Molecular Motors and a Spectrin Matrix Associate with Golgi Membranes In Vitro (original) (raw)
Abstract
Cytoplasmic dynein is a microtubule minus-end–directed motor that is thought to power the transport of vesicles from the TGN to the apical cortex in polarized epithelial cells. _Trans_-Golgi enriched membranes, which were isolated from primary polarized intestinal epithelial cells, contain both the actin-based motor myosin-I and dynein, whereas isolated Golgi stacks lack dynein but contain myosin-I (Fath, K.R., G.M. Trimbur, and D.R. Burgess. 1994. J. Cell Biol. 126:661–675). We show now that Golgi stacks in vitro bind dynein supplied from cytosol in the absence of ATP, and bud small membranes when incubated with cytosol and ATP. Cytosolic dynein binds to regions of stacks that are destined to bud because dynein is present in budded membranes, but absent from stacks after budding. Budded membranes move exclusively towards microtubule minus-ends in in vitro motility assays. Extraction studies suggest that dynein binds to a Golgi peripheral membrane protein(s) that resists extraction by ice-cold Triton X-100. In the presence of cytosol, these membrane ghosts can move towards the minus-ends of microtubules. Detergent-extracted Golgi stacks and TGN-containing membranes are closely associated with an amorphous matrix composed in part of spectrin and ankyrin. Although spectrin has been proposed to help link dynein to organellar membranes, we found that functional dynein may bind to extracted membranes independently of spectrin and ankyrin.
Enterocytes, like other polarized epithelial cells, possess two distinct plasma membrane domains that are required for their function in absorption and ion transport (Louvard et al., 1992). The protein and lipid components of the plasma membrane and the organization of the underlying cytoskeleton are distinct for these domains. The development and maintenance of the apical and basolateral domains requires the correct sorting of intracellular vesicles at the level of the TGN (Danielsen and Cowell, 1985; Griffiths and Simons, 1986; Bennett et al., 1988; Danielsen, 1995) and the vectorial transport of specific proteins and lipids to the plasma membrane. In enterocytes, delivery to the apical membrane can be direct, from the Golgi apparatus to the plasma membrane, or indirect, by transcytosis via the basolateral membrane and then on to the apical surface, whereas delivery to the basolateral surface is direct (Achler et al., 1989; Trahair et al., 1989).
Microtubule depolymerization studies suggest that the generation and maintenance of epithelial cell membrane polarity requires intact microtubules for both the direct and indirect pathways of delivery of newly synthesized proteins and lipids (recently reviewed by LaFont and Simons, 1996). The microtubules of polarized epithelia in vivo are collected into parallel bundles with their minus-ends in the apical cytoplasm and their plus-ends towards the basal cytoplasm (Sandoz et al., 1985; Drenckhahn and Dermietzel, 1988; Achler et al., 1989; Bacallao et al., 1989; Gilbert et al., 1991; Fath et al., 1994). There is also a transverse network of microtubules of mixed polarity in the apical cytoplasm subjacent to the terminal web (Bacallao et al., 1989). The Golgi apparatus in fully polarized MDCK cells is a convoluted tubular structure that extends between the nucleus and the apical cytoplasm (Bacallao et al., 1989) and immediately supranuclear in intestinal epithelial cells (Achler et al., 1989; Drenckhahn et al., 1996). Such orientation of microtubules relative to the Golgi apparatus would predict that a minus-end–directed motor, such as cytoplasmic dynein (Paschal and Vallee, 1987), powers the transport of newly synthesized materials from the Golgi apparatus to the apical plasma membrane, and a plus-end– directed kinesin-related motor (Vale et al., 1985) in the transport of basolaterally destined vesicles. Consistent with the minus-ends of microtubules being found in the apical cytoplasm of intestinal epithelia (Achler et al., 1989; Fath et al., 1994), we have found that apically targeted Golgi derived vesicles isolated from intestinal epithelial cells possess cytoplasmic dynein on their surface (Fath et al., 1994). Immunodepletion studies of dynein or kinesin from cytosol in in vitro transport assays with permeabilized MDCK (a canine kidney cell line) cells suggest that kinesin is required for basolateral transport along the longitudinal and transverse microtubules, whereas both dynein and kinesin are necessary for efficient apical transport (Lafont et al., 1994). The apically targeted vesicles may use dynein to move apically on the longitudinal microtubules, then kinesin and/or dynein to traverse the apical transverse network of microtubules to reach the apical cytoplasm.
Although evidence demonstrates roles for kinesin, cytoplasmic dynein and myosins in intracellular transport, (Bomsel et al., 1990; Corthésy-Theulaz et al., 1992; Lafont et al., 1994; Mermall et al., 1994; Govindan et al., 1995), less is known about how the activity of these motors is regulated or how motors associate with membranes. Observations that mitochondria move bidirectionally in axons (Morris and Hollenbeck, 1993), and other observations of the bidirectional movement of pigment granules in melanophores (Haimo and Thaler, 1994), suggest that both dynein and kinesin are present on the same vesicle, and that vesicular movement may be directed by regulating the activity of the motors (Thaler and Haimo, 1996).
Another possible mechanism for controlling vesicular movement is to regulate the binding of specific molecular motors to specific membranes through the interaction with a motor binding protein or receptor. An integral membrane protein, kinectin, which is localized to the ER, has been identified as a kinesin binding protein that may also bind cytoplasmic dynein (Toyoshima et al., 1992; Yu et al., 1992). Another protein that has been proposed to link dynein to membranes is the dynactin complex. This multisubunit complex activates dynein motility on membranes in in vitro motility assays (Gill et al., 1991; Schroer and Sheetz, 1991). One of the components of this complex, p150_Glued_, binds to dynein intermediate chains (IC)1 in vitro (Karki and Holzbaur, 1995; Vaughan and Vallee, 1995), and is codistributed with dynein on small membranes throughout the cytoplasm (Gill et al., 1991; Clark and Meyer, 1992; Paschal et al., 1993). There is also genetic evidence that dynein and the dynactin complex interact in vivo in the same pathway in Neurospora (reviewed by Holzbaur and Vallee, 1994). Our previous work has shown that Golgi stacks isolated from enterocytes lack dynein, whereas TGN-containing membranes possess dynein (Fath et al., 1994). In addition, ∼70% of the cytoplasmic dynein is soluble in these cells, with the remaining dynein bound to membranes or microtubules (Fath et al., 1994). These observations suggest that dynein binding to Golgi membranes may be regulated and specific, perhaps because of a dynein binding protein present on these membranes.
Although the dynactin complex may mediate the binding of dynein to its intracellular binding sites, because this complex is mostly soluble (Gill et al., 1991) with no evidence of a direct interaction with membranes, another protein(s) more intimately associated with the membrane may bind to dynactin. In the past few years, work from several laboratories has provided biochemical and morphological evidence for a matrix containing a homologue of erythroid β-spectrin (βIΣI or βIΣ*) and ankyrin that is associated with Golgi membranes in several polarized and nonpolarized cell types (Beck et al., 1994; Devarajan et al., 1996; Holleran et al., 1996; Beck et al., 1997). Spectrin and ankyrin are typically associated with the plasma membrane, where they are thought to play a role in the maintenance of membrane structure and organization (Bennett, 1990). Because the dynactin complex contains a short F-actin–like filament containing Arp1 (Schafer et al., 1994), the complex may bind to Golgi membranes via the actin binding site on spectrin (Brenner and Korn, 1979). Support for such an interaction comes from transfection studies. The overexpression of the dynactin complex component p50 causes Golgi apparatus fragmentation and dispersal (Burkhardt, J.K., C.J. Echeverri, and R.B. Vallee. 1995. Mol. Biol. Cell. 6:266a), however, the overexpression of Arp1 (centractin) causes the alignment of Golgi markers and spectrin along novel Arp1 filaments (Holleran et al., 1996).
In this report, we examined the binding of molecular motors to Golgi membranes isolated from polarized intestinal epithelial cells. We found that functional cytoplasmic dynein, but not kinesin, binds to a tightly bound Golgi peripheral membrane protein(s) selectively in regions of Golgi stacks that are destined to bud. Isolated Golgi stacks and TGN-containing membranes were closely associated with an amorphous matrix that resisted extraction with cold 1% Triton X-100 (TX-100). By immunoblotting, we found that this matrix contains the dynactin complex, myosin-I, spectrin and ankyrin, and in TGN-containing membranes, dynein. Although dynein may be tethered to Golgi membranes indirectly via spectrin and ankyrin, we found that dynein can bind to these membranes independently of these matrix components.
Materials and Methods
Isolation of Golgi Membranes
Golgi membranes were isolated from chicken intestinal epithelial cells as described previously (Fath and Burgess, 1993), with several modifications. Intestinal epithelial cells were homogenized in ice-cold 0.5 M sucrose-PKM buffer (100 mM potassium phosphate, pH 6.5, 5 mM MgCl2, and 3 mM KCl) with a hand-held tissue grinder (Tissue•Tearor; BioSpec Products, Inc., Bartlesville, OK) for 90 s at a setting of 2. The following steps were performed at 4°C. Nuclei and any intact cells were pelleted by a 10-min centrifugation at 600 gave. The postnuclear supernatant was layered onto a step gradient containing 1.3 M sucrose-PKM and 0.7 M sucrose-PKM, then centrifuged at 105,000 gmax (SW41 rotor; Beckman Instruments, Corp., Palo Alto, CA) for 60 min. The supernatant, termed cytosol, was frozen in liquid nitrogen, stored at −80°C, and then used as a source of dynein and other soluble proteins. Before use, the cytosol was thawed rapidly and clarified at 200,000 gmax for 30–40 min. Membranes that concentrated at the 0.7/1.3 M sucrose interface were collected and adjusted to 1.25 M sucrose-PKM. The membranes were overlaid with 1.1 M sucrose-PKM, 0.5 M sucrose-PKM, and then centrifuged at 90,000 gmax (SW41 rotor) for 90 min. Golgi membranes were collected at the 0.5/1.1 M interface, adjusted to 0.7 M sucrose-PKM, and then centrifuged at 10,000 gmax for 15 min to pellet Golgi stacks. Small TGN-containing membranes remaining in the supernatant (Fath et al., 1994) were collected by centrifugation at 259,000 gmax for 30 min. Membranes were resuspended in PEMS (10 mM Pipes pH 7.0, 1 mM EGTA, 2 mM MgCl2, and 0.25 M sucrose) with the addition of the protease inhibitors PMSF, aprotinin and leupeptin, frozen in liquid nitrogen, and then stored at −80°C.
In Vitro Golgi Stack Budding Assay
50 μl of Golgi stacks (∼500 μg/ml final concentration) were mixed with 10 μl of 10× budding buffer (250 mM Hepes, 15 mM Mg-Acetate, 250 mM KCl, 0.25 M sucrose, pH 6.7; Salamero et al., 1990), 10 μl of creatine-phosphokinase (0.8 mg/ml), 6.6 μl of 80 mM phosphocreatine, 10 μl of clarified cytosol (∼1–2 mg/ml final concentration), and 1 μl of 200 mM ATP. The final volume was adjusted to 100 μl by the addition of 0.25 M sucrose-PKM. In experiments not shown, by immunoblotting we found that actin, tubulin, dynein, and p150_Glued_pelleted in the absence of membranes when the final cytosol concentrations was increased to ⩾5 mg/ml. Therefore, these assays were performed at cytosol concentration ⩽2 mg/ml. In controls either lacking cytosol, or ATP and the ATP regenerating system, an equal volume of the appropriate buffer was used in their place. In some experiments, 4 U/ml apyrase (Sigma Chem. Co., St. Louis, MO) was used to deplete any ATP present in cytosol, or ADP was used in place of ATP. The mixture was incubated at 37°C for 15 min, chilled on ice, and centrifuged at 10,000 gave for 15 min through a 100-μl 0.5 M sucrose cushion. This pellet represents the stacks after budding. The resulting supernatant was then centrifuged at 259,000 gmax for 30 min at 4°C through a 100-μl 0.5 M sucrose cushion to pellet budded membranes. The supernatant and sucrose cushion were removed, the membrane pellets resuspended in SDS sample buffer, and then analyzed by SDS-PAGE and immunoblotting (Fath et al., 1994). The budded membranes were quantitated by scanning densitometry of immunoblots probed with alkaline phosphatase antibodies or by enzymatic assay (Weiser, 1973) that was adapted for 96-well microtiter plates.
Dynein Binding to Golgi Stacks
Dynein binding to Golgi stacks was performed as described for the in vitro budding assay, except that the ATP and the ATP regenerating system were omitted. In some experiments (see Fig. 1), dynein binding was assayed in conditions that were identical to those used in the in vitro motility assays described below with the omission of ATP. After incubation at 37°C, membranes were pelleted at 259,000 gmax (TLS-55; Beckman) for 30 min through a 75-μl cushion containing 0.5 M sucrose PKM at 4°C. For centrifugation, 11 × 34 mm polycarbonate tubes were used as rotor inserts to hold the 7 × 20 mm polycarbonate tubes, which contained the samples. The smaller tubes were surrounded by a cushion of 50 μl of water to prevent their distortion. The pellet was resuspended and analyzed by SDS-PAGE and immunoblotting.
Figure 1.

Cytosolic dynein, but not kinesin, binds to isolated Golgi stacks. (A) Isolated Golgi stacks, which lack dynein and kinesin, were incubated for 15 min at 37°C in motility buffers (in the absence of ATP) or with buffer including cytosol. A third tube contained cytosol, but no membranes. The samples were then chilled and pelleted through 0.5 M sucrose to separate stacks and bound material from cytosol. All of each pellet and one-half of the supernatant were immunoblotted with a mixture of dynein IC and kinesin heavy chain mAbs. Pelleted Golgi stacks incubated in buffer (Stacks) contained no cytosolic dynein (DIC, dynein IC) or kinesin (KHC, kinesin heavy chain). Soluble cytosolic kinesin and dynein remained in the supernatant in cytosol-alone controls (Cytosol). Pelleted stacks that were incubated with cytosol (Stacks + Cytosol) bound dynein, but not kinesin. (B) Cytosolic dynein binding to Golgi stacks was also demonstrated on flotation gradients. Golgi stacks and cytosol were incubated as described in A, then made 25% nycodenz. The samples were overlaid with 20% nycodenz/PEMS and PEMS. After centrifugation, immunoblots show that both Golgi cisternae, as indicated by the presence of α-mannosidase II, and cytosolic dynein were present at the 20% nycodenz/PEMS interface in samples containing both Golgi stacks and cytosol (Stacks + Cytosol). Dynein was not detected at this interface in samples containing Golgi stacks incubated in buffer (Stacks) or in cytosol alone controls (Cytosol).
Dynein binding was also assayed on flotation gradients. 11 μl of Golgi stacks (10 μg) were mixed with 5 μl of 10× budding buffer and 10 μl of clarified cytosol (55 μg). The final volume was adjusted to 50 μl by the addition of PEMS. In the cytosol alone controls, 11 μl PEMS replaced the Golgi stacks and in the Golgi stack only controls, 10 μl of 0.5 M sucrose/ PKM replaced the cytosol. After incubation at 37°C, samples were made 25% nycodenz by the addition of 50 μl of 50% nycodenz (Sigma) in PEMS. The samples were overlaid with 100 μl of 20% nycodenz/PEMS and 50 μl of PEMS in a 7 × 20 mm centrifuge tube and centrifuged at 259,000 gmax (TLS-55; Beckman) for 30 min at 4°C. The materials that floated to the interface of PEMS and 20% nycodenz interface were analyzed by SDS-PAGE and immunoblotting.
Motility and Directionality Assays
In vitro motility assays were performed on polar arrays of microtubules that were illuminated with a 100 W Hg lamp and viewed using differential interference microscopy (Nikon Microphot FX-A; Nikon, Melville, NY) equipped with a 60× (1.4 NA) planapochromatic objective and a DIC aplanatic achromat (1.4 NA) condenser. The image was projected using a 4× eyepiece onto a Hamamatsu C2400 CCD camera (Hamamatsu Corp., Bridgewater, NJ). Digitization, background subtraction and contrast enhancement were accomplished with a Hamamatsu ARGUS-20 image processor and the resultant images stored in real time on VHS videotape using a JVC HR-S5300 recorder. Microscope perfusion chambers (∼7 μl capacity) were formed with a no. 1 glass coverslip on glass slides with either double-sided adhesive tape or vacuum grease (Apiezon L; Apiezon Prod. Ltd., London, UK) spacers. Chamber fluids were exchanged by capillary action using filter paper. Salt-extracted sea urchin sperm axonemes or Chlamydomonas axonemes were adsorbed to the coverslip by inverting the slide cover slip down for 2–3 min, then the glass surfaces were coated with α-casein (19 mg/ml) for 5 min. The casein was washed out with several chamber volumes of motility buffer (25 mM Pipes, 0.5 mM MgCl2, 0.5 mM EGTA, pH 7.0). Then 3 μl of 13 mg/ml phosphocellulose-purified tubulin and 1.6 mM GTP were perfused into the chamber (approximately twofold dilution with motility buffer already in the chamber) and the extent of microtubule polymerization from the ends of the axonemes determined in the microscope. When sufficient polymerization from the plus ends of the axonemes was achieved, ∼2 min at 25°C, unpolymerized tubulin was removed by addition of the motility samples. The membrane motility samples were: 12.5 μl mixture containing 0.2–0.4 mg/ml Golgi stacks, 1–5 mg/ml clarified cytosol, 4 mM ATP, 20 μM taxol, 0.27 mg/ml α-casein, in motility buffer. Final concentration of Mg-ATP was 4 mM and temperature 21–25°C. In some experiments the Mg-ATP was replaced with 4 mM Mg-5′-adenylylimidodiphosphate (AMP-PNP) or contained 4 mM each of Mg-ATP and Mg-AMP-PNP. Some membranes were preincubated in complete motility buffers lacking ATP but containing 2 mM _N_-ethylmaleimide (NEM) for 15 min at room temperature. ATP was then added to the samples before viewing. Images were recorded using background subtraction and a rolling average of four video frames. Translocation rates were measured on traverses greater than 1 μm using ARGUS-20 software. Images were captured using an AV-equipped Macintosh (Apple Computer, Inc., Cupertino, CA) and processed in Adobe Photoshop (Adobe Systems Incorporated, Mountain View, CA).
Gel Electrophoresis, Immunoblotting, and Densitometry
Proteins were separated on 10% SDS-PAGE gels and electroblotted to PVDF membranes (Fath et al., 1994). Immunoblots were incubated with appropriate primary antibodies followed by incubation with peroxidase-labeled secondary antibodies and visualized using enhanced chemiluminescence reagents (Amersham Corp., Arlington Heights, IL) on X-OMAT AR film (Eastman-Kodak Co., Rochester, NY) and developed with GBX (Eastman-Kodak). Autoradiography films were digitized using transmitted light on a flatbed scanner (Envisions 24 Pro; UMAX) driven by Adobe Photoshop. To assure quantitative accuracy, the absence of digitized image saturation was shown by ensuring that all bands of interest had pixel values between 10 and 245 as determined in Photoshop. The linear response of the film and antibody binding was shown by immunoblotting a dilution series of purified dynein. Exposures within the linear range of the film were exported in TIFF format for quantitation using Scan Analysis software (BIOSOFT, Cambridge, UK). Molecular weight markers were prestained proteins in the SDS-7B kit from Sigma Chemical Co.
The following antibodies were used for immunoblotting: cytoplasmic dynein IC (mAb 70.1; Sigma Chemical Co., and mAb 74.1; Chemicon International Inc.); p150_Glued_(mAb 150.1; Gill et al., 1991); β-COP (mAb M3A5; Sigma Chemical Co.); TGN-38/41; (Luzio et al., 1990); brush border myosin-I (Fath et al., 1994); alkaline phosphatase (Chemicon, Intl. Inc., Temecula, CA); Arp1, (RPV; Lees-Miller et al., 1992); Na+K+-ATPase, (α-subunit; Nelson and Meshnock, 1986); affinity purified β-spectrin, (βspec-1; Beck et al., 1994); α-mannosidase II (Velasco et al., 1993); brain spectrin (Burridge et al., 1982); ankyrin (chicken RBC; Calbiochem-Novabiochem, La Jolla, CA); kinesin heavy chain (SUK4 mAb; Ingold et al., 1988).
Protease Digestion, Salt, Alkaline, and Detergent Extraction of Golgi Stacks
Approximately 300 μg/ml of Golgi stack protein in PEMS was digested with 1 μg/ml of papain at 37°C for 10 min. Digestion was stopped by the addition of a large excess of PMSF, leupeptin, and α-macroglobulin and chilling on ice. Controls were treated in a similar manner except that no papain was added. The stacks were centrifuged through a 0.5 M sucrose-PKM cushion to remove any papain, resuspended in PEMS, incubated with cytosol and analyzed for the presence or absence of dynein binding by immunoblotting.
Golgi stacks and TGN-containing membranes in PEMS were extracted with 0.6 M KI, 1.0 M NaCl or with 20 vol of 0.1 M Na2CO3 pH 11.5 on ice for 30 min. Membranes were also extracted with 1% (vol/vol) TX-100 (Surfact-Amps; Pierce, Rockford, IL) in PEMS on ice for 15 min. In some experiments, Golgi stacks were extracted on ice for 15 min with 1% (vol/ vol) TX-100, then 20 vol of 0.1 M Na2CO3, pH 11.5, were added and the samples incubated for an additional 30 min. The extracted membranes were collected by centrifugation through a 0.5 M sucrose-PKM cushion at 259,000 gmax for 30 min at 4°C. Pellets were rinsed and resuspended in PEMS. Samples were then either immunoblotted or incubated with cytosol as described for dynein binding and immunoblotting. Some detergent-extracted Golgi stacks were analyzed by in vitro motility assays (as described above) either in the presence or absence of added cytosol.
Wheat Germ Agglutinin (WGA)-Agarose Fractionation
50 μg of isolated Golgi stacks and TGN-containing membranes were incubated in a total volume of 400 μl PEMS with 100 μl of WGA-agarose (settled bead volume; Sigma Chemical Co.) for 16–18 h at 4°C with rotation. The beads were allowed to settle by gravity and the supernatant removed. The pellets were resuspended with 300 μl of PEMS and allowed to resettle by gravity. The supernatants were combined and centrifuged at 259,000 gmax for 30 min at 4°C to collect any membranes not binding to WGA-agarose. The distribution of specific proteins in the WGA-agarose bound and unbound fractions was calculated by quantitation of immunoblots. We could not determine the amount of total protein in the bound fraction because of WGA, therefore the amount of total protein in the WGA-agarose unbound fraction was compared with the amount in the unbound fraction after incubation with unconjugated CL-4B Agarose (Sigma Chemical Co.). The CL-4B Agarose served as a control to assess the amount of membrane binding to Agarose and for the amount of membranes trapped in the bead pellets. Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad Laboratories, Richmond, CA) using IgG as the protein standard.
Electron Microscopy
Isolated Golgi membranes were fixed in suspension with an equal volume of 2% glutaraldehyde-PEMS for 60 min on ice, then pelleted and processed for thin sectioning as described previously (Fath et al., 1994). Budded Golgi membrane pellets were negative stained as described previously (Fath et al., 1994).
Results
Cytosolic Dynein Binds to Golgi Stacks In Vitro
We have shown previously that cytoplasmic dynein is present on isolated TGN-containing membranes (possessing TGN 38/41, but lacking β-COP), whereas both kinesin and dynein are absent from isolated Golgi stacks (Fath et al., 1994). We wished to establish whether isolated stacks were competent to bind soluble dynein or kinesin supplied by cytosol. Therefore, isolated Golgi stacks were incubated with cytosol as a source of soluble motors in the absence of ATP at 37°C. The stacks were then separated from cytosol by pelleting at 259,000 g through a 0.5 M sucrose pad and assayed for dynein binding by immunoblotting with antibodies recognizing the 74-kD cytoplasmic dynein IC or for the kinesin heavy chain (Fig. 1). Although there was no detectable dynein on stacks as isolated (Fig. 1 A, Stacks P; and Fath et al., 1994), dynein pelleted with stacks after incubation with cytosol (Fig. 1 A, Stacks + Cytosol P). Quantitative densitometry of this immunoblot shows that ∼5% of the cytosolic dynein bound to membranes. Using purified 20 S rat testis cytoplasmic dynein as a reference standard on immunoblots (generous gift of Dr. David Asai, Purdue University), we calculated that the clarified cytosol, at a protein concentration of 7.85 mg/ml, contained 31 μg/ml dynein. In binding studies with Golgi stacks incubated with maximum workable concentrations of cytosol, we determined that dynein bound at 0.65 pmol/ μg stack protein. These calculations assume that the molecular mass of cytoplasmic dynein is 1,200 kD (Vallee et al., 1988). Such binding is consistent with the binding of purified cytoplasmic dynein to isolated synaptic vesicles (Lacey and Haimo, 1992) that saturates at 1 pmol/μg vesicle protein. In contrast, no dynein pelleted when cytosol at this concentration was centrifuged in the absence of stacks under similar buffer conditions (Fig. 1 A, Cytosol P). We were unable to perform complete saturation studies due to the fact that when cytosol was used at higher concentrations, microtubules polymerized and dynein and dynactin pelleted in the absence of membranes (data not shown). Attempts to inhibit microtubule polymerization with nocodazole did not prevent dynein and dynactin pelleting from higher concentrations of cytosol. Although there was abundant cytosolic kinesin (Fig. 1 A, Cytosol S), kinesin was not detected on stacks either in the presence or absence of cytosol (Fig. 1 A). These data suggest that cytosolic dynein, but not kinesin can bind to and pellet with Golgi stacks in in vitro binding assays done in the absence of ATP.
Cytosolic dynein was also shown to bind to and partition with Golgi stacks on discontinuous nycodenz flotation gradients. Nycodenz was used to form iso-osmotic gradients with low viscosity. Golgi stacks that were incubated with cytosol, as described for the pelleting assays, were made 25% nycodenz/PEMS, overlaid with 20% nycodenz/PEMS and PEMS. After centrifugation, the membranes that floated to the interface of PEMS and 20% nycodenz/PEMS were collected and immunoblotted for α-mannosidase II as a marker for Golgi cisternae (Velasco et al., 1993). Golgi stacks, being less dense then 20% nycodenz (Hammond and Helenius, 1994), floated to the interface of PEMS and 20% nycodenz/PEMS (Fig. 1 B). On the same gradients, dynein was detected at the interface of PEMS and 20% nycodenz/PEMS in the complete binding reactions (Stacks + Cytosol), but not in the incomplete reactions (Stacks or Cytosol ) alone. These data, in conjunction with the pelleting assays, suggests strongly that cytosolic dynein can bind to Golgi stacks in vitro.
Dynein Binds to Golgi Stacks in Regions That Are Destined to Bud in an In Vitro Budding Assay
To examine further the regulation of dynein binding to membranes, we adapted an in vitro Golgi budding assay that was developed for isolated rat liver Golgi membranes (Salamero et al., 1990). Golgi stacks were incubated in the presence or absence of cytosol and ATP at 37°C in buffers that promote budding. Electron microscopy confirmed the appearance of buds from the ends of stack membranes in this assay (Fig. 2 B). The post-budding stacks were separated from budded membranes and cytosol by pelleting the stacks through a sucrose pad at 10,000 g. The budded membranes, which remained in the supernatant, were subsequently separated from cytosol by pelleting at 259,000 g through sucrose. Negative stain electron microscopy of the budded membrane pellet shows that the budded membranes are comprised of 50–200-nm vesicles (Fig. 2 D) and are approximately the same size as the buds at the ends of Golgi stacks observed in thin sections (Fig. 2 B).
Figure 2.
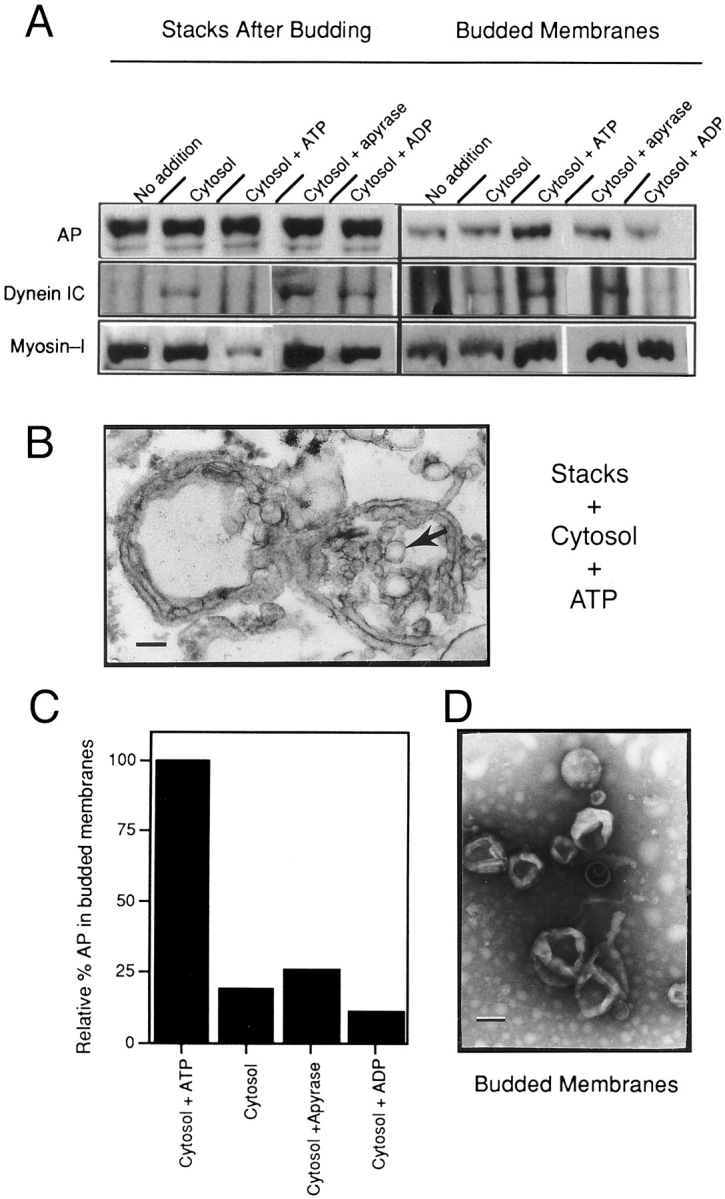
Cytoplasmic dynein binds to stacks in an in vitro Golgi stack budding assay and remains associated specifically with the budded membranes. (A) Golgi stacks were incubated in various conditions at 37°C, then pelleted through a 0.5-M sucrose pad at 10,000 g for 15 min (left). The budded membranes that were released from the stacks and remained in the low-speed supernatant were collected by centrifugation through a 0.5 M sucrose cushion at 259,000 g for 30 min (right). Pelleted stacks and budded membranes were immunoblotted for AP, dynein IC, and myosin-I. Golgi stacks were incubated in the presence or absence of cytosol and ATP or with the addition of apyrase to hydrolyze ATP, or ADP in place of ATP as indicated above each lane. (B) Thin section electron micrograph of Golgi stacks that were incubated with cytosol and ATP to initiate budding. Budded vesicles (arrow) are visible at the ends of the Golgi stacks. (C) The immunoblots in A suggested that ATP was required for efficient budding, therefore to quantify budding we measured the relative levels of the plasma membrane protein AP in budded membranes in various conditions. The level of AP present in the budded membranes in each condition as determined by quantitative immunoblotting is expressed relative to the levels in the complete budding mixture (Cytosol + ATP), which was set at 100%. The omission of ATP, the inclusion of apyrase or ADP, decreased the level of AP in budded membranes 4–5-fold. (D) Negative stain electron micrograph showing that budded membrane pellets contained 50–200-nm vesicles. Bars: 100 nm.
To establish biochemically the extent of budding in these reactions, we followed the disappearance of alkaline phosphatase (AP) from Golgi stacks after budding (10,000-g pellet) and its appearance on budded membranes (259,000-g pellet). AP was used as a marker for budding, because it is an abundant protein that is processed throughout the Golgi apparatus and targeted to the apical plasma membrane. We found that efficient budding required ATP; the omission of ATP, addition of apyrase to hydrolyze any cytosolic ATP, or the replacement of ATP with ADP decreased the amount of AP in the budded membrane pellet. We calculated the relative amount of AP-containing membranes in the budded fraction by quantitation of linear immunoblots of AP (Fig. 2 C) and by measuring AP enzymatic activity. Although there was a background level of AP in the 259,000-g pellet when stacks were incubated in buffer alone (Fig. 2 A), we calculated by either method that there was a 4–5-fold increase in AP in the 259,000-g pellet when in the presence of complete budding reactions (Fig. 2 A, Cytosol + ATP). The ATP requirement suggests that the increase in AP on budded membranes is the result of the active budding of vesicles from Golgi stacks.
To establish whether cytosolic dynein was bound to Golgi stacks and/or the budded membranes in this assay, stacks after budding and budded membranes were immunoblotted for dynein (Fig. 2 A). Dynein was not detectable on Golgi stacks that were incubated in buffer lacking cytosol; however, upon the addition of cytosol in the absence of ATP, dynein was present on Golgi stacks, which did not bud efficiently (as measured by AP levels; Fig. 2 C). The addition of ATP plus cytosol, which promotes efficient budding, resulted in the accumulation of dynein on the budded membranes but none on the stacks. This result suggests that dynein binds to regions of Golgi stacks destined to bud, and that upon addition of ATP, dynein remains associated with budded membranes. Most of the dynein remained associated with stacks under conditions that do not promote efficient budding, such as the removal of endogenous ATP with apyrase or the replacement of ATP with ADP (Fig. 2 A), or if a complete reaction is incubated on ice (data not shown).
The actin-based motor myosin-I is present on isolated Golgi stacks (Fath et al., 1994). When the same blots of the in vitro budding reaction were analyzed for the presence of myosin-I, it is interesting that most of the myosin-I was lost from the stacks after budding, and enriched in the budded membranes, along with dynein (Fig. 2 A). This result supports our hypothesis that both dynein and myosin-I are bound to apically targeted Golgi-derived membranes (Fath et al., 1994).
Cytosolic Dynein Binds Functionally to Golgi Stacks
To assess whether the cytosolic dynein that bound in vitro to Golgi membranes was functional, we performed in vitro motility assays with these membranes on polarized arrays of microtubules. In these assays, microtubules were polymerized from the ends of axonemes from sea urchin sperm or Chlamydomonas flagella in a microscope slide chamber. Then budded membranes containing dynein were perfused into the chamber either in the presence or absence of cytosol. Budded membranes bound to and translocated along microtubules as observed using video-enhanced differential interference contrast microscopy. We found that the addition of cytosol stimulated the frequency of moving membranes. Budded membranes in the presence of cytosol translocated at 1.64 ± 0.5 μm/s (mean ± SD; n = 81; 25°C). Motility assays were also performed using Golgi stacks. Stacks in the absence of cytosol occasionally bound to microtubules; however, they were never observed to move (data not shown). This inability to move is consistent with the lack of dynein and kinesin (Fig. 1 A; and Fath et al., 1994) on isolated stacks. In contrast, when stacks were preincubated with cytosol plus ATP for several minutes before perfusion under the coverslip, membranes bound to and moved along microtubules at 1.69 ± 0.4 μm/s (mean ± SD; n = 110; 25°C) (Fig. 3).
Figure 3.
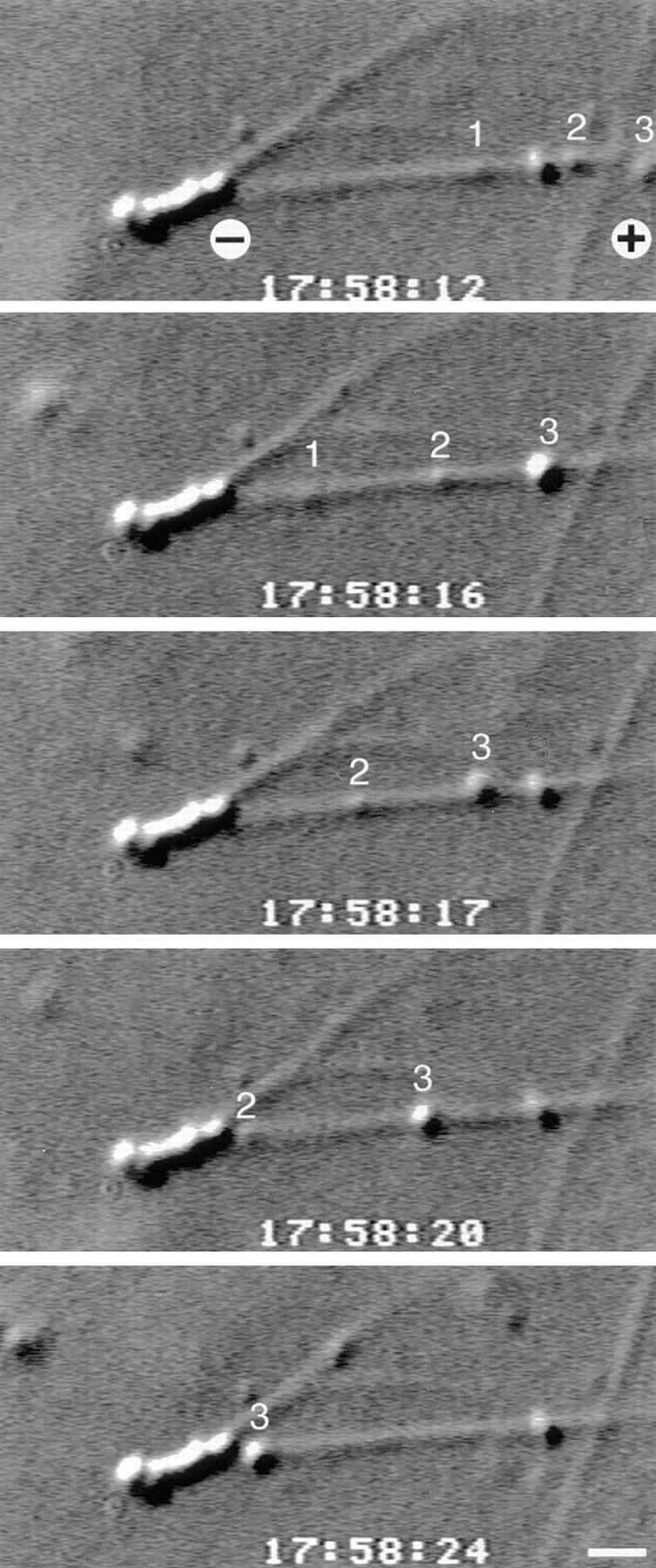
Golgi stacks incubated with cytosol move only towards microtubule minus-ends in in vitro motility assays. To determine whether the cytosolic dynein that bound to Golgi stacks was functional, Golgi membranes were incubated with cytosol and with microtubules that were polymerized from the ends of axonemes. This series of video frames shows three numbered Golgi membranes moving towards the minus-end of a microtubule. This microtubule was one of several that polymerized from the plus-end of a sea urchin sperm axoneme. Bar, 1 μm.
The microtubules in the motility chambers were polymerized off the ends of axonemes, therefore we could determine microtubule polarity and thus the directionality of membrane movements (Fig. 3). Only minus-end–directed movement was observed in the stacks plus cytosol preparations. For example, in two experiments we counted a total of 49 membranes moving towards the minus-ends of microtubules and zero plus-end–directed movements. The presence of minus-end–directed movements and absence of plus-end–directed movements suggests that dynein and not kinesin is the active motor in these assays.
Although no kinesin was detected binding to Golgi membranes, several kinesin-related proteins are minus-end– directed motors (see review by Endow, 1995). Because these or other kinesin-related proteins may not be recognized by the SUK4 mAb that we used, but could be present in cytosol, motility assays were done in the presence of the nonhydrolyzable ATP analogue AMP-PNP. This analogue, when present equimolar with ATP, inhibits the motor activity of all kinesins so far tested (Walker and Sheetz, 1993), but not dynein. Consistent with movement directed by dynein, Golgi membranes translocated on microtubules in motility assays in buffers containing 4 mM Mg-ATP and 4 mM Mg-AMP-PNP. We observed no microtubule-based membrane movements in buffers containing 4 mM Mg-AMP-PNP in the absence of ATP. Furthermore, AMP-PNP did not induce a pronounced rigor linkage of membranes to microtubules as would be expected if membranes contained kinesin (Schnapp et al., 1992). Because dynein, but not kinesin, is inactivated by 2 mM NEM (Vale et al., 1985), in another series of experiments we performed motility assays in the presence of NEM. Consistent with dynein-driven motility, we found that all movements were inhibited by inclusion of 2 mM NEM in our assays. Therefore, it is unlikely that a functional kinesin is bound to Golgi membranes in these preparations.
Cytosolic Dynein Binds to a Golgi Peripheral Membrane Protein That Is Extracted at Alkaline pH, but Resists Extraction with Cold TX-100
To establish whether cytosolic dynein bound to stacks via a Golgi surface protein, stacks were incubated with the protease papain. Digestion was stopped using protease inhibitors on ice and the membranes were washed free of papain by pelleting through sucrose. The stacks were resuspended, incubated with cytosol to allow cytosolic dynein binding, and repelleted through sucrose. Quantitation of immunoblots probed with a dynein IC antibody showed that papain-treated membranes bound only 25 ± 13% (mean ± SD; n = 5) as much dynein as did untreated membranes. These data suggest that a Golgi stack surface protein is necessary for dynein binding. It is unlikely that residual protease digested cytosolic dynein or other cytosolic factors required for dynein binding. Immunoblots detected no lower molecular weight immunoreactive bands indicative of dynein proteolysis and there was an equal amount of dynein IC in the supernatants containing cytosol after incubation in both the mock and protease-treated samples (data not shown).
To assess the nature of the association between a putative dynein binding protein(s) and Golgi membranes, isolated stacks were stripped of peripheral membrane proteins by incubation in high salt or 0.1 M sodium carbonate (pH 11.5) before monitoring dynein binding from added cytosol (Fig. 4 A). Cytosolic dynein bound to control stacks and stacks from which weakly bound peripheral membrane proteins were stripped with 0.6 M KI (Fig. 4 B) or 1.0 M NaCl (data not shown). In contrast, cytosolic dynein did not bind to stacks that had all peripheral membrane proteins stripped at pH 11.5 (Fig. 4 B). Therefore, it is likely that a tightly bound Golgi stack peripheral membrane protein is required for dynein binding.
Figure 4.

Cytosolic dynein binds to a Golgi peripheral membrane protein. (A) To examine the mode of association of a putative dynein binding protein(s) with Golgi membranes, isolated stacks were incubated with PEMS buffer (Control), 0.6 M KI, 0.1 M Na2CO3 pH 11.5, or with ice-cold 1% TX-100. The stacks were separated from extracted proteins by centrifugation through a 0.5 M sucrose pad, then analyzed by immunoblotting for dynein IC, p150_Glued_ and Arp1(Extracted Golgi Stacks). (B) Extracted stacks were also incubated with cytosol and immunoblotted for dynein IC to determine the effects of extraction on dynein binding (Extracted Golgi Stacks + Cytosol). Cytosolic dynein bound to stacks that were extracted with 0.6 M KI or 1% TX-100, but not to stacks that were extracted at pH 11.5. Endogenous p150_Glued_ and Arp1 were stripped from the membrane at alkaline pH; cytosolic Arp1 rebound to these membranes to nearly control levels, whereas only a small amount of p150_Glued_ rebound. TX-100 extracted Arp1, but not p150_Glued_, from stack membranes; cytosolic Arp1 rebound to these membranes to control levels.
It has been proposed that the dynactin complex may link dynein either directly or indirectly to organelle membranes (Vaughan and Vallee, 1995; Echeverri et al., 1996; Holleran et al., 1996). As a first step in assessing whether the dynactin complex may link dynein to components of the Golgi membrane, we immunoblotted Golgi membranes to determine whether the dynactin complex was present. We found that two members of the dynactin complex, p150_Glued_ (Gill et al., 1991) and Arp1 (actin-related protein 1; Lees-Miller et al., 1992), were present on isolated Golgi stacks (Fig. 4 A). The p150_Glued_ mAb recognized only a 150/135-kD doublet as is commonly seen in other organisms (Holzbaur and Vallee, 1994). The Arp1 antibody was specific for Arp1 and did not recognize conventional actin (data not shown). Because the putative Golgi stack dynein receptor is a tightly bound peripheral membrane protein, we wished to establish whether the dynactin complex, which has been shown to be a peripheral membrane complex in other cells, is also peripherally associated with Golgi membranes. Therefore, stacks were incubated with high salt or pH 11.5 to strip them of peripheral membrane proteins. Immunoblots showed that little Arp1 was solubilized by 0.6 M KI (Fig. 4 A) or 1 M NaCl (data not shown). By contrast, 0.6 M KI (Fig. 4 A) or 1 M NaCl (data not shown) released the majority of the p150_Glued_ 135-kD isoform with a lesser amount of the p150_Glued_ 150-kD isoform being extracted. When stacks were stripped of tightly associated peripheral membrane proteins with the more stringent pH 11.5, both Arp1 and p150_Glued_ were nearly entirely released from the membrane (Fig. 4 A). Thus, the dynactin complex behaves as a Golgi stack peripheral membrane protein.
Next we wished to determine whether the presence of the dynactin complex correlated with the ability of Golgi stacks to bind cytosolic dynein. Therefore, we immunoblotted salt or alkaline-treated stacks after incubation with cytosol to ascertain whether the complex was present on membranes to which cytosolic dynein bound (control and KI-stripped) and absent from those to which dynein did not bind (alkaline-stripped). These immunoblots showed that cytosolic dynein bound to control and KI-stripped stacks, which contain both p150_Glued_ and Arp1 (Fig. 4 B). Although both p150_Glued_ and Arp1 were stripped from Golgi stack membranes at alkaline pH (Fig. 4 A), cytosolic Arp1 rebound to these stacks at levels nearly that of unextracted stacks (Fig. 4 B). We also found that a small amount of cytosolic p150_Glued_ rebound to the alkaline-stripped stacks, with the 150-kD isoform being more prevalent. However, we cannot determine whether the Arp1 and p150_Glued_ rebinding to the stripped membrane was native. Because cytosolic dynein did not bind to alkaline-stripped stacks that contained Arp1, these data suggest that Arp1 alone is not sufficient for dynein binding and further suggest that a peripheral membrane protein, perhaps the 135-kD p150_Glued_ component of the dynactin complex, is required for dynein binding to membranes.
Golgi stacks were also extracted with 1% TX-100 on ice (releasing 75% of total protein, see below) and analyzed for the presence of dynactin complex and for cytosolic dynein binding (Fig. 4 A). The detergent released most of the Arp1 from the membrane, while little, if any, of the p150_Glued_ was extracted. Immunoblots showed that cytosolic dynein could bind to stacks that were solubilized with cold 1% TX-100 (Fig. 4 B). Cytosolic dynein binding to detergent-extracted stacks was physiologically relevant as shown by two methods. First, the cytosolic dynein that bound to the stack ghosts was functional and could move these membranes on microtubules in in vitro motility assays (Fig. 5 A) at rates of 1.1 ± 0.2 μm/s (mean ± SD; n = 12; at 25°C); rates that were indistinguishable from the rates of unextracted membranes at 1.2 ± 0.5 μm/s (mean ± SD; n = 8; at 25°C) from the same preparation. In the absence of added cytosol, these membrane ghosts neither bound to nor moved on microtubules in these assays. Second, as with the native Golgi stacks, the dynein binding protein(s) on Triton membrane ghosts was extracted at alkaline pH. Cytosolic dynein did not bind to Golgi stacks that had been sequentially extracted with TX-100 and pH 11.5 (Fig. 5 B). These two independent assays suggest that cytosolic dynein is binding to the same peripheral membrane protein(s) in intact and detergent-extracted Golgi stacks.
Figure 5.
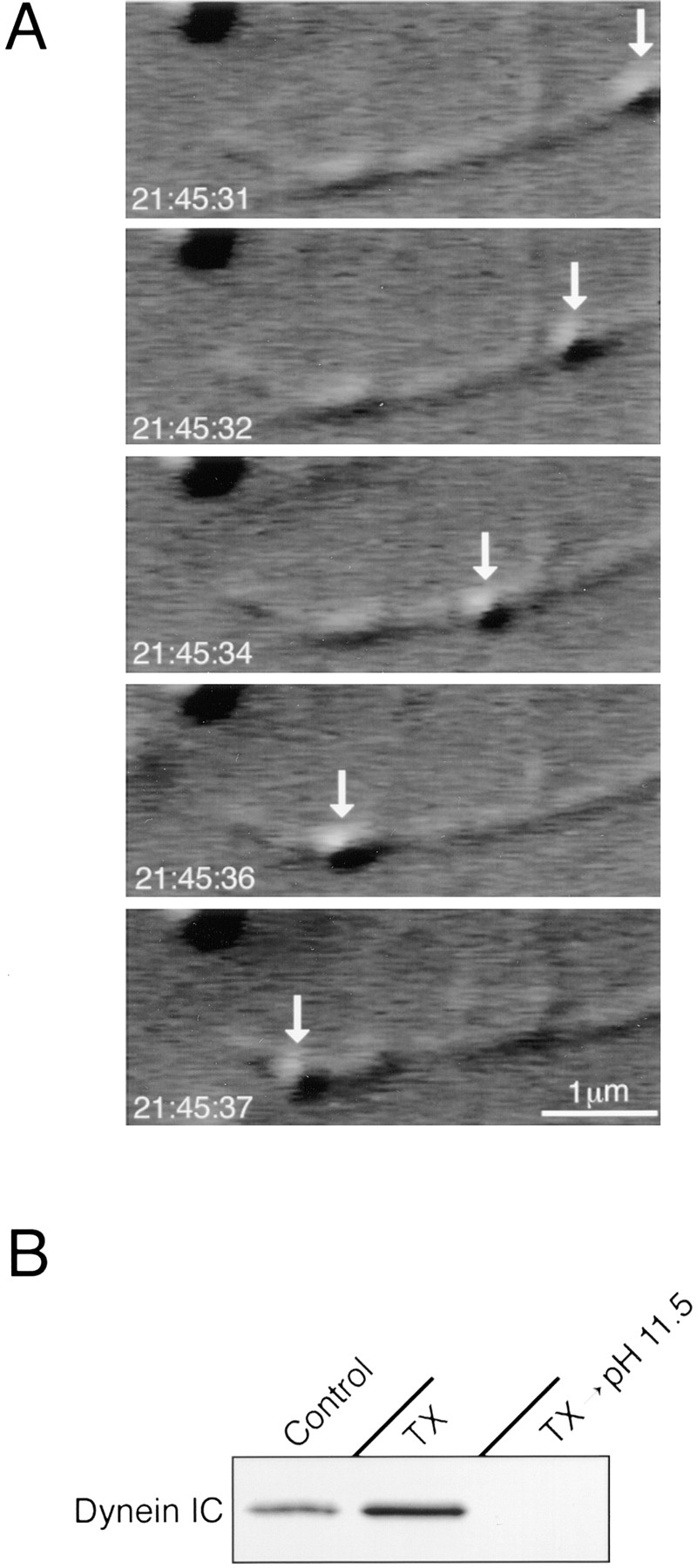
Cytosolic dynein binding to TX-100-extracted Golgi stacks is physiologically relevant. (A) Cytosolic dynein that bound to detergent-extracted Golgi stacks was functional in in vitro motility assays. Golgi stacks were extracted with 1% TX-100 on ice, pelleted and resuspended in buffer. In a motility chamber, the detergent ghosts were incubated with cytosol and with microtubules that were polymerized from the ends of axonemes. This series of video frames shows a membrane ghost (arrow) moving along a microtubule. (B) Dynein binding protein(s) are extracted from Golgi stack detergent ghosts at alkaline pH. Golgi stacks were incubated on ice with PEMS (Control); 1% TX-100 (TX); or 1% TX-100 followed by 0.1 M Na2CO3 (TX → pH 11.5). The membranes were collected by centrifugation, resuspended in PEMS, incubated with cytosol to allow dynein binding, and repelleted through 0.5 M sucrose. This immunoblot shows that cytosolic dynein could bind to both unextracted and TX-100–extracted Golgi membranes, but not to stacks that were sequentially extracted with TX-100 and Na2CO3.
Identification of a Golgi Matrix
In our motility studies, Golgi stack detergent ghosts appeared intact in the light microscope. To examine these structures in more detail, Golgi stacks were extracted with cold 1% TX-100, then separated from solubilized protein by sedimentation onto a sucrose pad and analyzed by electron microscopy. As we have shown previously (Fath et al., 1994), isolated enterocyte Golgi stacks contain many archetypal Golgi apparatus profiles (Fig. 6). We found that distinct cisternae remained after TX-100 extraction, although the detergent vesiculated many of the stacks. These cisternae were clearly formed of a bilayer consistent with glycosphingolipid-rich Golgi membrane microdomains being resistant to cold TX-100 (Brown and Rose, 1992).
Figure 6.
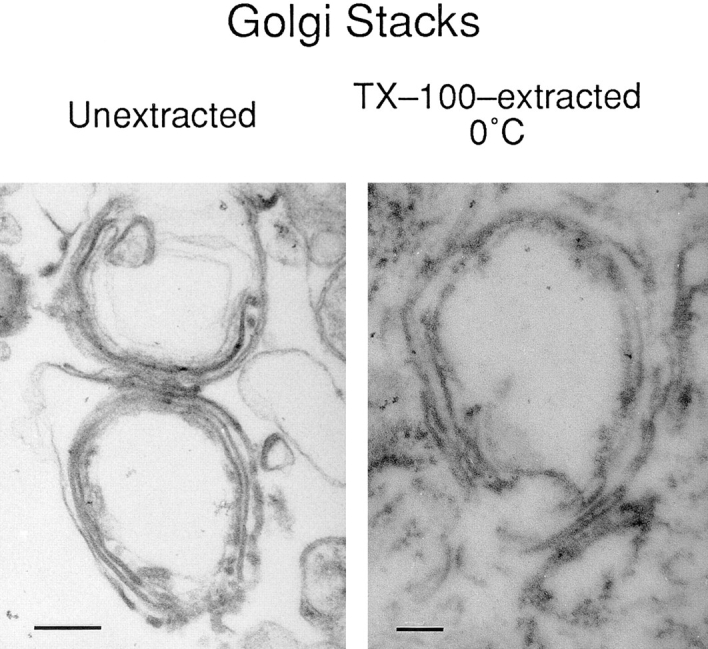
Ultrastructural identification of a Golgi matrix. Unextracted Golgi fraction contained many classical Golgi stack profiles as seen by thin-section electron microscopy. Although ∼75% of the total protein was extracted by ice-cold 1% TX-100 (Fig. 7), a matrix remained that preserved the shape of the Golgi cisternae. Bars: (unextracted) 250 nm; (extracted) 100 nm.
In addition to a bilayer, the detergent-resistant membrane cisternae (and TGN-containing membranes, data not shown) were covered with a dense material or matrix (Fig. 6 B). To begin characterization of the nature of this matrix that was intimately associated with the Golgi membrane detergent ghosts, Golgi stacks or TGN-containing membranes were extracted for 15 min with 1% TX-100 on ice. The extracts were then separated into soluble and insoluble fractions by ultracentrifugation. The supernatants and pellets were analyzed by immunoblotting, which was quantitated by densitometry. The basolateral integral membrane protein Na+K+-ATPase was used to monitor the efficacy of extraction. In stacks (lacking dynein) and TGN-containing membrane fractions (possessing dynein) (Fath et al., 1994), >75% of total protein (as determined by protein assay) and 72% of Na+K+-ATPase were solubilized by TX-100 (Fig. 7). Because TX-100 Golgi membrane ghosts can move on microtubules in motility assays, pellets from both membrane fractions were also immunoblotted for molecular motors. We found that dynein (on TGN-containing membranes), p150_Glued_ and myosin-I were insoluble in TX-100. In agreement with the blots shown on Fig. 4, we found that 64% of Arp1 was extracted by TX-100, in contrast to p150_Glued_, which was completely unextracted by cold TX-100 (Fig. 7).
Figure 7.
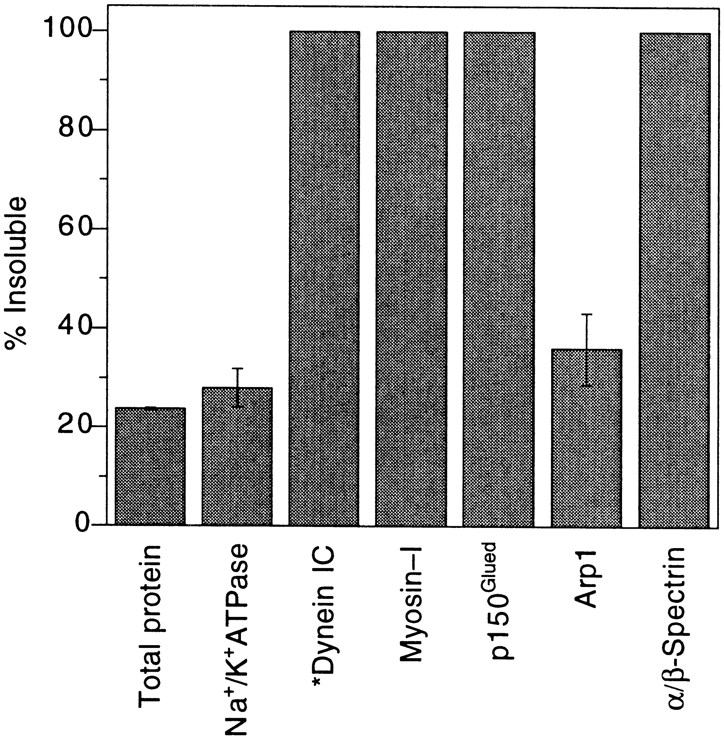
Characterization of Golgi membrane protein solubility in cold TX-100. Golgi membranes were extracted on ice for 15 min with 1% TX-100 and collected by centrifugation. Equivalent volumes of supernatants and pellets were analyzed by immunoblotting, and the percentage of detergent insoluble protein was determined by quantitative scanning densitometry. Except for dynein*, which is absent from stacks, the data are representative of both stacks and TGN-containing membranes, because there were no significant differences between the two membrane fractions. The percentage of total protein was determined by protein assay. Each data point is an average of three or more independent experiments (± SD).
Spectrin and Ankyrin Are Associated with a Golgi Matrix
We wished to establish whether the matrix, identified both ultrastructurally and biochemically, that remains associated with the Golgi membrane detergent ghosts, was composed of members of the recently identified Golgi spectrin matrix (Beck et al., 1994; Devarajan et al., 1996). Therefore, we first immunoblotted intact, isolated Golgi stacks and TGN-containing membranes to determine whether spectrin and ankyrin are present on the unextracted membranes. For spectrin, we used either the affinity-purified βspec-1 polyclonal antibody that recognizes βIΣ*-spectrin (Beck et al., 1994) or a polyclonal antibody that was raised against bovine brain fodrin (α/β-spectrin; Burridge et al., 1982). For ankyrin, we immunoblotted with a polyclonal antibody raised against chicken erythrocyte ankyrin. Both spectrin antibodies recognized spectrin in the stack and TGN-containing membrane fractions as an ∼220–230-kD polypeptide(s) (Fig. 8 A). Due to the decreased resolution at high molecular masses on the 10% SDS–polyacrylamide gels used in this study, it is not possible to assign exact molecular masses, nor is it possible to discern whether both α-and β-spectrin are detected by the fodrin antibody. Hereafter, we will call the polypeptide(s) that react with the fodrin antibody α/β-spectrin. In avians, a common 240-kD α-spectrin is found in all spectrin heterodimers (see review by Bement and Mooseker, 1996). The ankyrin antibody recognized an ∼225–240-kD polypeptide in both membrane fractions (Fig. 8 A), which comigrated with a similar-sized polypeptide from chicken RBC membrane ghosts (data not shown). There was also consistently an ∼160-kD ankyrin immunoreactive peptide that may be constitute a second ankyrin isoform (Beck et al., 1997; Devarajan et al., 1996) or an ankyrin proteolytic breakdown product.
Figure 8.

Ankyrin and spectrin are Golgi peripheral membrane proteins. (A) Isolated Golgi stacks were immunoblotted with polyclonal antibodies recognizing ankyrin or spectrin to determine whether these proteins are associated with the Golgi apparatus. We found that spectrin was associated with Golgi membranes using antibodies either recognizing α/β-spectrin (Control) or βIΣ*-spectrin (not shown). Ankyrin was also bound to Golgi stacks (Control). To examine the mode of association of ankyrin and spectrin with Golgi membranes, isolated membranes were incubated with PEMS buffer (Control), 0.6 M KI, 0.1 M Na2CO3 pH 11.5, or cold 1% TX-100. The membranes were separated from extracted proteins by centrifugation through a 0.5 M sucrose pad, then analyzed by immunoblotting. These blots indicate that both ankyrin and spectrin are tightly bound peripheral membrane proteins that are resistant to extraction with KI and cold 1% TX-100, but are released from the membrane at alkaline pH. (B) We also immunoblotted the Golgi membranes for ankyrin and spectrin following incubation with cytosol. In the presence of added cytosol, spectrin and ankyrin are present on control and 0.6 M KI-extracted membranes, but are absent from membranes that were extracted at pH 11.5 or with cold 1% TX-100.
To demonstrate that spectrin and ankyrin were associated with Golgi membranes and not with potential contaminating plasma membranes, stacks and TGN-containing membranes were incubated with WGA conjugated to agarose. Fractionation on WGA-agarose leads to a differential partitioning of Golgi- and plasma membrane–derived vesicles because the lectin will bind to plasma membranes that vesiculate right-side out, but not to intact Golgi- derived membranes whose lectin binding sites are lumenal, and thus inaccessible (Persson et al., 1991; Beck et al., 1994). We found that 87% of the Golgi stack fraction βIΣ*-spectrin, and 80% α/β-spectrin was associated with membranes that did not bind to the lectin-coated beads. In the TGN-containing membrane fractions, 80% of βIΣ*spectrin, 70% of α/β-spectrin, and 92% of the ankyrin was associated with membranes that did not bind to the WGA-agarose. These data suggest that spectrin and ankyrin are associated with Golgi membranes and not with any possible contaminating plasma membranes.
To determine the mode of association of spectrin and ankyrin with the Golgi membrane, isolated membranes were extracted with 0.6 M KI or at pH 11.5. We found that α/β-spectrin and ankyrin were strongly associated peripheral membrane proteins that remained bound to the membrane in the presence of KI, but were stripped from the membrane at alkaline pH (Fig. 8 A). To determine whether spectrin and ankyrin are associated with the detergent-stable Golgi matrix, Golgi membranes were also solubilized with cold 1% TX-100 and immunoblotted for these components. We found that both α/β-spectrin and ankyrin are part of the Triton Golgi membrane ghosts (Fig. 8 A).
Based on their in vitro binding properties, it has been proposed that dynein may attach to intracellular membranes by binding to the dynactin complex, which in turn is bound to a membrane spectrin-based skeleton (Holleran et al., 1996; Vallee and Sheetz, 1996). Although dynein may be tethered to Golgi membranes indirectly through such a matrix, we found that functional cytosolic dynein may also bind to Golgi membranes by peripheral membrane proteins other than a spectrin/ankyrin membrane skeleton. In our Golgi extraction studies, ankyrin and spectrin are associated with cold 1% TX-100 membrane ghosts (Fig. 8 A). Quite surprisingly, spectrin and the ∼225–240-kD ankyrin were solubilized when these ghosts were incubated with cytosol (Fig. 8 B), conditions which also allow for cytosolic dynein binding (Fig. 4 B). The lower molecular weight ankyrin immunoreactive polypeptide was released to a lesser extent. The absence of spectrin and ankyrin from these membranes did not reflect the nonspecific removal of all membrane proteins because this immunoblot, which was reprobed here, was previously shown to contain both p150_Glued_ and Arp1 (Fig. 4). In that motility supporting cytosolic dynein bound to these Triton-treated membranes, we propose that functional dynein can also bind to Golgi membranes independent of an ankyrin/spectrin matrix.
Discussion
In this study we found that Golgi stacks isolated from polarized intestinal epithelial cells, which lack dynein as isolated, can bind soluble dynein from cytosol. When Golgi stacks are incubated with cytosol plus ATP, 50–200-nm vesicles are shed or budded from the stacks. These budded membranes contain the apical plasma membrane enzyme AP, possess TGN 38/41, lack β-COP, and are highly enriched for dynein relative to the stacks from which they budded. Thus, we propose that there is a regulated binding of dynein to regions of Golgi stacks that are destined to bud. Cytosolic dynein binds to the Golgi membrane via a tightly bound peripheral membrane protein(s), for this binding moiety is sensitive to protease digestion on intact membranes and is extracted from the membrane by alkaline pH, but not by high salt nor by cold TX-100. The bound dynein is functional and can move Golgi membranes towards the minus-ends of microtubules in in vitro motility assays.
We have shown previously that the actin-based motor myosin-I is found on isolated Golgi stacks and TGN-containing membranes, whereas dynein is found only on the TGN-containing membranes, and absent from stacks as isolated (Fath et al., 1994). Thus, the binding of dynein to the Golgi apparatus and Golgi-derived membranes may be regulated both spatially and temporally. Therefore, we began a series of studies focusing on the in vitro binding of cytosolic dynein to Golgi stacks by using an in vitro budding assay (Salamero et al., 1990). We monitored the extent of budding by quantifying the release of membranes containing AP from stacks. We chose AP because it is an abundant apical plasma membrane protein in polarized epithelial cells. In complete budding reactions containing both ATP and cytosol, there is efficient release of 50–200-nm vesicles containing dynein and AP (Fig. 2). There is, however, little dynein (Table I and Fig. 2 A) remaining bound to the stacks after budding. These results suggest that dynein binds to regions of the Golgi stacks that are destined to bud and, more importantly, suggest that dynein binding is a late event in the budding process, occurring when vesicles are budding from the TGN. By quantifying immunoblots, we found that there is a fivefold de-enrichment of β-COP and a fivefold enrichment of TGN 38/41 in the budded membranes in comparison to stacks (Table I). The partitioning of these proteins suggests that budded membranes are not enriched in intracisternal Golgi vesicles (containing β-COP); instead they are enriched in vesicles budded from the TGN. These data also argue that buds form preferentially from the small amount of TGN 38/41 present in the stack preparation. Our results are consistent with the results obtained by Salamero et al. (1990), that budding occurs predominantly from the TGN, and argue that the budded membranes are not simply the result of Golgi stack fragmentation or vesiculation. Thus, a motor thought to be required for the movement of apically targeted membranes may bind to and bud with these membranes from the TGN. It is also possible that this bound dynein may assist in the budding process by providing the force required to pull the membrane into the shape of a bud.
Table I.
Comparison of the Relative Levels of Dynein, TGN 38/41 and β-COP in Golgi Membrane Fractions
| Golgi stacks | Stacks after budding | Budded membranes | |
|---|---|---|---|
| Dynein IC* | 0 | 0 | 1 |
| TGN 38/41 | 0.22 | 0.10 | 1 |
| β-COP | 1 | 0.83 | 0.16 |
Isolated Golgi stacks rarely bound and never moved on microtubules in in vitro motility studies. The lack of motility is consistent with our immunoblot results that kinesin and dynein are absent from isolated Golgi stacks. In the presence of cytosol and ATP, membranes in the Golgi stack fraction and isolated budded membranes moved along microtubules with an average velocity of 1.6 μm/s. These movements are consistent with the binding of dynein that we observe by immunoblotting by being microtubule minus-end–directed (Fig. 3) and inhibited by 2 mM NEM. It is unlikely that these movements are driven by a microtubule minus-end–directed kinesin-like protein. First, although kinesin was present in cytosol, no kinesin could be detected on Golgi stacks (Fig. 1 A). Second, membrane transport continued in buffers containing an equimolar mixture of AMP-PNP and ATP. When equimolar with ATP, AMP-PNP inhibits the motor activity of all kinesins examined to date (Walker and Sheetz, 1993).
Although cytosolic dynein remained tightly bound to Golgi membranes and could pellet with these membranes through 0.5 M sucrose in buffers identical to those used in motility assays, we found that membranes washed free of cytosol moved only infrequently in motility assays. Because dynein (and the dynactin complex) could bind tightly to these membranes, it is unlikely that the continuous presence of cytosol is necessary for supplying additional dynein to replace dynein that might be lost from the membrane. We also found that TGN-containing membranes that contain dynein as isolated (Fath et al., 1994) can translocate on microtubules in motility assays in the absence of cytosol, however, the frequency of movement also greatly increased when cytosol was added with the membranes (data not shown). In that we cannot compare the amount of dynein that is bound in the presence of cytosol with the amount that remains bound in the absence of cytosol, it is possible that sufficient dynein is lost from the membrane to decrease the frequency of membrane movements. A similar decrease in minus-end motility by vesicles isolated from squid axoplasm that were separated from cytosol on flotation gradients has been seen previously (Muresan et al., 1996).
The finding that cytoplasmic dynein IC can bind p150_Glued_ in vitro (Karki and Holzbaur, 1995; Vaughan and Vallee, 1995) and that the dynactin complex stimulates the in vitro motility of membranes (Schroer and Sheetz, 1991), suggests that dynactin may mediate the binding of dynein to intracellular membranes. Although purified cytoplasmic dynein can bind to artificial phospholipid vesicles (Lacey and Haimo, 1994), lipid-bound dynein is presumably not a functional motor without other components such as the dynactin complex (Gill et al., 1991). We found that Golgi stacks that are capable of binding cytosolic dynein contain at least two members of the dynactin complex, p150_Glued_ and Arp1 (Fig. 4 A). Being peripheral membrane proteins, both p150_Glued_ and Arp1 were stripped from the membrane at alkaline pH. When these stripped membranes were incubated with cytosol, nearly control levels of Arp1 rebound, while only a small portion of p150_Glued_ rebound (Fig. 4 B). We found that the 150-kD isoform of p150_Glued_ rebound to higher levels than did the 135-kD isoform. Because the incubated membranes were pelleted through a 0.5 M sucrose cushion, it is possible that both components of the dynactin complex bound to membranes, but a weakly bound p150_Glued_ was stripped off by the sucrose. These data are in agreement with the apparently weak binding of cytosolic p150_Glued_ and the stronger binding of cytosolic Arp1 to KI-stripped membranes that were isolated from squid axoplasm (Muresan, V., and B.J. Schnapp. 1996. Mol. Biol. Cell. 7:402a). We also found that when Golgi stacks were extracted with cold 1% TX-100, conditions allowing dynein binding, Arp1 but not p150_Glued_, was released from the membrane ghosts (Fig. 4 A). These data suggest that members of the dynactin complex may interact with the Golgi membrane independently. Moreover, because detectable levels of dynein did not bind to alkali-stripped Golgi membranes lacking the 135-kD isoform of p150_Glued_, these data are consistent with the hypothesis that p150_Glued_ may be required for dynein binding.
Recently, it has been proposed that dynein and dynactin may be linked indirectly to intracellular membranes via an organelle membrane skeleton that contains spectrin and ankyrin (see Introduction). Ankyrin and spectrin have been localized to the Golgi complex by immunofluorescence in a number of polarized and unpolarized cell lines (Beck et al., 1994; Devarajan et al., 1996; Holleran et al., 1996) and by immunoblotting of isolated rat liver Golgi membranes (Beck et al., 1994). We found that Golgi stacks and TGN-containing membranes that were isolated from polarized intestinal epithelia also contain a spectrin and ankyrin matrix. We have detected Golgi spectrin with an antibody specific for an erythroid β-spectrin (βspec-1) and with a polyclonal antibody that recognizes fodrin (α- and nonerythroid β-spectrin). On the 10% SDS–polyacrylamide gels that we used in this study, it was difficult to discern whether the fodrin antibody recognized α-spectrin, which is found in all avian spectrin heterodimers, or the nonerythroid β-spectrin. Spectrin and ankyrin are tightly bound peripheral membrane proteins that resist extraction from Golgi membranes by 0.6 M KI and cold 1% TX-100, but are extracted at alkaline pH (Fig. 8). We found, however, that spectrin and ankyrin were released from detergent-extracted Golgi membranes by incubation with cytosol. Because cytosolic dynein bound to these membrane ghosts, these results suggest that functional dynein can bind to Golgi detergent ghosts independent of a spectrin matrix. Moreover, these results suggest that a component of cytosol may release this matrix from detergent-extracted membranes. In contrast, spectrin and ankyrin were not released from Golgi membranes that were preincubated in buffer or with 0.6 M KI before adding cytosol.
What may be a role for a matrix on the cytoplasmic face of the Golgi complex? We found that although >75% (Fig. 7) of total Golgi protein is extracted with ice-cold 1% TX-100, these membrane ghosts were closely associated with an amorphous framework (Fig. 6). These structures suggest that the characteristic Golgi morphology may be maintained in part by a Golgi matrix in combination with glycosphingolipid-rich membrane domains (which are not extracted by cold TX-100), because no curved, stacked structures were evident after extraction with the detergent _n_-octylglucoside (data not shown and Garcia et al., 1993). This framework may be similar to a matrix that has been identified biochemically as a detergent and salt resistant matrix from purified rat liver Golgi stacks, which has been proposed to establish a functional polarity of the Golgi stack (Nakamura et al., 1995) or to maintain Golgi structure.
This matrix may also contain in part members of the Golgi-associated coats (e.g., clathrin and COPs) that are thought to have roles in regulating Golgi membrane traffic. There is a growing list of vesicle coats that have been associated with secretory processes (Schekman and Orci, 1996; Robinson, 1997). Support for a role of a spectrin and ankyrin matrix as a Golgi coat comes from studies in which the Golgi apparatus is disrupted by drug treatment. When the Golgi apparatus structure and function are abolished with the drug brefeldin A, βIΣ*-spectrin, β-COP (Beck et al., 1994), and ankyrin (Beck et al., 1997) become diffusely distributed in the cytoplasm. In contrast, when the Golgi apparatus structure, but not function, is disrupted by depolymerizing microtubules with nocodazole, β-spectrin and ankyrin remain associated with the dispersed Golgi membranes. These data suggest that spectrin may be important in Golgi biosynthetic processing and/or packaging.
In addition to maintaining Golgi apparatus structural integrity and regulating carrier vesicle formation, a Golgi matrix may also possess the binding sites for the specific molecular motors required for delivery of the vesicle to the plasma membrane. In this scenario, motors bind to the budding vesicle via a component of a surface matrix (Holleran et al., 1996; Vallee and Sheetz, 1996). The organelle with its surface skeleton and attached motor then translocate to the plasma membrane along cytoskeletal tracks. Our observations that motor components, such as dynein, p150_Glued_ and myosin-I, are present in a detergent insoluble fraction associated with TGN-containing membranes is consistent with the idea that a Golgi matrix organizes or couples the placement of motors with budding membranes. A matrix could therefore organize not only the structure, but the recruitment or placement of specific receptors at certain sites such as regions of the Golgi stacks that are destined to bud. Consistent with a directed placement, it has been reported that the movement of Golgi membrane tubules along microtubules in vitro occurs at the tips of these tubules (Allan and Vale, 1994).
The association of dynein with the Golgi complex of both fibroblasts and enterocytes in which the Golgi apparatus is nearer microtubule minus- or plus-ends, respectively, suggests that dyneins may have multiple roles in Golgi function. The production of isoform-specific dynein antibodies (Vaisberg et al., 1996) may now allow us to begin to explore the roles for dynein in different cellular compartments. We are also only beginning to understand how dynein-based motility may be regulated. Data suggest that dynein-based transport may be directed by regulating the binding of the motor to the membrane by the phosphorylation of dynein heavy- (Lin and Collins, 1992) or light-intermediate chain (Niclas et al., 1996) and by regulating motor activity through differential phosphorylation of the dynein heavy chain (Dillman and Pfister, 1994). Undoubtably, the binding of dynein (and other molecular motors) to membranes is regulated at multiple levels. As we begin to discover the components of the binding machinery, we will begin to understand further how motors are targeted to selected membranes and how multiple motors on the same organelle are regulated in different regions of the cytoplasm.
Acknowledgments
We thank Dr. T. Schroer (Johns Hopkins Univ.) for mAb 150.1; Dr. K. Howell (Univ. of Colorado) for TGN 38/41 antiserum; Drs. W.J. Nelson and K. Beck for Na+K+-ATPase and βIΣ*antiserum (Stanford Univ.); Dr. Kelley Moremen (University of Georgia) for α-mannosidase II antiserum; and Dr. D. Helfman for the antiserum against Arp1 (Cold Spring Harbor Laboratory). We thank Dr. David Asai for purified rat testis dynein (Purdue University). Purified tubulin and sea urchin axonemes were a gift from Dr. T. Salmon (University of North Carolina). We thank Dr. L. Cassimeris (Lehigh University) for Chlamydomonas axonemes and for advice on motility assays. We also thank Dr. S. Gilbert (University of Pittsburgh) for purified tubulin and Tom Harper for assistance with electron microscopy.
Footnotes
1. Abbreviations used in this paper: AP, alkaline phosphatase; AMP-PNP, 5′-adenylylimidodiphosphate; IC, intermediate chains; NEM, _N_-ethylmaleimide; TX-100, Triton X-100.
Address all correspondence to David R. Burgess, Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260. Tel.: (412) 624-4350. Fax: (412) 624-4759. E-mail: dburg+@pitt.edu
References
- Achler C, Filmer D, Merte C, Drenckhahn D. Role of microtubules in polarized delivery of apical membrane proteins to the brush border of the intestinal epithelium. J Cell Biol. 1989;109:179–189. doi: 10.1083/jcb.109.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan V, Vale R. Movement of membrane tubules along microtubules in vitro: evidence for specialized sites of motor attachment. J Cell Sci. 1994;107:1885–1897. doi: 10.1242/jcs.107.7.1885. [DOI] [PubMed] [Google Scholar]
- Bacallao R, Antony C, Dotti C, Karsenti E, Stelzer EHK, Simons K. The subcellular organization of Madin-Darby Canine Kidney cells during the formation of a polarized epithelium. J Cell Biol. 1989;109:2817–2832. doi: 10.1083/jcb.109.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck KA, Buchanan JA, Malhotra V, Nelson WJ. Golgi spectrin: identification of an erythroid β-spectrin homologue associated with the Golgi complex. J Cell Biol. 1994;127:707–723. doi: 10.1083/jcb.127.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck KA, Buchanan J, Nelson WJ. Golgi membrane skeleton: identification, localization and oligomerization of a 195 kD ankyrin isoform associated with the Golgi complex. J Cell Sci. 1997;110:1239–1249. doi: 10.1242/jcs.110.10.1239. [DOI] [PubMed] [Google Scholar]
- Bement, W.M., and M.S. Mooseker. 1996. The cytoskeleton of the intestinal epithelium: components, assembly, and dynamic rearrangements. In Cytoskeleton in Specialized Tissues and in Pathological States. Vol. 3. J.E. Hesketh and I.F. Pryme editors. JAI Press Inc., Greewich, CT. 359–404.
- Bennett V. Spectrin: a structural mediator between diverse plasma membrane proteins and the cytoplasm. Curr Opin Cell Biol. 1990;2:51–56. doi: 10.1016/s0955-0674(05)80030-4. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Wandinger-Ness A, Simons K. Release of putative exocytic transport vesicles from perforated MDCK cells. EMBO (Eur Mol Biol Organ) J. 1988;7:4075–4085. doi: 10.1002/j.1460-2075.1988.tb03301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsel M, Parton R, Kuznetsov SA, Schroer TA, Gruenberg J. Microtubule- and motor dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell. 1990;62:719–731. doi: 10.1016/0092-8674(90)90117-w. [DOI] [PubMed] [Google Scholar]
- Brenner SL, Korn ED. Spectrin-actin interaction. J Biol Chem. 1979;254:8620–8627. [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Burridge K, Kelly T, Mangeat P. Nonerythrocyte spectrins: actin-membrane attachment proteins occurring in many cell types. J Cell Biol. 1982;95:478–486. doi: 10.1083/jcb.95.2.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SW, Meyer DI. Centractin is an actin homologue associated with the centrosome. Nature. 1992;359:246–250. doi: 10.1038/359246a0. [DOI] [PubMed] [Google Scholar]
- Corthésy-Theulaz I, Pauloin A, Pfeffer S. Cytoplasmic dynein participates in the centrosomal localization of the Golgi complex. J Cell Biol. 1992;118:1333–1345. doi: 10.1083/jcb.118.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsen EM. Involvement of detergent-insoluble complexes in the intracellular transport of intestinal brush border enzymes. Biochemistry. 1995;34:1596–1605. doi: 10.1021/bi00005a016. [DOI] [PubMed] [Google Scholar]
- Danielsen M, Cowell GM. Biosynthesis of intestinal microvillar proteins: evidence for an intracellular sorting taking place in, or shortly after, exit from the Golgi complex. Eur J Biochem. 1985;152:493–499. doi: 10.1111/j.1432-1033.1985.tb09223.x. [DOI] [PubMed] [Google Scholar]
- Devarajan P, Stabach PR, Mann AS, Ardito T, Kashgarian M, Morrow JS. Identification of a small cytoplasmic ankyrin (AnkG119) in the kidney and muscle that binds βIΣ* spectrin and associates with the Golgi apparatus. J Cell Biol. 1996;133:819–830. doi: 10.1083/jcb.133.4.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillman JF, Pfister KK. Differential phosphorylation in vivo of cytoplasmic dynein associated with anterogradely moving organelles. J Cell Biol. 1994;127:1671–1681. doi: 10.1083/jcb.127.6.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenckhahn D, Dermietzel R. Organization of the actin filament cytoskeleton in the intestinal brush border: a quantitative and qualitative immunoelectron microscope study. J Cell Biol. 1988;107:1037–1048. doi: 10.1083/jcb.107.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenckhahn, D., T. Jöns, B. Püschel, and F. Schmitz. 1996. Role of the cytoskeleton in the development of epithelial polarity. In Role in Cell Physiology. Vol. 2. J.E. Hesketh, and I.F. Pryme, editors. JAI Press Inc., Greenwich, CT. 141–165.
- Echeverri CJ, Paschal BM, Vaughan KT, Vallee RB. Molecular characterization of 50 kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J Cell Biol. 1996;132:617–633. doi: 10.1083/jcb.132.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endow SA. Determinants of motor polarity in the kinesin proteins. Biophys J. 1995;68:271–274s. [PMC free article] [PubMed] [Google Scholar]
- Fath KR, Burgess DR. Golgi-derived vesicles from developing epithelial cells bind actin filaments and possess myosin-I as a cytoplasmically oriented peripheral membrane protein. J Cell Biol. 1993;120:117–127. doi: 10.1083/jcb.120.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath KR, Trimbur GM, Burgess DR. Molecular motors are differentially distributed on Golgi membranes from polarized epithelial cells. J Cell Biol. 1994;126:661–675. doi: 10.1083/jcb.126.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M, Mirre C, Quaroni A, Reggio H, Le Bivic A. GPI-anchored proteins associate to form microdomains during their intracellular transport in Caco-2 cells. J Cell Sci. 1993;104:1281–1290. doi: 10.1242/jcs.104.4.1281. [DOI] [PubMed] [Google Scholar]
- Gilbert T, Le Bivic A, Quaroni A, Rodriguez-Boulan E. Microtubular organization and its involvement in the biogenetic pathways of plasma membrane proteins in Caco-2 intestinal epithelial cells. J Cell Biol. 1991;113:275–288. doi: 10.1083/jcb.113.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Schroer TA, Szilak I, Steuer ER, Sheetz MP, Cleveland DW. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J Cell Biol. 1991;115:1639–1650. doi: 10.1083/jcb.115.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan B, Bowser R, Novick P. The role of Myo2, a yeast class V myosin, in vesicular transport. J Cell Biol. 1995;128:1055–1068. doi: 10.1083/jcb.128.6.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Simons K. The transGolgi network: sorting at the exit site of the Golgi complex. Science. 1986;234:438–443. doi: 10.1126/science.2945253. [DOI] [PubMed] [Google Scholar]
- Haimo LT, Thaler CD. Regulation of organelle transport: lessons from color change in fish. Bioessays. 1994;16:727–733. [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran EA, Tokito MK, Karki S, Holtzbaur ELF. Centractin (ARP1) associates with spectrin revealing a potential mechanism to link dynactin to intracellular organelles. J Cell Biol. 1996;135:1815–1829. doi: 10.1083/jcb.135.6.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzbaur ELF, Vallee RB. Dyneins: molecular structure and cellular function. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Ingold AL, Cohn SA, Scholey JM. Inhibition of kinesin-driven microtubule motility by monoclonal antibodies to kinesin heavy chains. J Cell Biol. 1988;107:2657–2667. doi: 10.1083/jcb.107.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S, Holzbaur ELF. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J Biol Chem. 1995;270:28806–28811. doi: 10.1074/jbc.270.48.28806. [DOI] [PubMed] [Google Scholar]
- Lacey ML, Haimo LT. Cytoplasmic dynein is a vesicle protein. J Biol Chem. 1992;267:4793–4798. [PubMed] [Google Scholar]
- Lacey ML, Haimo LT. Cytoplasmic dynein binds to phospholipid vesicles. Cell Motil Cytoskel. 1994;28:205–212. doi: 10.1002/cm.970280304. [DOI] [PubMed] [Google Scholar]
- Lafont F, Burkhardt JK, Simons K. Involvement of microtubule motors in basolateral and apical transport in kidney cells. Nature. 1994;372:801–803. doi: 10.1038/372801a0. [DOI] [PubMed] [Google Scholar]
- LaFont F, Simons K. The role of microtubule-based motors in the exocytic transport of polarized cells. Cell Dev Biol. 1996;7:343–355. [Google Scholar]
- Lees-Miller JP, Helfman DM, Schroer TA. A vertebrate actin- related protein is a component of a multisubunit complex involved in microtubule-based vesicle motility. Nature. 1992;359:244–246. doi: 10.1038/359244a0. [DOI] [PubMed] [Google Scholar]
- Lin SXH, Collins CA. Immunolocalization of cytoplasmic dynein to lysosomes in cultured cells. J Cell Sci. 1992;101:125–137. doi: 10.1242/jcs.101.1.125. [DOI] [PubMed] [Google Scholar]
- Louvard, D., M. Kedinger, and H.P. Hauri. 1992. The differentiating intestinal epithelial cell: establishment and maintenance of functions through interactions between cellular structures. In Annual Review of Cell Biology. Vol. 8. G. Palade, editor. Annual Reviews, Inc., Palo Alto. 157–195. [DOI] [PubMed]
- Luzio JP, Brake B, Banting G, Howell KE, Braghetta P, Stanley KK. TGN 38: identification, sequencing and expression of an integral membrane protein of the trans-Golgi network. Biochemical J. 1990;270:97–102. doi: 10.1042/bj2700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermall V, McNally JG, Miller KG. Transport of cytoplasmic particles catalysed by an unconventional myosin in living Drosophilaembryos. Nature. 1994;369:560–562. doi: 10.1038/369560a0. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Muresan V, Godek CP, Reese TS, Schnapp BJ. Plus-end motors override minus-end motors during transport of squid axon vesicles on microtubules. J Cell Biol. 1996;135:383–397. doi: 10.1083/jcb.135.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ, Meshnock PJ. Dynamics of membrane skeleton (Fodrin) organization during development of polarity in Madin-Darby Canine Kidney epithelial cells. J Cell Biol. 1986;103:1751–1765. doi: 10.1083/jcb.103.5.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclas J, Allan VJ, Vale RD. Cell cycle regulation of dynein association with membranes modulates microtubule-based organelle transport. J Cell Biol. 1996;133:585–593. doi: 10.1083/jcb.133.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, Vallee RB. Retrograde transport by the microtubule-associated protein MAP 1C. Nature. 1987;330:181–183. doi: 10.1038/330181a0. [DOI] [PubMed] [Google Scholar]
- Paschal BM, Holzbaur ELF, Pfister KK, Clark S, Meyer DI, Vallee RB. Characterization of a 50kD polypeptide in cytoplasmic dynein preparations reveals a complex with p150Gluedand a novel actin. J Biol Chem. 1993;268:15318–15323. [PubMed] [Google Scholar]
- Persson A, Johansson B, Olsson H, Jergil B. Purification of rat liver plasma membranes by wheat-germ-agglutinin affinity partitioning. Biochem J. 1991;273:173–177. doi: 10.1042/bj2730173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS. Coats and vesicle budding. Trends Cell Biology. 1997;7:99–102. doi: 10.1016/S0962-8924(96)10048-9. [DOI] [PubMed] [Google Scholar]
- Salamero J, Sztul ES, Howell KE. Exocytic transport vesicles generated in vitro from the trans-Golgi network carry secretory and plasma membrane proteins. Proc Natl Acad Sci USA. 1990;87:7717–7721. doi: 10.1073/pnas.87.19.7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoz D, Lainé M-C, Nicolas G. Distribution of microtubules within the intestinal terminal web as revealed by quick-freezing and cryosubstitution. Eur J Cell Biol. 1985;39:481–484. [PubMed] [Google Scholar]
- Schafer DA, Gill SR, Cooper JA, Heuser JE, Schroer TA. Ultrastructural analysis of the dynactin complex: An actin-related protein is a component of a filament that resembles F-actin. J Cell Biol. 1994;126:403–412. doi: 10.1083/jcb.126.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Schnapp BJ, Reese TS, Bechtold R. Kinesin is bound with high affinity to squid axon organelles that move to the plus-end of microtubules. J Cell Biol. 1992;119:389–399. doi: 10.1083/jcb.119.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA, Sheetz MP. Two activators of microtubule-based vesicle transport. J Cell Biol. 1991;115:1309–1388. doi: 10.1083/jcb.115.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler, C.D., and L.T. Haimo. 1996. Microtubules and microtubule motors: mechanisms of regulation. In International Review of Cytology. Vol. 164. K.W. Jeon and J. Jarvik, editors. Academic Press, Inc., San Diego, CA. 269–327. [DOI] [PubMed]
- Toyoshima I, Yu H, Steuer ER, Sheetz MP. Kinectin, a major kinesin-binding protein on ER. J Cell Biol. 1992;118:1121–1131. doi: 10.1083/jcb.118.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trahair JF, Neutra MR, Gordon JI. Use of transgenic mice to study the routing of secretory proteins in intestinal epithelial cells: analysis of human growth hormone compartmentalization as a function of cell type and differentiation. J Cell Biol. 1989;109:3231–3242. doi: 10.1083/jcb.109.6.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisberg EA, Grissom PM, McIntosh JR. Mammalian cells express three distinct dynein heavy chains that are localized to different cytoplasmic organelles. J Cell Biol. 1996;133:831–842. doi: 10.1083/jcb.133.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale R, Schnapp B, Mitchison T, Steuer E, Reese T, Sheetz M. Different axoplasmic proteins generate movement in opposite directions along microtubules in vitro. Cell. 1985;43:623–632. doi: 10.1016/0092-8674(85)90234-x. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Sheetz MP. Targeting of motor proteins. Science. 1996;271:1539–1544. doi: 10.1126/science.271.5255.1539. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Wall JS, Paschal BM, Shpetner HS. Microtubule- associated protein 1C from brain is a two-headed cytosolic dynein. Nature. 1988;332:561–563. doi: 10.1038/332561a0. [DOI] [PubMed] [Google Scholar]
- Vaughan, K.T., and R.B. Vallee. 1995. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150_Glued. J. Cell Biol._ 131:1507–1516. [DOI] [PMC free article] [PubMed]
- Velasco A, Hendricks L, Moremen KW, Tulsiani DRP, Touster O, Farquhar MG. Cell type-dependent variations in the subcellular distribution of α-mannosidase I and II. J Cell Biol. 1993;122:39–51. doi: 10.1083/jcb.122.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RA, Sheetz MP. Cytoplasmic microtubule-associated motors. Annu Rev Biochem. 1993;62:429–451. doi: 10.1146/annurev.bi.62.070193.002241. [DOI] [PubMed] [Google Scholar]
- Weiser MM. Intestinal epithelial cell surface membrane glycoprotein synthesis. I. An indicator of cellular differentiation. J Biol Chem. 1973;248:2536–2541. [PubMed] [Google Scholar]
- Yu H, Toyoshima I, Steuer ER, Sheetz MP. Kinesin and cytoplasmic dynein binding to brain microsomes. J Biol Chem. 1992;267:20457–20464. [PubMed] [Google Scholar]