Pak-Family Kinases Regulate Cell and Actin Polarization Throughout the Cell Cycle of Saccharomyces cerevisiae (original) (raw)
Abstract
During the cell cycle of the yeast Saccharomyces cerevisiae, the actin cytoskeleton and cell surface growth are polarized, mediating bud emergence, bud growth, and cytokinesis. We have determined whether p21-activated kinase (PAK)-family kinases regulate cell and actin polarization at one or several points during the yeast cell cycle. Inactivation of the PAK homologues Ste20 and Cla4 at various points in the cell cycle resulted in loss of cell and actin cytoskeletal polarity, but not in depolymerization of F-actin. Loss of PAK function in G1 depolarized the cortical actin cytoskeleton and blocked bud emergence, but allowed isotropic growth and led to defects in septin assembly, indicating that PAKs are effectors of the Rho–guanosine triphosphatase Cdc42. PAK inactivation in S/G2 resulted in depolarized growth of the mother and bud and a loss of actin polarity. Loss of PAK function in mitosis caused a defect in cytokinesis and a failure to polarize the cortical actin cytoskeleton to the mother-bud neck. Cla4–green fluorescent protein localized to sites where the cortical actin cytoskeleton and cell surface growth are polarized, independently of an intact actin cytoskeleton. Thus, PAK family kinases are primary regulators of cell and actin cytoskeletal polarity throughout most or all of the yeast cell cycle. PAK-family kinases in higher organisms may have similar functions.
Keywords: p21-activated kinase, actin, polarity, cell cycle, Cdc42
Polarization is the process whereby cells establish and maintain specialized subdomains of the plasma membrane ( Drubin and Nelson 1996; Keller and Simons 1997). Cell polarization involves the vectorial delivery of lipids, proteins, and extracellular matrix or cell wall components to specific locations on the plasma membrane. In the budding yeast Saccharomyces cerevisiae, polarization requires the targeted delivery of secretory vesicles by mechanisms requiring the actin cytoskeleton but not microtubules ( Novick and Schekman 1979; Field and Schekman 1980; Novick and Botstein 1985; Bretscher et al. 1994; Finger and Novick 1998).
In budding yeast, polarization of cortical actin patches and actin cables is regulated during the cell cycle, which is thought to control where cell surface growth occurs ( Drubin and Nelson 1996; Botstein et al. 1997; Karpova et al. 1998). Early in G1, actin patches and cables are oriented randomly and the mother grows isotropically. Late in G1, actin cables orient along the mother-bud axis, usually terminating at actin patches clustered at a site marked for bud emergence ( Karpova et al. 1998). As secretory vesicles are delivered to this site, the bud emerges. As the bud grows, actin patches and cables polarize to the bud tip, allowing apical growth of the bud. Before anaphase, the actin cytoskeleton of the bud depolarizes, resulting in a random distribution of cortical patches and subsequent isotropic growth of the bud. The timing of the switch from apical to isotropic bud growth determines the shape of the bud ( Lew and Reed 1993). Premature switching results in unusually spherical buds, and delayed switching leads to markedly elongated buds. After anaphase, patches are transiently depolarized in both the mother and bud. Patches subsequently cluster at the mother-bud neck where cytokinesis ensues. After cytokinesis, actin patches are depolarized in both the mother and daughter, and isotropic growth occurs. In contrast to patches and cables, an actomyosin ring forms at the mother-bud neck during mitosis and contracts to facilitate cytokinesis ( Bi et al. 1998; Shannon and Li 1999).
Polarization of the yeast actin cytoskeleton in G1 is regulated by Cdc42, a member of the Rho subfamily of monomeric GTPases ( Adams et al. 1990; Johnson and Pringle 1990; Johnson 1999). Cdc42 localizes to the incipient bud site where actin patches cluster and cables orient late in G1. Conditional mutations that inactivate Cdc42 or its guanine nucleotide exchange factor Cdc24 result in a failure to polarize actin patches and cables to the incipient bud site. Accordingly, bud emergence and polarized cell growth are blocked; however, isotropic cell surface growth and the nuclear division cycle continue.
Ste20 and Cla4, a pair of p21-activated kinase (PAK)1 homologues, may be effectors in polarization pathways regulated by Cdc42. Ste20 and Cla4 interact with activated Cdc42 ( Peter et al. 1996; Benton et al. 1997; Leberer et al. 1997). Overexpression of Ste20 suppresses the growth defect of a cdc42-1 temperature-sensitive mutation ( Eby et al. 1998), whereas loss of Ste20 and Cla4 arrests cell growth ( Cvrckova et al. 1995). Ste20 localizes to sites of polarized cell surface growth ( Peter et al. 1996; Leberer et al. 1997), as does Cdc42. Purified Ste20 kinase can correct the actin nucleation defect of permeabilized cdc42-1 mutants in vitro ( Eby et al. 1998).
Whether Ste20 and Cla4 are effectors of Cdc42 that control cell polarity in G1 or other phases of the cell cycle remains unclear. Inactivation of Ste20 and Cla4 arrests cell growth, but it does not recapitulate the uniformly unbudded phenotype of cdc42-1 mutants. For example, a _ste20_Δ cla4-75 temperature-sensitive mutant arrests as budded cells with wide necks in which actin patches and cables are polarized to the bud tip ( Cvrckova et al. 1995), and repression of STE20 expression in a _cla4_Δ mutant does not arrest cell growth exclusively in G1 ( Eby et al. 1998). Furthermore, Cla4 may function late in the cell cycle because its kinase activity peaks near mitosis ( Benton et al. 1997). Nevertheless, these findings do not rule out that Ste20 and Cla4 are effectors of Cdc42. The cell division defect of _ste20_Δ cla4-75 mutants could be an indirect consequence of the failure to form a normal neck early in the cell cycle. Furthermore, Ste20 and Cla4 may function at several points in the cell cycle, including acting as effectors of Cdc42 in G1.
Here we have tested the hypothesis that PAK-family kinases are important regulators of cell polarity during G1 or other parts of the yeast cell cycle. By mapping the points in the cell cycle when Ste20 and Cla4 execute their functions, we provide evidence that PAK-family kinases are required for actin and cell polarization throughout much, or perhaps all, of the yeast cell cycle.
Materials and Methods
Plasmids and Strains
The following procedure was used to construct an integrating plasmid encoding a tagged form of Cla4 that would be degraded rapidly by the N-end rule pathway following a shift to 37°C. The open reading frame (ORF) of the temperature-sensitive cla4-75 allele (YCp_TRP1-cla4-75_; Cvrckova et al. 1995) was amplified with flanking HindIII sites by using Pfu polymerase, and inserted into the HindIII site of the integrating plasmid pPW66R ( Dohmen et al. 1994), producing p_cla4-td_ (temperature-sensitive degron). This procedure produced an in-frame fusion between a hemagglutinin (HA)-tagged, thermolabile form of dihydrofolate reductase (DHFR) and the NH2 terminus of the temperature-sensitive Cla4 protein; expression of the fusion protein is driven from the inducible CUP1 promoter. Cleavage of p_cla4-td_ with NcoI was used to direct integration of the promoter fusion protein cassette at the URA3 locus.
All yeast strains used were isogenic derivatives of W303 (Table ), except the cdc42-1 mutant. Standard genetic techniques were performed according to standard methods ( Sherman 1991); disruptions were confirmed by PCR. CLA4 was disrupted in W303 by one-step gene deletion, resulting in KBY209. A ste20_Δ cla4_Δ double mutant (KBY211) expressing the temperature-sensitive cla4-75 allele on YCp_TRP1-cla4-75 ( Cvrckova et al. 1995) was obtained as a segregant from a cross between KBY209 carrying YCp_TRP1-cla4-75 and a _MAT_α _ste20_Δ mutant (KBY210) carrying a GAL-STE20 plasmid (pRD56) ( Eby et al. 1998). The pRD56 plasmid was eliminated from KBY211 by selection on 5-fluoroorotic acid (5-FOA). The cla4-td allele was integrated at the URA3 locus of wild-type cells (SWY518, a W303 derivative), producing KBY213, and the URA3 locus of a cla4_Δ ste20_Δ mutant (KBY211) carrying YC_pTRP1-cla4-75, producing KBY212. Integration was confirmed by the ability of cells to lose YCp_TRP1-cla4-75, by a temperature-sensitive growth phenotype, and by detection of the DHFR-HA-Cla4 protein. A green fluorescent protein (GFP)-Tub1 fusion (encoded by plasmid pAFS91) ( Straight et al. 1997) was integrated at the HIS3 locus of a _ste20_Δ cla4-td mutant (KBY212) and an isogenic wild-type strain (KBY213), producing KBY216 and KBY215, respectively; integration was confirmed by observing fluorescently labeled microtubules.
Table 1.
Yeast Strains
| Strain | Genotype | Reference |
|---|---|---|
| SWY518 | MAT a ura3 his3 trp1 leu2 can1 [YCp_TRP1_-_cla4-75_] | Srinivasa et al. 1998 |
| KBY209 | MAT a cla4::LEU2 ura3 his3 trp1 leu2 ade2 Gal+ | This study |
| KBY210 | _MAT_α ste20::ADE2 ura3 his3 trp1 leu2 ade2 Gal+ | This study |
| KBY211 | _MAT_α cla4::LEU2 ste20::ADE2 ura3 his3 trp1 leu2 ade2 Gal+ [YCp_TRP1-cla4-75_] | This study |
| KBY212 | _MAT_α cla4::LEU2 ste20::ADE2 URA3::cla4-td his3 trp1 leu2 ade2 Gal+ | This study |
| KBY213 | MAT a URA3::cla4-td his3 trp1 leu2 can1 | This study |
| KBY214 | MAT a CLA4::CLA4-GFP ura3 his3 trp1 leu2 can1 KanR | This study |
| KBY215 | MAT a ADE2::URA3 HIS3::GFP-TUB1 ura3 trp1 leu2 Gal+ | This study |
| KBY216 | _MAT_α cla4::LEU2 ste20::ADE2 URA3::cla4-td HIS3::GFP-TUB1 trp1 leu2 ade2 Gal+ | This study |
| 186 | _MAT_α cdc42-1 ura3-52 his3 trp1 Gal− | Eby et al. 1998 |
Immunoblotting
Cells were grown overnight in synthetic media lacking uracil with or without 500 μM Cu-acetate, diluted into 15 ml of the same media, and grown 4 h at 23°C to an OD600 = 0.5. For temperature-shift experiments, cells were diluted into media lacking uracil and copper at 37 or 23°C for various time periods. Cells were harvested, washed in media lacking copper, and lysed according to a NaOH extraction method ( Yaffe and Schatz 1984). Total protein was quantified by the method of Lowry. Proteins resolved by SDS-PAGE were transferred to nitrocellulose membranes, blocked, probed with the 12CA5 (anti-HA) mAb and a monoclonal goat anti–mouse HRP-coupled secondary antibody (Cappell Laboratories), and visualized by enhanced chemiluminescence (ECL) (Amersham Pharmacia Biotech). Protein expression was quantified by analyzing immunoblots with NIH Image.
Cell Synchronization
G1 cells were isolated using a modification of published methods ( Ayscough et al. 1997). Cells were plated as a lawn on synthetic media containing dextrose and lacking tryptophan for 3 d at room temperature, suspended in 10 ml of media lacking glucose, and sedimented through 1 M sorbitol in selective synthetic media lacking glucose. Unbudded cells remained in the supernatant fraction. Yields of unbudded cells were 85–95% of total cells. Cells in the supernatant fraction were collected by filtration and suspended for 1–2 h at 37°C in synthetic media lacking tryptophan and glucose and then transferred to rich media containing yeast extract, bactopeptone, and dextrose (YPD) at 37°C for the duration of the experiment. At various time points, cells were viewed by Nomarski microscopy or fixed in 3.7% formaldehyde and stained with 0.66 μM rhodamine-phalloidin ( Amatruda et al. 1992). 4,6-diamidino-2-phenylindole (DAPI) (Molecular Probes) was included in mounting media for fixed cells.
For mitotic arrest assays, cells were grown in YPD containing 1% DMSO to facilitate solubilization of nocodazole. After addition of nocodazole (15 μg/ml final concentration), cells were incubated with shaking for at least 3 h at 23°C, resulting in 70–90% of cells arrested with large buds and unsegregated nuclei. Cells were pelleted, washed once in selective synthetic media lacking glucose at 37°C, which inhibited recovery from cell cycle arrest, and suspended in the same media at 37°C for 1–2 h. Cells were collected by filtration and washed at 37°C with at least 3 ml of selective synthetic media lacking glucose to remove residual nocodazole. Cells were suspended in 3 ml of 37°C YPD to resume growth. Cells were fixed with 3.7% formaldehyde at 20-min intervals. Fixed cells were subjected to Zymolyase (ICN) treatment to determine if cell separation had occurred. Cells in phosphate buffer were added to 1 M sorbitol containing 0.2 mg/ml Zymolyase and incubated at 37°C for 15 min.
Immunofluorescence, GFP Tagging, and Imaging
Septin localization was determined by staining cells with anti-Cdc11 antibodies according to published immunofluorescence protocols ( Pringle et al. 1991; Amatruda et al. 1992). In brief, cells were collected at various times after shift to the nonpermissive temperature, fixed, processed, and stained with anti-Cdc11 polyclonal antibody (1:3 dilution; gift of M. Longtine, Oklahoma State University, Stillwater, OK) and FITC-conjugated goat anti–rabbit secondary antibodies (1:500 dilution; Cappell Laboratories). Fusions encoding Cla4-GFP and Abp1-GFP were constructed by using PCR and integrative transformation to append the GFP-S65T coding region to the 3′ end of the genomic CLA4 and ABP1 ORFs. The GFP reading frame was preceded by a Gly8 linker. The fusions, verified by PCR, were fully functional as judged by phenotypic analysis. Images of cells expressing various GFP fusion proteins were captured by using a DAGE cooled CCD camera mounted on an Olympus BX-60 or Olympus IX 70 inverted microscope equipped with a UPlanApo 100× objective. All other immunofluorescence and Nomarski images were acquired on a DAGE cooled CCD camera mounted on an Olympus BH-2 microscope equipped with a DPlanApo100UV 100× objective.
Video Microscopy
Movies of actin patches labeled with Abp1-GFP were obtained according to published methods ( Waddle et al. 1996). For each time point, 9–10 z-plane images covering the entire cell were superimposed to yield a single two-dimensional image. Images were collected with a LG-3 framegrabber (Scion Corporation) and a DAGE ISIT camera mounted on an Olympus BX-60 microscope with a UPlanApo 100× objective. Nomarski movies were made by capturing images at 5-min intervals on an Olympus BH-2 microscope. Thermostatic controls were used to maintain the stage at desired temperatures.
Results
Previous studies have revealed terminal phenotypes resulting from inactivation of Ste20 and Cla4 ( Cvrckova et al. 1995; Peter et al. 1996; Leberer et al. 1997; Eby et al. 1998), but they have not established when these PAK homologues execute their functions during the cell cycle. This point required further study because the terminal phenotype of a mutant can be caused by activation of a checkpoint early in the cell cycle that arrests cells at a point later in the cell cycle ( Weinert and Hartwell 1988, Weinert and Hartwell 1993; Weinert et al. 1994). Therefore, our goal was to determine when in the cell cycle Ste20 and Cla4 execute their functions. This would test the hypothesis that these PAK-family kinases function as effectors of Cdc42 to polarize cells during G1 or other phases of the yeast cell cycle.
To define when during the cell cycle the activity of Ste20 and Cla4 is required, we needed a conditional means of inactivating Cla4 rapidly and completely in a cell that lacks Ste20. Inactivation of both kinases was necessary, because either one is sufficient to allow cell growth and division. To inactivate Cla4 in a cell that lacks Ste20, we constructed a temperature-sensitive degron allele ( Dohmen et al. 1994) by fusing a thermolabile form of DHFR to the NH2 terminus of Cla4 encoded by the existing cla4-75 temperature-sensitive allele ( Cvrckova et al. 1995). Temperature-induced degradation of the DHFR-Cla4 fusion should result in rapid loss of Cla4 function. To facilitate protein detection, the DHFR-Cla4 fusion was tagged with the HA epitope and expressed from the inducible CUP1 promoter. The DHFR-Cla4 expression cassette was integrated at the ura3 locus in a _cla4_Δ _ste20_Δ mutant that carried a plasmid-borne copy of the cla4-75 allele.
The DHFR-Cla4 fusion functioned in the expected manner. At permissive temperature and basal expression levels (absence of added copper), the DHFR-Cla4 fusion expressed from the chromosome was functional because cells could readily lose the plasmid carrying the cla4-75 allele. Cells that had lost the cla4-75 plasmid (referred to subsequently as the _ste20_Δ cla4-td mutant, for temperature-sensitive degron) had a normal morphology and growth rate at the permissive temperature (23°C) (data not shown). As anticipated, the _ste20_Δ cla4-td mutant did not grow at nonpermissive temperature (37°C) under basal or inducing expression conditions (data not shown; see below), indicating that DHFR-Cla4 had been inactivated.
Consistent with these phenotypic effects, the cla4-td allele encoded a protein that was degraded upon shift to nonpermissive temperature ( Fig. 1). After inducing protein expression with copper and shifting cells to 37°C in the absence of copper, the steady-state levels of the DHFR-Cla4 fusion declined rapidly. In contrast, steady state levels of DHFR-Cla4 remained constant at permissive temperature (23°C) under otherwise identical conditions. All subsequent experiments were performed under basal expression conditions, which appeared to provide normal levels of Cla4 function.
Figure 1.

Temperature-dependent degradation of DHFR-Cla4. Cells expressing an HA epitope-tagged form of a DHFR-Cla4 fusion protein were grown at permissive temperature (23°C) and either maintained at permissive temperature or shifted to the nonpermissive temperature (37°C) for the indicated times. DHFR-HA-Cla4 was detected by immunoblotting.
Bud Emergence Requires Ste20 and Cla4
To determine whether Ste20 and Cla4 are effectors of Cdc42 in G1, we asked whether loss of Ste20 and Cla4 recapitulates phenotypes of cdc42-1 mutants: arrest as unbudded cells with depolarized actin patches and the continuation of the nuclear division cycle ( Adams et al. 1990). Because various CLA4 alleles could have different effects, we compared wild-type cells with _ste20_Δ mutants that expressed either the cla4-75 temperature-sensitive allele or the novel cla4-td allele. Furthermore, Ste20 and Cla4 may function at several points in the cell cycle. Therefore, a cell synchronization protocol was used to determine whether Cla4 executes an essential function in G1 when Ste20 is absent. Cells were arrested early in G1 by nutrient deprivation at permissive temperature (23°C) ( Ayscough et al. 1997), shifted to the nonpermissive temperature (37°C) for 1 or 2 h (_ste20_Δ cla4-td or _ste20_Δ cla4-75 mutants, respectively), released from the nutritional block at the nonpermissive temperature, and monitored for the kinetics of bud emergence ( Fig. 2). Wild-type cells treated in this way recovered from nutritional arrest and resumed budding efficiently. In contrast, the _ste20_Δ cla4-td double mutant failed to undergo bud emergence even 22 h after release from the nutritional block. Similar to cdc42-1 mutants, the _ste20_Δ cla4-td mutant cells enlarged over time (data not shown), indicating that isotropic growth occurred. The _ste20_Δ cla4-td mutant did not lose viability at nonpermissive temperature because cells did not stain with methylene blue, and they resumed budding when returned to permissive temperature (data not shown). These results indicated that Ste20 and Cla4 are required for bud emergence, consistent with the hypothesis that they function as effectors of Cdc42 in G1.
Figure 2.

Dependence of bud emergence on Ste20 and Cla4 function. Wild-type cells (W303), a _ste20_Δ cla4-75 mutant (KBY211), and a _ste20_Δ cla4-td mutant (KBY212) were synchronized in G1 at permissive temperature (23°C) by nutritional deprivation, shifted to nonpermissive temperature (37°C), and released from the nutritional block at nonpermissive temperature (see Materials and Methods). Bud emergence was scored microscopically and expressed as the fraction of the cell population that had resumed growth and budded. Cells with buds and wide necks were scored as budded cells. (A) Budding kinetics of wild-type cells (indicated by squares) and a _ste20_Δ cla4-td mutant (indicated by triangles), and (B) wild-type cells (indicated by squares) and a _ste20_Δ cla4-75 mutant (indicated by triangles) are shown.
In contrast to the unbudded phenotype of the _ste20_Δ cla4-td mutant, an isogenic _ste20_Δ cla4-75 double mutant was able to bud at nonpermissive temperature with kinetics and efficiency similar to wild-type cells ( Fig. 2). However, the _ste20_Δ cla4-75 mutant formed cells with wide necks, and cell division did not occur (data not shown). This phenotype was identical to the reported terminal phenotype of _ste20_Δ cla4-75 double mutants ( Cvrckova et al. 1995). Because these phenotypes are less severe than those of the _ste20_Δ cla4-td mutant, the cla4-td allele appeared to inactivate Cla4 function more completely.
To rule out that the requirement of Ste20 and Cla4 for bud emergence was due to the synchronization process, we analyzed the phenotypes of asynchronous cultures shifted to the nonpermissive temperature (Table ). Cultures of the _ste20_Δ cla4-td mutant displayed an increase in the proportion of unbudded cells, consistent with the hypothesis that Ste20 and Cla4 are required for bud emergence. Those of the _ste20_Δ cla4-75 mutant accumulated budded cells with a wide-neck morphology.
Table 2.
Effects of CLA4 Mutations on Cell Morphology and Actin Polarization
| Time (h) | Actin polarization | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Morphology | Wild-type | _ste20_Δ cla4-td | |||||||||
| Wild-type | _ste20_Δ cla4-td | _ste20_Δ cla4-75 | Unbudded | Sm. bud | Unbudded | Sm. bud | |||||
| % Unbudded | % Unbudded | % Peanut | % P | % D | % P | % D | % P* | % D | % P* | % D | |
| 0 | 14 ± 3 | 22 ± 2 | 0 | ||||||||
| 2 | 8 ± 6 | 62 ± 8 | 22 ± 1 | ||||||||
| 4 | 8 ± 2 | 55 ± 7 | 28 ± 1 | 27 | 73 | 94 | 6 | 13 | 87 | 13 | 87 |
| 6 | 15 ± 8 | 54 ± 14 | 49 ± 1 |
If Ste20 and Cla4 are important effectors of Cdc42 in G1, loss of these PAK homologues should result in other phenotypes characteristic of cdc42-1 mutants: depolarization of the actin cytoskeleton while allowing the nuclear division cycle to proceed. To examine actin polarization, we used cells that were synchronized in G1 by nutritional deprivation, shifted to the nonpermissive temperature, released from the nutritional block at the nonpermissive temperature, and stained at various times with rhodamine-phalloidin ( Fig. 3). Wild-type cells and the _ste20_Δ cla4-75 mutant formed buds and polarized actin patches and cables to the bud. In contrast, the _ste20_Δ cla4-td mutant did not bud and never exhibited fully polarized actin patches or cables. However, cortical actin patches and cables were present, indicating that loss of PAK function did not result in wholesale depolymerization of F-actin structures. Similarly, when an asynchronous culture of the _ste20_Δ cla4-td mutant was analyzed, the cortical actin cytoskeleton of unbudded as well as budded cells was depolarized (Table ).
Figure 3.

Effects of Ste20 and Cla4 inactivation on actin cytoskeletal organization in G1. Cells were arrested in G1, shifted to the nonpermissive temperature, released from the nutritional block at the nonpermissive temperature, fixed at the indicated times, and stained with rhodamine-phalloidin and DAPI. Wild-type cells (a–f), a _ste20_Δ cla4-75 mutant (g–l), a _ste20_Δ cla4-td mutant (m–r), and a cdc42-1 mutant (s and t) were analyzed.
To further determine whether actin polarization was defective in the _ste20_Δ cla4-td mutant, we asked whether the cortical actin cytoskeleton polarized transiently. Movies were made by acquiring images at 2-min intervals of cells expressing Abp1-GFP, a component of cortical actin patches. During the course of the experiment (80–90 min) there was no evidence of transient polarization of actin patches in the _ste20_Δ cla4-td mutant at nonpermissive temperature (data not shown). Furthermore, actin patch motility was unperturbed as indicated by making real time movies of cells expressing Abp1-GFP (data not shown), ruling out a role for Ste20 and Cla4 in this process. Therefore, loss of Ste20 and Cla4 results in a severe loss of actin polarity in G1.
To examine the nuclear division cycle, we expressed GFP-tagged tubulin (GFP-Tub1) in wild-type cells and the _ste20_Δ cla4-td mutant. Cells were incubated at permissive or nonpermissive temperature, and microtubule morphology in only unbudded cells was scored periodically over 3 h. As expected, unbudded wild-type cells always displayed astral microtubules attached to a single spindle pole body, indicating that the nuclear division cycle had not progressed beyond G1 ( Fig. 4 A; data not shown). In contrast, most (70%) of the unbudded _ste20_Δ cla4-td cells analyzed at the nonpermissive temperature had microtubule phenotypes characteristic of nuclear cell cycle progression ( Fig. 4 A; data not shown). Most of these cells had duplicated spindle pole bodies separated by a short spindle, whereas some cells (10%) possessed longer mitotic spindles and separated spindle pole bodies typical of cells in anaphase or telophase. As expected from these results, we also found that the kinetics of nuclear division was delayed in the _ste20_Δ cla4-td mutant, as indicated by the rate of appearance of binucleate cells ( Fig. 4 B). The delay in nuclear division could be due to a delay in DNA replication or to the activation of a checkpoint, because results that will be presented in a later section indicate that Ste20 and Cla4 do not regulate nuclear division per se. Indeed, a checkpoint activated by morphogenesis or actin cytoskeletal defects early in the cell cycle has been described by Lew, Snyder, and colleagues ( Lew and Reed 1993, Lew and Reed 1995a, Lew and Reed 1995b; McMillan et al. 1998; Barral et al. 1999). Therefore, we conclude that Ste20 and Cla4 are required for bud emergence and actin polarization early in the cell cycle, indicating that PAK homologues are effectors of Cdc42.
Figure 4.

Effect of Ste20 and Cla4 inactivation on nuclear division. A shows spindle phenotypes in wild-type cells (a and b) and a _ste20_Δ cla4-td mutant (c–f) expressing GFP-tubulin (GFP-Tub1) after incubation at restrictive temperature for 6 h. Wild-type cells showed spindle phenotypes appropriate for various stages of the cell cycle. Unbudded _ste20_Δ cla4-td mutant cells displayed unduplicated spindle pole bodies (c) like unbudded wild-type cells, but more frequently displayed spindle phenotypes inappropriate for unbudded cells including short spindles (d), anaphase spindles (e), and postmitotic segregated spindle pole bodies (f), indicating that the nuclear division cycle continued in unbudded cells. B shows the kinetics of nuclear division after wild-type cells (indicated by squares) and a _ste20_Δ cla4-td mutant (indicated by circles) were released from a G1 block at nonpermissive temperature, as described in Fig. 2. Nuclear division was scored over time by staining cells with DAPI.
Ste20 and Cla4 Are Required for Polarized Growth of Budded Cells
Because shifting an asynchronous culture of the _ste20_Δ cla4-td mutant to the nonpermissive temperature did not arrest cells exclusively with an unbudded phenotype (Table ), we hypothesized that Ste20 and Cla4 are required for progression through later phases of the cell cycle. To test this hypothesis, we used time-lapse video microscopy of single cells to determine whether _ste20_Δ cla4-td cells that have budded (S/G2 phase) can complete bud growth and divide at the nonpermissive temperature ( Fig. 5). Wild-type and mutant cells were shifted to 37°C for 2 h, mounted on media-containing agar pads, and maintained at nonpermissive temperature. Cells with small or medium-sized buds were identified, and images were acquired at 5-min intervals for 2 h. In all wild-type cells examined (6/6), growth was polarized or asymmetric, because buds grew whereas mothers did not, resulting in a decrease in the ratio of mother to bud volume over time ( Fig. 5). Subsequently, wild-type cells divided, completing a round of cell division over the course of the experiment. In striking contrast, the _ste20_Δ cla4-td mutant displayed a profound defect in polarized growth of the bud (7/7 cells examined). Bud growth was much slower than in wild-type cells because both the mother and bud increased in volume, resulting in a constant ratio of mother to bud volume ( Fig. 5), as expected for completely isotropic or depolarized growth. Consistent with a depolarized growth phenotype, cortical actin patches in small budded cells were depolarized (distributed equally between mother and bud) in the _ste20_Δ cla4-td mutant (Table ). Therefore, Cla4 and Ste20 are required for polarized growth of the bud.
Figure 5.

Loss of polarized bud growth in mutants lacking Ste20 and Cla4. Asynchronous wild-type (upper panels) or _ste20_Δ cla4-td (lower panels) cells were incubated 2 h at 37°C before mounting on an agarose pad containing synthetic media. The ratio of mother to bud volume for a typical mother-bud pair (labeled with an asterisk, calculated assuming a spherical shape) is shown. Wild-type cells (6/6 cells) grew in a polarized manner, since the bud grew relative to the mother, thereby decreasing the mother bud ratio, whereas the mother did not grow appreciably (7% in 2 h for the cell shown). However, in the _ste20_Δ cla4-td mutant, cell growth occurred isotropically (7/7 cells). Both the mother and bud grew (mother and bud grew 31% and 30%, respectively, in 110 min in the cells shown), thereby maintaining a constant mother to bud volume ratio.
Cell Division Requires Ste20 and Cla4
Polarization of the actin cytoskeleton to the mother-bud neck and the formation and contraction of an actin ring accompany cell division. Therefore, we determined whether Ste20 and Cla4 are required for these processes. Wild-type or _ste20_Δ cla4-td mutant cells were grown at permissive temperature to allow bud emergence to occur. Cells were arrested at permissive temperature early in mitosis by depolymerizing microtubules with nocodazole. Arrested cells were shifted to the nonpermissive temperature for 1 h to inactivate Cla4, released from the nocodazole block at the nonpermissive temperature, and scored for the ability to divide as indicated by decreases in the proportion of cells exhibiting a large budded morphology ( Fig. 6 A). Whereas wild-type cells divided when released from the nocodazole block, the _ste20_Δ cla4-td mutant showed a strong block or delay in cell division. To confirm that cell division had not occurred, we used cell wall digestion to distinguish cells that had failed to divide from cells that had divided but failed to separate. This treatment resulted in only a slight decrease in the number of large budded cells (data not shown), indicating that the _ste20_Δ cla4-td mutant was defective in the completion of mitosis or cytokinesis rather than cell separation. The cell division defect was accompanied by the failure to polarize actin patches to the mother-bud neck ( Fig. 6 B). However, we found that nuclear division was unaffected, as revealed by DAPI staining (in wild-type and _ste20_Δ cla4-td mutants, binucleate cells appeared within 30 min after release from the nocodazole block; data not shown). Because this result indicated that Ste20 and Cla4 are not required for nuclear division, the delay in nuclear division in unbudded _ste20_Δ cla4-td cells described in a previous section is probably due to the activation of a checkpoint. Therefore, we conclude that Cla4 is required late in the cell cycle to promote actin patch polarization to the neck and the completion of cytokinesis. This is consistent with the observation that Cla4 kinase activity peaks near mitosis ( Benton et al. 1997).
Figure 6.

Dependence of cell division on Ste20 and Cla4 function. Cells were arrested before mitosis by nocodazole treatment at permissive temperature (23°C), shifted to the nonpermissive temperature (37°C) for 1–2 h and released from the nocodazole block at the nonpermissive temperature. Cell division was scored microscopically by the disappearance of large budded cells over time. A shows results obtained using wild-type cells (indicated by squares) and a _ste20_Δ cla4-td mutant (indicated by triangles). B shows the morphology and actin distribution of the wild-type cells and the _ste20_Δ cla4-td mutant after release from the nocodazole block at the nonpermissive temperature. C shows results obtained using wild-type cells (indicated by squares) and a _ste20_Δ cla4-75 mutant (indicated by triangles).
Previous studies have shown that _ste20_Δ cla4-75 mutants arrest growth as large budded cells with wide necks separating the mother and bud, resulting in a cytokinesis defect. However, it had not been determined whether this defect is a direct consequence of a failure to execute events at the time of mitosis or cytokinesis, or an indirect consequence of the failure to form a normal neck early in the cell cycle that results in arrest in mitosis. An indirect block could occur due to a structural defect in the neck that prevents cytokinesis, or to the activation of a checkpoint.
We tested these hypotheses by subjecting the _ste20_Δ cla4-75 mutant to the same nocodazole block and release protocol used above to analyze the _ste20_Δ cla4-td mutant. After release of the mitotic block at nonpermissive temperature, the _ste20_Δ cla4-75 mutant underwent cell division at a wild-type rate ( Fig. 6 C). Therefore, the _ste20_Δ cla4-75 mutant was able to execute functions required at the time of cell division, provided that a proper bud neck had formed. This provided a further indication that the cla4-75 mutation partially inactivates Cla4 function. Therefore, we suggest that the previously reported cell division defect of the _ste20_Δ cla4-75 mutant results from a structural defect in the mother-bud neck or from the activation of a checkpoint in response to the abnormal neck formed early in the cell cycle.
Septin Localization
Septins (CDC3, CDC10, CDC11, and CDC12 gene products) are assembled in G1 into a filament network that forms a double ring structure or collar at the mother-bud neck ( Haarer and Pringle 1987; Kim et al. 1991). Previous studies have indicated that septins are severely mislocalized in _ste20_Δ cla4-75 double mutants, possibly explaining the wide-neck morphology and cytokinesis defect of these cells ( Cvrckova et al. 1995).
To address this question further, we compared septin localization in wild-type cells, and in _ste20_Δ mutants that carried a _cla4_-75 or cla4-td allele. In contrast to previous reports, we found that septins were localized to the mother-bud neck in the _ste20_Δ cla4-75 mutant at the nonpermissive temperature ( Fig. 7 A). Only when cells were grown in rich media at nonpermissive temperature was a septin localization defect observed. However, even under these conditions septins localized to the mother-bud neck in 93% of the _ste20_Δ cla4-75 cells, although the staining pattern was not as sharp as in wild-type cells, suggesting a mild defect in septin organization. The remaining 7% of the cells mislocalized septins to the bud tip, similar to previous reports. Regardless of the septin localization phenotype, the _ste20_Δ cla4-75 mutant always displayed a wide-neck morphology, indicating that severe mislocalization of septins is not required to observe the wide-neck phenotype. Furthermore, septin localization was relatively normal in the _ste20_Δ cla4-td mutant at the nonpermissive temperature, suggesting that Ste20 and Cla4 are not essential for septin stability.
Figure 7.

Effect of Ste20 and Cla4 inactivation on septin stability and assembly. A shows the effects on septin stability. Asynchronous cultures of wild-type cells (a–f) and a _ste20_Δ cla4-75 mutant (g–l) were incubated at the nonpermissive temperature for 6 h; the _ste20_Δ cla4-td mutant (m–r) was incubated 2 h at the nonpermissive temperature. Cells were fixed and stained with anti-Cdc11 polyclonal antibodies and an FITC-labeled secondary antibody. Cells at different stages of the cell cycle are shown. B shows the effects on septin assembly. The kinetics of septin assembly in wild-type cells and the _ste20_Δ cla4-td mutant were determined after release from a G1 block at nonpermissive temperature. The proportion of the cell population that stained with anti-Cdc11 polyclonal antibodies and an FITC-labeled secondary antibody was scored.
Other evidence also suggests that the wide-neck morphology and cytokinesis defect of _ste20_Δ cla4-75 mutants are not due entirely to loss of septin function or assembly. First, septin mutants have narrower necks than _ste20_Δ cla4-75 mutants ( Longtine et al. 1998). Second, unlike _ste20_Δ cla4-75 double mutants, septin-null mutants can form microcolonies containing 80 or more cells, suggesting that several rounds of cell division occur in the absence of septins (M. Longtine, personal communication). Third, we never observed a septin localization defect as severe as that in mutants lacking Nim1-homologous kinases (Gin4, Hsl1, and Kcc4) ( Barral et al. 1999), which localize to the septin ring and control its stability.
Although our results indicated that Ste20 and Cla4 are relatively unimportant to maintain the stability of septins, these kinases could facilitate septin assembly in G1. To address this possibility, we used nutritional arrest to enrich for G1 wild-type and _ste20_Δ cla4-td mutant cells at permissive temperature (23°C). Cells were shifted to the nonpermissive temperature (37°C), immediately released from the nutritional block, and analyzed for septin localization over time. The _ste20_Δ cla4-td mutant displayed a significant defect in septin assembly ( Fig. 7 B). Therefore, it appears that Cla4 facilitates septin assembly rather than stability.
Cla4 Localization
The preceding results suggest that in the absence of Ste20, Cla4 is required for cell and actin polarization throughout most or all of the cell cycle, and that Cla4 facilitates septin assembly. If Cla4 directly regulates cell and actin polarization, it should be localized to sites of polarized growth independently of an intact actin cytoskeleton. Similarly, if Cla4 regulates septin localization, it may associate with the neck or neck filaments, as do Nim1-related kinases that regulate septin stability ( Barral et al. 1999). To test these predictions, we tagged the chromosomal CLA4 locus at the 3′ end of its ORF with the GFP coding region, resulting in the production of a functional Cla4-GFP fusion protein. Cla4-GFP localization was examined under conditions where the actin cytoskeleton was intact or depolymerized ( Fig. 8). In cells with small buds, Cla4-GFP was concentrated in a crescent at the tips and sides of the bud. In cells with medium-sized buds, Cla4-GFP was detected as dots or patches distributed over the cortex of the bud. At wild-type expression levels, Cla4-GFP was undetectable in unbudded cells or in the bud at later points in the cell cycle, and it was never observed in the mother. Cla4-GFP was not observed to localize to neck filaments or the mother-bud neck, which is consistent with the ability of Cla4 to facilitate the assembly rather than the stability of septins. Cla4-GFP localization to the tips of small buds or the cortex of medium buds was not disrupted when latrunculin was used to depolymerize the actin cytoskeleton. This result is in contrast to the actin-dependent polarized localization of proteins such as Myo2 and Sec4 that are thought to function downstream of actin by delivering secretory vesicles to sites of polarized growth ( Pruyne et al. 1998). Therefore, throughout much of the cell cycle Cla4-GFP localizes to sites of polarized cell growth.
Figure 8.
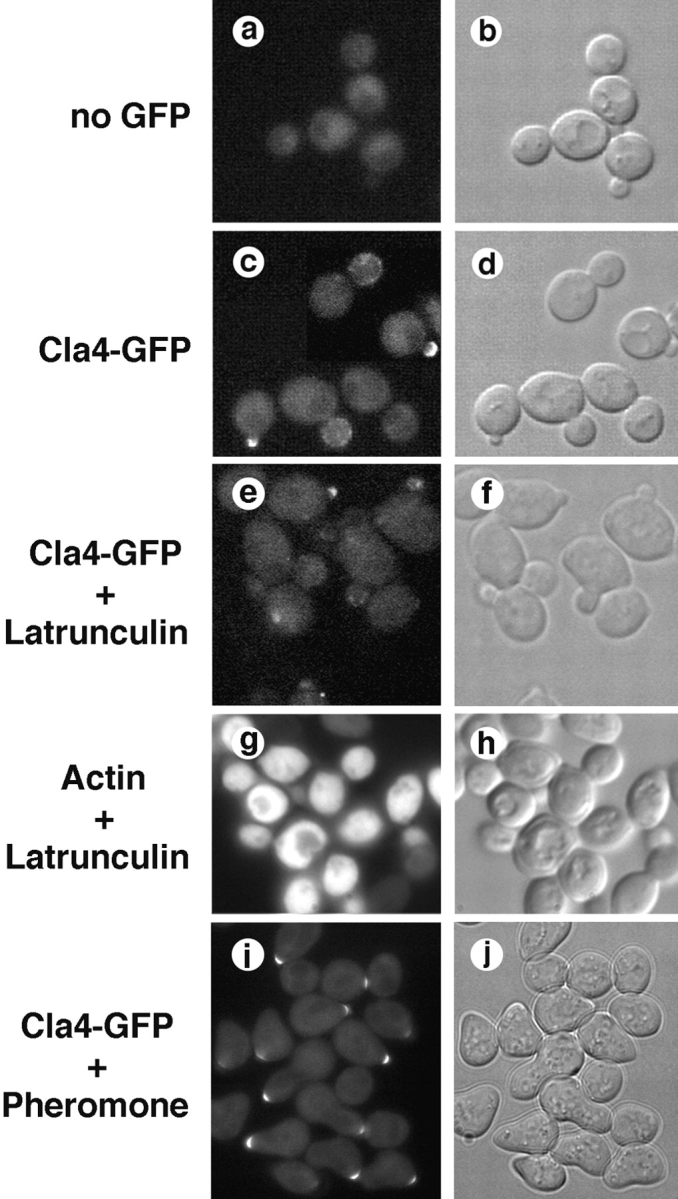
Localization of Cla4-GFP to sites of polarized cell surface growth. Cla4-GFP expressed from the chromosomal CLA4 locus was localized by fluorescence microscopy. Cla4-GFP was detected at the apex of small buds (c), and in dots distributed over the cortex of larger buds (c); depolymerization of the actin cytoskeleton by treating cells with latrunculin (10 min) did not affect the localization of Cla4-GFP (e). Cla4-GFP localized to the tips of polarized cell-surface projections in cells treated 2 h with mating pheromone (α-factor) (i).
Interestingly, Cla4-GFP also localized to the tips of polarized cell surface projections formed when cells were stimulated with mating pheromone ( Fig. 8). Whether this suggests a previously unappreciated role for Cla4 in mating is unknown, but it reinforces the conclusion that Cla4-GFP becomes concentrated to sites of polarized cell growth. Taken together, these results and those of previous studies indicate that Cdc42, Ste20, and Cla4 function in a pathway that transmits signals that polarize the actin cytoskeleton to promote bud emergence and polarized cell surface growth.
Discussion
Throughout much of the cell cycle the actin cytoskeleton of budding yeast is highly polarized, mediating bud emergence, polarized bud growth, and efficient cytokinesis. Here we have shown that cell and actin cytoskeletal polarization requires the PAK homologues Ste20 and Cla4. In cells lacking Ste20, inactivation of Cla4 at various points in the cell cycle reveals that these PAK homologues are required for polarization of the actin cytoskeleton and cell growth during bud emergence, polarized growth of the bud with respect to the mother, and completion of cytokinesis. To our knowledge, Ste20 and Cla4 are the first signaling molecules that have been shown to be required for cell and actin polarization throughout much or all of the yeast cell cycle. Thus, PAK-family kinases appear to be primary regulators of yeast cell polarity.
Regulation of Actin Polarization and Bud Emergence in G1
We have found that Ste20 or Cla4 is required to polarize the actin cytoskeleton and initiate bud emergence. Whereas mutants lacking either kinase can carry out these processes, loss of Ste20 and Cla4 blocks these events, displaying phenotypes like those of cdc42-1 mutants ( Adams et al. 1990). Because results presented here and elsewhere indicate that Cla4 and Ste20 interact and colocalize with Cdc42 at sites of polarized growth ( Adams et al. 1990; Peter et al. 1996; Benton et al. 1997; Leberer et al. 1997), these PAK homologues function as direct signaling effectors of Cdc42 in pathways that promote bud emergence and actin polarization in G1. In contrast, Ste20 and Cla4 are not required for isotropic growth or progression of the nuclear division cycle, indicating that they have primary roles in cell and actin polarization.
Several observations indicate that Ste20 and Cla4 promote bud emergence by executing functions that are at least partially distinct from those carried out by the Cdc42 binding proteins Gic1 and Gic2. Whereas _ste20_Δ _cla4_Δ double mutants are inviable, _gic1_Δ _gic2_Δ double mutants are viable, except at elevated temperature where they display bud emergence and growth defects ( Brown et al. 1997; Chen et al. 1997). Furthermore, _gic1_Δ _gic2_Δ double mutants are nonconditionally inviable when CLA4 is also deleted ( Chen et al. 1997). Finally, overexpression of STE20 or CLA4 suppresses the conditional growth defect of _gic1_Δ _gic2_Δ double mutants ( Chen et al. 1997). Although Ste20 and Cla4 clearly do not require Gic1 or Gic2 to promote bud emergence, there could be functional interactions between these two classes of Cdc42 effectors. For example, PAKs and Gic1/2 could regulate each other's localization, stability, activity, interaction with Cdc42, or association with downstream targets. However, Ste20 and Cla4 may have unique targets, possibly including Bem1. Bem1 is involved in bud emergence, localizes to sites of polarized growth, associates with Ste20 immunoprecipitates, and is phosphorylated in vivo ( Chenevert et al. 1992; Leeuw et al. 1995; Ayscough et al. 1997).
By what mechanisms do Ste20 and Cla4 promote bud emergence? Evidence suggests that these PAK homologues may trigger bud emergence at least in part by stimulating actin filament assembly at the site of bud emergence. In unpolarized cells early in G1, F-actin assembly occurs independently of Cdc42 and its effectors because mutations that inactivate Cdc42 or its effectors do not lead to wholesale depolymerization of F-actin structures ( Adams et al. 1990; Brown et al. 1997; Chen et al. 1997; Eby et al. 1998; this paper). Recruitment of Cdc42 and its effectors, including Ste20 and Cla4, to the presumptive bud site may stimulate the rate of actin filament assembly locally, because activated forms of purified Cdc42 or Ste20 stimulate actin filament assembly in an in vitro assay using permeabilized cdc42-1 cells ( Li et al. 1995; Eby et al. 1998). Furthermore, permeabilized _cla4_Δ or _ste20_Δ mutants show defects in this in vitro assay ( Eby et al. 1998). Stimulation of actin assembly at the bud site could bias the accumulation of new patches and cables to this location at the expense of distal sites on the cell cortex. Several observations are consistent with this hypothesis. Actin monomers dissociate rapidly from patches and cables ( Ayscough et al. 1997). The number of patches or total filamentous actin does not increase in polarized cells ( Waddle et al. 1996; Karpova et al. 1998). A relatively small, invariant pool of actin monomer is present throughout the cell cycle ( Karpova et al. 1995), and inactivation of Ste20 and Cla4 does not affect the relative levels of F- and G-actin (our unpublished results).
Cdc42 and its effectors may stimulate localized actin assembly and cell polarization by targeting the Arp2/3 complex. Indeed, an arp3 temperature-sensitive mutant loses actin polarity at the nonpermissive temperature ( Winter et al. 1997). In vertebrates, the Arp2/3 complex is required for Cdc42 to stimulate actin assembly in cell free extracts ( Ma et al. 1998). Moreover, Cdc42 and PIP2 synergistically activate N-WASP, a mammalian protein that stimulates actin filament assembly by the Arp2/3 complex ( Rohatgi et al. 1999). However, the yeast WASP homologue (Bee1/Las17) ( Li 1997) may not be a direct target of Cdc42. Bee1 lacks a consensus Cdc42 binding domain and a _bee1_Δ mutant does not display a defect in bud emergence ( Li 1997). Instead, _bee1_Δ mutants are partially defective in bud growth and cytokinesis, accumulate secretory vesicles, and have a disorganized actin cytoskeleton ( Li 1997). Accordingly, Cdc42 may use Bee1-dependent and -independent mechanisms to stimulate actin assembly or polarization.
The polarization complex organized by Cdc42 in yeast could also function by capturing actin cables and patches, tethering them near the incipient bud site. However, tethering of the actin cytoskeleton to the polarization complex appears to be flexible and of relatively low affinity. This hypothesis is based on observations that polarized actin patches move at rates similar to unpolarized actin patches ( Doyle and Botstein 1996; Waddle et al. 1996), and that polarized patches can move away from or escape the presumptive bud site ( Waddle et al. 1996). Tethering molecules that link actin cables and patches to the polarization complex remain to be identified.
Cdc42 and its effectors may promote polarization and bud emergence by executing functions in addition to polarizing the actin cytoskeleton. We suggest this because bud emergence can occur in mutants (such as tor2) ( Schmidt et al. 1996) that apparently have lost actin polarization in G1. In tor2 mutants, Cdc42 and it effectors may promote bud emergence by facilitating the docking or fusion of secretory vesicles delivered by actin patches and cables that localize by chance to the incipient bud site. This would be consistent with the essential role of F-actin in bud emergence ( Ayscough et al. 1997). In this regard, two myosin I homologues (Myo3 and Myo5) have been proposed to be targets of Ste20 and Cla4. Mutants lacking Myo3 and Myo5 have a disorganized actin cytoskeleton and accumulate intracellular vesicles ( Goodson and Spudich 1995; Goodson et al. 1996). Furthermore, sites in Myo3 and Myo5 that are phosphorylated by Ste20 in vitro are required for function in vivo ( Wu et al. 1997). However, although _myo3_Δ _myo5_Δ double mutants have severe growth defects, they can carry out bud emergence ( Goodson et al. 1996), indicating that there are other targets of Ste20 and Cla4 involved in this process.
Coordination of Bud Emergence and Neck Morphogenesis
The level of Cla4 activity appears to be critical for cells to coordinate the processes of bud emergence and neck morphogenesis. We suggest this because incubation of _ste20_Δ cla4-75 double mutants at restrictive temperature allows actin polarization to the incipient bud site and subsequent bud emergence to occur, but results in the formation of cells with abnormally wide necks. In contrast, more complete inactivation of Cla4, as occurs in the _ste20_Δ cla4-td double mutant, completely blocks bud emergence.
Cla4 may coordinate neck morphogenesis and bud emergence by regulating septin and actin organization. Whereas our results indicate that Cla4 facilitates septin assembly during G1, it is less clear whether Cla4 also stabilizes septins once they are assembled. On the one hand, Cla4 may be involved in septin stabilization because it activates Gin4, one of three Nim1-homologous kinases that have been suggested to maintain neck filament organization after bud emergence ( Longtine et al. 1998; Barral et al. 1999). However, we find that inactivation of Cla4 in a _ste20_Δ mutant does not lead to wholesale disassembly of septins. Perhaps in the absence of Cla4, other kinases activate Nim1 kinases and promote septin stability. Neck morphogenesis may also be regulated by the ability of Cla4 to polarize the actin cytoskeleton. We suggest this because incomplete loss of Cla4 function (in _ste20_Δ cla4-75 mutants) causes a partial defect in actin polarization (less tightly clustered actin patches) (Holly, S.P., unpublished data). This in turn could result in the delivery of vesicles over a larger region of the cell cortex and the formation of a wide neck.
Regulation of Apical and Isotropic Bud Growth
Previous studies have suggested that Cla4 is required to inhibit apical growth of the bud, resulting in a switch to isotropic bud growth during mitosis. This hypothesis is based on the finding that _cla4_Δ mutants are hyperpolarized and have long buds, suggesting that the switch to isotropic bud growth is delayed. Cla4 has been proposed to promote the apical to isotropic switch by activating Gin4 ( Tjandra et al. 1998) and possibly other Nim1 homologues, which in turn repress apical bud growth ( Benton et al. 1997; Carroll et al. 1998; Longtine et al. 1998; Barral et al. 1999).
Our results indicate that Cla4 is also required for isotropic growth of the bud. In the absence of Ste20, inactivation of Cla4 in G2/M leads to a complete loss of polarization, allowing both the mother and bud to grow. Cla4 may promote isotropic growth of the bud by directing the localization or assembly of actin patches over the bud cortex. Indeed, we find that Cla4-GFP localizes in a punctate pattern over the entire cortex of the bud in G2/M, while being undetectable in the mother. Furthermore, loss of Ste20 and Cla4 in budded cells results in the distribution of actin patches over the cortex of the bud and the mother.
Regulation of Cell Division
By analyzing synchronized cells lacking Ste20, we have found that loss of Cla4 early in mitosis blocks cell division. Because nuclear division is unaffected, Ste20 and Cla4 are likely to promote cell division directly. Indeed, we find that inactivation of Ste20 and Cla4 is accompanied by a failure to polarize actin patches to the neck before cytokinesis. Potentially, Ste20 and Cla4 could also regulate the formation or contraction of the actin ring at the mother-bud neck, which involves myosin II (MYO1 gene product) and an IQ-GAP homologue (IQG1/CYK1 gene product) ( Epp and Chant 1997; Bi et al. 1998; Shannon and Li 1999).
In conclusion, because PAK-family kinases in yeast are required for cell polarization throughout much or all of the cell cycle, members of this family of protein kinases are likely to be critical regulators of cell polarization and actin organization in other eukaryotes. Although many functions of mammalian PAKs remain to be established, there is a variety of evidence linking them to signaling pathways that regulate the organization of the actin cytoskeleton ( Sells et al. 1997; Abo et al. 1998; Daniels et al. 1998; Zhao et al. 1998). Defining the mechanisms whereby yeast and mammalian PAKs regulate cell polarization and actin organization will require the identification of specific substrates. Based on our studies of Ste20 and Cla4, PAKs are likely to have several targets that regulate cell polarization during various steps in the cell cycle and in response to extracellular signals.
Acknowledgments
We thank J. Cooper and T. Karpova (Washington University, St. Louis, MO), M. Longtine, K. Nasmyth (Institute of Molecular Pathology, Vienna, Austria), and J. Pringle (University of North Carolina, Chapel Hill, NC) for providing plasmids, strains, and antibodies. We thank J. Cooper and T. Karpova for providing advice over the course of this project and comments on the manuscript, and an anonymous reviewer for valuable suggestions.
This work was supported by grants from the U.S. Public Health Service (GM44592) and the American Cancer Society. K.J. Blumer is an Established Investigator of the American Heart Association.
Footnotes
1.used in this paper: DAPI, 4,6-diamidino-2-phenylindole; DHFR, dihydrofolate reductase; GFP, green fluorescent protein; HA, hemagglutinin; ORF, open reading frame; PAK, p21-activated kinase; YPD, rich media containing yeast extract, bactopeptone, and dextrose
References
- Abo A., Qu J., Cammarano M.S., Dan C., Fritsch A., Baud V., Belisle B., Minden A. PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:6527–6540 . doi: 10.1093/emboj/17.22.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams A.E., Johnson D.I., Longnecker R.M., Sloat B.F., Pringle J.R. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae . J. Cell Biol. 1990;111:131–142 . doi: 10.1083/jcb.111.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatruda J.F., Gattermeir D.J., Karpova T.S., Cooper J.A. Effects of null mutations and overexpression of capping protein on morphogenesis, actin distribution, and polarized secretion in yeast. J. Cell Biol. 1992;119:1151–1162 . doi: 10.1083/jcb.119.5.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough K.R., Stryker J., Pokala N., Sanders M., Crews P., Drubin D.G. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol. 1997;137:399–416 . doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral Y., Parra M., Bidlingmaier S., Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13:176–187 . doi: 10.1101/gad.13.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton B.K., Tinkelenberg A., Gonzalez I., Cross F.R. Cla4p, a Saccharomyces cerevisiae Cdc42p-activated kinase involved in cytokinesis, is activated at mitosis. Mol. Cell Biol. 1997;17:5067–5076 . doi: 10.1128/mcb.17.9.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E., Maddox P., Lew D.J., Salmon E.D., McMillan J.N., Yeh E., Pringle J.R. Involvement of an actomyosin contractile ring in Saccharomyces cerevisiae cytokinesis. J. Cell Biol. 1998;142:1301–1312 . doi: 10.1083/jcb.142.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D., Amberg D., Mulholland J., Huffaker T., Adams A., Drubin D., Stearns T. The yeast cytoskeleton. In: Pringle J.R., Broach J.R., Jones E.W., editors. The Molecular and Cellular Biology of the Yeast Saccharomyces. Vol. 3. Cold Spring Harbor Laboratory Press; Plainview, NY : 1997. pp. 1–90. [Google Scholar]
- Bretscher A., Drees B., Harsay E., Schott D., Wang T. What are the basic functions of microfilaments? Insights from studies in budding yeast. J. Cell Biol. 1994;126:821–825 . doi: 10.1083/jcb.126.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.L., Jaquenoud M., Gulli M.P., Chant J., Peter M. Novel Cdc42-binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes Dev. 1997;11:2972–2982 . doi: 10.1101/gad.11.22.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll C.W., Altman R., Schieltz D., Yates J.R., Kellogg D. The septins are required for the mitosis-specific activation of the Gin4 kinase. J. Cell Biol. 1998;143:709–717 . doi: 10.1083/jcb.143.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.C., Kim Y.J., Chan C.S.M. The Cdc42 GTPase-associated proteins Gic1 and Gic2 are required for polarized cell growth in Saccharomyces cerevisiae . Genes Dev. 1997;11:2958–2971 . doi: 10.1101/gad.11.22.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenevert J., Corrado K., Bender A., Pringle J., Herskowitz I. A yeast gene (BEM1) necessary for cell polarization whose product contains two SH3 domains. Nature. 1992;356:77–79 . doi: 10.1038/356077a0. [DOI] [PubMed] [Google Scholar]
- Cvrckova F., De Virgilio C., Manser E., Pringle J.R., Nasmyth K. Ste20-like protein kinases are required for normal localization of cell growth and for cytokinesis in budding yeast. Genes Dev. 1995;9:1817–1830 . doi: 10.1101/gad.9.15.1817. [DOI] [PubMed] [Google Scholar]
- Daniels R.H., Hall P.S., Bokoch G.M. Membrane targeting of p21-activated kinase 1 (PAK1) induces neurite outgrowth from PC12 cells. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:754–764 . doi: 10.1093/emboj/17.3.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohmen R.J., Wu P., Varshavsky A. Heat-inducible degrona method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276 . doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- Doyle T., Botstein D. Movement of yeast cortical actin cytoskeleton visualized in vivo. Proc. Natl. Acad. Sci. USA. 1996;93:3886–3891 . doi: 10.1073/pnas.93.9.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin D.G., Nelson W.J. Origins of cell polarity. Cell. 1996;84:335–344 . doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Eby J.J., Holly S.P., van Drogen F., Grishin A.V., Peter M., Drubin D.G., Blumer K.J. Actin cytoskeleton organization regulated by the PAK family of protein kinases. Curr. Biol. 1998;8:967–970 . doi: 10.1016/s0960-9822(98)00398-4. [DOI] [PubMed] [Google Scholar]
- Epp J.A., Chant J. An IQGAP-related protein controls actin-ring formation and cytokinesis in yeast. Curr. Biol. 1997;7:921–929 . doi: 10.1016/s0960-9822(06)00411-8. [DOI] [PubMed] [Google Scholar]
- Field C., Schekman R. Localized secretion of acid phosphatase reflects the pattern of cell surface growth in Saccharomyces cerevisiae . J. Cell Biol. 1980;86:123–128 . doi: 10.1083/jcb.86.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger F.P., Novick P. Spatial regulation of exocytosislessons from yeast. J. Cell Biol. 1998;142:609–612 . doi: 10.1083/jcb.142.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson H.V., Spudich J.A. Identification and molecular characterization of a yeast myosin I. Cell Motil. Cytoskelet. 1995;30:73–84 . doi: 10.1002/cm.970300109. [DOI] [PubMed] [Google Scholar]
- Goodson H.V., Anderson B.L., Warrick H.M., Pon L.A., Spudich J.A. Synthetic lethality screen identifies a novel yeast myosin I gene (MYO5)myosin I proteins are required for polarization of the actin cytoskeleton. J. Cell Biol. 1996;133:1277–1291 . doi: 10.1083/jcb.133.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarer B.K., Pringle J.R. Immunofluorescence localization of the Saccharomyces cerevisiae CDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol. Cell. Biol. 1987;7:3678–3687 . doi: 10.1128/mcb.7.10.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.I. Cdc42an essential rho-type GTPase controlling eukaryotic cell polarity. Microbiol. Mol. Biol. Rev. 1999;63:54–105 . doi: 10.1128/mmbr.63.1.54-105.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.I., Pringle J.R. Molecular characterization of CDC42, a Saccharomyces cerevisiae gene involved in the development of cell polarity. J. Cell Biol. 1990;111:143–152 . doi: 10.1083/jcb.111.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova T.S., Tatchell K., Cooper J.A. Actin filaments in yeast are unstable in the absence of capping protein or fimbrin. J. Cell Biol. 1995;131:1483–1493 . doi: 10.1083/jcb.131.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova T.S., McNally J.G., Moltz S.L., Cooper J.A. Assembly and function of the actin cytoskeleton of yeastrelationships between cables and patches. J. Cell Biol. 1998;142:1501–1517 . doi: 10.1083/jcb.142.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller P., Simons K. Post-Golgi biosynthetic trafficking. J. Cell Sci. 1997;110:3001–3009 . doi: 10.1242/jcs.110.24.3001. [DOI] [PubMed] [Google Scholar]
- Kim H.B., Haarer B.K., Pringle J.R. Cellular morphogenesis in the Saccharomyces cerevisiae cell cyclelocalization of the CDC3 gene product and the timing of events at the budding site. J. Cell Biol. 1991;112:535–544 . doi: 10.1083/jcb.112.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberer E., Wu C., Leeuw T., Fourest-Lieuvin A., Segall J.E., Thomas D.Y. Functional characterization of the Cdc42p binding domain of yeast Ste20p protein kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:83–97 . doi: 10.1093/emboj/16.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuw T., Fourest-Lieuvin A., Wu C., Chenevert J., Clark K., Whiteway M., Thomas D.Y., Leberer E. Pheromone response in yeastassociation of Bem1p with proteins of the MAP kinase cascade and actin. Science. 1995;270:1210–1213 . doi: 10.1126/science.270.5239.1210. [DOI] [PubMed] [Google Scholar]
- Lew D.J., Reed S.I. Morphogenesis in the yeast cell cycleregulation by Cdc28 and cyclins. J. Cell Biol. 1993;120:1305–1320 . doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew D.J., Reed S.I. A cell cycle checkpoint monitors cell morphogenesis in budding yeast J. Cell Biol. 129 1995. 739 749a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew D.J., Reed S.I. Cell cycle control of morphogenesis in budding yeast Curr. Opin. Genet. Dev. 5 1995. 17 23b [DOI] [PubMed] [Google Scholar]
- Li R. Bee1, a yeast protein with homology to Wiscott-Aldrich syndrome protein, is critical for the assembly of the cortical actin cytoskeleton. J. Cell Biol. 1997;136:649–658 . doi: 10.1083/jcb.136.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Zheng Y., Drubin D.G. Regulation of cortical actin cytoskeleton assembly during polarized cell growth in budding yeast. J. Cell Biol. 1995;128:599–615 . doi: 10.1083/jcb.128.4.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M.S., Fares H., Pringle J.R. Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J. Cell Biol. 1998;143:719–736 . doi: 10.1083/jcb.143.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Rohatgi R., Kirschner M.W. The Arp2/3 complex mediates actin polymerization induced by the small GTP-binding protein Cdc42. Proc. Natl. Acad. Sci. USA. 1998;95:15362–15367 . doi: 10.1073/pnas.95.26.15362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan J.N., Sia R.A.L., Lew D.J. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J. Cell Biol. 1998;142:1487–1499 . doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P., Schekman R. Secretion and cell-surface growth are blocked in a temperature-sensitive mutant of Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. USA. 1979;76:1858–1862 . doi: 10.1073/pnas.76.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P., Botstein D. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell. 1985;40:405–416 . doi: 10.1016/0092-8674(85)90154-0. [DOI] [PubMed] [Google Scholar]
- Peter M., Neiman A.M., Park H.O., van Lohuizen M., Herskowitz I. Functional analysis of the interaction between the small GTP binding protein Cdc42 and the Ste20 protein kinase in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:7046–7059 . [PMC free article] [PubMed] [Google Scholar]
- Pringle J.R., Adams A.E., Drubin D.G., Haarer B.K. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–602 . doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Pruyne D.W., Schott D.H., Bretscher A. Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 1998;143:1931–1945 . doi: 10.1083/jcb.143.7.1931. [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M.W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231 . doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Schmidt A., Kunz J., Hall M.N. TOR2 is required for organization of the actin cytoskeleton in yeast. Proc. Natl. Acad. Sci. USA. 1996;93:13780–13785 . doi: 10.1073/pnas.93.24.13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells M.A., Knaus U.G., Bagrodia S., Ambrose D.M., Bokoch G.M., Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 1997;7:202–210 . doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Shannon K.B., Li R. The multiple roles of Cyk1p in the assembly and function of the actomyosin ring in budding yeast. Mol. Biol. Cell. 1999;10:283–296 . doi: 10.1091/mbc.10.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21 . doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Srinivasa S.P., Bernstein L.S., Blumer K.J., Linder M.E. Plasma membrane localization is required for RGS4 function in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 1998;95:5584–5589 . doi: 10.1073/pnas.95.10.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight A.F., Marshall W.F., Sedat J.W., Murray A.W. Mitosis in living budding yeastanaphase A but no metaphase plate. Science. 1997;277:574–578 . doi: 10.1126/science.277.5325.574. [DOI] [PubMed] [Google Scholar]
- Tjandra H., Compton J., Kellogg D. Control of mitotic events by the Cdc42 GTPase, the Clb2 cyclin and a member of the PAK kinase family. Curr. Biol. 1998;8:991–1000 . doi: 10.1016/s0960-9822(07)00419-8. [DOI] [PubMed] [Google Scholar]
- Waddle J.A., Karpova T.S., Waterston R.H., Cooper J.A. Movement of cortical actin patches in yeast. J. Cell Biol. 1996;132:861–870 . doi: 10.1083/jcb.132.5.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T.A., Hartwell L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae . Science. 1988;241:317–322 . doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- Weinert T.A., Hartwell L.H. Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics. 1993;134:63–80 . doi: 10.1093/genetics/134.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T.A., Kiser G.L., Hartwell L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;8:652–665 . doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- Winter D., Podtelejnikov A.V., Mann M., Li R. The complex containing actin-related proteins Arp2 and Arp3 is required for the motility and integrity of yeast actin patches. Curr. Biol. 1997;7:519–529 . doi: 10.1016/s0960-9822(06)00223-5. [DOI] [PubMed] [Google Scholar]
- Wu C., Lytvyn V., Thomas D.Y., Leberer E. The phosphorylation site for Ste20p-like protein kinases is essential for the function of myosin-I in yeast. J. Biol. Chem. 1997;272:30623–30626 . doi: 10.1074/jbc.272.49.30623. [DOI] [PubMed] [Google Scholar]
- Yaffe M.P., Schatz G. Two nuclear mutations that block mitochondrial protein import in yeast. Proc. Natl. Acad. Sci. USA. 1984;81:4819–4823 . doi: 10.1073/pnas.81.15.4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.S., Manser E., Chen X.Q., Chong C., Leung T., Lim L. A conserved negative regulatory region in alphaPAKinhibition of PAK kinases reveals their morphological roles downstream of Cdc42 and Rac1. Mol. Cell. Biol. 1998;18:2153–2163. doi: 10.1128/mcb.18.4.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]