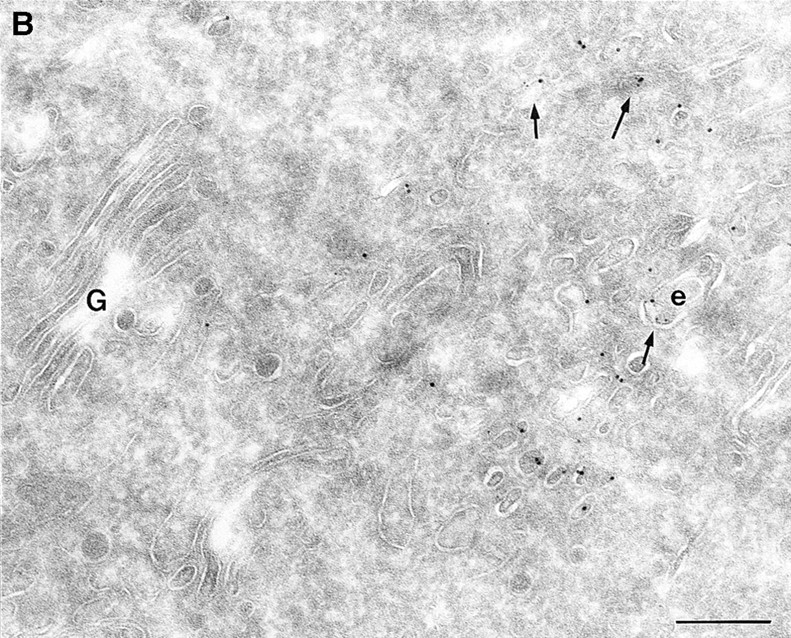
(A and B) HRP distribution in cells expressing ARF6(Q67L) and ARF6(T27N). Cells expressing ARF6(Q67L) (A) and ARF6(T27N) (B) were incubated with HRP for 60 min, fixed, and then processed for ultrathin cryosections. Sections were first labeled with anti-ARF6 antibody followed by protein A–conjugated gold particles (10 nm) and then with an anti-HRP antibody followed by protein A–gold particles (5 nm). (A) Typically, in ARF6 (Q67L)-expressing cells, HRP labeling was restricted to small vesicles at the periphery (e). Membrane invaginations were observed even at very low levels of ARF6(Q67L) expression, implicit by low numbers of gold particles on the membrane. (B) A small population of ARF6(T27N)-positive vesicles were also labeled with HRP (arrows). i, invaginations; e, endosomes; p, plasma membrane. Bar, 200 nm.
Discussion
In this study we have undertaken a characterization of membrane rearrangements induced on expression of wild-type ARF6 and its mutant derivatives defective in either GTP binding [ARF6(T27N)] or hydrolysis [ARF6(Q67L)]. Previously, we have shown that ARF6 localizes to the plasma membrane and intracellular compartments (D'Souza-Schorey et al., 1995; Peters et al., 1995). Now we show that at relatively low levels of expression, ARF6 in CHO cells, is localized almost exclusively to an intracellular pericentriolar compartment. This compartment is morphologically reminiscent of a recycling endosomal compartment that has been previously described (Yashimoro et al., 1984; Griffiths et al., 1989; Connolly et al., 1994; Ghosh and Maxfield, 1995). The recycling compartment appears as a collection of vesicles and tubules at the pericentriolar region of the cell and is distinct from the larger sorting endosomes and the Golgi complex and is enriched in recycling receptors such as the transferrin and LDL receptor. The transfer of recycling membrane from the pericentriolar region to the plasma membrane is likely to involve the formation of fusogenic recycling intermediates, although these have not been characterized. The colocalization of ARF6 with Tfn-Rs in vesicles and tubules in the pericentriolar region suggests that most of the intracellular ARF6 resides in the recycling endosomal compartment. The recycling compartment has also been characterized as a tubular network located around the pericentriolar region of other cell types including AtT20 cells and HEp2 cells (Hopkins et al., 1990; Tooze and Hollinshead, 1991). Whether such a compartment serves as a passive vehicle for the transport of molecules to the cell surface, or whether there may be other functions associated with the recycling compartment has been a subject of investigation. The isolation of a CHO mutant cell line (12-4) in which the exit of receptors from the recycling compartment is delayed and that of bulk membrane is normal, suggests that the recycling compartment may have a sorting function (Presley et al., 1993). Furthermore, there is mounting evidence for communication between the TGN and the recycling endosomal compartment (Geuze and Morre, 1991; Futter, 1995; Leitinger et al., 1995; Sariola et al., 1995; Ulrich, 1996).
Our findings on the subcellular distribution of ARF6 in CHO cells are in marked contrast to observations made by Cavenagh et al. (1996), who reported that ARF6 was localized exclusively to the plasma membrane of CHO cells. In their study, early endosomes were identified by endocytosis of HRP for 10 min. We have compared the intracellular distribution of ARF6 relative to that of HRP. Interestingly, when we determined the distribution of HRP after a short 5-min exposure to the ligand and that of wild-type ARF6, no colocalization was observed indicating that ARF6 was not present on newly endocytosed vesicles. With a more prolonged exposure however, HRP does appear to have access to only a small proportion of ARF6-positive vesicles around the pericentriolar region of the cell that likely represent recycling vesicles that are destined toward the plasma membrane. Based on our findings, it is unlikely that ARF6-positive compartments would be loaded with HRP under the experimental conditions used by Cavenagh et al. (1996), and even if it did, the fraction of ARF6-positive vesicles that contain HRP is so minor that it may have escaped detection by Western blot analysis. Furthermore, the latter study used sucrose density gradients and free flow electrophoresis for separation of endosomes from Golgi complex, ER, and plasma membrane fractions. It is possible that recycling endocytic vesicles that fuse with the plasma membrane may cofractionate with the plasma membrane fraction, when using a separation procedure based on the electrophoretic mobility of membranes.
The colocalization of ARF6 with Tfn-Rs and cellubrevin at the pericentriolar region, strongly supports the contention that ARF6 localizes to the recycling endosomal compartment. Cellubrevin has been localized to Tfn-containing vesicles in fibroblasts (McMohan et al., 1993). In addition, tetanus toxin cleavage of cellubrevin decreases the rate of Tfn release in CHO cells (Galli et al., 1994), a phenotype shared by the dominant negative mutant of ARF6, ARF6 (T27N). The presence of peripherally distributed Tfn-R– and cellubrevin-positive compartments that are devoid of the ARF6 label may likely represent Tfn-R endosomes on the inward leg of the Tfn cycle. We also attempted to compare the distribution of Rab4 and Rab11 with ARF6; unfortunately we were not able to localize endogenous Rab proteins. It would be interesting to determine whether ARF6 would function downstream of Rab 11 in the endosomal recycling pathway. Also of interest are the striking differences in the morphological characteristics of the ARF6 (T27N) compartment in the different cell types examined. The distinct “coat” structure seen on the cytoplasmic phase of the ARF6(T27N)-containing vesicle in HEK 293 cells was not as apparent in CHO cells. Why such differences exist between cell types is unclear at present. The ARF6(T27N)-coated vesicles do not label with antibodies directed against clathrin or β-COP (Peters et al., 1995). The presence of 100-nm nonclathrin-coated buds on Tfn-R–positive endosomes also have been described in A431 cells (Stoorvogel et al., 1996). The recycling of coated vesicles and its subsequent fusion with the plasma membrane would require “uncoating,” a process that may be triggered by activation/nucleotide exchange of ARF6. In this regard, studies in sec7 mutant Saccharomyces cerevisiae cells have indicated that in individual ARFs may perform distinct functions in vesicular coat dynamics, namely coat recruitment (budding) and vesicle uncoating, which precede vesicle docking and fusion (Deitz et al., 1996).
More recently, ARF6 has been shown to cofractionate along with chromaffin granules in adrenal chromaffin cells and has been implicated in regulated exocytosis (Galas et al., 1997), although a detailed morphological characterization of ARF6 in these cells has not been reported. Hence, it appears that the subcellular localization of ARF6 may be cell type specific. Therefore, the question that arises is what is the underlying function of ARF6 in various cell types. It is possible that ARF6 serves to regulate the outward flow of membrane traffic and thereby modulate the shape and structural organization of the plasma membrane. In CHO cells, using quantitative fluorescence microscopy it has been demonstrated that C6-NBD-SM (_N-_[N- (7-nitro-2,1,3-benzoxadiolol-4)-ε-aminihexanoyl] sphingosyl phosphorycholine), a bulk plasma membrane marker, transits the endocytic system in a manner that is kinetically and morphologically identical to that of recycling receptors (Mayor et al., 1993). Thus, in these cells, ARF6 may also serve to regulate the exit of bulk membrane. Conceivably, the rates of such a process would be dramatically altered during cellular processes that would require a remodeling of the plasma membrane such as diapedesis, cell spreading, and metastasis.
Based on our observations, it is tempting to speculate how ARF6 could control membrane recycling via its nucleotide cycle (Fig. 9). We propose that nucleotide exchange on ARF6 drives the uncoating/transport and subsequent fusion of ARF6-positive intracellular recycling vesicles with the plasma membrane. At the surface, the hydrolysis of bound GTP would allow either (a) the internalization of membrane-bound ARF6 via the endocytic pathway or (b) the release of ARF6 from the membrane into the cytosol. In the latter case, cytosolic ARF6:GDP would have to be recruited onto the recycling endosomal compartment and with subsequent nucleotide exchange on ARF6, another cycle follows. Although we did not observe any colocalization of ARF6 with HRP loaded early endocytic structures, at this point we cannot rule out the possibility that ARF6 could indeed localize to incoming vesicles that are not accessible to HRP and that later on by the process of endosome fusion become HRP and Tfn-R positive.
Figure 9.

Model for the ARF6:GTP/GDP cycle in CHO cells. Nucleotide exchange on ARF6 drives the transport and subsequent fusion of recycling vesicles with the plasma membrane. Hydrolysis of bound GTP at the cell surface allows the internalization of ARF6 via membrane vesicles inaccessible to HRP or the release of ARF6 from the membrane into the cytosol. Cytosolic ARF6 would have to be recruited back onto the recycling compartment (R.C.) for another nucleotide cycle to ensue.
The model described above explains the phenotypes observed by expression of ARF6 mutants. On expression of ARF6(T27N), the mutant defective in nucleotide exchange, an accumulation of vesicles at the perinuclear region of the cell and an inhibition of recycling of ligands to the cell surface was observed (D'Souza-Schorey et al., 1995). On expression of ARF6(Q67L), the mutant defective in GTP hydrolysis, an accumulation of membrane at the cell surface was observed along with the depletion of intracellular endosomal compartments (Peters et al., 1995; this study). The lack of coated vesicle formation along the ARF6- induced membrane invaginations may explain, in part, the decrease in efficiency of clathrin-dependent endocytosis observed in these cells. Reconstitution of transferrin recycling in streptolysin O–permeabilized CHO cells has shown that fusion of recycling endosomal vesicles with the plasma membrane does not require GTP hydrolysis, is brefeldin A insensitive, and is stimulated by GTPγs at suboptimal concentrations of cytosol (Martys et al., 1996). Furthermore, it has been reported that Tfn-R–positive recycling vesicles in the perinuclear region of the cell do not exhibit a tubular morphology on treatment with the drug brefeldin A, as do the early endosomal compartments from which they are derived (Daro et al., 1996). These reports are consistent with our observations that ARF6 distribution is brefeldin A insensitive (Peters et al., 1995) and that its redistribution to the plasma membrane does not require GTP hydrolysis. The stimulation of vesicle fusion with the plasma membrane by GTPγS, besides endosomal membrane recycling, is also characteristic of regulated secretion (Gomperts et al., 1990). The lack of requirement of GTP hydrolysis for fusion of endosomal recycling vesicles and of secretory vesicles during regulated secretion with the plasma membrane distinguishes these processes from constitutive exocytosis from the TGN to the cell surface since the latter is inhibited by GTPγS (Miller and Moore, 1991). It is possible that ARF6 could promote the generation of fusogenic lipids at the plasma membrane. ARF6 had been shown to stimulate phospholipase D (PLD) activity in vitro (Massenburg et al., 1995). PA a product of PLD metabolism stimulates the synthesis of phosphatidyl inositol 4,5,bisphosphate (PIP2; Loyens and Anderson, 1996). Thus, via its effects on phospholipid metabolism, the maintenance of ARF6 in the GTP state would result in the accumulation of charged lipids such as PIP2 at the cell periphery. PIP2 has also been shown to stimulate actin polymerization; indeed actin polymerization at the cell surface is induced on expression of ARF6(Q67L), the GTP-bound mutant of ARF6 (Radhakrishna et al., 1996; D'Souza-Schorey et al., 1997). We are currently establishing in vitro assays to investigate these possibilities.
It is clear from our studies that ARF6 localizes to an intracellular tubulovesicular membrane recycling compartment in CHO cells. Furthermore, our findings indicate that the ability of ARF6 to cycle between its GTP- and GDP-bound conformation is critical for maintaining the integrity of peripheral membrane organization and trafficking.
Acknowledgments
This work was supported in part by grants from the Dutch Cancer Society (RUU 95-1098) to P.J. Peters, the Leukemia Research Foundation to C. D'Souza-Schorey, and the National Institutes of Health to P.D. Stahl. C. D'Souza-Schorey is a Leukemia Society of America Special Fellow.
Abbreviations used in this paper
ARF
ADP-ribosylation factor
Tfn-R
transferrin receptor
HA
hemagglutinin
MPR
cation-dependent mannose-6-phosphate receptor
LAMP
lysosome-associated membrane protein
PLD
phospholipase D
PIP2
phosphatidylinositol 4,5, bisphosphate
SNARE
soluble NSF attachment protein (SNAP) receptor
VAMP
vesicle-associated membrane protein
Footnotes
We sincerely thank Hans Geuze and Willem Stoorvogel for critical comments on the manuscript and for insightful and helpful discussions during these studies. We also thank, Judith Klumpermann, Viola Oorschot, and Janice Griffiths for helpful suggestions with labeling procedures; Elizabeth Wolffe for help with generation of recombinant vaccinia virus; Guangpu Li for helpful discussions; Jeanette Leusen for reading the manuscript; and Tom van Rijn, George Posthuma, Mauritz Niekerk, and Rene Schriwanek for excellent technical assistance with photography and handling of EM micrographs.
References
- Anusbel, F.M., R. Brent, R. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1987. Current Protocols in Molecular Biology. John Wiley and Sons, New York. 16.16–16.17.
- Barbieri MA, Li G, Colombo MI, Stahl PD. Rab5 and early acting endosomal GTPase, supports endosome fusion without GTP hydrolysis. J Biol Chem. 1994;269:18720–18724. [PubMed] [Google Scholar]
- Bowman AL, Kahn RA. ARFs: policeman of membrane protein traffic. Trends Biochem Sci. 1995;20:147–150. doi: 10.1016/s0968-0004(00)88991-4. [DOI] [PubMed] [Google Scholar]
- Brown HA, Gutowski S, Moomaw CR, Slaughter C, Sternweiss PC. ADP-ribosylation factor, a small GTP-dependent regulatory protein stimulates phospholipase D activity. Cell. 1993;75:1137–1144. doi: 10.1016/0092-8674(93)90323-i. [DOI] [PubMed] [Google Scholar]
- Bucci C, Parton R, Mather IM, Stunnenberg H, Simon K, Hoflack B, Zerial M. The small GTPase Rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–728. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- Cavenagh MM, Whitney JA, Carroll K, Zhang C-J, Bowman AL, Rosenwald AG, Mellman I, Kahn RA. Intracellular distribution of ARF proteins in mammalian cells. J Biol Chem. 1996;271:21767–21774. doi: 10.1074/jbc.271.36.21767. [DOI] [PubMed] [Google Scholar]
- Cockroft S, Thomas GMH, Fensome A, Geny B, Cunningham E, Gout I, Hiles I, Totty NF, Truong O, Hsuan JJ. Phospholipase D: a downstream effector of ARF in granulocytes. Science. 1994;263:523–526. doi: 10.1126/science.8290961. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Futter C, Gibson A, Hopkins CR, Cutler DF. Transport into and out of the Golgi complex studied by transfecting cells with cDNAs encoding horseradish peroxidase. J Cell Biol. 1994;127:641–652. doi: 10.1083/jcb.127.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Li G, Colombo MI, Stahl PD. A regulatory role for ARF6 in receptor-mediated endocytosis. Science. 1995;267:1175–1178. doi: 10.1126/science.7855600. [DOI] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Boshans RL, McDonough M, Stahl P, Van Aelst L. A role for POR1 a Rac1 interacting protein in ARF6 mediated cytoskeletal rearrangements. EMBO (Eur Mol Biol Organ) J. 1997;17:5445–5454. doi: 10.1093/emboj/16.17.5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daro E, van der Sluijs P, Galli T, Mellman I. Rab4 and cellubrevin define early endosome polulations on the pathway of transferrin receptor recycling. Proc Natl Acad Sci USA. 1996;93:9559–9564. doi: 10.1073/pnas.93.18.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitz SB, Wu C, Silve S, Howell K, Melancon P, Kahn R, Franzusoff A. Human ARF4 expression rescues sec7mutant yeast cells. Mol Cell Biol. 1996;16:3275–3284. doi: 10.1128/mcb.16.7.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Klausner RD. ARF: A key regulatory switch in membrane traffic and organelle structure. Curr Opin Cell Biol. 1994;6:527–532. doi: 10.1016/0955-0674(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Dunn KW, McGraw TE, Maxfield FR. Iterative fractionation of recycling receptors from lysosomally destined ligands in an early sorting endosome. J Cell Biol. 1989;109:3303–3314. doi: 10.1083/jcb.109.6.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1988;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futter CE, Connolly CN, Cutler DF, Hopkins CR. Newly synthesized transferrin receptors can be detected in the endosome before they appear on the cell surface. J Biol Chem. 1995;270:10999–11003. doi: 10.1074/jbc.270.18.10999. [DOI] [PubMed] [Google Scholar]
- Galas M-C, Helms JB, Vitale N, Thierese D, Aunis D, Bader M-F. Regulated exocytois in chromaffin cells. A potential role for a secretory granule associated ARF6 protein. J Biol Chem. 1997;272:2788–2798. doi: 10.1074/jbc.272.5.2788. [DOI] [PubMed] [Google Scholar]
- Galli T, Chilcote T, Mundgil O, Binz T, Nieman H, Camilli P. Tetanus-toxin-mediated cleavage of cellubrevin impairs exocytosis of transferrin receptor-containing vesicles in CHO cells. J Cell Biol. 1994;125:1015–1024. doi: 10.1083/jcb.125.5.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuze H, Morre DJ. Trans-Golgi reticulum. J Electron Microsc. 1991;17:24–34. doi: 10.1002/jemt.1060170105. [DOI] [PubMed] [Google Scholar]
- Geuze, H.J., J.W. Slot, G.J.A.M. Strous, H.F. and A.L. Schwartz. 1983. Intracellular site of asialogycoprotein receptor-ligand uncoupling: double label immunoelectron microscopy during receptor endocytosis. Cell. 32:277–287. [DOI] [PubMed]
- Geuze HJ, Slot JW, Schwartz AL. Membranes of sorting organelles display lateral heterogeniety in receptor distribution. J Cell Biol. 1987;128:549–561. doi: 10.1083/jcb.104.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh RN, Maxfield FR. Evidence for nonvectorial retrograde transferrin trafficking in early endosomes of HEp2 cells. J Cell Biol. 1995;128:549–561. doi: 10.1083/jcb.128.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts BD. A GTP-binding protein mediating exocytosis. Annu Rev Physiol. 1990;52:591–606. doi: 10.1146/annurev.ph.52.030190.003111. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Back R, Marsh M. A quantitative analysis of the endocytic pathway in baby hamster kidney cells. J Cell Biol. 1989;109:2703–2720. doi: 10.1083/jcb.109.6.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Griffiths G, Howell KE. Characterization of the early endosome and putative carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J Cell Biol. 1989;108:1301–1316. doi: 10.1083/jcb.108.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins CR, Gibson A, Shipman M, Miller K. Movement of internalized ligand-receptor complexes along a continous endoplasmic reticulum. Nature. 1990;349:335–339. doi: 10.1038/346335a0. [DOI] [PubMed] [Google Scholar]
- Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and transferrin receptors in human carcinoma cells. J Cell Biol. 1983;125:1265–1274. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins CR, Gibson A, Shipman M, Strickland DK, Trowbridge IS. In migrating fibroblasts, recycling receptors are concentrated in narrow tubules in the pericentriolar area, and then routed to the plasma membrane of the leading lamella. J Cell Biol. 1994;125:1265–1274. doi: 10.1083/jcb.125.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AO, Gosh RN, Dunn KW, Garrippa R, Pack J, Mayor S, Maxfield FR, McGraw TE. Transferrin receptor containing SDY QRL motif of TGN38 causes a reorganization of the recycling compartment but is not targeted to the TGN. J Cell Biol. 1996;135:1749–1762. doi: 10.1083/jcb.135.6.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA, Gilman AG. The protein cofactor necessary for ADP-ribosylation of Gs by cholera toxin is itself a GTP-binding protein. J Biol Chem. 1986;261:7906–7911. [PubMed] [Google Scholar]
- Kahn RA, Yucel JK, Malhotra V. ARF signalling: a potential role for phospholipase D in membrane traffic. Cell. 1995;75:1045–1048. doi: 10.1016/0092-8674(93)90314-g. [DOI] [PubMed] [Google Scholar]
- Kistakis N, Brown HA, Waters MG, Sternweiss PC, Roth MG. Evidence that phospholipase D mediates ADP ribosylation factor-dependent formation of Golgi coated vesicles. J Cell Biol. 1996;108:2169–2181. doi: 10.1083/jcb.134.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamaze C, Schmid SL. The emergence of clathrin-independent pinocytic pathways. Curr Opin Cell Biol. 1995;7:573–580. doi: 10.1016/0955-0674(95)80015-8. [DOI] [PubMed] [Google Scholar]
- Leitinger B, Hille-Rehfeld A, Speiss M. Biosynthetic transport of the asialoglycoprotein receptor H1 to the cell surface occurs via endosomes. Proc Natl Acad Sci USA. 1995;92:10109–10113. doi: 10.1073/pnas.92.22.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscovitch M, Cantley LC. Signal transduction and membrane traffic: the PITP/phophoinositide connection. Cell. 1995;81:659–662. doi: 10.1016/0092-8674(95)90525-1. [DOI] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Slot JW. Improving structural integrity of cryosections for immunogold labeling. J Histochem. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- Loijens JC, Anderson RA. Type I phosphatidylinositol 4-phosphate 5 kinase are distinct members of this novel lipid kinase family. J Biol Chem. 1996;271:32937–32943. doi: 10.1074/jbc.271.51.32937. [DOI] [PubMed] [Google Scholar]
- Marsh EW, Leopold PL, Jones NL, Maxfield FR. Oligomerized transferrin receptors are selectively retained by a luminal sorting signal in a long-lived endocytic recycling compartment. J Cell Biol. 1995;129:1509–1522. doi: 10.1083/jcb.129.6.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martys JL, Shevell T, McGraw TE. Studies of transferrin recycling reconstituted in streptolysin O permeabilized Chinese hamster ovary cells. J Biol Chem. 1995;270:25976–25984. doi: 10.1074/jbc.270.43.25976. [DOI] [PubMed] [Google Scholar]
- Massenburg D, Han J-S, Liyanage M, Patton WA, Rhee SG, Moss J, Vaughan M. Activation of rat brain phospholipase D by ADP-ribosylation factors 1,5, and 6: Separation of ADP ribosylation factor dependent and oleate dependent enzymes. Proc Natl Acad Sci USA. 1995;91:11718–11722. doi: 10.1073/pnas.91.24.11718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, Presley JF, Maxfield FR. Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J Cell Biol. 1993;121:1257–1269. doi: 10.1083/jcb.121.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw TE, Greenefield L, Maxfield FR. Functional expression of human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J Cell Biol. 1987;105:207–214. doi: 10.1083/jcb.105.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMohan HT, Ushkaryov YA, Edelmann L, Link E, Binz T, Niemann H, Jahn R, Sudhof T. Cellubrevin is a ubiquitous tetanus toxin substrate homologous to a putative synaptic vesicle fusion protein. Nature. 1993;364:346–349. doi: 10.1038/364346a0. [DOI] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Miller SG, Moore HP. Reconstitution of constitutive secretion using semi-intact cells: regulation by GTP but not calclium. J Cell Biol. 1991;112:39–54. doi: 10.1083/jcb.112.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss J, Vaughan M. Structure and function of ARF proteins: activators of cholera toxin and critical components of intracellular vesicular transport processes. J Biol Chem. 1995;266:2606–2614. doi: 10.1074/jbc.270.21.12327. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Neefjes JJ, Oorschot V, Ploegh HL, Geuze HG. MHC class II molecules segregate from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature. 1991;349:669–679. doi: 10.1038/349669a0. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Mayor S, Dunn KW, Johnson LS, McGraw TE, Maxfield FR. The End2mutation in CHO cells slows the exit of transferrin receptors from the recycling compartment but bulk membrane recycling is unaffected. J Cell Biol. 1993;122:1231–1241. doi: 10.1083/jcb.122.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishna H, Klausner RD, Donaldson JG. Aluminum fluoride stimulates surface protrusions in cells overexpressing the ARF6 GTPase. J Cell Biol. 1996;134:935–946. doi: 10.1083/jcb.134.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo, G., M.J. Kleijmeer, G. Posthuma, J.W. Slot, and H.J. Geuze. 1997. Immunogold labeling of ultrathin cryosections: application in immunology. In Handbook of Experimental Immunology. 5th edition, Vol. 4. 208:1–11.
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Sandvig K, van Deurs B. Endocytosis without clathrin. Trends Cell Biol. 1994;4:275–277. doi: 10.1016/0962-8924(94)90211-9. [DOI] [PubMed] [Google Scholar]
- Sariola M, Saraste J, Kuismanen EK. Communication of post-Golgi elements with the early endocytic pathway: regulation of endoproteolytic cleavage of Semiliki Forest virus p62 precursor. J Cell Sci. 1995;108:2465–2575. doi: 10.1242/jcs.108.6.2465. [DOI] [PubMed] [Google Scholar]
- Stoorvogel W, Strous GJ, Geuze HJ, Schwartz AL. Late endosomes derive from early endosomes by maturation. Cell. 1991;65:417–427. doi: 10.1016/0092-8674(91)90459-c. [DOI] [PubMed] [Google Scholar]
- Stoorvogel W, Oorschot V, Geuze H. A novel class of clathrin-coated vesicles budding from endosomes. J Cell Biol. 1996;132:21–23. doi: 10.1083/jcb.132.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA, Bushnell A, Silverstein SC. Tubular lysosome morphology and distribution within macrophages depend on the integrity of cytoplasmic microtubules. Proc Natl Acad Sci USA. 1987;84:1921–1925. doi: 10.1073/pnas.84.7.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze J, Hollinshead M. Tubular early networks in AtT20 and other cells. J Cell Biol. 1991;115:635–653. doi: 10.1083/jcb.115.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya M, Price SR, Tsai S-C, Moss J, Vaughan M. Molecular identification of ADP-ribosylation factor mRNAs and their expression in mammalian cells. J Biol Chem. 1991;266:2772–2777. [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs P, Hull M, Webster P, Male P, Goud B, Mellman I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell. 1992;70:729–740. doi: 10.1016/0092-8674(92)90307-x. [DOI] [PubMed] [Google Scholar]
- van Deurs B, Holm PK, Kayser L, Sandvig K, Hansen SH. Multivesicular bodies in Hep-2 cells are maturing endosomes. Eur J Cell Biol. 1993;61:208–224. [PubMed] [Google Scholar]
- Yashimoro DJ, Tokyo B, Fluss SR, Maxfield FR. Segragation of transferrin to a mildly acidic (pH 6.5) Para-Golgi compartment in a recycling pathway. Cell. 1984;37:789–800. doi: 10.1016/0092-8674(84)90414-8. [DOI] [PubMed] [Google Scholar]
- White S, Miller K, Hopkins C, Trowbridge IS. Monoclonal antibodies against defined epitopes of the human transferrin receptor cytoplasmic tail. Biochim Biophys Acta. 1992;1136:28–34. doi: 10.1016/0167-4889(92)90081-l. [DOI] [PubMed] [Google Scholar]