Genetic Analysis of Collagen Q: Roles in Acetylcholinesterase and Butyrylcholinesterase Assembly and in Synaptic Structure and Function (original) (raw)
Abstract
Acetylcholinesterase (AChE) occurs in both asymmetric forms, covalently associated with a collagenous subunit called Q (ColQ), and globular forms that may be either soluble or membrane associated. At the skeletal neuromuscular junction, asymmetric AChE is anchored to the basal lamina of the synaptic cleft, where it hydrolyzes acetylcholine to terminate synaptic transmission. AChE has also been hypothesized to play developmental roles in the nervous system, and ColQ is also expressed in some AChE-poor tissues. To seek roles of ColQ and AChE at synapses and elsewhere, we generated ColQ-deficient mutant mice. ColQ −/− mice completely lacked asymmetric AChE in skeletal and cardiac muscles and brain; they also lacked asymmetric forms of the AChE homologue, butyrylcholinesterase. Thus, products of the ColQ gene are required for assembly of all detectable asymmetric AChE and butyrylcholinesterase. Surprisingly, globular AChE tetramers were also absent from neonatal ColQ −/− muscles, suggesting a role for the ColQ gene in assembly or stabilization of AChE forms that do not themselves contain a collagenous subunit. Histochemical, immunohistochemical, toxicological, and electrophysiological assays all indicated absence of AChE at ColQ −/− neuromuscular junctions. Nonetheless, neuromuscular function was initially robust, demonstrating that AChE and ColQ do not play obligatory roles in early phases of synaptogenesis. Moreover, because acute inhibition of synaptic AChE is fatal to normal animals, there must be compensatory mechanisms in the mutant that allow the synapse to function in the chronic absence of AChE. One structural mechanism appears to be a partial ensheathment of nerve terminals by Schwann cells. Compensation was incomplete, however, as animals lacking ColQ and synaptic AChE failed to thrive and most died before they reached maturity.
Keywords: acetylcholine, acetylcholinesterase, butyrylcholinesterase, collagen, neuromuscular junction
Acetylcholinesterase (AChE)1 is concentrated in the synaptic cleft of the skeletal neuromuscular junction. Acetylcholine released from motor nerve terminals is rapidly inactivated by the synaptic AChE, thereby limiting the action of the neurotransmitter. The importance of the enzyme is demonstrated by the effects of blocking its activity. Inhibition of AChE, for example by nerve gas, causes prolonged activation of acetylcholine receptors (AChRs), leading to desensitization of the receptors, respiratory paralysis, and death. Partial inhibition of AChE, for example by overexposure to insecticides, results in excessive influx of calcium through the AChR's ion channel, which leads to local necrotic myopathy (Leonard and Salpeter, 1979; Wecker et al., 1986). On the other hand, inhibitors of AChE are therapeutically useful to patients with diseases of inadequate neurotransmission in which levels of AChRs are decreased (such as acquired autoimmune myasthenia gravis; Engel, 1994a) or insufficient acetylcholine is released (such as familial infantile myasthenia; Engel, 1994b).
Although the function of AChE is apparently simple, its patterns of structure and distribution are remarkably complex. First, the transcript of the AChE gene is subject to alternative splicing and its protein products can associate covalently with each other and with noncatalytic subunits (for review see Massoulié et al., 1993, 1998). Second, the oligomers can occur in cytoplasmic, membrane-bound and extracellular matrix–associated forms (for review see Massoulié and Bon, 1982; Toutant and Massoulié, 1988). Third, AChE is found not only at neuromuscular junctions, but also at cholinergic interneuronal synapses, in some noncholinergic neurons, and in some nonneural cells such as erythrocytes (Massoulié et al., 1993). Fourth, a related enzyme, butyrylcholinesterase (BuChE), capable of hydrolyzing acetylcholine, is expressed in some of the same sites as AChE, including the neuromuscular junction (for review see Chatonnet and Lockridge, 1989). Based in part on their complex patterns of expression in embryos and adults, AChE and BuChE have been hypothesized to play a variety of noncholinergic and even noncatalytic roles in embryos and adults (for review see Layer and Willbold, 1995).
Much of the AChE at the neuromuscular junction occurs in forms that have been called asymmetric. Asymmetric forms of AChE were initially identified in Torpedo and Electrophorus electric organ; they were defined and distinguished from the remaining (globular) forms by virtue of their anomalous sedimentation properties in sucrose density gradients. The asymmetry was shown to reflect the association of catalytic AChE subunits with a tail that was rod-shaped, collagenase-sensitive, and rich in amino acids characteristic of collagens (hydroxyproline and hydroxylysine; Lwebuga-Mukasa et al., 1976; Rosenberry and Richardson, 1977). Three asymmetric forms of AChE were characterized, in which one, two, or three tetramers of catalytic subunits (now called AChET) were disulfide-bonded to a single collagenous triple helical tail. These forms are called A4, A8, and A12, to indicate the total number of catalytic subunits per oligomer (Massoulié and Bon, 1982).
Hall (1973) showed that asymmetric forms of AChE were selectively associated with synapse-containing regions of rodent skeletal muscle, whereas globular forms were more uniformly distributed. Later, it was found that synaptic AChE was stably associated with the basal lamina that runs between the motor nerve terminal and the postsynaptic membrane at the neuromuscular junction (McMahan et al., 1978; Sanes and Hall, 1979). The collagen tail of asymmetric AChE is likely to be critical for anchoring the enzyme to the basal lamina, perhaps by association with proteoglycans (Bon et al., 1978; Vigny et al., 1983; Brandan et al., 1985; Deprez and Inestrosa, 1995; Rossi and Rotundo, 1996; Rotundo et al., 1997). However, the relationship of asymmetric AChE to the synapse is not completely understood in that some synaptic AChE may be globular (Anglister et al., 1994) and some asymmetric AChE is found extrasynaptically (Carson et al., 1979; Younkin et al., 1982; Sketelj and Brzin, 1985).
Structural and functional analyses of the asymmetric and synaptic forms of AChE were delayed by the difficulty of isolating its collagenous subunit. Recently, however, cDNAs encoding an AChE-associated collagenous subunit called Q (ColQ) were molecularly cloned from rat muscle (Krejci et al., 1997), based on homology to a previously isolated Q subunit from Torpedo (Krejci et al., 1991). Antibodies to recombinant ColQ recognize native asymmetric AChE and BuChE, and coexpression of cDNAs encoding ColQ and AChET generates asymmetric AChE in heterologous cells (Krejci et al., 1997). Assembly of asymmetric AChE involves interaction of a proline-rich attachment domain (PRAD) in ColQ with a tryptophan amphiphilic tetramerization domain in AChET (Bon et al., 1997; Krejci et al., 1997; Simon et al., 1998). Alternatively spliced products of the ColQ gene that encode a PRAD but no collagenous domain have been described; they may organize other AChE isoforms or interact with other proteins. In fact, ColQ RNA is expressed in many tissues with little or no asymmetric AChE, suggesting that it may have additional roles (Krejci et al., 1997).
Here, we have used homologous recombination in embryonic stem (ES) cells to inactivate the ColQ gene in mice, thereby allowing us to assess roles of the ColQ protein in vivo. Using a combination of biochemical, histological, and electrophysiological methods, we answered the following questions. Does asymmetric AChE in muscle require ColQ protein for assembly or accumulation? Are only asymmetric forms of AChE ColQ-dependent? How much of the AChE concentrated at the neuromuscular junction is ColQ-dependent? Does asymmetric BuChE require the ColQ gene product? How does loss of ColQ affect the structure and function of the neuromuscular junction? Is AChE or ColQ required for proper neuromuscular development? Is ColQ required for the structure or function of nonmuscle tissues?
Materials and Methods
Generation of Mutant Mice
Genomic clones containing the ColQ gene were isolated by screening a 129sv strain mouse genomic library (Stratagene) with cDNAs encoding rat ColQ (Krejci et al., 1997). For targeting vector PRAD1 (Fig. 1 a), a 0.8-kb EcoRI-HindIII fragment containing the exon encoding the PRAD domain as well as adjacent intronic sequences was replaced by a PGK-neo cassette (Tybulewicz et al., 1991). For targeting vector PRAD2 (Fig. 1 a), a 0.6-kb HindIII fragment containing the PRAD exon was replaced by a cassette containing neo plus the Escherichia coli lacZ gene, for monitoring gene expression. The PRAD2 vector also contained a diphtheria toxin-A gene for negative selection (Yagi et al., 1990). Both constructs were transfected into R1 ES cells (Nagy et al., 1993) by electroporation. Homologous recombinants were identified by PCR and confirmed by probing genomic Southern blots with an 0.8-kb fragment of the ColQ gene that was entirely external to the targeting vector (Fig. 1 a). Chimeric mice from one PRAD1 and two PRAD2 ES cell clones gave rise to heterozygous and then homozygous mutant mice. The PRAD2 vector contained a lacZ gene, but no β-galactosidase activity was detected in heterozygotes, either histochemically or immunohistochemically.
Figure 1.

Generation of a ColQ mutant. (a) The targeted alleles. The top line shows schematic of the ColQ protein, including the amino-terminal PRAD domain that links ColQ to the catalytic subunit of AChE. The second line shows gene structure, with exons indicated by dark boxes and introns by lines. The third and fourth lines show the PRAD1 and PRAD2 vectors, in which the exon encoding the PRAD domain was deleted. The fifth and sixth lines, labeled recombinant, show predicted structures of the mutant alleles. Location of probe used for Southern blots is indicated. (b) Southern blots of wild-type ES cells and homologous recombinant PRAD1 and PRAD2 clones. In the recombinants, the 3.2-kb EcoRI fragment from the wild-type gene is shifted to 4.6 kb. (c) PCR assay used to genotype ColQ mutants. (d) Weight of ColQ −/− mice (bottom line) and littermates (top line). Mutant homozygotes are smaller than littermates by P5, and weigh less than half as much as littermates by P20. Bars show mean ± SEM. n = 4–14.
Histology
For immunohistochemistry, sternomastoid and tibialis anterior muscles were embedded in Tissue-Tek OCT compound (Sakura Finetek USA), frozen in liquid nitrogen–cooled isopentane, and sectioned at 10 μm in a cryostat. Sections were blocked for 1 h with 2% BSA and 5% normal goat serum in PBS, incubated with primary antibodies for 1–2 h, washed, and then reincubated with secondary antibodies for another 1–2 h. Antibodies to the following antigens were used: ColQ (Krejci et al., 1997), AChE (a gift of Terrone Rosenberry, Mayo Clinic), α-sarcoglycan, β-dystroglycan, utrophin (Novocastra Laboratories Ltd.), rapsyn (Phillips et al., 1991), and agrin (a gift of Z. Hall, University of California, San Francisco). FITC-conjugated Vicia villosa agglutinin-B4 (VVA-B4) was obtained from Sigma Chemical Co. and rhodamine-α-bungarotoxin was obtained from Molecular Probes.
Cholinesterase activity was detected by the histochemical method of Karnovsky and Roots (1964) on sections fixed with 2% paraformaldehyde in PBS. To distinguish AChE from BuChE, the reaction mixture was supplemented with 10−4 M tetraisopropylpyrophosphoramide (iso-OMPA), a selective inhibitor of BuChE, or with 5 × 10−5 M 1:5-bis (4-allyldimethylammoniumphenyl)-pentan-3-one dibromide (BW284c51), a selective inhibitor of AChE (Austin and Berry, 1953). Where indicated, sections were treated with 1% Triton X-100 for 60 min at room temperature or highly purified bacterial collagenase (800 U/ml, type VII; Sigma Chemical Co.) for 60 min at 37°C before fixation and staining.
For electron microscopy, mice were perfused with lactated Ringer's solution followed by 4% paraformaldehyde and 4% glutaraldehyde in 100 mM cacodylate buffer, pH 7.2. Sternomastoid muscles were dissected and fixed overnight at 4°C. The endplate-rich region of the muscle was refixed in 1% OsO4 in cacodylate buffer, dehydrated, and embedded in Araldite. Thin sections were stained with uranyl acetate and lead citrate.
Biochemistry
Mice were killed and tissues were homogenized in a glass Potter homogenizer in 1 ml of low-salt (LS) buffer, which contained 50 mM Tris, pH 7.0, 40 mM MgCl2, 1% Triton X-100, 2 mM benzamidine, 40 μg/ml leupeptin, and 25 μg/ml pepstatin. The homogenates were centrifuged for 20 min at 60,000 rpm, the supernatant was removed, and the pellet was homogenized in a second aliquot of LS buffer and centrifuged again. The two resulting supernatants comprised the detergent-soluble (DS) extracts. The pellet was then homogenized with 300 μl of high-salt (HS) buffer, which consisted of LS buffer plus 0.8 M NaCl. After centrifugation, the supernatant (HS extract) and the pellet were both saved.
Samples of the DS and HS extracts were analyzed by sedimentation in 5–20% sucrose gradients containing 50 mM Tris, pH 7.0, 40 mM MgCl2, 1% Brij96, and 0.4 M NaCl. Centrifugation was in an SW41 rotor for 16 h at 39,000 rpm. About 75 fractions of 150 μl were collected from the bottom of the tube. 60-μl aliquots were transferred to a microtiter plate. Cholinesterase activity was assayed colorimetrically, using acetylcholine as substrate (Ellman et al., 1961). To assay AChE, iso-OMPA (10−4 M) was included to inhibit BuChE. To assay BuChE, BW284c51 (10−6 M) was included to inhibit AChE. Alkaline phosphatase (6.1 S) and β-galactosidase (16 S) from E. coli were included as internal sedimentation standards, and their activity profiles were used to establish a linear relationship between the fraction numbers and Svedberg units (Simon et al., 1998). All reagents were from Sigma Chemical Co.
Electrophysiology
ColQ −/− mice or control littermates were killed and immediately exsanguinated. Hemi-diaphragms with their associated phrenic nerves were isolated. The two hemi-diaphragms were separated and each was mounted in a 2-ml bath lined with Rhodorsil (Rhône-Poulenc) and superfused with saline of the following composition: 154 mM NaCl, 5 mM KCl, 2 mM CaC12, 1 mM MgCl2, 5 mM Hepes, pH 7.4, and 11 mM glucose. The solution was bubbled with pure O2.
The membrane potential and miniature endplate potentials (mepps) were recorded from endplate regions at room temperature (22–24°C) with intracellular microelectrodes filled with 3 M KCl (resistance of 16–26 MΩ), using conventional techniques and an Axoclamp-2A system (Axon Instruments). Recordings were made continuously from the same endplate before and throughout application of the cholinesterase inhibitors tested. Data were digitized and analyzed using a computer equipped with an analogue and digital I/O interface board (DT2821; Data Translation) and a program kindly provided by Dr. John Dempster (University of Strathclyde, Scotland). Mepps were analyzed individually for amplitude and time course.
Results
Generation of a ColQ Mutant
Clones encoding the ColQ gene were isolated from a genomic library and used to produce two targeting vectors, PRAD1 and PRAD2 (Fig. 1 a). Both vectors deleted the PRAD domain of ColQ, which forms the binding site for the catalytic (AChET) subunits of AChE (Bon et al., 1997). Both targeting vectors gave rise to homologous recombinant ES cells, germline chimeras, heterozygotes, and homozygotes (Fig. 1, b and c). Phenotypes of PRAD1 and PRAD2 homozygotes were identical, so both are called ColQ −/− and described together here.
Homozygous ColQ −/− mice were born in expected numbers, and were externally indistinguishable from heterozygous (ColQ +/−) and wild-type siblings at birth. Around 5 d after birth (postnatal day [P]5), however, ColQ −/− mice began to exhibit a tremor when moving. The tremor persisted throughout life. The homozygotes remained active, responsive, and capable of feeding, but failed to thrive, and grew at a slower rate than littermates (Fig. 1 d). About half of the ColQ −/− mice died at about the time of weaning (P21) and around two-thirds of the survivors died over the next few weeks. 10–20% of ColQ −/− mice lived into adulthood (>3 mo) but remained much smaller than littermates.
ColQ Mutants Lack Synaptic AChE
Sections of skeletal muscle from ColQ −/− mutants and littermates were doubly stained with an antiserum to recombinant ColQ plus rhodamine-α-bungarotoxin, which binds to AChRs and thereby marks synaptic sites. In controls, ColQ was concentrated at synaptic sites and was undetectable extrasynaptically (Fig. 2, a and a′). In ColQ −/− mutants, AChR-rich synaptic sites were nearly normal in size, shape, and distribution (see below) but, as expected, bore no ColQ (Fig. 2, b and b′).
Figure 2.

ColQ −/− endplates lack AChE. Sections of skeletal muscle from ColQ +/− and ColQ −/− littermates were double-labeled with rhodamine-α-bungarotoxin plus antibodies to either the catalytic (AChET) or collagenous (ColQ) subunit of AChE. Homozygous mutant endplates lacked not only ColQ but also all detectable AChET. Bar, 10 μm.
The ColQ subunit is believed to anchor AChE to the basal lamina at the neuromuscular junction. To obtain definitive evidence on this point, we stained sections of mutant and control muscle with antibodies specific for the AChE catalytic subunit. This antibody recognizes both of the known alternatively spliced products (T and H) of the mammalian AChE gene (Duval et al., 1992). AChE was abundant at all synaptic sites in controls, but undetectable in homozygotes (Fig. 2, c and d).
This result suggested that ColQ is required for accumulation of all synaptic AChE. An alternative explanation, however, was that the association of AChE with synaptic membranes or matrix had merely been weakened in the absence of its collagenous tail, in which case AChE might have been present at synapses in vivo but lost during preparation of sections. Therefore, we performed two additional tests to seek AChE at synaptic sites in ColQ −/− muscle in situ. First, we injected fasciculin-2, a selective inhibitor of AChE (Rodriguez-Ithurralde et al., 1983) into ColQ −/− mice and their littermates. This inhibitor was chosen because it penetrates the central nervous system poorly, and is therefore believed to exert its effects predominantly on peripheral AChE. Fasciculin-2 (3–6 μg/g, injected intraperitoneally) caused tremor followed by flaccid paralysis in controls. In mutants, in contrast, effects of fasciculin were minor, and injected animals were reflexive and capable of righting. Second, we recorded mepps from muscle fibers of ColQ −/− mice and littermate controls (Fig. 3). The decay times of mepps were variable but on average slower in homozygotes than in controls (data not shown), presumably because AChE terminates neurotransmitter action at normal neuromuscular junctions (Whathey et al., 1979), whereas diffusion does so in ColQ −/− muscle. More importantly, addition of fasciculin greatly slowed the decay of mepps in normal muscle, but had no significant effect on the time course of mepps in ColQ −/− muscle (from 3.57 ± 0.18 to 3.47 ± 0.07 ms in four experiments; P > 0.2). Together, these toxicological and physiological tests support the idea that ColQ −/− mutants lack functional synaptic AChE.
Figure 3.

Mepps recorded in muscle fibers from 40-d-old ColQ +/− (top) and ColQ −/− (bottom) mice. Addition of the AChE inhibitor fasciculin-2 (320 nM) to the bath increased the amplitude and prolonged the decay time constant of mepps in control muscle but had no significant effect on the size or shape of mepps in ColQ −/− muscle. Each trace represents the average of 50 mepps. The decay phases of averaged mepps were fitted by a single exponential from 10% to 90% of the maximal amplitude (vertical lines). The difference in mepp amplitudes between ColQ +/− and ColQ −/− muscles is due to differences in muscle fiber diameter and input resistance.
ColQ Is Required for Assembly of All Asymmetric and Some Globular AChE in Muscle
We used sucrose density gradient centrifugation to assess the molecular forms of AChE present in ColQ −/− muscles. Globular forms are extracted into detergent-containing buffers at low ionic strength, whereas asymmetric (collagen-tailed forms) are extracted only at high ionic strength (Bon et al., 1978). Therefore, we first homogenized muscles with detergent-containing LS buffers, then reincubated the pellet with HS buffers to selectively solubilize asymmetric forms.
Control (ColQ +/+ and ColQ +/−) muscles contain three asymmetric forms, corresponding to a single helical trimer of ColQ chains attached to one, two, or three tetramers of catalytic subunits (A4, A8, and A12, respectively; Massoulié and Bon, 1982). In contrast, no asymmetric forms were detectable in ColQ −/− muscle (Fig. 4, b and c). Because some asymmetric AChE is difficult to solubilize from tissue (Rossi and Rotundo, 1993), we also assayed the pellet remaining after HS and detergent extraction. AChE activity in insoluble fractions was 14% of that in the HS extract for ColQ +/+ muscles (n = 4), and 30% of that in the HS extract for ColQ +/− muscles. However, AChE activity was undetectable in the insoluble fraction from ColQ −/− muscles. Similar results were obtained with muscles from neonates (P0) and at P20. Thus, all asymmetric AChE in muscle requires ColQ for its assembly and/or accumulation.
Figure 4.


Molecular forms of cholinesterase in neonatal ColQ mutant muscles. DS and HS pools of cholinesterase, which correspond to globular and asymmetric forms, respectively, were separated on sucrose gradients. Fractions were incubated in the presence of the BuChE inhibitor iso-OMPA (a and b) or the AChE inhibitor BW284c51 (d and e), to selectively assay AChE or BuChE, respectively. a–e show profiles from representative gradients. In each case, ColQ −/−, ColQ +/−, and ColQ +/+ littermates were assayed in parallel, and with identical incubation times. High background levels in e reflect low specific activity, which necessitated overnight incubation of samples. c shows average AChE activities from all experiments, expressed as total activity in International Units (IU) per muscle. In the absence of ColQ, muscles are devoid of asymmetric forms of AChE and BuChE. Levels of G4 na AChE are also dramatically reduced. At P20, asymmetric AChE and BuChE were also completely absent in ColQ −/−, but G4 AChE was present.
Surprisingly, loss of AChE in ColQ −/− mice was not limited to the asymmetric forms. At birth, three major peaks of AChE activity were distinguishable in detergent extracts of control muscle, corresponding to amphiphilic monomers and dimers (G1 a and G2 a) and nonamphiphilic tetramers (G4 na; Massoulié et al., 1998). G1 a and G2 a were retained in neonatal ColQ −/− muscles, but the G4 na peak was almost entirely absent (Fig. 4 a). In addition, the pool of globular tetramers extracted by HS was absent from ColQ −/− neonates (Fig. 4 b). At P20, however, amphiphilic and nonamphiphilic tetramers (G4 a and G4 na) as well as G1 a and G2 a forms were present in ColQ −/− muscles (data not shown). Thus, ColQ-independent mechanisms for assembly or retention of G4 do exist (see Discussion).
ColQ Is Required for Assembly of Asymmetric BuChE
BuChE is homologous to AChE, hydrolyzes acetylcholine, and is present at the neuromuscular junction (Chatonnet and Lockridge, 1989; Chapron et al., 1997). Like AChE, BuChE catalytic monomers assemble into both globular and asymmetric multimers, and the asymmetric multimers bear a collagenase-sensitive subunit (Vigny et al., 1978; Toutant et al., 1985). To determine whether ColQ serves as the collagen tail for BuChE, we assayed sucrose density gradients in the presence of a selective inhibitor of AChE, BW284c51. Asymmetric and globular forms of BuChE were detectable in control muscle (Fig. 4, d and e). As reported previously, S values of BuChE differed from those of AChE (Vigny et al., 1978; Krejci et al., 1997), providing assurance that the activity seen in the presence of BW284c51 did not reflect incomplete inhibition of AChE. Moreover, activity was inhibited by the selective inhibitor of BuChE, iso-OMPA (data not shown). Globular isoforms of BuChE were retained in ColQ −/− mutants, but no asymmetric BuChE was detectable (Fig. 4, d and e). Thus, the ColQ gene appears to encode the collagenous tails of both AChE and BuChE.
Lacking antibodies to BuChE, we used the histochemical stain of Karnovsky and Roots (1964) to localize BuChE in control and mutant muscles. This method relies on hydrolysis of the acetylcholine analogue, acetylthiocholine, which is cleaved by both AChE and BuChE. At P20, reaction product was readily detectable at ColQ −/− synaptic sites, although levels were substantially reduced compared with controls (Fig. 5, a and b). The AChE inhibitor BW284c51 had no effect on the staining of ColQ −/− synaptic sites, but decreased staining in controls to approximately the level seen in mutants (Fig. 5, c and d). This result provides independent evidence that AChE is depleted from ColQ −/− neuromuscular junctions. In contrast, the BuChE inhibitor iso-OMPA decreased staining of control synaptic sites only slightly, but completely abolished staining of ColQ −/− synaptic sites (Fig. 5, e and f). Thus, the synaptic acetylcholine-hydrolyzing activity in mutants is largely or entirely BuChE. Reaction product was undetectable in controls even after prolonged incubation when a mixture of BW284c51 plus iso-OMPA was added with the substrate, indicating that AChE and BuChE together account for most or all of the ACh-hydrolyzing activity at control synapses (Fig. 5, g and h).
Figure 5.
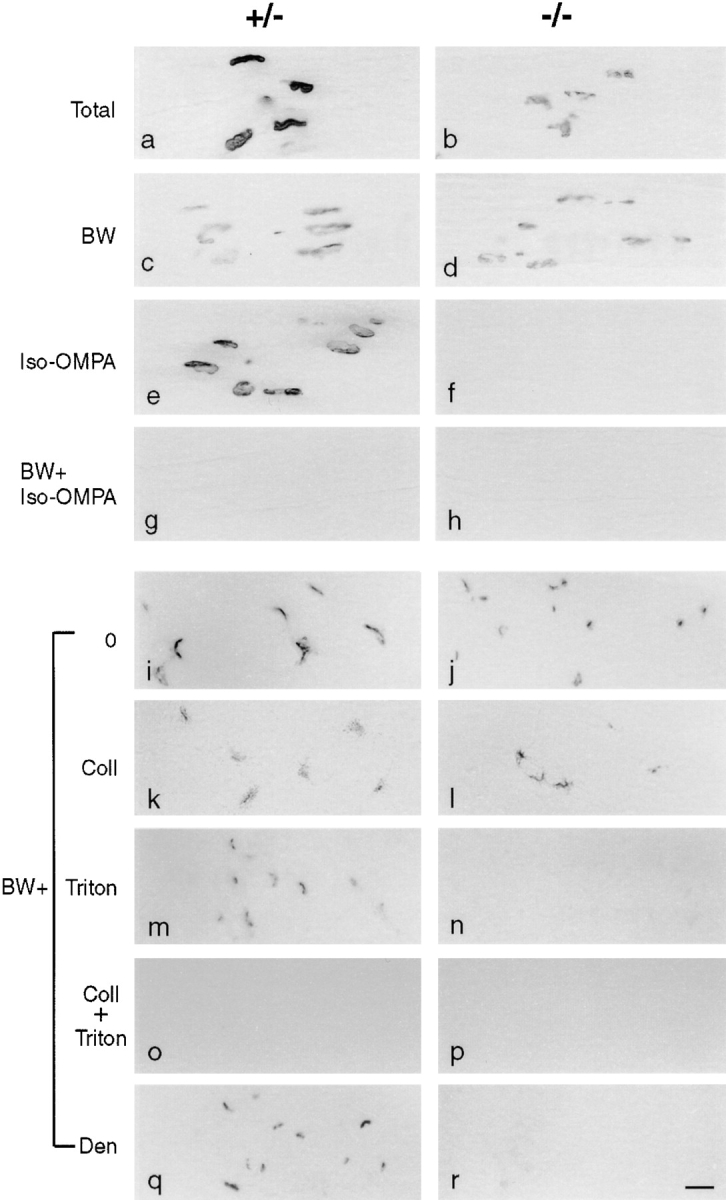
Sections of muscle from P20 ColQ −/− and ColQ +/− littermates stained for cholinesterase activity by the Karnovsky method. a–h are longitudinal sections; i–r are cross-sections. (a and b) Levels of cholinesterase activity are much higher at control than ColQ −/− endplates. (c, d, i, and j) BW284c51 (BW), a selective inhibitor of AChE, has no effect on synaptic cholinesterase activity in ColQ −/− muscle, but decreases activity in controls to a level equivalent to that in the mutant. (e and f) Iso-OMPA, a selective inhibitor of BuChE, has only a slight effect on cholinesterase activity in controls but abolishes all activity in mutants. (g and h) No activity is detectable in either muscle in the presence of both BW284c51 and iso-OMPA. These results indicate that both AChE and BuChE are present at control endplates, but only BuChE at ColQ −/− endplates. (k and l) Incubation of sections with collagenase to release asymmetric enzyme before staining for BuChE (BW+) reduces activity in controls but has no effect at ColQ −/− synapses. (m and n) After incubation with Triton X-100 to release membrane-bound enzyme, some BuChE activity persists at control synapses but is abolished at ColQ −/− synapses. (o and p) No BuChE activity is detectable in controls or mutants after sequential treatment with Triton and collagenase. (q and r) 3 d after nerve section, when nerve terminals have degenerated, BuChE persists at control synaptic sites but is lost from mutant synaptic sites. Bar, 20 μm.
To obtain information on the molecular forms and cellular localization of synaptic BuChE, we treated cross-sections in various ways then stained them in the presence of BW284c51. In both control and ColQ −/− muscle, reaction product was tightly associated with synaptic sites (Fig. 5, i and j). Pretreatment of sections with either collagenase, which releases matrix-associated enzyme, or with the detergent Triton X-100, which releases membrane bound or intracellular enzyme, reduced staining of synaptic sites in control muscles (Fig. 5, k and m). Combined treatment with collagenase and Triton X-100 removed all detectable BuChE (Fig. 5, o and p). Thus, both matrix- and membrane-associated BuChE are present at control synapses. In ColQ −/− muscle, collagenase treatment had no effect on the level of BuChE (Fig. 5 l) but detergent treatment abolished all staining (Fig. 5 n), confirming the absence of matrix-associated BuChE and showing that residual activity is membrane-associated. Denervation, which causes loss of nerve terminals and rapid remodeling of terminal Schwann cells, led to complete loss of this membrane-associated BuChE from ColQ −/− synapses but had little effect on controls (Fig. 5, q and r). Together, these results suggest that both membrane- and matrix-associated forms of BuChE are normally concentrated at the neuromuscular junction. The BuChE of the synaptic cleft is predominantly matrix-associated, asymmetric, and requires ColQ for assembly or localization. Membrane-associated BuChE that persists in the absence of ColQ, in contrast, is likely to be associated with nerve terminals or terminal Schwann cells, although we cannot exclude the possibility that it is postsynaptic.
Although the synapse-associated BuChE in ColQ −/− muscles did not appear to be localized to the synaptic cleft, it might have been near enough to hydrolyze released acetylcholine. We tested this possibility by determining whether iso-OMPA affected the decay of mepps recorded from ColQ −/− neuromuscular junctions. No significant effect was seen (τ = 2.70 ± 0.15 ms in normal saline and 2.98 ± 0.49 ms in the presence of 10 μM iso-OMPA; n = 4; P > 0.2). Therefore, although there is synapse-associated BuChE in ColQ −/− mice, it does not appear to provide a mechanism to compensate for loss of AChE.
Synaptic Structure Is Abnormal in ColQ Mutants
To assess the size and shape of neuromuscular junctions in ColQ −/− muscle, thick longitudinal sections were stained with rhodamine-α-bungarotoxin. In controls, the AChR-rich membrane is a roughly circular plaque at birth, then passes through a perforated-plaque stage (Steinbach, 1981) and eventually matures into a pretzel-like array of distinct AChR-rich branches by P20 (Fig. 6 a). The precise geometry of branches is unique at each synaptic site, but the general pattern is stereotyped. In mutant homozygotes, in contrast, synaptic geometry was variable. Some synaptic sites were smaller than controls but roughly normal in appearance (Fig. 6 b), some retained the immature appearance characteristic of ∼1-wk-old controls (Fig. 6 c), and some appeared fragmented (Fig. 6 d). Approximately 40% of the neuromuscular junctions were normal in geometry, another 40% were fragmented, and the remaining 20% were immature.
Figure 6.

Geometry of synapses in ColQ −/− mice. Longitudinal sections of P20 control (a) and ColQ −/− muscle were stained with rhodamine-α-bungarotoxin. In controls, the postsynaptic membrane contains a set of interconnected AChR-rich gutters. In mutants, some synaptic sites are relatively normal in appearance (b), other appears immature (c), and still others appear fragmented (d). Bar, 10 μm.
Electron microscopy revealed two structural abnormalities at synaptic sites in ColQ −/− muscles. First, the cytoplasm beneath the postsynaptic membrane was often riddled with holes, consistent with a localized degenerative response (Fig. 7 b). Such local degeneration has been observed after acute inhibition of AChE (Laskowski et al., 1977), and is believed to result from entry of Ca2+ through excessively activated AChRs (Leonard and Salpeter, 1979). Consistent with this interpretation, signs of necrosis were seldom observed in nonsynaptic regions of ColQ −/− muscle, and no signs of muscle fiber degeneration and regeneration (such as central nuclei, which are diagnostic of immature or regenerated fibers) were detectable. Second, nerve terminals were sometimes partially enwrapped by processes of Schwann cells (Fig. 7 d). Such enwrapment has been seen in several pathological situations, and can occur after muscle damage, or as a direct consequence of abnormalities in the synaptic cleft (Duchen et al., 1974; Jirmanová, 1975; Noakes et al., 1995; Patton et al., 1998). Because Schwann cell processes are impermeable to neurotransmitter, their intrusion into the synaptic cleft might represent one adaptive mechanism by which ColQ −/− synapses limit activation of AChRs.
Figure 7.
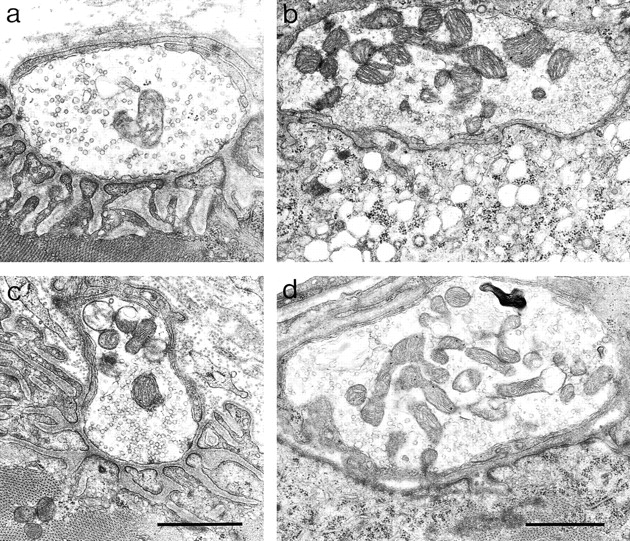
Ultrastructure of synapses in ColQ −/− mice. Electron micrographs from P20 control (a), P20 ColQ −/− (b), and P180 ColQ −/− (c and d) muscles. In controls, nerve terminals lie atop a folded postsynaptic membrane and are capped by processes of Schwann cells. In ColQ −/− mutants, the subsynaptic cytoplasm appears necrotic at some synapses (b), and some nerve terminals are enwrapped by Schwann cells (d). Even at 6 mo of age, however, some mutant synapses appear normal (c). Bar, 1 μm.
Interestingly, the incidence of subsynaptic necrosis and Schwann cell enwrapment of nerve terminals varied as a function of age. At P20, nearly two-thirds of synaptic sites encountered (80/125) showed clear signs of necrosis. By 6 mo of age, however, <5% of synaptic profiles (2/71) showed necrosis, and many were indistinguishable from controls (compare Fig. 7, a and c). This difference raises the possibility that damage occurring early is repaired and that compensatory mechanisms, such as Schwann cell enwrapment, prevent damage from recurring. Indeed, the incidence of nerve terminals partially or completely enwrapped by Schwann cell processes increased somewhat, from one-third (42/125) at P20 to over one-half (39/71) at 6 mo of age. This difference suggests either that Schwann cell enwrapment is progressive, or that individuals with more complete enwrapment are more likely to survive. Necrosis and Schwann cell wrapping were seen in only 1 and 4 of 83 control endplates, respectively.
Finally, we stained sections of muscle from ColQ −/− mice and littermate controls with a panel of antibodies to proteins present at synaptic sites. These included the transmembrane proteins α-sarcoglycan and β-dystroglycan, which are present throughout the muscle fiber but concentrated in the postsynaptic membrane; rapsyn and utrophin, which are selectively associated with the subsynaptic cytoplasm; and agrin, which is concentrated in synaptic basal lamina (Sanes and Lichtman, 1999). The distribution of all of these markers was qualitatively similar in control and homozygous mutant mice (Fig. 8, a–j).
Figure 8.

Molecular architecture of synapses in ColQ −/− mice. Sections from ColQ −/− and control muscle were doubly stained with rhodamine-α-bunagrotoxin (a′–l′) plus antibodies to α-sarcoglycan (a and b), β-dystroglycan (c and d), rapsyn (e and f), utrophin (g and h), or agrin (i and j), or with the lectin VVA-B4 (k and l). No qualitative differences were apparent between mutant and control synapses. Bar, 10 μm.
We also stained sections with the lectin VVA-B4. VVA-B4 binds to glycoconjugates that terminate with a β-_N_-acetylgalactosaminyl residue and selectively stains the synaptic cleft at the neuromuscular junction (Scott et al., 1988). We showed previously that VVA-B4 binds to asymmetric but not globular forms of AChE in muscle, but presented indirect evidence that other VVA-B4–binding proteins were also present in the synaptic cleft (Scott et al., 1988; Martin and Sanes, 1995). VVA-B4 stained synaptic sites nearly as intensely in ColQ −/− muscle as in control muscle (Fig. 8, k and l), providing direct evidence for the existence of additional synaptic VVA-B4–binding moieties.
ColQ Is Required for Assembly of All Asymmetric AChE
Asymmetric forms of AChE are most abundant in skeletal muscle, but are also present in several other tissues (Toutant and Massoulié, 1988). To determine whether asymmetric AChE in nonmuscle tissues requires ColQ for assembly, we analyzed HS extracts of brain and heart. Low levels of asymmetric AChE were detectable in both tissues from control mice but in neither tissue from ColQ −/− mice (Fig. 9).
Figure 9.

ColQ is required for assembly of asymmetric forms of AChE in heart and brain. HS extracts of heart (a) or brain (b) of 2-mo-old mice were fractionated on sucrose gradients and assayed for AChE. Only portions of the gradient corresponding to 8–20S material are shown. No asymmetric AChE was detected in ColQ −/− tissue.
We also made a preliminary survey of tissue structure in the major organs of ColQ −/− mice, but found no abnormalities.
Discussion
Previous biochemical studies have shown that the ColQ protein is a collagenous subunit of asymmetric AChE, and suggested that it is critical for accumulation of AChE in the synaptic cleft of the neuromuscular junction. Analysis of ColQ −/− mutant mice has now allowed us to ascertain, in situ, the roles that ColQ plays in the assembly and anchoring of AChE and in the development and function of the neuromuscular junction. Specifically, we have been able to answer the seven questions posed in the Introduction.
First, no asymmetric AChE is present in muscles that lack ColQ, presumably because the ColQ protein forms the collagen tail that endows asymmetric forms with their distinguishing feature. This result was expected but not inevitable. For example, ColQ might have been only one of multiple collagenous subunits, a possibility suggested by immunological studies showing a structural change in the collagen tail during posthatching development in birds (Tsim et al., 1988b). Alternatively, a synaptic collagen that normally plays other roles (Miner and Sanes, 1994, 1996) might have been capable of compensating for lack of ColQ in mutants.
Second, muscles from newborn ColQ −/− mice lack not only all asymmetric AChE, but also the nonamphiphilic tetrameric form of AChE, G4 na. This observation was unexpected because the G4 na molecules have been assumed to be homotetramers of AChET catalytic subunits. Possible explanations for this result include the following. G4 na molecules may be derived from collagen-tailed molecules by proteolysis of the collagen tail, either in vivo or after extraction. Assembly of the tetramer might require transient association of AChET with ColQ. Assembly might require an alternatively spliced product of the ColQ gene that contains the amino-terminal PRAD domain, but not the collagenous domain. Such alternatively spliced products do in fact occur (Krejci et al., 1997), but have not yet been characterized. Some of the G4 na may comprise tetramers associated with a single full-length collagen chain rather than a triple helix; such molecules sediment around 10S (Krejci, E., unpublished results). In any event, muscles of ColQ −/− mice did contain both amphiphilic and nonamphiphilic tetramers (G4 a and G4 na) at P20. These tetramers might have been assembled without any organizer or might contain an alternate organizing subunit such as the still incompletely characterized P subunit (Massoulié et al., 1993). Thus, although the PRAD of ColQ can induce tetramerization of AChET (Bon et al., 1997; Simon et al., 1998), and may do so in vivo at P0, there are clearly additional PRAD-independent mechanisms for forming tetramers.
Third, no detectable AChE is present at synaptic sites in ColQ −/− muscles. Histochemical, immunochemical, toxicological, and electrophysiological assays all gave similar results in this regard. The lack of synaptic AChE could reflect loss of collagen-tailed AChE and/or loss of G4 forms. We favor the interpretation that all synaptic AChE is collagen-tailed because all of the histochemically detectable AChE can be removed by collagenase treatment of sections (data not shown) or tissue (Hall and Kelly, 1971). In addition, all globular forms (G1 a, G2 a, G4 a, and G4 na) are present in ColQ −/− mice at P20, yet synaptic AChE is absent at all ages tested. Globular AChE is associated with isolated basal lamina from frog muscles (Anglister et al., 1994), but this may result from the alterations produced in the experimental system of muscle and nerve degeneration.
Fourth, ColQ is required for assembly or accumulation of asymmetric BuChE as well as AChE. In birds, collagen-tailed molecules containing both AChE and BChE subunits have been detected in muscles of hatchlings (Tsim et al., 1988a), suggesting that both enzymes use a common tail. Likewise, Krejci et al. (1997) showed that antibodies to ColQ interact with both AChE and BuChE as assessed by sucrose density centrifugations. Taken together with these biochemical data, our genetic results strongly support the conclusion that the ColQ gene produces the collagen tails for both AChE and BuChE.
Fifth, the function of the neuromuscular junction is impaired in the absence of ColQ. However, the defects are not as severe as might be expected, considering the dramatic effects of acutely administering extremely selective blockers of peripheral AChE such as fasciculin. In particular, movements of ColQ −/− mice are relatively normal, while fasciculin-treated normal mice are paralyzed. Moreover, the postsynaptic necrosis observed in ColQ −/− weanlings, a likely result of excessive calcium influx through AChRs (Leonard and Salpeter, 1979), is largely absent from those mutants that survive to later ages. These observations suggest that neuromuscular junctions compensate for the absence of AChE. Compensation does not appear to involve persistence or upregulation of BuChE: asymmetric BuChE is lost in the mutant and the BuChE inhibitor, iso-OMPA, does not affect the time course of mepps. Instead, ultrastructural studies suggest a simple structural mechanism to limit the amount of neurotransmitter that reaches the postsynaptic membrane: invasion of the synaptic cleft by Schwann cell processes at ColQ −/− synapses. Enwrapment of nerve terminals by Schwann cell processes is, in fact, actively regulated by another component of synaptic basal lamina, laminin-11 (Patton et al., 1998). ColQ might exert a similar effect. In addition, compensatory changes in quantal size and quantal content may occur; such homeostatic changes have been documented in response to several chemical or genetic perturbations of the efficacy of neuromuscular transmission in both vertebrates and invertebrates (reviewed in Landmesser, 1998). Currently, we are using electrophysiological methods to seek such alterations in ColQ −/− mice, with the aim of learning how the synapse combines structural and physiological compensations to adapt to the challenge posed by AChE deficiency.
Sixth, initial formation of the neuromuscular junction proceeds in the absence of ColQ and AChE. AChE promotes the extension of neurites from cultured motoneurons by a noncatalytic mechanism (Sternfeld et al., 1998; see also Small et al., 1995), and overexpression of AChE in transgenic frogs and mice perturbs formation of neuromuscular junctions in vivo (Seidman et al., 1994; Andres et al., 1997; Sternfeld et al., 1998). Our results imply that the developmental roles of ColQ and AChE at the synapse are either subtle or masked by compensatory mechanisms. However, it remains possible that expression of AChE at other sites is crucial for synaptogenesis. Moreover, at later stages, there are clear defects in the maturation of ColQ −/− neuromuscular junctions: some remain immature in geometry and others appear fragmented. These abnormalities could reflect a direct requirement for AChE or ColQ in synaptic maturation or maintenance, but might also be secondary consequences of subsynaptic necrosis or alterations in activity levels. Further experiments will be required to distinguish these alternatives.
Seventh, ColQ is required for assembly or accumulation of asymmetric AChE in all tissues tested, but tissues other than skeletal muscle are not obviously defective in structure or function. However, it is premature to conclude that ColQ plays no roles in these tissues. For example, asymmetric AChE is concentrated in synaptic layers of the retina (Reiss et al., 1996); analyses of these synapses have not yet been undertaken. In addition, it is tempting to speculate that in AChE-poor tissues, ColQ or other PRAD-containing products of the ColQ gene serve to anchor proteins other than AChE to membranes or matrix. Identification of such proteins will guide the search for subtle phenotypes in ColQ −/− mice.
Finally, it is interesting to note the parallels between ColQ −/− mice and humans with AChE-deficiency syndrome (CMS-Ic). This syndrome was described by Engel et al. (1977) over 20 years ago, and was reported just this year to result from defects in the human COLQ gene (Donger et al., 1998; Ohno et al., 1998). Some patients appear to be protein nulls, while others have mutations in the carboxy terminus of ColQ that permit formation of asymmetric AChE but prevent its stable association with the basal lamina of the synaptic cleft. All of the COLQ patients are severely myasthenic, as are the ColQ −/− mice, making the mice a valid model for assessing pathogenic mechanisms and therapeutic interventions.
Acknowledgments
We thank Mia Nichol, Carol Borgmeyer, Jimmy Gross, and Sherry Weng for assistance.
This work was supported by grants from the National Institutes of Health and Centre National de la Recherche Scientifique, the Direction des Recherches et Etudes Techniques, the Association Française contre les Myopathies, and Human Frontier Science Program Organization. G. Feng was a fellow of the Jane Coffin Childs Memorial Fund for Medical Research.
Abbreviations used in this paper
AChE
acetylcholinesterase
AChR
acetylcholine receptor
BuChE
butyrylcholinesterase
ColQ
collagen Q
DS
detergent-soluble
ES cells
embryonic stem cells
HS
high-salt
LS
low-salt
mepps
miniature endplate potentials
PRAD
proline-rich attachment domain
VVA-B4
Vicia villosa agglutinin-B4
Footnotes
G. Feng and E. Krejci contributed equally to this work.
References
- Andres C, Beeri R, Friedman A, Lev-Lehman E, Henis S, Timberg R, Shani M, Soreq H. Acetylcholinesterase-transgenic mice display embryonic modulations in spinal cord choline acetyltransferase and neurexin I beta gene expression followed by late-onset neuromotor deterioration. Proc Natl Acad Sci USA. 1997;94:8173–8178. doi: 10.1073/pnas.94.15.8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglister L, Haesaert B, McMahan UJ. Globular and asymmetric acetylcholinesterase in the synaptic basal lamina of skeletal muscle. J Cell Biol. 1994;125:183–196. doi: 10.1083/jcb.125.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin L, Berry WK. Two selective inhibitors of cholinesterase. Biochem J. 1953;54:695–700. [PMC free article] [PubMed] [Google Scholar]
- Bon S, Massoulié J. Quaternary association of acetylcholinesterase. I. Oligomeric associations of T subunits with and without the amino-terminal domain of the collagen tail. J Biol Chem. 1997;272:3007–3015. doi: 10.1074/jbc.272.5.3007. [DOI] [PubMed] [Google Scholar]
- Bon S, Coussen F, Massoulié J. Quaternary associations of acetylcholinesterase. II. The polyproline attachment domain of the collagen tail. J Biol Chem. 1997;272:3016–3021. doi: 10.1074/jbc.272.5.3016. [DOI] [PubMed] [Google Scholar]
- Brandan E, Maldonado M, Garrido J, Inestrosa NC. Anchorage of collagen-tailed acetylcholinesterase to the extracellular matrix is mediated by heparan sulfate proteoglycans. J Cell Biol. 1985;101:985–992. doi: 10.1083/jcb.101.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson S, Bon S, Vigny M, Massoulie J, Fardeau M. Distribution of acetylcholinesterase molecular forms in neural and non-neural sections of human muscle. FEBS Lett. 1979;97:348–352. doi: 10.1016/0014-5793(79)80119-2. [DOI] [PubMed] [Google Scholar]
- Chapron J, De La Porte S, Delepine L, Koenig J. Schwann cells modify expression of acetylcholinesterase and butyrylcholinesterase at rat neuromuscular junctions. Eur J Neurosci. 1997;9:260–270. doi: 10.1111/j.1460-9568.1997.tb01396.x. [DOI] [PubMed] [Google Scholar]
- Chatonnet A, Lockridge O. Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem J. 1989;260:625–634. doi: 10.1042/bj2600625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deprez PN, Inestrosa NC. Two heparin-binding domains are present on the collagenic tail of asymmetric acetylcholinesterase. J Biol Chem. 1995;270:11043–11046. doi: 10.1074/jbc.270.19.11043. [DOI] [PubMed] [Google Scholar]
- Donger C, Krejci E, Serradell AP, Eymard B, Bon S, Nicole S, Chateau D, Gary F, Fardeau M, Massoulié J, Guicheney P. Mutation in the human acetylcholinesterase-associated collagen gene, COLQ, is responsible for congenital myasthenic syndrome with endplate acetylcholinesterase deficiency (type Ic) Am J Hum Genet. 1998;63:967–975. doi: 10.1086/302059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen LW, Excell BJ, Patel R, Smith B. Changes in motor endplates resulting from muscle fibre necrosis and regeneration. A light and electron microscopic study of the effects of the depolarizing fraction (cardiotoxin) of Dendroaspis jamesonivenom. J Neurol Sci. 1974;21:391–417. doi: 10.1016/0022-510x(74)90041-0. [DOI] [PubMed] [Google Scholar]
- Duval N, Massoulié J, Bon S. H and T subunits of acetylcholinesterase from Torpedo, expressed in COS cells, generate all types of globular forms. J Cell Biol. 1992;118:641–653. doi: 10.1083/jcb.118.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Engel, A.G. 1994a. Acquired autoimmune myasthenia gravis. In Myology. A.G. Engel and C. Franzini-Armstrong, editors. McGraw Hill, New York. 1769–1798.
- Engel, A.G. 1994b. Myasthenic syndromes. In Myology. A.G. Engel and C. Franzini-Armstrong, editors. McGraw Hill, New York. 1798–1835.
- Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with endplate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol. 1977;1:315–330. doi: 10.1002/ana.410010403. [DOI] [PubMed] [Google Scholar]
- Hall ZW. Multiple forms of acetylcholinesterase and their distribution in endplate and non-endplate regions of rat diaphragm muscle. J Neurobiol. 1973;4:343–361. doi: 10.1002/neu.480040404. [DOI] [PubMed] [Google Scholar]
- Hall ZW, Kelly RB. Enzymatic detachment of endplate acetylcholinesterase from muscle. Nat New Biol. 1971;232:62–63. doi: 10.1038/newbio232062a0. [DOI] [PubMed] [Google Scholar]
- Jirmanová I. Ultrastructure of motor endplates during pharmacologically-induced degeneration and subsequent regeneration of skeletal muscle. J Neurocytol. 1975;4:141–155. doi: 10.1007/BF01098779. [DOI] [PubMed] [Google Scholar]
- Karnovsky MJ, Roots L. A “direct-coloring” thiocholine method for cholinesterases. J Histochem Cytochem. 1964;12:219–221. doi: 10.1177/12.3.219. [DOI] [PubMed] [Google Scholar]
- Krejci E, Coussen F, Duval N, Chatel JM, Legay C, Puype M, Vandekerckhove J, Cartaud J, Bon S, Massoulié J. Primary structure of a collagenic tail peptide of Torpedoacetylcholinesterase: co-expression with catalytic subunit induces the production of collagen-tailed forms in transfected cells. EMBO (Eur Mol Biol Organ) J. 1991;10:1285–1293. doi: 10.1002/j.1460-2075.1991.tb08070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci E, Thomine S, Boschetti N, Legay C, Sketelj J, Massoulié J. The mammalian gene of acetylcholinesterase-associated collagen. J Biol Chem. 1997;272:22840–22847. doi: 10.1074/jbc.272.36.22840. [DOI] [PubMed] [Google Scholar]
- Landmesser LT. Synaptic plasticity: keeping synapses under control. Curr Biol. 1998;8:R564–R567. doi: 10.1016/s0960-9822(07)00362-4. [DOI] [PubMed] [Google Scholar]
- Laskowski MB, Olson WH, Dettbarn WD. Initial ultrastructural abnormalities at the motor endplate produced by a cholinesterase inhibitor. Exp Neurol. 1977;57:13–33. doi: 10.1016/0014-4886(77)90041-3. [DOI] [PubMed] [Google Scholar]
- Layer PG, Willbold E. Novel functions of cholinesterases in development, physiology and disease. Prog Histochem Cytochem. 1995;29:1–94. doi: 10.1016/s0079-6336(11)80046-x. [DOI] [PubMed] [Google Scholar]
- Leonard JP, Salpeter MM. Agonist-induced myopathy at the neuromuscular junction is mediated by calcium. J Cell Biol. 1979;82:811–819. doi: 10.1083/jcb.82.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lwebuga-Mukasa JS, Lappi S, Taylor P. Molecular forms of acetylcholinesterase from Torpedo californica:their relationship to synaptic membranes. Biochemistry. 1976;15:1425–1434. doi: 10.1021/bi00652a012. [DOI] [PubMed] [Google Scholar]
- Martin PT, Sanes JR. Role for a synapse-specific carbohydrate in agrin-induced clustering of acetylcholine receptors. Neuron. 1995;14:743–754. doi: 10.1016/0896-6273(95)90218-x. [DOI] [PubMed] [Google Scholar]
- Massoulié J, Bon S. The molecular forms of cholinesterase and acetylcholinesterase in vertebrates. Ann Rev Neurosci. 1982;5:57–106. doi: 10.1146/annurev.ne.05.030182.000421. [DOI] [PubMed] [Google Scholar]
- Massoulié J, Pezzementi L, Bon S, Krejci E, Vallette FM. Molecular and cellular biology of cholinesterases. Prog Neurobiol. 1993;41:31–91. doi: 10.1016/0301-0082(93)90040-y. [DOI] [PubMed] [Google Scholar]
- Massoulié J, Anselmet A, Bon S, Krejci E, Legay C, Morel N, Simon S. Acetylcholinesterase: C-terminal domains, molecular forms and functional localization. J Physiol. 1998;92:183–190. doi: 10.1016/s0928-4257(98)80007-7. [DOI] [PubMed] [Google Scholar]
- McMahan UJ, Sanes JR, Marshall LM. Cholinesterase is associated with the basal lamina at the neuromuscular junction. Nature. 1978;271:172–174. doi: 10.1038/271172a0. [DOI] [PubMed] [Google Scholar]
- Miner JH, Sanes JR. Collagen IV α3, α4, and α5 chains in rodent basal laminae: sequence, distribution, association with laminins, and developmental switches. J Cell Biol. 1994;127:879–891. doi: 10.1083/jcb.127.3.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen α3(IV): implications for Alport syndrome. J Cell Biol. 1996;135:1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin β2. Nature. 1995;374:258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- Ohno K, Brengman J, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (COLQ) of the asymmetric enzyme. Proc Natl Acad Sci USA. 1998;95:9654–9659. doi: 10.1073/pnas.95.16.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton BL, Chiu AY, Sanes JR. Synaptic laminin prevents glial entry into the synaptic cleft. Nature. 1998;393:698–701. doi: 10.1038/31502. [DOI] [PubMed] [Google Scholar]
- Phillips WD, Maimone MM, Merlie JP. Mutagenesis of the 43-kD postsynaptic protein defines domains involved in plasma membrane targeting and AChR clustering. J Cell Biol. 1991;115:1713–1723. doi: 10.1083/jcb.115.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss Y, Kroger S, Grassi J, Tsim KWK, Willbold E, Layer PG. Extracellular and asymmetric forms of acetylcholinesterase are expressed on cholinergic and noncholinergic terminal neuropil of the developing chick retina. Cell Tissue Res. 1996;286:13–22. doi: 10.1007/s004410050670. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ithurralde D, Silveira R, Barbeito L, Dajas F. Fasciculin, a powerful anticholinesterase polypeptide from Dendroaspis angusticepsvenom. Neurochem Int. 1983;5:267–274. doi: 10.1016/0197-0186(83)90028-1. [DOI] [PubMed] [Google Scholar]
- Rosenberry TL, Richardson JM. Structure of 18S and 14S acetylcholinesterase. Identification of collagen-like subunits that are linked by disulfide bonds to catalytic subunits. Biochemistry. 1977;16:3550–3558. doi: 10.1021/bi00635a008. [DOI] [PubMed] [Google Scholar]
- Rossi SG, Rotundo RL. Localization of “non-extractable” acetylcholinesterase to the vertebrate neuromuscular junction. J Biol Chem. 1993;268:19152–19159. [PubMed] [Google Scholar]
- Rossi SG, Rotundo RL. Transient interactions between collagen-tailed acetylcholinesterase and sulfated proteoglycans prior to immobilization on the extracellular matrix. J Biol Chem. 1996;271:1979–1987. doi: 10.1074/jbc.271.4.1979. [DOI] [PubMed] [Google Scholar]
- Rotundo RL, Rossi SG, Anglister L. Transplantation of quail collagen-tailed acetylcholinesterase molecules onto the frog neuromuscular synapse. J Cell Biol. 1997;136:367–374. doi: 10.1083/jcb.136.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Hall ZW. Antibodies that bind specifically to synaptic sites on muscle fiber basal lamina. J Cell Biol. 1979;83:357–370. doi: 10.1083/jcb.83.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Ann Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Scott LJ, Bacou F, Sanes JR. A synapse-specific carbohydrate at the neuromuscular junction: association with both acetylcholinesterase and a glycolipid. J Neurosci. 1988;8:932–944. doi: 10.1523/JNEUROSCI.08-03-00932.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman S, Aziz-Aloya RB, Timberg R, Loewenstein Y, Velan B, Shafferman A, Liao J, Norgaard-Pedersen B, Brodbeck U, Soreq H. Overexpressed monomeric human acetylcholinesterase induces subtle ultrastructural modifications in developing neuromuscular junctions of Xenopus laevisembryos. J Neurochem. 1994;62:1670–1681. doi: 10.1046/j.1471-4159.1994.62051670.x. [DOI] [PubMed] [Google Scholar]
- Simon S, Krejci E, Massoulié J. A four-to-one association between peptide motifs: four C-terminal domains from cholinesterase assemble with one proline-rich attachment domain (PRAD) in the secretory pathway. EMBO (Eur Mol Biol Organ) J. 1998;17:6178–6187. doi: 10.1093/emboj/17.21.6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sketelj J, Brzin M. Asymmetric molecular forms of acetylcholinesterase in mammalian skeletal muscles. J Neurosci Res. 1985;14:95–103. doi: 10.1002/jnr.490140109. [DOI] [PubMed] [Google Scholar]
- Small DH, Reed G, Whitefield B, Nurcombe V. Cholinergic regulation of neurite outgrowth from isolated chick sympathetic neurons in culture. J Neurosci. 1995;15:144–151. doi: 10.1523/JNEUROSCI.15-01-00144.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JH. Developmental changes in acetylcholine receptor aggregates at rat skeletal neuromuscular junctions. Dev Biol. 1981;84:267–276. doi: 10.1016/0012-1606(81)90394-8. [DOI] [PubMed] [Google Scholar]
- Sternfeld M, Ming G, Song H, Sela K, Timberg R, Poo M, Soreq H. Acetylcholinesterase enhances neurite growth and synapse development through alternative contributions of its hydrolytic capacity, core protein, and variable C termini. J Neurosci. 1998;18:1240–1249. doi: 10.1523/JNEUROSCI.18-04-01240.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toutant, J.P., and J. Massoulié. 1988. Cholinesterases: tissue and cellular distribution of molecular forms and their physiological regulation. In Handbook of Experimental Pharmacology. Volume 86. V.P. Whittaker, editor. Springer-Verlag. 225–265.
- Toutant JP, Massoulié J, Bon S. Polymorphism of pseudocholinesterase in Torpedo marmoratatissues: comparative study of the catalytic and molecular properties of this enzyme with acetylcholinesterase. J Neurochem. 1985;44:580–592. doi: 10.1111/j.1471-4159.1985.tb05452.x. [DOI] [PubMed] [Google Scholar]
- Tsim KWK, Randall WR, Barnard EA. An asymmetric form of muscle acetylcholinesterase contains three subunit types and two enzymic activities in one molecule. Proc Natl Acad Sci USA. 1988a;85:1262–1266. doi: 10.1073/pnas.85.4.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsim KW, Randall WR, Barnard EA. Synaptic acetylcholinesterase of chicken muscle changes during development from a hybrid to a homogeneous enzyme. EMBO (Eur Mol Biol Organ) J. 1988b;7:2451–2456. doi: 10.1002/j.1460-2075.1988.tb03091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Vigny M, Gisiger V, Massoulié J. “Nonspecific” cholinesterase and acetylcholinesterase in rat tissues: molecular forms, structural and catalytic properties, and significance of the two enzyme systems. Proc Natl Acad Sci USA. 1978;75:2588–2592. doi: 10.1073/pnas.75.6.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigny M, Martin GR, Grotendorst GR. Interactions of asymmetric forms of acetylcholinesterase with basement membrane components. J Biol Chem. 1983;258:8794–8798. [PubMed] [Google Scholar]
- Wecker L, Mrak RE, Dettbarn WD. Evidence of necrosis in human intercostal muscle following inhalation of an organophosphate insecticide. Fund Appl Toxicol. 1986;6:172–174. doi: 10.1016/0272-0590(86)90273-3. [DOI] [PubMed] [Google Scholar]
- Whathey JC, Nass NM, Lester HA. Numerical reconstruction of the quantal event at nicotinic synapses. Biophys J. 1979;27:145–164. doi: 10.1016/S0006-3495(79)85208-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Ikawa Y, Yoshida K, Shigetani Y, Takeda N, Mabuchi I, Yamamoto T, Aizawa S. Homologous recombination at c-fynlocus of mouse embryonic stem cells with use of diphtheria toxin A-fragment gene in negative selection. Proc Natl Acad Sci USA. 1990;87:9918–9922. doi: 10.1073/pnas.87.24.9918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younkin SG, Rosenstein C, Collins PL, Rosenberry TL. Cellular localization of the molecular forms of acetylcholinesterase in rat diaphragm. J Biol Chem. 1982;257:13630–13637. [PubMed] [Google Scholar]