Adenovirus E4orf4 protein induces PP2A-dependent growth arrest in Saccharomyces cerevisiae and interacts with the anaphase-promoting complex/cyclosome (original) (raw)
Abstract
Adenovirus early region 4 open reading frame 4 (E4orf4) protein has been reported to induce p53-independent, protein phosphatase 2A (PP2A)–dependent apoptosis in transformed mammalian cells. In this report, we show that E4orf4 induces an irreversible growth arrest in Saccharomyces cerevisiae at the G2/M phase of the cell cycle. Growth inhibition requires the presence of yeast PP2A-Cdc55, and is accompanied by accumulation of reactive oxygen species. E4orf4 expression is synthetically lethal with mutants defective in mitosis, including Cdc28/Cdk1 and anaphase-promoting complex/cyclosome (APC/C) mutants. Although APC/C activity is inhibited in the presence of E4orf4, Cdc28/Cdk1 is activated and partially counteracts the E4orf4-induced cell cycle arrest. The E4orf4–PP2A complex physically interacts with the APC/C, suggesting that E4orf4 functions by directly targeting PP2A to the APC/C, thereby leading to its inactivation. Finally, we show that E4orf4 can induce G2/M arrest in mammalian cells before apoptosis, indicating that E4orf4-induced events in yeast and mammalian cells are highly conserved.
Keywords: adenovirus; E4orf4; PP2A; anaphase-promoting complex/cyclosome; yeast
Introduction
Protein phosphatase 2A (PP2A),* one of the major protein serine/threonine phosphatases in the cell, plays a role in several cellular processes, including metabolism, transcription, RNA splicing, translation, cell cycle progression, morphogenesis, signal transduction, development, and transformation (Mumby and Walter, 1993; Wera and Hemmings, 1995). The predominant form of PP2A in cells is a heterotrimer consisting of three subunits. The 36-kD catalytic C subunit and the 63-kD regulatory A subunit (PR65) form the core enzyme, and the B subunit binds the core enzyme to form the holoenzyme. The A and C subunits both exist as two isoforms (α and β), which are closely related, whereas the B subunit is variable and its multiple isoforms belong to at least three unrelated gene families, B/B55/PR55, B′/B56/PR61, and B″/PR72/PR130 (Kamibayashi et al., 1994; McCright and Virshup, 1995; Csortos et al., 1996). The cellular B subunits can also be replaced by viral proteins, such as the SV40 small t antigen and the polyomavirus small and middle T antigens (Mumby, 1995).
In Saccharomyces cerevisiae, two closely related genes, PPH21 and PPH22, redundantly encode the major PP2A catalytic subunit (Sneddon et al., 1990). TPD3 encodes the only A subunit, and two distinct B subunits, encoded by CDC55 and RTS1, are homologous to mammalian B and B′, respectively (Healy et al., 1991; van Zyl et al., 1992; Shu et al., 1997). Mutation of both PPH21 and PPH22 eliminates most of the PP2A activity in the cell and drastically reduces growth. Strains lacking PPH21, PPH22, and a third related gene, PPH3, are completely inviable (Ronne et al., 1991). Two other PP2A-like phosphatase catalytic subunits exist, encoded by SIT4 (Sutton et al., 1991) and PPG1 (Posas et al., 1993), which perform nonredundant functions in the cells.
Mutations of CDC55 yield defects in cytokinesis and result in abnormal cell morphology at low temperature, whereas mutation of RTS1 results in growth defects at high temperature (Healy et al., 1991; Shu et al., 1997). PP2A was proposed to play a role in activation of Clb–Cdc28 kinase complexes for progression from G2 to mitosis (Lin and Arndt, 1995). The effect of Cdc55 on cellular morphogenesis is also mediated through Cdc28, and it was proposed that PP2A, regulated by Cdc55, affects the activity of the Cdc28 regulators Mih1 and Swe1 (Minshull et al., 1996; Wang and Burke, 1997; Yang et al., 2000). CDC55 was also implicated as a component of the spindle checkpoint pathway: _cdc55_Δ mutants are nocodazole sensitive (Wang and Burke, 1997), and in the presence of a defective spindle, Cdc55-deficient cells are still able to separate their sister chromatids and to leave mitosis even though cyclin B destruction is prevented, possibly by promoting inactivation of Cdc28 through inhibitory phosphorylation (Minshull et al., 1996).
Adenovirus early region 4 open reading frame 4 (E4orf4) protein is a multifunctional viral regulator. Our work, as well as work from other laboratories, has shown that the E4orf4 protein downregulates expression of genes that have been activated by E1A and cAMP (Müller et al., 1992; Kleinberger and Shenk, 1993), induces hypophosphorylation of various viral and cellular proteins (Müller et al., 1992; Kanopka et al., 1998), regulates alternative splicing of adenovirus mRNAs (Kanopka et al., 1998), and induces p53-independent apoptosis in transformed cells (Lavoie et al., 1998; Marcellus et al., 1998; Shtrichman and Kleinberger, 1998). Induction of apoptosis has been reported to be caspase independent in CHO cells (Lavoie et al., 1998), but it is caspase dependent in 293T cells (Livne et al., 2001). We have previously shown that E4orf4 interacts with the PP2A holoenzyme through a direct association with the Bα/B55 subunit (Kleinberger and Shenk, 1993). Interaction with PP2A complexes that include the Bα/B55 subunit is required for induction of apoptosis by E4orf4 (Shtrichman et al., 1999). The presence of an active PP2A also contributes to other functions performed by the E4orf4 protein, including down regulation of transcription (Kleinberger and Shenk, 1993; Bondesson et al., 1996; Whalen et al., 1997) and alternative splicing (Kanopka et al., 1998). Recently, we demonstrated that E4orf4 associates not only with PP2A-Bα, but also with several isoforms of PP2A-B′. However, only the interaction of E4orf4 with a PP2A holoenzyme population that includes the Bα subunit is required for induction of apoptosis (Shtrichman et al., 2000).
A key unresolved question is the identity of the targets of the PP2A-Bα–E4orf4 complex involved in induction of apoptosis. To gain insight into the cellular pathway induced by the PP2A-Bα–E4orf4 complex, we expressed E4orf4 in the yeast S. cerevisiae. Although yeast do not possess some of the cellular machinery required for the apoptotic process, such as caspases and Bcl-2 family members, this organism can serve as a powerful tool for apoptosis research (Matsuyama et al., 1999). Here, we report that in yeast, expression of E4orf4 induces irreversible growth arrest through a mechanism that requires Cdc55 and Tpd3, the yeast homologues of the B and A subunits of PP2A. The cells arrested in mitosis and exhibited enhanced accumulation of reactive oxygen species (ROS), a feature of apoptosis in metazoan cells. Our data suggest that E4orf4 acts via modulation of the activity of the anaphase-promoting complex/cyclosome (APC/C) ubiquitination complex. Furthermore, we show that E4orf4 can induce G2/M arrest before apoptosis in mammalian cells, suggesting that E4orf4-induced events are conserved in yeast and mammals.
Results
E4orf4 induces irreversible growth arrest in yeast
Since E4orf4 binds the PP2A-B subunit, and yeast and mammalian PP2A-B subunits share an extensive homology (53% identity and 67% similarity) (Healy et al., 1991), we tested whether expression of E4orf4 may affect yeast cell growth in a PP2A-dependent manner. Wild-type E4orf4 and the A3 mutant, which did not bind an active PP2A and did not induce apoptosis in mammalian cells (Shtrichman et al., 1999), were cloned in a yeast vector under the control of the GAL1,10 promoter. On galactose plates, expression of E4orf4, but not of mutant A3, prevented yeast growth, whereas no growth defect was apparent on _GAL1,10_-repressing glucose plates (Fig. 1A). This growth inhibition depended on Cdc55, because in a _cdc55_Δ strain, growth was unaffected by E4orf4 (Fig. 1 B). Reintroduction of the CDC55 gene into the _cdc55_Δ strain reinstated E4orf4-induced toxicity in these cells (Fig. 1 B). A yeast strain lacking the RTS1 gene did not lose the ability to respond to E4orf4 (Fig. 1 C), indicating that, as in mammalian cells, Cdc55/B but not Rts1/B′, is required for E4orf4-induced toxicity. Deletion of the TPD3 gene, homologous to mammalian PP2A-A, also resulted in loss of the cellular response to E4orf4 (Fig. 1 D). The altered response to E4orf4 did not result from changes in levels of E4orf4 expression (Fig. 1 E). The E4orf4-expressing plasmid was introduced into yeast strains lacking each of the PP2A-like catalytic subunits: Pph21, Pph22, Pph3, Sit4, and Ppg1. Each of these deletion strains maintained the response to E4orf4 expression (Table I), suggesting a redundancy in the catalytic subunit required for the response to E4orf4.
Figure 1.
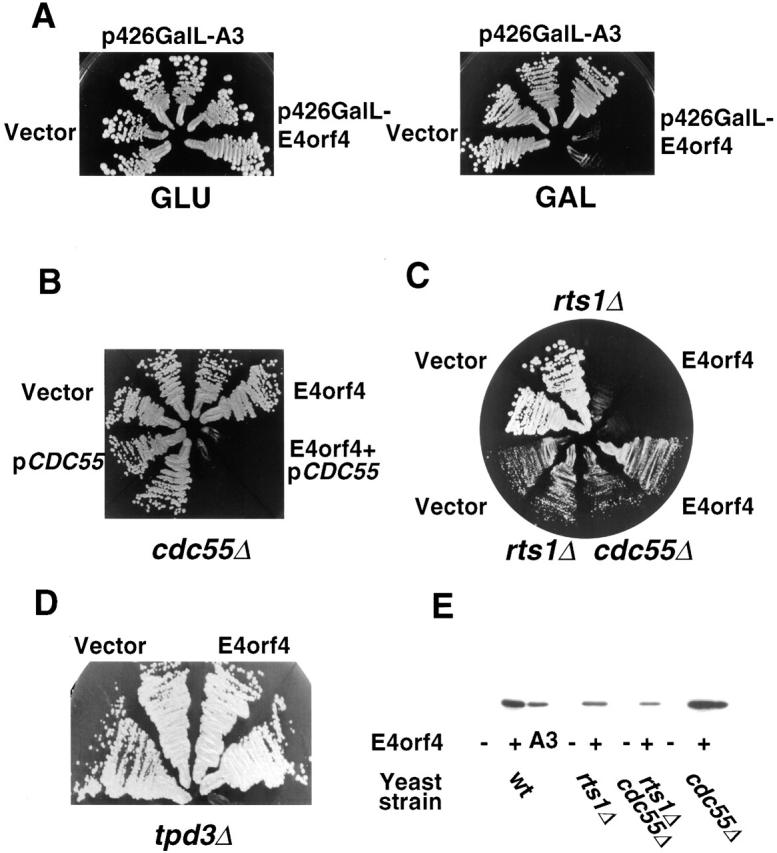
E4orf4 inhibits growth in S. cerevisiae in a PP2A-dependent manner. W303 cells (A) or mutant cells (B–D) transformed with the indicated plasmids were plated on galactose (B–D) or on glucose versus galactose (A) and allowed to grow for 2 d. (E) Proteins were prepared from the yeast cells used in A–D, and E4orf4 levels were analyzed by Western blot. A3, the E4orf4 A3 mutant.
Table I. Growth of various yeast mutants in the presence of E4orf4.
| Yeast strain | Plasmid | |
|---|---|---|
| p426-GalL-E4orf4 | pDAD-E4orf4 | |
| W303 | − | ++ |
| cdc55_Δ_ | +++ | ND |
| _cdc55_Δ | − | − |
| rts1_Δ_ | − | ND |
| cdc55_Δ_ rts1_Δ_ | +++ | ND |
| tpd3_Δ_ | +++ | ND |
| pph21_Δ_ | − | ND |
| pph22_Δ_ | − | ND |
| pph3_Δ_ | − | ND |
| sit4-36 | - | ND |
| ppg1_Δ_ | − | ND |
| cdc28-4 | ND | ± |
| cdc28-1N | ND | − |
| clb1_Δ_ clb2-v1 clb3_Δ_ clb4_Δ_ | ND | − |
| mih1_Δ_ | ND | − |
| swe1_Δ_ | − | ++ |
| W303 <_GAL-SWE1_> | ND | − |
| swe1_Δ_ mih1_Δ_ | − | ++ |
| CDC28F19 | − | ++ |
| cdc34-2 | ND | ++ |
| cdc53-1 | ND | ++ |
| cdc15-2 | ND | ++ |
| sic1_Δ_ | ND | ++ |
| cdc20-1 | ND | − |
| cdh1_Δ_ | ND | − |
| pds1_Δ_ | − | ++ |
| cdc16-1 | ND | − |
| CDC16-6A CDC23-A CDC27-5A | − | ND |
| mad1_Δ_ | − | ND |
| mad2_Δ_ | − | ND |
| mad3_Δ_ | − | ND |
| bub1_Δ_ | − | ++ |
| bub2_Δ_ | − | ND |
We next investigated whether the E4orf4-induced growth arrest was reversible. E4orf4 expression was induced by galactose in liquid medium, and growth was monitored by a general count using a hemacytometer, and by a viable count. Fig. 2A shows that until 6 h after induction there was no change in the apparent growth rate of cells expressing E4orf4 versus control cells. However, at later times, E4orf4-expressing cells showed a slower growth and stopped growing by 10 h after induction. If the cells were diluted to 3 × 106 cells/ml in fresh galactose-containing medium at that time, the E4orf4-containing cells did not regain exponential growth, whereas control cells did (Fig. 2 B). Residual cell growth seen at late time after E4orf4 induction may reflect selection in liquid medium of cells carrying fewer copies of the multicopy 2μ plasmid and expressing less E4orf4. Indeed, Western blot analysis indicated that at 24 and 48 h after induction, E4orf4 levels in the population were lower, compared with 3 and 6 h after induction (results not shown). When E4orf4-expressing cells were transferred to glucose medium, the GAL promoter was shut off and E4orf4 protein levels decreased (results not shown). Nonetheless, cell viability, measured as the ability to produce colonies on glucose plates, dropped rapidly within a few hours of growth in the galactose-containing medium (Fig. 2 C). These results indicate that E4orf4-induced growth arrest is irreversible.
Figure 2.

E4orf4-induced growth arrest is irreversible and occurs both in wild-type and rho-0 yeast cells. ▪, cells containing vector plasmid; •, cells expressing E4orf4. (A) Cells were transferred from raffinose to galactose at time 0. Aliquots were collected at various time points after induction, and cells were counted microscopically. (B) The experiment was done as in A, but at 9 and 24 h after induction cells were diluted to 3 × 106/ml in medium containing galactose and allowed to continue growing. (C) At the time points shown in A, 1,000 cells were plated on glucose plates. Colonies were counted after 2 d, and the number of colonies at time 0 was defined as 100%. (D) A similar experiment as described in C was performed, except a doxycycline-regulatable promoter was used, E4orf4 expression was induced by removal of doxycycline at time 0, and rho-0 cells (□ and ○) were compared with wild-type cells (▪ and •). Every experiment shown is one of a series of three that yielded similar results.
It has been reported that cell death induced in yeast by mammalian proapoptotic genes, such as Bax, is accompanied by changes in cell membranes and DNA degradation, typical of mammalian apoptosis (Zha et al., 1996). We tested whether E4orf4 induced similar changes. However, no alterations in trypan blue exclusion were observed at several time points after induction of E4orf4 expression, and no DNA degradation was detected on agarose gels as late as 48 h after induction (results not shown). Furthermore, it has been previously reported that Bax induction in yeast inhibited cell growth in all cells, but it did not lead to death in cells lacking functional mitochondria (petite) (Greenhalf et al., 1996). We investigated whether E4orf4 expression in petite (rho-0) cells resulted in cell death, measured by colony formation. E4orf4 was expressed from a tetracyclin-regulatable promoter (Gari et al., 1997), since petite cells did not grow well on galactose. E4orf4 induction by doxycycline removal was slower compared with its induction by galactose, and 6 h after induction, its expression reached levels comparable to those seen upon galactose induction 3 h after induction (results not shown). Cells were replated on plates containing doxycycline at various times after induction. Fig. 2 D shows that loss of viability upon induction of E4orf4 was similar in wild type and in petite yeast cells. The toxic effect appeared later than with galactose-induced E4orf4, probably due to the slower rate of E4orf4 induction by doxycycline removal. Thus, this experiment demonstrates that E4orf4-induced toxicity is independent of functions encoded by mitochondrial DNA. The cellular events induced by E4orf4 appear to differ from those induced by Bax expression.
E4orf4 enhances cellular ROS levels
It has been recently reported that generation and accumulation of oxygen radicals is a key event in apoptotic pathways both in yeast and in mammalian cells (Hockenbery et al., 1993; Kane et al., 1993; Slater et al., 1995; Madeo et al., 1999). We tested whether E4orf4 induced enhanced ROS levels in yeast cells. E4orf4 expression was induced with galactose for 6 h, and cells were then stained with 1 μg/ml dihydroethidium for 10 min. Dihydroethidium is oxidized specifically by superoxide ions to fluorescent ethidium (Budd et al., 1997). Fig. 3(left) shows an example of E4orf4-expressing cells, which accumulated ROS, as compared with control cells. Fluorescence levels in the cells were measured by FACS® analysis, and Fig. 3 (right) demonstrates that 26% of E4orf4-expressing cells were stained above background levels, whereas only 3.3% of vector-containing cells fluoresced above background. Staining with dihydrorhodamine 123, a compound that is oxidized to the fluorescent chromophore rhodamine 123 by ROS (Schultz et al., 1996), led to similar results (data not shown).
Figure 3.

ROS accumulate in E4orf4-expressing cells. Cells carrying pGalL-E4orf4 or the vector plasmid were grown for 6 h in galactose medium and were then stained for 10 min with 1 μg/ml dihydroethidium (DHE). Fluorescence was visualized through a rhodamine filter on a Zeiss Axioscop, at a magnification of 600×. The proportion of dihydroxyethidium-stained cells was measured by FACS® analysis (right).
E4orf4 induces G2/M arrest
Microscopic examination of cells expressing E4orf4 showed striking morphological changes: 8 h after induction, cells were greatly enlarged, and many displayed abnormal buds. To determine whether E4orf4 affects the cell cycle, DNA content of cells expressing E4orf4 from the GAL promoter was analyzed by FACS®. Fig. 4A shows that within 4.5 h of galactose induction, E4orf4-expressing cells started to accumulate in G2/M. To determine at what stage of mitosis the cells arrested, we visualized the spindles with green fluorescent protein (GFP)–tubulin. Fig. 4 B shows that cells were arrested at several mitotic stages. Most cells were arrested either with short preanaphase spindles, or with extended telophase spindles (49 and 40%, respectively; n = 100). Some of these cells had undergone a second round of budding. After longer induction times, chains of unseparated buds became visible, and anucleated cells started to accumulate (not shown).
Figure 4.

E4orf4-expressing cells accumulate in different stages of mitosis. (A) Cells carrying either the p426GalL-E4orf4 plasmid or the p426GalL vector were grown to midlog phase in glucose, washed once, and resuspended in galactose. Samples were removed every 90 min after induction by galactose, and cellular DNA content was measured by FACS® analysis. (B) Cells expressing a GFP–Tub1 fusion, and carrying either the p426GalL-E4orf4 plasmid or the p426GalL vector, were induced with galactose for 8 h. Cells were harvested, fixed with 4% paraformaldehyde, stained with DAPI, and visualized by light microscopy (left), by fluorescence in the DAPI channel (middle), or in the FITC channel to detect GFP–tubulin fluorescence (right) at a 1,000× magnification.
E4orf4 expression is synthetically lethal with reduction in Cdc28 activity
To pinpoint the cell cycle target of E4orf4, specific cell cycle mutants were assayed for synthetic effects with E4orf4 expression. To that end, an alternative expression vector was used, expressing lower levels of E4orf4 upon galactose induction (pDAD-E4orf4). Comparison between levels of E4orf4 expression from the pDAD and p426-GalL vectors is shown in Fig. 5A. When pDAD-E4orf4 was introduced into wild-type yeast cells, only a weak effect on yeast growth was detected, manifested by a slightly smaller colony size, compared with control cells (Fig. 5 B). However, mutants of the main yeast cell cycle kinase, Cdc28, were significantly more sensitive to E4orf4 expression. Temperature-sensitive CDC28 mutants, such as cdc28-4 and cdc28-1N (Surana et al., 1991), as well as a mitotic cyclin–defective strain (_clb1_Δ _clb2ts clb3_Δ _clb4_Δ), were unable to grow even in the presence of the low levels of E4orf4 expressed from the pDAD vector (Table I). In contrast, cell cycle mutants specifically affected in the G1/S transition, such as cdc34 or cdc53, or mutants affected in mitotic exit, such as cdc15 or _sic1_Δ, were not supersensitive to E4orf4 (Table I), confirming that E4orf4 acts specifically at mitosis. The kinase Swe1 (the S. cerevisiae homologue of mammalian Wee1) and the phosphatase Mih1 (the S. cerevisiae homologue of Cdc25), respectively, inhibit and activate Cdc28 activity (Russell et al., 1989; Booher et al., 1993). _mih1_Δ cells were more sensitive to E4orf4 expression (Fig. 5 B), and overexpression of Swe1 was synthetically lethal with E4orf4 expression (Table I), suggesting that E4orf4 may act via activation of the inhibitory kinase Swe1. However, deletion of SWE1 did not suppress the toxicity of E4orf4 (Table I). Furthermore, although Swe1 and Mih1 both act on Tyr19 of Cdc28, expression of the constitutively active Cdc28-F19 mutant in wild-type cells did not suppress the toxicity of E4orf4 (Table I), suggesting that in these cells the mechanism of action of E4orf4 does not involve modulation of phosphorylation of Cdc28 on Tyr19.
Figure 5.
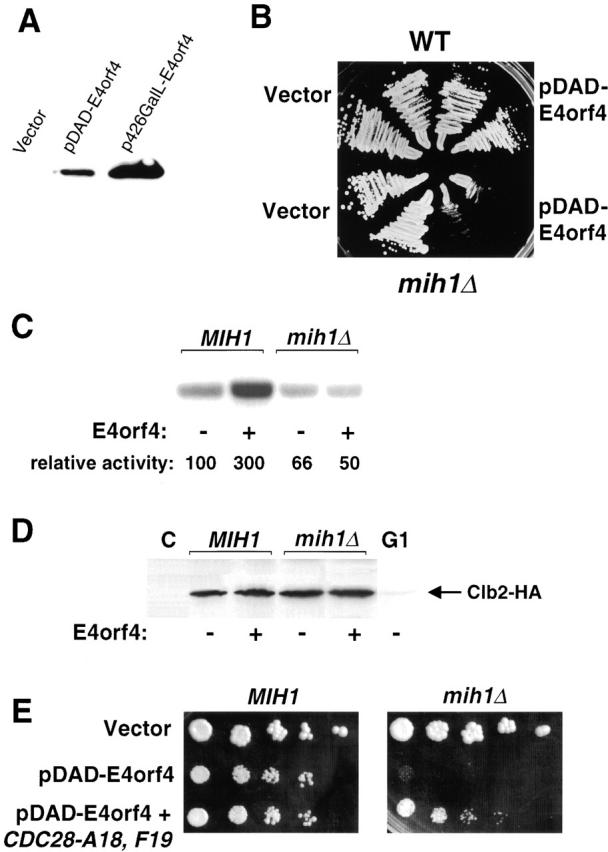
Functional interaction of E4orf4 with Mih1. (A) To compare E4orf4 expression levels from the pDAD and p426-GalL vectors, cells were induced with galactose for 3 h, and E4orf4 levels were determined by Western blot. (B) Wild-type or _mih1_Δ cells carrying the indicated plasmids were grown on galactose plates for 2 d. (C) KY630 (MIH1) or KY631 (_mih1_Δ) cells carrying a CLB2-3xHA construct integrated at CLB2 and transformed with either vector or p426GalL-E4orf4 were induced for 6 h with galactose. Clb2-associated kinase activity in the extracts was determined using Histone H1 as substrate. The amount of Histone H1 phosphorylation was measured with a phosphorimager. (D) Clb2-3xHA levels in the extracts described in C were determined by Western blotting. C, a no tag control; G1, an extract of _CLB2-3xHA_-expressing mutant cells arrested at restrictive temperature in G1. (E) MIH1 and _mih1_Δ cells were transformed with the indicated plasmids, and serial dilutions were plated dropwise on galactose plates.
To further investigate the E4orf4 effect on Cdc28 activity, Cdc28-Clb2 kinase activity was compared in cells containing p426-GalL-E4orf4 or the empty vector 6 h after induction with galactose. As seen in Fig. 5 C, enhanced kinase activity was observed in wild-type cells in the presence of E4orf4, whereas no differences in kinase activity were seen in an _mih1_Δ mutant. The levels of Clb2 protein were similar in control cells and in cells expressing E4orf4 (Fig. 5 D). These results indicate that E4orf4 expression leads to an Mih1-dependent increase in specific Cdc28-Clb2 kinase activity. Thus, the hyperactivation of Cdc28 by E4orf4 may be a secondary E4orf4 effect that partially counteracts the main E4orf4 inhibitory effect on cell cycle progression. The inability of the _mih1_Δ strain to hyperactivate Cdc28 by Tyr19 dephosphorylation might explain its supersensitivity to E4orf4. To test this hypothesis, the nonphosphorylatable Cdc28-A18F19 mutant was introduced in the _mih1_Δ strain. The resulting strain reverted to a level of E4orf4 sensitivity similar to the wild-type strain (Fig. 5 E), indicating that inability to dephosphorylate Cdc28 is the reason for the hypersensitivity of the _mih1_Δ mutant to E4orf4.
E4orf4-expressing cells are supersensitive to benomyl
Cdc55 has been identified as a component of the spindle assembly checkpoint (Wang and Burke, 1997). Mutations in genes that affect the kinetochore/spindle checkpoint result in cells that are more sensitive to the microtubule-depolymerizing drug benomyl than wild-type cells, and deletion of CDC55 causes enhanced benomyl sensitivity (Minshull et al., 1996; Wang and Burke, 1997). We tested whether E4orf4 affects benomyl sensitivity of cells. Yeast cells expressing E4orf4, mutant A3, or the empty vector were serially diluted and plated on galactose plates containing sublethal concentrations of benomyl. Fig. 6shows that cells expressing low E4orf4 levels are highly sensitive to benomyl, compared with control cells. Cells that express mutant E4orf4 (A3), even at high levels, are no more sensitive to benomyl than are the control cells. Thus, E4orf4, through its interaction with PP2A, may functionally interact with the spindle assembly checkpoint, or it may directly inhibit the target of this checkpoint. Several yeast strains containing mutations in genes involved in spindle checkpoint control were tested for a synthetic effect with E4orf4. However, these genes, including MAD1, -2, -3 and Bub1 and -2 did not manifest such an effect in the absence of benomyl (Table I).
Figure 6.

E4orf4-expressing cells are supersensitive to benomyl. Cells expressing low levels (pDAD-E4orf4) or high levels (p426-GalL-E4orf4) of E4orf4 or the E4orf4A3 mutant were grown in liquid medium containing glucose, serially diluted, and grown on galactose-containing plates, with or without 15 μg/ml benomyl, for 2 d.
The APC/C is a target of the E4orf4 pathway
The APC/C ubiquitination complex is required, with its Cdc20 subunit, for the metaphase-to-anaphase transition, and with another subunit, Hct1/Cdh1, it is involved in mitotic exit. We found that APC/C mutants, as well as the cdc20-1 and cdc20-3 mutants, and the cdh1/hct1 deletion, are supersensitive to E4orf4 (Table I). It was reported that the exit from mitosis involves a biphasic inactivation of the Cdc28-Clb2 kinase and degradation of Clb2. The first phase is dependent on Cdc20 and starts at the metaphase-to-anaphase transition. The second phase occurs in telophase and requires activation of the Cdc20 homologue, Hct1/Cdh1 (Visintin et al., 1997; Baumer et al., 2000; Yeong et al., 2000). To characterize the effect of E4orf4 on APC/C activity, we followed accumulation and degradation of substrates of both APC/C complexes. To measure degradation rates by APC/CCdh1, the substrate Ase1 was used, since APCCdh1 activity is highest in G1, and ectopic Clb2 expression was incompatible with maintenance of G1 arrest (Juang et al., 1997; Visintin et al., 1997). As shown in Fig. 7A, Ase1 was significantly stabilized in the presence of E4orf4, its half-life increasing from 2.5 to 7 min. Pds1, an inhibitor of the metaphase-to-anaphase transition, is degraded by APCCdc20 (Visintin et al., 1997). In E4orf4-expressing cells, Pds1 accumulated to significantly higher levels than in control cells, suggesting that Pds1 degradation is also inhibited by E4orf4 (Fig. 7 B). To directly assay the effect of E4orf4 on Pds1 degradation by pulse–chase, we artificially arrested the cells with α-factor in G1, a cell cycle phase where Pds1 had been shown to be unstable (Cohen-Fix et al., 1996). Under these conditions, however, no difference was seen in Pds1 degradation between control and E4orf4-expressing cells—in both cases, Pds1 was degraded with a half-life of 3 min (results not shown). Since Pds1 degradation normally occurs at the metaphase-to-anaphase transition, its ectopic degradation in G1-arrested cells may reflect an alternative mechanism that is different from its normal mechanism of degradation. Therefore, an attempt was made to arrest the cells in metaphase by expressing nondegradable Pds1 (Cohen-Fix and Koshland, 1999). However, cell cycle arrest could not be maintained during the relatively long period of time required for E4orf4 accumulation. Thus, we could not reliably determine directly the effect of E4orf4 on Pds1 stability.
Figure 7.

Effect of E4orf4 on degradation of APC/C substrates. (A) Degradation of Ase1-3xMyc in cells expressing E4orf4 from the p426-GalL-E4orf4 plasmid was measured by pulse–chase analysis. Cells arrested in G1 with α-factor were induced for 6 h with galactose. FACS® analysis of the cell population at the time of the chase indicates that the G1 arrest was maintained throughout the induction. The graph shows the result of the quantitation of the Ase1 band by phosphorimager. (B) Accumulation of Pds1-3xMyc in control versus p426-GalL-E4orf4–containing cells was measured by Western blotting after 6 h of galactose induction. Cells arrested by nocodazole (NOC) served as control.
E4orf4 physically interacts with the APC/C and recruits PP2A to the complex
One possibility for explaining the effect of E4orf4 on APC/C activity is that it directly targets PP2A to the APC/C. We tested this hypothesis by assaying whether E4orf4 physically interacts with the APC/C. HA-tagged Cdc16 was expressed in E4orf4 (wild type or the A3 mutant)–expressing versus control cells, and cell extracts were prepared 5 h after induction with galactose. E4orf4 was immunoprecipitated from the various samples, and the presence of the HA-tagged APC/C subunit Cdc16 in the immune complexes was detected by Western blot analysis. Fig. 8A demonstrates that wild-type E4orf4 coimmunoprecipitated with Cdc16, whereas the A3 mutant associated with HA-Cdc16 more weakly. The A3 mutant did not bind Tpd3 at all, similar to our findings in mammalian cells (Shtrichman et al., 2000). No HA-Cdc16 was immunoprecipitated in the absence of E4orf4 proteins. These results indicate that E4orf4 specifically associates with APC/C in yeast cells. We next tested whether E4orf4 expression leads to enhanced recruitment of PP2A to the APC/C. HA-Cdc16 was expressed in wild-type yeast cells or in _tpd3_Δ cells, with wild-type E4orf4 or with the empty vector. Immunoprecipitation reactions were carried out with antibodies to Tpd3, and the presence of HA-Cdc16 in the immune complexes was detected by a Western blot. Fig. 8 B demonstrates that Cdc16 was present in a complex with PP2A in cell extracts expressing E4orf4. No interaction above background levels, determined by a similar immunoprecipitation from _tpd3_Δ cell extracts, was detected in the absence of E4orf4. These results could suggest that in the absence of E4orf4, PP2A and the APC/C do not interact; alternatively, PP2A and the APC/C may normally interact, but this interaction may be too weak to be detected by immunoprecipitation in the absence of E4orf4.
Figure 8.
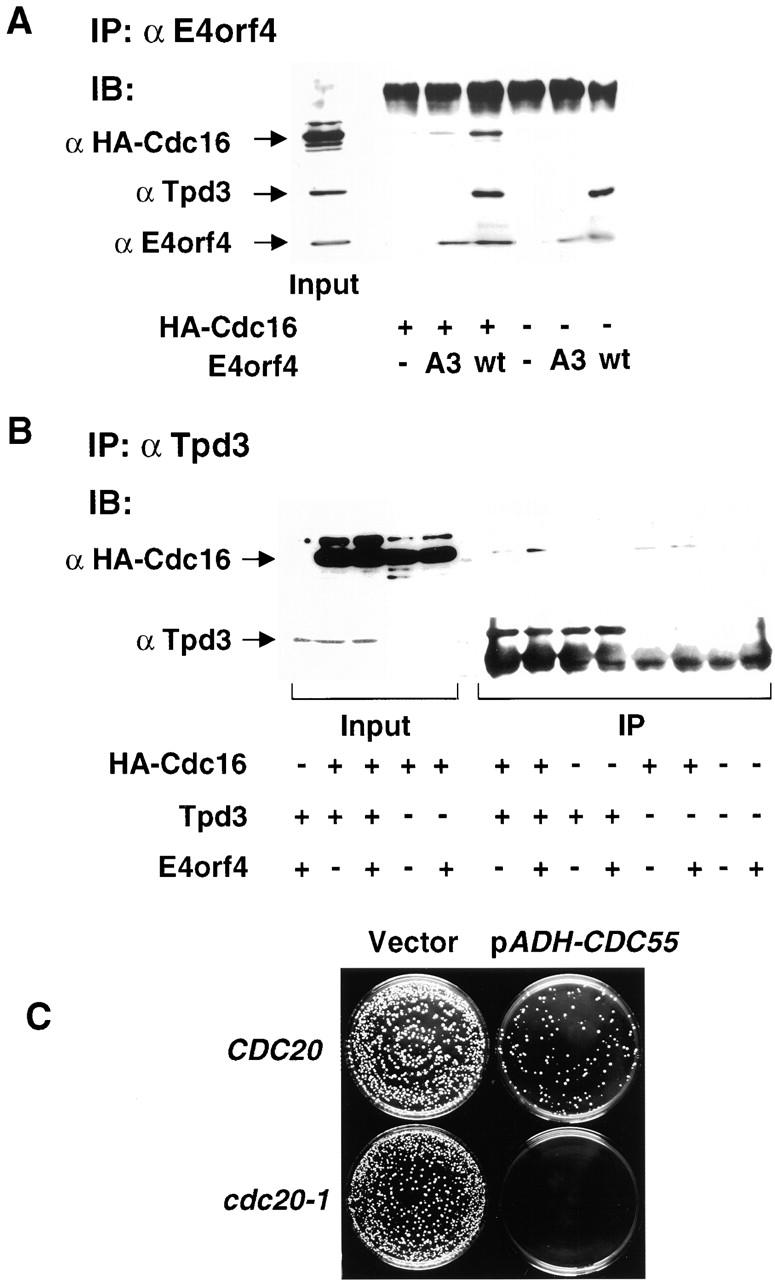
E4orf4 targets PP2A to the APC/C. (A) Cells expressing wild-type E4orf4 or the A3 mutant from the galactose-inducible promoter, and HA-tagged Cdc16, were induced with galactose for 5 h. Proteins were immunoprecipitated with antibodies to E4orf4, and the presence of HA-Cdc16, Tpd3, and E4orf4 in the immune complexes was detected by a Western blot. (B) W303 and _tpd3_Δ cells expressing wild-type E4orf4 and the HA-tagged Cdc16 were induced as in A. Proteins were immunoprecipitated with antibodies to Tpd3, and the presence of HA-Cdc16 and Tpd3 in the immune complexes was detected by a Western blot. (C) CDC20 and cdc20-1 cells were transformed with pADH-CDC55 or a vector plasmid and plated on SC-LEU to select for transformants.
To test for functional interactions between PP2A and the APC/C, the pADH-CDC55 plasmid, overexpressing Cdc55, was transformed into wild-type yeast cells or into cdc20-1 cells. As seen in Fig. 8 C, although wild-type cells were efficiently transformed with the pADH-CDC55 plasmid, no transformants were obtained in the cdc20-1 background, suggesting that increased PP2A-Cdc55 activity is lethal in this background. Conversely, it has been previously reported that deletion of CDC55 partially suppresses the temperature sensitivity of the cdc20-1 mutant (Wang and Burke, 1997). Together, these data suggest that even in the absence of E4orf4, PP2A-Cdc55 acts as a negative regulator of the APC/CCdc20. Thus, the results presented in Fig. 8 suggest that E4orf4 may be merely stabilizing an existing interaction between these two protein complexes.
E4orf4 can induce G2/M arrest in mammalian cells
The work presented here demonstrated that yeast cells expressing E4orf4 accumulated in G2/M, whereas previous work in mammalian cells indicated that E4orf4 expression led to apoptosis. Upon transient transfection of E4orf4 into various mammalian cell lines, no cell cycle effects were detected. However, in a stably transfected 293-derived cell line, expressing E4orf4 from a tetracycline-regulated promoter, G2/M arrest was observed 24 h after induction with doxycycline (Fig. 9) . No such arrest was observed upon treatment of a parallel control cell line. 48 h after induction, the G2/M arrest was released, and cells with sub-G1 DNA content undergoing apoptosis started to accumulate. These results indicate that, under certain physiological conditions, E4orf4 can initially induce cell cycle arrest, from which the cells eventually escape and undergo apoptosis.
Figure 9.

Induction of G2/M arrest in 293 cells expressing E4orf4 from a tetracycline-inducible promoter. 293 cells expressing E4orf4 from a tetracycline- inducible promoter (E4orf4-positive cell line) and control cells (E4orf4-negative cell line) were induced with increasing concentrations of doxycycline. FACS® analysis was performed for each point.
Discussion
In this work, we showed that adenovirus E4orf4, which induces apoptosis in transformed mammalian cells through a mechanism involving its interaction with PP2A complexes containing the B, but not the B′ subunit, causes irreversible growth arrest in S. cerevisiae. This growth arrest requires yeast PP2A complexes containing Cdc55, but not Rts1, and occurs at a specific cell cycle stage. These observations, and our recent findings indicating that nontoxic E4orf4 mutants selected in yeast could not induce apoptosis in mammalian cells (Afifi et al., 2001), underscore the high conservation of the E4orf4 targets between yeast and mammalian cells. Thus, as a first step towards elucidating the E4orf4-induced apoptotic pathway in mammalian cells, we took advantage of the wealth of available yeast cell cycle mutants to map the interaction of E4orf4 with the yeast cell cycle machinery.
Genetic interactions between E4orf4 and Cdc28
Genetic interactions suggested that reducing the activity of the Cdc28 kinase resulted in hypersensitivity to E4orf4. Thus, the growth arrest was more severe in cdc28 or mitotic cyclin mutants, and in mutants lacking the activating Mih1 phosphatase or overexpressing the inhibitory Swe1 kinase (Fig. 5; Table I). Absence of the Swe1 kinase, or of the Swe1 target on Cdc28 (Cdc28-F19) did not affect sensitivity to E4orf4. Strikingly however, in the presence of E4orf4, Clb2-associated kinase was hyperactivated in an Mih1-dependent manner (Fig. 5). Thus, E4orf4 may partially counteract its own cell cycle inhibitory effect by stimulating dephosphorylation of Cdc28-Y19 by Mih1. The hypersensitivity of the mih1 mutant would be due to its inability to respond to this stimulation. Indeed, the hypersensitivity was overcome by expression of a constitutively active Cdc28-A18F19 mutant in the _mih1_Δ cells (Fig. 5 E).
E4orf4 inhibits APC/C activity
Morphologically, E4orf4-arrested cells appear to be in preanaphase and telophase stages (Fig. 4) in wild type, as well as in _mih1_Δ cells, however the proportion of short preanaphase spindles was higher in the _mih1_Δ mutant (data not shown). Biochemical examination of the E4orf4-expressing cells indicates that they are defective in Ase1 degradation and accumulate high levels of Pds1 as well (Fig. 7). These results suggest that E4orf4 specifically inhibits APCCdh1 and may also inhibit APCCdc20. This is supported by the hypersensitivity of both cdc20ts mutants and _cdh1_Δ mutants to E4orf4, as well as by the dual morphology of the arrested cells. The lack of an effect of E4orf4 on Pds1 degradation in G1-arrested cells does not support an inhibition of APCCdc20; however, whereas Pds1 degradation normally occurs at the metaphase-to-anaphase transition, it could for technical reasons only be assayed in G1-arrested cells (Visintin et al., 1997). Thus, it is possible that under these artificial conditions, the effect of E4orf4 on APCCdc20 activity is lost. An arrest of the cells in metaphase by expression of a nondegradable Pds1 is transient (Cohen-Fix and Koshland, 1999) and does not provide enough time for optimal E4orf4 expression. In any case, it should also be noted that since _pds1_Δ cells are still sensitive to E4orf4 (Table I), there must be another substrate of APCCdc20 that is required for E4orf4-induced arrest. The supersensitivity of E4orf4-expressing cells to benomyl (Fig. 6) is also best explained by inhibition of APCCdc20. Benomyl, which transiently triggers the spindle assembly checkpoint by inhibiting APCCdc20 (Fang et al., 1998; Hwang et al., 1998), might cause permanent arrest when the APC is already partially inhibited.
E4orf4 targets PP2A to the APC/C
The observations that E4orf4 coimmunoprecipitates with APC/C subunits and that PP2A associates with the APC/C in E4orf4-expressing cells (Fig. 8) suggest that E4orf4 functions by directly targeting PP2A to the APC/C. Several yeast APC/C subunits were shown to be phosphorylated (Peters et al., 1996). Furthermore, phosphorylation of the APC/C by Cdk1 was shown to be required for its activation both in metazoans (Lahav-Baratz et al., 1995; Shteinberg et al., 1999) and in yeast (Rudner and Murray, 2000), either directly or indirectly via activation of the Cdc5/Polo kinase (Charles et al., 1998). Thus, E4orf4 may inhibit the APC by inducing its dephosphorylation by PP2A. We suggest that APC/C activity in E4orf4-expressing cells results from the net effect of phosphorylation by Cdc28 and dephosphorylation by E4orf4/PP2A. Therefore, mutants having reduced Cdc28 activity are hypersensitive to E4orf4. This model is summarized in Fig. 10.
Figure 10.

A schematic model for the mechanism of E4orf4 interference with cell cycle regulation. APC/C activity in E4orf4- expressing cells results from the net effect of activation by Cdc28 and inactivation by E4orf4/PP2A (see text for details).
The identity of the specific phosphorylation sites on the APC/C targeted by E4orf4 still eludes us. It appears that the phosphorylation sites described previously in Cdc16, Cdc23, and Cdc27 (Rudner and Murray, 2000) are not the substrates of the E4orf4–PP2A complex, since substitution of these sites to alanine residues has only a weak effect on APC/C function (Rudner and Murray, 2000), and do not render the yeast cells resistant to E4orf4 toxicity (Table I). Furthermore, although it was reported that APC/C phosphorylation at these sites affects only the activity of the APC/CCdc20 complex (Rudner et al., 2000), our results suggest that E4orf4, through its association with both PP2A and the APC/C, affects the APC/CCdh1 complex as well. It should be noted that Cdh1 itself is activated, rather than repressed, by dephosphorylation; however, this dephosphorylation is carried out by a distinct phosphatase, Cdc14 (Visintin et al., 1998; Zachariae et al., 1998).
The inhibitory effect of PP2A on APC/C activity in the presence of E4orf4 could represent a novel interaction induced by the viral protein; alternatively, E4orf4 could be merely enhancing a physiological interaction of PP2A with the APC/C. We found that overexpression of Cdc55 was synthetically lethal with the cdc20-1 mutation even in the absence of E4orf4 (Fig. 8 C). Conversely, Wang and Burke (1997) reported that deletion of CDC55 partially suppressed the cdc20-1 temperature sensitivity. Therefore, we suggest that one of the normal functions of PP2A is to inhibit the APC/C. Deletion of the PP2A regulator CDC55 would result in derepression of APC/C activity, counterbalancing the reduction of Cdc20 activity of the cdc20-1 mutant, thereby resulting in suppression of the mutant phenotype. This may also provide an additional explanation for the loss of spindle assembly checkpoint activity detected in the _cdc55_Δ mutant by Minshull et al. (1996). These authors attributed the loss of the checkpoint to increased inhibitory phosphorylation of Cdc28 on Tyr19. However, even in the presence of the Cdc28-V18F19 mutant, a reduction in Clb2 levels, and a concomitant reduction in Clb2-associated kinase activity, was detectable in the _cdc55_Δ cells (Minshull et al., 1996). Therefore, we suggest that derepression of APC/C activity in the _cdc55_Δ mutant may additionally make inactivation of the APC/C by the checkpoint pathway less effective, contributing to loss of checkpoint activity.
The similarity between E4orf4-initiated pathways in yeast and mammalian cells
In a 293-derived cell line, E4orf4 induces G2/M arrest before induction of apoptosis (Fig. 9). In a previous report, Wersto et al. (1998) demonstrated that, in the absence of the E1 region of adenovirus, the E4 region of the virus (excluding E4orf6) can induce G2 growth arrest and elevation in cyclins A and B and p34cdc2 kinase protein levels. The finding of elevated levels of mitotic cyclins in E4-arrested mammalian cells could result from interference with the degradation of mitotic cyclins (Wersto et al., 1998). Thus, the similar effect of E4orf4 on mammalian and yeast cells could reflect a conservation not only of the PP2A–E4orf4 interaction, but also of the interaction of PP2A–E4orf4 with its target, the APC/C. This possibility is currently being investigated. Many viral proteins cause cell cycle dysregulation, either as their primary function to create optimal conditions for viral replication, or as a by-product of their primary function. However, whether the effect of E4orf4 on the cell cycle reflects one of its natural functions or whether it is a consequence of its ectopic expression outside the viral life cycle is not clear yet.
Our initial motivation in expressing E4orf4 in yeast was to gain more information on the pathway of apoptosis induction by E4orf4 in transformed mammalian cells. Although apoptosis has mainly been described in multicellular organisms, in recent years there have been several observations of apoptosis-like phenomena in unicellular eukaryotes, including S. cerevisiae (Madeo et al., 1997). Despite the fact that none of the genes involved in the basic apoptotic machinery in metazoans, such as Bcl-2 or caspases, were identified in yeast, introduction of proapoptotic genes, such as BAX or BAK into yeast, resulted in cytotoxicity with apoptotic-like phenotypes (Greenhalf et al., 1996; Zha et al., 1996; Ink et al., 1997; Tao et al., 1997). Furthermore, the generation of ROS plays a role in the apoptotic process both in metazoans and in yeast (Hockenbery et al., 1993; Kane et al., 1993; Slater et al., 1995; Madeo et al., 1999). Thus, yeast has become a useful model system for the study of parts of the apoptotic pathway (Matsuyama et al., 1999).
We found that yeast cells expressing E4orf4 irreversibly stopped proliferating and exhibited one of the hallmarks of apoptosis, namely, accumulation of ROS. E4orf4 expression also leads to accumulation of ROS in mammalian cells. However, this similarity in phenotype does not necessarily reflect a similarity in mechanism, especially since yeast cells lack the death receptor pathway that is recruited by E4orf4 in mammalian cells to induce ROS accumulation (Livne et al., 2001). Other regulators shown to be involved in E4orf4-induced apoptosis in mammalian cells are also lacking in yeast, such as the Src family of kinases (Lavoie et al., 2000). However, E4orf4 induces both caspase-dependent and caspase-independent cell death pathways in mammalian cells (Lavoie et al., 1998; Livne et al., 2001). The cell cycle arrest induced by E4orf4 in yeast may be mechanistically relevant to at least one of these two apoptotic pathways.
Perturbations in cell cycle regulation and induction of apoptosis
We found that in a 293-derived cell line, the E4orf4-induced G2/M arrest is followed by apoptosis (Fig. 9). Several compounds—PKC inhibitors (Begemann et al., 1998), taxol (Jordan et al., 1996), and viral genes such as HIV-1 Vpr (Stewart et al., 1997)—have been reported to induce both G2/M arrest and apoptosis in mammalian cells. Conversely, several reports demonstrated that induction of apoptosis by various stimuli required CDC2 or CDK2 activity (Shi et al., 1994; Meikrantz and Schlegel, 1996; Yao et al., 1996). Thus, perturbations in cell cycle control can lead to apoptosis. Furthermore, a specific link between APC/C inhibition and apoptosis has been previously established in a report showing that the APC/C Cdc27 subunit was cleaved by caspases in Jurkat cells induced to undergo apoptosis by Fas, resulting in APC/C inhibition and cyclin A and B stabilization (Zhou et al., 1998). CDC2 kinase activity was further induced in these cells due to caspase cleavage and inactivation of Wee1, resulting in reduced tyrosine phosphorylation at the inhibitory site. In these cells, similar to our findings in yeast expressing E4orf4, the APC/C is inhibited and Cdks are stimulated. It is possible that this combination of APC/C inhibition and Cdk stimulation contributes to induction of the apoptotic process in mammalian cells, and that this is one of the pathways through which E4orf4 selectively kills transformed cells. This notion, which can now be tested directly, may eventually result in novel therapeutic strategies.
Materials and methods
Yeast strains, media, and mammalian cell lines
Yeast were grown either in YPD (1% yeast extract, 2% bacto peptone, 0.015% l-tryptophan, 2% glucose) or in synthetic complete medium (Sherman et al., 1982). For induction with galactose, cells were grown in synthetic medium with 2% raffinose overnight. They were then diluted to A 600 0.3 and allowed to grow another 2 h before addition of galactose to 2%. Alternatively, cells were grown in 2% glucose to midlog phase, washed once, and resuspended in 2% galactose. For induction by doxycycline removal, cells were grown overnight in medium containing 2% glucose and 5 μg/ml doxycycline. They were then diluted to A 600 0.3 and allowed to grow another 2 h in the same medium before being washed twice in water and transferred to medium without doxycycline.
All yeast strains used in this study are listed in Table II, along with their sources. All the strains are isogenic with strain W303. Derivation of a rho-0 strain was carried out as described (Sherman et al., 1982). Strain KY520 was built by crossing strain CY5580 (a, leu2, ura3, cdc55::LEU2) (Wang and Burke, 1997) with W303. Similarly, KY679 was obtained by crossing Y1366 (a, his3_Δ_1, lys2-1, leu2-3, 112 ura3-52, tpd3_Δ::LEU2_) (van Zyl et al., 1992) with W303. Strains KY596 and KY600 were obtained by transformation with mih1::LEU2 (Russell et al., 1989) and swe1::LEU2 (Booher et al., 1993) deletion plasmids, respectively. Strain KY620 was obtained by crossing a swe1_Δ::LEU2_ with a mih1_Δ::LEU2_ strain. Presence of the two markers was deduced from tetrad analysis and confirmed by backcross. Strains KY630, KY631 were obtained by integrating the YiP CLB2-3xHA plasmid (L. Johnston, National Institute for Medical Research, London, UK) into the W303 and KY620 strains at CLB2.
Table II. Yeast strains.
| Strain | Genotype | Reference or source |
|---|---|---|
| W303-1A | a ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 GAL+ | R. Rothsteina |
| 10131-7C | a ura3-52 leu2-3 sit4-36 GAL+ | G. Fink |
| H314 | α ura3-1 leu2-3,112 trp1-1 ade2-1 pph22-d1::HIS3 | Ronne et al., 1991 |
| H339 | ura3-1 leu2-3,112 trp1-1 ade2-1 pph3::LEU2 | Ronne et al., 1991 |
| H341 | a ura3-1 leu2-3,112 trp1-1 ade2-1 pph21-d1::HIS3 | Ronne et al., 1991 |
| rts1-null | α ura3-1 leu2-3,112 his3 trp1-1 rts1::HIS3 | Shu et al., 1997 |
| rts1-null,cdc55-null | α ura3-1 leu2-3,112 his3 trp1-1 rts1::HIS3 cdc55::TRP1 | Shu et al., 1997 |
| _ppg1_Δ | a ura3-1 leu2-3,112 his3-11 trp1-1 ppg1::TRP1 | Posas et al., 1993 |
| K1989 | a cdc28-4 ura3 trp1 | K. Nasmyth |
| K1993 | a cdc15-2 ura3 trp1 leu2 | K. Nasmyth |
| A364 | a cdc16-1 leu2 trp1 | A. Amon |
| A368 | a cdc28-1N ura3 leu2 trp1 | A. Amon |
| A460 | a cdc20-1 ura3 leu2 trp1 his3 | A. Amon |
| A544 | a clb1D clb3::TRP1 clb4::HIS3 clb2-v1(ts) ura3 | A. Amon |
| A698 | a ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 bar1D sic1::HIS3 | A. Amon |
| W321 | a ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 hct1D1::LEU2 | Schwab et al., 1997 |
| MTY670 | a ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 cdc34-2 | M. Tyersb |
| MTY740 | a ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 cdc53-1 | M. Tyers |
| PS694 | α ura3-52 leu2 trp1-1 CDC28-F19::TRP1 | P. Sorgerc |
| K7375 | a pds1::URA3, ura3::URA3 tetOs, leu2::LEU2 tetR-GFP | Alexandru et al., 1999 |
| KH123 | a ura3-52 leu2-3,112 trp1 can1 ade2 his3-11,15 mad1D1::HIS3 | Hardwick and Murray, 1995 |
| KH141 | a ura3-52 leu2-3,112 trp1 can1 ade2 his3-11,15 mad2D::URA3 | Hardwick and Murray, 1995 |
| KH125 | a ura3-52 leu2-3,112 trp1 can1 ade2 his3-11,15 mad3D1::LEU2 | Hardwick and Murray, 1995 |
| KH127 | a ura3-52 leu2-3,112 trp1 can1 ade2 his3-11,15 bub1D::HIS3 | Hardwick and Murray, 1995 |
| KH128 | a u_ra3-52 leu2-3,112 trp1 can1 ade2 his3-11,15 bub2D::URA3_ | Hardwick and Murray, 1995 |
| ADR2032 | a CDC16-6A::TRP1 CDC23-A-HA CDC27-5A:KANR bar1D | Rudner and Murray, 2000 |
| KY520 | α ura3-1 leu2-3,112 trp1-1 GAL+ cdc55::LEU2 | This work |
| KY547 | a ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 rho-0 | This work |
| KY596 | a ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 mih1::LEU2 | This work |
| KY600 | a ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 swe1::LEU2 | This work |
| KY620 | α ura3-1 leu2-3,112 trp1-1 ade2-1 his3-11,15 swe1::LEU2 mih1::LEU2 | This work |
| KY630 | W303 CLB2-3xHA::URA3 | This work |
| KY631 | KY620 CLB2-3xHA::URA3 | This work |
| KY679 | a ura3-1 leu2-3 tpd3::LEU2 | This work |
A 293-derived cell line expressing E4orf4 from a tetracycline-inducible promoter was prepared by introducing E4orf4 cDNA cloned in the pUHG10-3 vector (Gossen and Bujard, 1992) and pBabe-Puro (Morgenstern and Land, 1990) into the 293 Tet-On cell line (CLONTECH Laboratories) and selection for puromycine-resistant colonies. A cell line selected in the same way but not expressing E4orf4 is used as a negative control.
Plasmids
E4orf4 and mutant A3 were subcloned into the BamHI-ClaI sites of pCM190 (Gari et al., 1997), into the BamHI-EcoRI sites of p414 GALL (Mumberg et al., 1994), or into the BglII-EcoRI sites of pDAD2, a yeast 2μ plasmid containing the GAL1,10 promoter (Kornitzer et al., 1994). PDS1 was cloned by PCR under the CUP1 promoter of plasmid KB354 (Meimoun et al., 2000) and fused to the triple Myc epitope tag. Other plasmids have been previously described: YCp50-CDC55 (Nickels and Broach, 1996), pDB20(HA-CDC55) (Zhao et al., 1997), GAL10:SweI (Booher et al., 1993), HA-Cdc16 (Hwang and Murray, 1997), GAL-Ase1-Mycx3 (Juang et al., 1997).
Detection of ROS accumulation
Free intracellular radicals were detected with dihydroethidium or with dihydrorhodamine 123 (Sigma-Aldrich). Dihydroethidium was added at 1 μg/ml of cell culture, and cells were viewed through a rhodamine optical filter after a 10-min incubation. Dihydrorhodamine 123 was added at 5 μg/ml of cell culture, and cells were viewed through a rhodamine filter after a 2-hr incubation. For FACS® analysis, cells were incubated with dihydroethidium as described and analyzed using FACSCalibur® (Becton Dickinson).
Histone H1 kinase assay
∼3 × 108 cells treated as indicated were pelleted, washed once, resuspended in 0.1 ml extraction buffer (20 mM Tris-Cl, pH 7.5, 100 mM NaCl, 10 mM EDTA, 1% Triton X-100, 5% glycerol, 1 mM each of NaF, Na-pyrophosphate, NaVO3, β-glycerolphosphate, EGTA, and the protease inhibitors PMSF, TPCK, TLCK, leupeptin, pepstatin, and chymostatin at 20–50 μg/ml), and broken by vortexing in the presence of glass beads. Extract containing 0.15 mg protein was incubated 1 h on ice with 1 μg of α-HA antibody, followed by incubation 1 h with protein A–sepharose beads (Amersham Pharmacia Biotech). The beads were washed three times in kinase assay buffer (25 mM MOPS, pH 7.2, 15 mM MgCl2, 5 mM EGTA, 1 mM dithiothreitol, 60 mM β-glycerolphosphate, 0.1 mM NaVO3). Reactions were performed at 30°C for 30 min in 10 μM kinase assay buffer containing 0.5 mM cold ATP, 2 μCi γ[32P]ATP, and 5 μg histone HI. The reaction products were separated by polyacrylamide electrophoresis and histone HI phosphorylation was quantitated with a phosphorimager.
Protein degradation assay
Ase1 and Pds1 degradation were measured in _bar1_Δ cells arrested in G1 with α-factor. Overnight cultures grown in raffinose were diluted in raffinose medium with 1 μM α-factor. When more than 90% of the cells had reached G1 arrest, typically within 3–4 h, E4orf4 expression was induced by the addition of 3% galactose for 7 h. Pulse–chase analysis was then performed as described (Meimoun et al., 2000). Maintenance of G1 arrest was confirmed by subjecting an aliquot of the culture taken at the time of the chase to FACS® analysis.
Immunoblot analysis and coimmunoprecipitations
For immunoblots, yeast cells were collected at various times after induction and lysed in the presence of 1.85 M NaOH and 7.4% β-mercaptoethanol. Proteins were TCA precipitated and resuspended in protein loading buffer (125 mM Tris-HCl, pH 6.8, 1% SDS, 10% glycerol). For visualization of E4orf4, proteins were chromatographed on 15% SDS polyacrylamide gels and subjected to Western blotting and detection by specific antibodies as previously described (Shtrichman et al., 1999).
For coimmunoprecipitations, yeast extracts were prepared by bead beating cells for 4 min at 4°C in lysis buffer (250 mM NaCl, 50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 0.5% Nonidet P-40, 0.1% Triton X-100, 50 mM NaF, 100 μM sodium vanadate, 100 nM okadaic acid [Calbiochem], complete protease inhibitor cocktail [Roche Molecular Biochemicals]). The lysates were spun to separate beads and debris from the clear lysate. The beads were washed twice more in the lysis buffer, and immunoprecipitations were carried out in the same lysis buffer. Antibodies to E4orf4 were covalently coupled to protein sepharose beads (Shtrichman et al., 1999). Antibodies to the HA tag were from Babco, and were used at 1:600. Lysates, antibodies, and protein A beads were rotated for 3 h at 4°C, and then washed three times in lysis buffer.
Antibodies used in this work were anti-HA (Babco), anti-E4orf4 (Shtrichman and Kleinberger, 1998), anti-Tpd3 (from J.R. Broach, Princeton University, Princeton, NJ), and anti-Myc monoclonal (9E10).
Acknowledgments
We are grateful to the following scientists for gifts of plasmids and yeast strains: D.J. Burke (University of Virginia, Charlottesville, VA), R. Hallberg (Syracuse University, Syracuse, NY), J.R. Broach, H. Ronne (Ludwig Institute for Cancer Research, Uppsala, Sweden), J. Arino (Universitat Autonoma de Barcelona, Barcelona, Spain), G. Fink (Whitehead Institute, Cambridge, MA), A. Amon (Massachusetts Institute of Technology), A.W. Murray (Harvard University, Cambridge, MA), K. Nasmyth (Research Institute of Molecular Pathology, Vienna, Austria), W. Zacchariae (Max Planck Institute, Dresden, Germany), A.A. DePaoli-Roach (Indiana University, Indianapolis, IN), S. Kron (University of Chicago, Chicago, IL), D. Pellman (Dana Farber Cancer Institute, Boston, MA), and L. Johnston. We thank Shavit Gonen-Fein for technical assistance and S. Selig for critical reading of the manuscript.
This work was supported by grants from the Israel Science Foundation, founded by the Israel Academy of Sciences and Humanities, the Israeli Ministry of Sciences, and from the Fund for the Promotion of Research at the Technion (to D. Kornitzer and T. Kleinberger).
Footnotes
*
Abbreviations used in this paper: APC/C, anaphase-promoting complex/cyclosome; E4orf4, early region 4 open reading frame 4; GFP, green fluorescent protein; PP2A, protein phosphatase 2A; ROS, reactive oxygen species.
References
- Afifi, R., R. Sharf, R. Shtrichman, and T. Kleinberger. 2001. Selection of apoptosis-deficient adenovirus E4orf4 mutants in S. cerevisiae. J. Virol. 75:4444–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandru, G., W. Zachariae, A. Schleiffer, and K. Nasmyth. 1999. Sister chromatid separation and chromosome re-duplication are regulated by different mechanisms in response to spindle damage. EMBO J. 18:2707–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumer, M., G.H. Braus, and S. Irniger. 2000. Two different modes of cyclin Clb2 proteolysis during mitosis in Saccharomyces cerevisiae. FEBS Lett. 468:142–148. [DOI] [PubMed] [Google Scholar]
- Begemann, M., S.A. Kashimawo, R.M. Lunn, T. Delohery, Y.J. Choi, S. Kim, D.F. Heitjan, R.M. Santella, P.B. Schiff, J.N. Bruce, and I.B. Weinstein. 1998. Growth inhibition induced by Ro 31-8220 and calphostin C in human glioblastoma cell lines is associated with apoptosis and inhibition of CDC2 kinase. Anticancer Res. 18:3139–3152. [PubMed] [Google Scholar]
- Bondesson, M., K. Ohman, M. Mannervik, S. Fan, and G. Akusjarvi. 1996. Adenovirus E4 open reading 4 protein autoregulates E4 transcription by inhibiting E1A transactivation of the E4 promoter. J. Virol. 70:3844–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher, R.N., R.J. Deshaies, and M.W. Kirschner. 1993. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 12:3417–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd, S.L., R.F. Castilho, and D.G. Nicholls. 1997. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 415:21–24. [DOI] [PubMed] [Google Scholar]
- Charles, J.F., S.L. Jaspersen, R.L. Tinker-Kulberg, L. Hwang, A. Szidon, and D.O. Morgan. 1998. The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr. Biol. 8:497–507. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix, O., and D. Koshland. 1999. Pds1p of budding yeast has dual roles: inhibition of anaphase initiation and regulation of mitotic exit. Genes Dev. 13:1950–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Fix, O., J.M. Peters, M.W. Kirschner, and D. Koshland. 1996. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 10:3081–3093. [DOI] [PubMed] [Google Scholar]
- Csortos, C., S. Zolnierowicz, E. Bako, S.D. Durbin, and A.A. DePaoli-Roach. 1996. High complexity in the expression of the B′ subunit of protein phosphatase 2A0. J. Biol. Chem. 271:2578–2588. [DOI] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari, E., L. Piedrafita, M. Aldea, and E. Herrero. 1997. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in S. cerevisiae. Yeast. 13:837–848. [DOI] [PubMed] [Google Scholar]
- Gossen, M., and H. Bujard. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA. 89:5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalf, W., C. Stephan, and B. Chaudhuri. 1996. Role of mitochondria and c-terminal membrane anchor of Bcl-2 in Bax induced growth arrest and mortality in S. cerevisiae. FEBS Lett. 380:169–175. [DOI] [PubMed] [Google Scholar]
- Hardwick, K.G., and A.W. Murray. 1995. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J. Cell Biol. 131:709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy, A.M., S. Zolnierowicz, A.E. Stapleton, M. Goebel, A.A. De-Paoli-Roach, and J.R. Pringle. 1991. CDC55, a S. cerevisiae gene involved in cellular morphogenesis: identification, characterization, and homology to the B subunit of mammalian type 2A protein phosphatase. Mol. Cell. Biol. 11:5767–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery, D.M., Z.N. Oltvai, X.-M. Yin, C.L. Milliman, and S.J. Korsmeyer. 1993. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 75:241–251. [DOI] [PubMed] [Google Scholar]
- Hwang, L.H., and A.W. Murray. 1997. A novel yeast screen for mitotic arrest mutants identifies DOC1, a new gene involved in cyclin proteolysis. Mol. Biol. Cell. 8:1877–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, L.H., L.F. Lau, D.L. Smith, C.A. Mistrot, K.G. Hardwick, E.S. Hwang, A. Amon, and A.W. Murray. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 279:1041–1044. [DOI] [PubMed] [Google Scholar]
- Ink, B., M. Zornig, B. Baum, N. Hajibagheri, C. James, T. Chittenden, and G. Evan. 1997. Human Bak induces cell death in S. pombe with morphological changes similar to those with apoptosis in mammalian cells. Mol. Cell. Biol. 17:2468–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, M.A., K. Wendell, S. Gardiner, W.B. Derry, H. Copp, and L. Wilson. 1996. Mitotic block induced in HeLa cells by low concentrations of Paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 56:816–825. [PubMed] [Google Scholar]
- Juang, Y.L., J. Huang, J.M. Peters, M.E. McLaughlin, C.Y. Tai, and D. Pellman. 1997. APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science. 275:1311–1314. [DOI] [PubMed] [Google Scholar]
- Kamibayashi, C., R. Estes, R.L. Lickteig, S.-I. Yang, C. Craft, and M. Mumby. 1994. Comparison of heterotrimeric protein phosphatase 2A containing different B subunits. J. Biol. Chem. 269:20139–20148. [PubMed] [Google Scholar]
- Kane, D.J., T.A. Sarafian, R. Anton, H. Hahn, E.B. Gralla, J.S. Valentine, T. Ord, and D.E. Bredesen. 1993. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 262:1274–1277. [DOI] [PubMed] [Google Scholar]
- Kanopka, A., O. Muhlemann, S. Petersen-Mahrt, C. Estmer, C. Ohrmalm, and G. Akusjarvi. 1998. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature. 393:185–187. [DOI] [PubMed] [Google Scholar]
- Kleinberger, T., and T. Shenk. 1993. Adenovirus E4orf4 protein binds to protein phosphatase 2A, and the complex downregulates E1A-enhanced junB transcription. J. Virol. 67:7556–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornitzer, D., B. Raboy, R.G. Kulka, and G.R. Fink. 1994. Regulated degradation of the transcription factor Gcn4. EMBO J. 13:6021–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav-Baratz, S., V. Sudakin, J.V. Ruderman, and A. Hershko. 1995. Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc. Natl. Acad. Sci. USA. 92:9303–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie, J.N., M. Nguyen, R.C. Marcellus, P.E. Branton, and G.C. Shore. 1998. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J. Cell Biol. 140:637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie, J.N., C. Champagne, M.-C. Gingras, and A. Robert. 2000. Adenovirus E4 open reading frame 4–induced apoptosis involves dysregulation of Src family kinases. J. Cell Biol. 150:1037–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, F.C., and K.T. Arndt. 1995. The role of Saccharomyces cerevisiae type 2A phosphatase in the actin cytoskeleton and in entry into mitosis. EMBO J. 14:2745–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livne, A., R. Shtrichman, and T. Kleinberger. 2001. Caspase activation by adenovirus E4orf4 protein is cell line-specific and is mediated by the death receptor pathway. J. Virol. 75:789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo, F., E. Frohlich, and K.-U. Frohlich. 1997. A yeast mutant showing diagnostic markers of early and late apoptosis. J. Cell Biol. 139:729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo, F., E. Frohlich, M. Ligr, M. Grey, S.J. Sigrist, D.H. Wolf, and K.-U. Frohlich. 1999. Oxygen stress: a regulator of apoptosis in yeast. J. Cell Biol. 145:757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellus, R.C., J.N. Lavoie, D. Boivin, G.C. Shore, G. Ketner, and P.E. Branton. 1998. The early region 4 orf4 protein of human adenovirus type 5 induces p53-independent cell death by apoptosis. J. Virol. 72:7144–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama, S., S. Nouraini, and J.C. Reed. 1999. Yeast as a tool for apoptosis research. Curr. Opin. Microbiol. 2:618–623. [DOI] [PubMed] [Google Scholar]
- McCright, B., and D.M. Virshup. 1995. Identification of a new family of protein phosphatase 2A regulatory subunits. J. Biol. Chem. 270:26123–26128. [DOI] [PubMed] [Google Scholar]
- Meikrantz, W., and R. Schlegel. 1996. Suppression of apoptosis by dominant negative mutants of cyclin-dependent protein kinases. J. Biol. Chem. 271:10205–10209. [DOI] [PubMed] [Google Scholar]
- Meimoun, A., T. Holtzman, Z. Weissman, H.J. McBride, D.J. Stillman, G.R. Fink, and D. Kornitzer. 2000. Degradation of the transcription factor Gcn4 requires the kinase Pho85 and the SCF(CDC4) ubiquitin-ligase complex. Mol. Biol. Cell. 11:915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull, J., A. Straight, A.D. Rudner, A.F. Dernburg, A. Belmont, and A.W. Murray. 1996. Protein phosphatase 2A regulates MPF activity and sister chromatid cohesion in budding yeast. Curr. Biol. 6:1609–1620. [DOI] [PubMed] [Google Scholar]
- Morgenstern, J.P., and H. Land. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18:3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, U., T. Kleinberger, and T. Shenk. 1992. Adenovirus E4orf4 protein reduces phosphorylation of c-fos and E1A proteins while simultaneously reducing the level of AP-1. J. Virol. 66:5867–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg, D., R. Muller, and M. Funk. 1994. Regulatable promoters of S. cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 22:5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumby, M. 1995. Regulation by tumor antigens defines a role for PP2A in signal transduction. Semin. Cancer Biol. 6:229–237. [DOI] [PubMed] [Google Scholar]
- Mumby, M.C., and G. Walter. 1993. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 73:673–699. [DOI] [PubMed] [Google Scholar]
- Nickels, J.T., and J.R. Broach. 1996. A ceramide-activated protein phosphatase mediates ceramide-induced G1 arrest of Saccharomyces cerevisiae. Genes Dev. 10:382–394. [DOI] [PubMed] [Google Scholar]
- Peters, J.M., R.W. King, C. Hoog, and M.W. Kirschner. 1996. Identification of BIME as a subunit of the anaphase-promoting complex. Science. 274:1199–1201. [DOI] [PubMed] [Google Scholar]
- Posas, F., J. Clotet, M.T. Muns, J. Corominas, A. Casamayor, and J. Arino. 1993. The gene PPG encodes a novel protein phosphatase involved in glycogen accumulation. J. Biol. Chem. 268:1349–1354. [PubMed] [Google Scholar]
- Ronne, H., M. Carlberg, G.-Z. Hu, and J.O. Nehlin. 1991. Protein phosphatase 2A in Saccharomyces cerevisiae: effects on cell growth and bud morphogenesis. Mol. Cell. Biol. 11:4876–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner, A.D., and A.W. Murray. 2000. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 149:1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner, A.D., K.G. Hardwick, and A.W. Murray. 2000. Cdc28 activates exit from mitosis in budding yeast. J. Cell Biol. 149:1361–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, P., S. Moreno, and S.I. Reed. 1989. Conservation of mitotic controls in fission and budding yeasts. Cell. 57:295–303. [DOI] [PubMed] [Google Scholar]
- Schultz, J.B., M. Weller, and T. Klockgether. 1996. Potassium deprivation-induced apoptosis of cerebellar granule neurons: a sequential requirement for new mRNA and protein synthesis, ICE-like protease activity, and reactive oxygen species. J. Neurosci. 16:4696–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, M., A.S. Lutum, and W. Seufert. 1997. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 90:683–693. [DOI] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and J.B. Hicks. 1982. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. 186 pp.
- Shi, L., W.K. Nishioka, J. Th'ng, E.M. Bradbury, D.W. Litchfield, and A.H. Greenberg. 1994. Premature p34cdc2 activation required for apoptosis. Science. 263:1143–1145. [DOI] [PubMed] [Google Scholar]
- Shteinberg, M., Y. Protopopov, T. Listovsky, M. Brandeis, and A. Hershko. 1999. Phosphorylation of the cyclosome is required for its stimulation by Fizzy/cdc20. Biochem. Biophys. Res. Commun. 260:193–198. [DOI] [PubMed] [Google Scholar]
- Shtrichman, R., and T. Kleinberger. 1998. Adenovirus type 5 E4 open reading frame 4 protein induces apoptosis in transformed cells. J. Virol. 72:2975–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman, R., R. Sharf, H. Barr, T. Dobner, and T. Kleinberger. 1999. Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc. Natl. Acad. Sci. USA. 96:10080–10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman, R., R. Sharf, and T. Kleinberger. 2000. Adenovirus E4orf4 protein interacts with both Bα and B′ subunits of protein phosphatase 2A, but E4orf4-induced apoptosis is mediated only by the interaction with Bα. Oncogene. 19:3757–3765. [DOI] [PubMed] [Google Scholar]
- Shu, Y., H. Yang, E. Hallberg, and R. Hallberg. 1997. Molecular genetic analysis of Rts1p, a B′ regulatory subunit of S. cerevisiae protein phosphatase 2A. Mol. Cell. Biol. 17:3242–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater, A.F.G., C. Stefan, I. Nobel, D.J. van den Dobbelsteen, and S. Orrenius. 1995. Signalling and oxidative stress in apoptosis. Toxicol. Lett. 82/83:149–153. [DOI] [PubMed] [Google Scholar]
- Sneddon, A.A., P.T.W. Cohen, and M.J.R. Stark. 1990. Saccharomyces cerevisiae protein phosphatase 2A performs an essential cellular function and is encoded by two genes. EMBO J. 9:4339–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, S.A., B. Poon, J.B.M. Jowett, and I.S.Y. Cen. 1997. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 71:5579–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana, U., H. Robitsch, C. Price, T. Schuster, I. Fitch, A.B. Futcher, and K. Nasmyth. 1991. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell. 65:145–161. [DOI] [PubMed] [Google Scholar]
- Sutton, A., D. Immanuel, and K.T. Arndt. 1991. The SIT4 protein phosphatase functions in late G1 for progression into S phase. Mol. Cell. Biol. 11:2133–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, W., C. Kurschner, and J.L. Morgan. 1997. Modulation of cell death in yeast by the Bcl-2 family of proteins. J. Biol. Chem. 272:15547–15552. [DOI] [PubMed] [Google Scholar]
- van Zyl, W., W. Huang, A.A. Sneddon, M. Stark, S. Camier, M. Werner, C. Marck, A. Sentenac, and J.R. Broach. 1992. Inactivation of the protein phosphatase 2A regulatory subunit A results in morphological and transcriptional defects in S. cerevisiae. Mol. Cell. Biol. 12:4946–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin, R., S. Prinz, and A. Amon. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 278:460–463. [DOI] [PubMed] [Google Scholar]
- Visintin, R., K. Craig, E.S. Hwang, S. Prinz, M. Tyers, and A. Amon. 1998. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell. 2:709–718. [DOI] [PubMed] [Google Scholar]
- Wang, Y., and D.J. Burke. 1997. Cdc55p, the B-type regulatory subunit of protein phosphatase 2A, has multiple functions in mitosis and is required for the kinetochore/spindle checkpoint in Saccharomyces cerevisiae. Mol. Cell. Biol. 17:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wera, S., and B.A. Hemmings. 1995. Serine/threonine protein phosphatases. Biochem. J. 311:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wersto, R.P., E.R. Rosenthal, P.K. Seth, N.T. Eissa, and R.E. Donahue. 1998. Recombinant, replication-defective adenovirus gene transfer vectors induce cell cycle dysregulation and inappropriate expression of cyclin proteins. J. Virol. 72:9491–9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen, S.G., R.C. Marcellus, A. Whalen, N.G. Ahn, R.P. Ricciardi, and P.E. Branton. 1997. Phosphorylation within the transactivation domain of adenovirus E1A protein by mitogen-activated protein kinase regulates expression of early region 4. J. Virol. 71:3545–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., W. Jiang, M. Gentry, and R.L. Hallberg. 2000. Loss of a protein phosphatase 2A regulatory subunit (Cdc55p) elicits improper regulation of Swe1p degradation. Mol. Cell. Biol. 20:8143–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, S., K.A. McKenna, S.J. Sharkis, and A. Bedi. 1996. Requirement of p34cdc2 kinase for apoptosis mediated by the Fas/APO-1 receptor and interleukin 1 β-converting enzyme-related proteases. Cancer Res. 56:4551–4555. [PubMed] [Google Scholar]
- Yeong, F.M., H.H. Lim, C.G. Padmashree, and U. Surana. 2000. Exit from mitosis in budding yeast: biphasic inactivation of the Cdc28-Clb2 mitotic kinase and the role of Cdc20. Mol. Cell. 5:501–511. [DOI] [PubMed] [Google Scholar]
- Zachariae, W., M. Schwab, K. Nasmyth, and W. Seufert. 1998. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 282:1721–1724. [DOI] [PubMed] [Google Scholar]
- Zha, H., H.A. Fisk, M.P. Yaffe, N. Mahajan, B. Herman, and J.C. Reed. 1996. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell. Biol. 16:6494–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y., G. Boguslawski, R.S. Zitomer, and A.A. DePaoli-Roach. 1997. S. cerevisiae homologs of mammalian B and B′ subunits of protein phosphatase 2A direct the enzyme to distinct cellular functions. J. Biol. Chem. 272:8256–8262. [DOI] [PubMed] [Google Scholar]
- Zhou, B.-B., H. Li, J. Yuan, and M.W. Kirschner. 1998. Caspase-dependent activation of cyclin-dependent kinases during Fas-induced apoptosis in Jurkat cells. Proc. Natl. Acad. Sci. USA. 95:6785–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]