The 193-Kd Vault Protein, Vparp, Is a Novel Poly(Adp-Ribose) Polymerase (original) (raw)
Abstract
Mammalian vaults are ribonucleoprotein (RNP) complexes, composed of a small ribonucleic acid and three proteins of 100, 193, and 240 kD in size. The 100-kD major vault protein (MVP) accounts for >70% of the particle mass. We have identified the 193-kD vault protein by its interaction with the MVP in a yeast two-hybrid screen and confirmed its identity by peptide sequence analysis. Analysis of the protein sequence revealed a region of ∼350 amino acids that shares 28% identity with the catalytic domain of poly(ADP-ribose) polymerase (PARP). PARP is a nuclear protein that catalyzes the formation of ADP-ribose polymers in response to DNA damage. The catalytic domain of p193 was expressed and purified from bacterial extracts. Like PARP, this domain is capable of catalyzing a poly(ADP-ribosyl)ation reaction; thus, the 193-kD protein is a new PARP. Purified vaults also contain the poly(ADP-ribosyl)ation activity, indicating that the assembled particle retains enzymatic activity. Furthermore, we show that one substrate for this vault-associated PARP activity is the MVP. Immunofluorescence and biochemical data reveal that p193 protein is not entirely associated with the vault particle, suggesting that it may interact with other protein(s). A portion of p193 is nuclear and localizes to the mitotic spindle.
Keywords: vaults, ribonucleoprotein particle, poly(ADP-ribose) polymerase, poly(ADP-ribose), mitotic spindle
Vaults have a mass of 13 MD, making them the largest RNP complex found in the cytoplasm of mammalian cells (Rome et al. 1991; Kickhoefer et al. 1996). Initially identified in preparations of clathrin-coated vesicles from rat liver, vaults were named for their distinctive lobular morphology (Kedersha and Rome 1986). They have since been identified in many other eukaryotes, including mice, bullfrogs, rabbits, Xenopus, sea urchins, and Dictyostelium (Kedersha et al. 1990; Hamill and Suprenant 1997). Vertebrate vaults are composed of a small RNA and three proteins of 100 (formerly 104), 193 (formerly 192), and 240 (formerly 210) kD in size. The vault-associated RNA (vRNA)1 has been cloned from several species, including human, mouse, rat, and bullfrog (Kickhoefer et al. 1993, Kickhoefer et al. 1998). vRNA length varies from 86 to 141 bases with some species containing multiple related RNAs. Mammalian vRNA sequences share ∼80% identity and can be folded into a similar predicted secondary structure. However, the vRNA is not a structural component of the vault particle as it makes up <5% of the vault mass, and its degradation does not result in the gross alteration of vault structure. The 100-kD subunit, termed the major vault protein (MVP), constitutes >70% of the particle mass. Its cDNA has been cloned from human, rat, Dictyostelium, and electric ray, and its sequence is highly conserved both at the gene and protein level (Vasu et al. 1993; Kickhoefer and Rome 1994; Scheffer et al. 1995; Vasu and Rome 1995; Herrmann et al. 1997). Vaults have a unique barrel shape that consists of two halves, with each half capable of opening into a flower-like structure with eight petals surrounding a central ring. Each petal is formed by 6 copies of MVP with 96 copies of MVP in the intact vault particle (Kedersha et al. 1991).
Their ubiquitous distribution and highly conserved morphology throughout eukaryotes suggests that vault function is essential, and that the structure of the particle must be important for its function. Although the cellular role of the vault particle has remained elusive, several findings support the notion that vaults may have a transport function. A reconstruction of the vault particle to 31 Å resolution has been completed recently (Kong et al. 1999). This reconstruction showed little internal density, suggesting that the purified vault particle is hollow on the inside, consistent with a carrier and/or sequestration function. We have quantitated the number of vaults in numerous cell types and estimate that there are 10,000–100,000 vaults per cell (Kickhoefer et al. 1998). Although the majority of vaults are localized to the cytoplasm, some have consistently been found in the nuclear fraction (Chugani et al. 1993). This nuclear vault fraction is resistant to removal by high salt and detergent washing, indicating that vault association with nuclei may be specific. Confocal microscopy and immunofluorescence labeling indicate that the intact particle is excluded from the nuclear lumen in mammalian cells. In purified rat liver nuclei, vaults have been immunolocalized to the nuclear membrane at or near nuclear pore complexes (Chugani et al. 1993). Due to the similarities in structure, mass, and symmetry, we have proposed that vaults may be a nuclear pore complex plug and/or transporter, or that they can interact with the nuclear pore complex (Chugani et al. 1991). Additional evidence for vaults as carriers comes from a study on the estrogen receptor in which increased levels of vaults were found in association with estrogen receptors in nuclear extracts (Abbondanza et al. 1998). In adult sea urchin coelomocytes, which are cells responsible for cellular immunity, MVP is localized to the nucleus and appears to be concentrated in the nucleolus (Hamill and Suprenant 1997). In Torpedo electric ray, vaults are highly enriched in the electromotor system where they are transported to the nerve terminal (Herrmann et al. 1996, Herrmann et al. 1999). Vaults have been found to be upregulated in some multidrug-resistant cancer cell lines (Scheffer et al. 1995; Kickhoefer et al. 1998). One mechanism for vault function in this process may be through binding either directly or indirectly to drugs, or by impeding the progress of the drugs to the nucleus or other sites of drug action. These findings are consistent with the idea of vault movement throughout the cytoplasm acting as a carrier and potentially influencing the nucleus.
To complete our characterization of the vault components, we have focused our attention on the higher molecular weight vault proteins p193 and p240. We have recently determined that the p240 vault protein is identical to the mammalian telomerase-associated protein 1 (TEP1) (Kickhoefer et al. 1999). TEP1 was first identified based on its homology to the RNA-binding domain of Tetrahymena p80 (Harrington et al. 1997; Nakayama et al. 1997). The role of TEP1 in the telomerase complex has not yet been defined. The sharing of the TEP1 protein by vaults and telomerase suggests that TEP1 may play a common role in some aspect of RNP structure, function, or assembly. Here we describe the identification and characterization of the 193-kD vault protein by its interaction with MVP in a yeast two-hybrid screen and by peptide sequence analysis. The cDNA encodes a 1724 amino acid (aa) sequence which contains a BRCA1 COOH terminus (BRCT) domain, a region homologous to the catalytic domain of poly(ADP-ribose) polymerase (PARP), and a region similar to the inter–α-trypsin inhibitor protein. Expression of the putative p193 catalytic domain has allowed assessment of this domain as a functional PARP. We also show that p193 has poly(ADP-ribose) activity and that it ADP-ribosylates the MVP in purified vaults.
Materials and Methods
Two-Hybrid Screening in Yeast
An NH2-terminal truncated MVP (bases 259–2754) was subcloned in two steps into the EcoR1 and XbaI restriction sites of pEG202 (kindly provided by Dr. Roger Brent, Molecular Science Institute, Berkeley, CA). The resultant plasmid, plex-MVP, and the reporter plasmid pSH-18 were then transformed into yeast cells of the EGY48 strain (Trp−Leu−His−Ura−, LacZ−). These cells were then transformed with a HeLa cell acid fusion cDNA library in the pJG7-4 expression vector (constructed by J. Gyuris, Mitotix, Cambridge, MA, and kindly provided by Dr. Roger Brent), and about 1 million transformants were plated onto dropout media lacking Trp, His, and Ura containing glucose. Positive clones were selected by replica plating onto dropout media lacking Trp, His, Ura, and Leu containing galactose and X-gal. About 256 clones were selected in the initial screen, but upon rescreening only 6 clones were able to coactivate the lexA-responsive LEU2 and lacZ reporter genes of EGY48 on galactose containing selection media. Putative interactor plasmids were rescued by transformation into KC8 cells. The six putative clones were tested for the specificity of interaction by retransformation into EGY48 along with the reporter plasmid pSH-18 and either the plex-MVP or plex-bicoid as bait plasmids. Three clones specifically interacted with plex-MVP only (the other three interacted with both baits suggesting their interaction was nonspecific). The three interactor clones (8, 15, and 21) were sequenced and determined to be independent overlapping clones of p193. All of the interactor clones contain the 3′ terminus of p193 beginning at bases 4515 (clone 15), 4633 (clone 21), and 4791 (clone 8). In vitro binding assays using glutathione _S_-transferase (GST) fusion proteins and in vitro translated MVP were carried out as described (Ausubel et al. 1995).
Peptide Sequence Analysis
Vaults were purified from monkey liver as described previously (Kedersha and Rome 1986; Kong et al. 1999). Purified vaults were fractionated onto four 6% SDS polyacrylamide gels, stained with copper, and the appropriate bands were excised. An estimated 26 pmol of the 193-kD vault protein was sent to Dr. William S. Lane (Harvard Microchemistry Facility, Cambridge, MA). Peptide sequences were determined on a Finnigan TSQ-7000 Triple Quadrapole Mass Spectrometer. Previously, NH2-terminal sequence analysis on p193 protein purified from bovine spleen vaults and transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories) was carried out by Dr. Audree Fowler (UCLA Protein Microsequencing Facility, University of California, Los Angeles School of Medicine, Los Angeles, CA). Although the degenerate peptide sequence was not useful for cloning, the sequence verified the NH2 terminus determined by 5′ rapid amplification of cDNA ends (RACE).
Cloning of p193 Full-Length cDNA
To isolate the cDNA encoding p193, a human cDNA library (kindly provided by Dr. Owen Witte) was screened as described previously (Kickhoefer et al. 1993). A total of 500,000 recombinants were screened with a randomly primed probe to the interactor clone 15 (EcoR1/XhoI, bases 4515–5490). 71 positive clones were identified. Restriction analysis determined the longest clone to be a 3-kb EcoR1 fragment (bases 2492–5490). Reverse transcription followed by PCR was used to isolate bases 663–2492. The first strand cDNA was synthesized using SuperScript II (Life Technologies, Inc.) and random primers (16-mers, kindly provided by Dr. Dohn Glitz, University of California, Los Angeles) from total HeLa cell RNA. The following primers were used for reverse transcription PCR: p193RB5 (5′-CCCCCGAATTCGTGGATGTCTTGCAGATATTTAGAGTT-3′), p193+2225 (5′-TTGGGAGATAGGCAGCAGACAAACCGATGT-3′), p193RT5 (5′-CCTTATAAGCCCCTGGACATCAC-ACCACCTCC-3′), and p193-RBD3 (5′-CCCGGATCCGGCCTTGGTGCTGCTGGAA-3′). The PCR products were digested with either EcoRI (bases 1330–2492, amplified with p193RB5 and P193+2225) or Eco RV and BamHI (bases 663–1940, amplified with p193RT5 and p193RBD3), and cloned into the corresponding sites in pBluescript SK+ (Stratagene). The 5′ end of the cDNA clone (bases 1–663) was obtained by 5′ RACE according to the manufacturer's instructions (Life Technologies, Inc.) except poly(A)+ RNA from 293 cells was used in place of total RNA. The gene-specific primers were GSP1 (5′-TCTGCCCAAATCATCTCTACTAAA-3′) and GSP2 (5′-GAGTGCTTGAATTCATGACTTCCTCC-3′). The amplified RACE product was digested with EcoRV (base 663) and SalI (a site from the abridged universal anchor primer) and was cloned into the corresponding sites in pBluescript SK+. A complete p193 cDNA clone was assembled from various restriction fragments. The NH2 terminus was tagged with VSVG (a 14 aa sequence, YTDIEMNRLGK, from vesicular stomatitis virus glycoprotein) and subcloned into the expression vector, pSVL (Amersham Pharmacia Biotech). COS cells were transiently transfected with the lipid reagent DMRIE (Life Technologies, Inc.) following the manufacturer's guidelines.
Northern Analysis
A multiple tissue Northern blot containing 2 μg of poly(A)+ human RNA was purchased from Origene and hybridized following their protocol with a randomly primed p193 probe (bases 385–880). The blot was stripped twice and hybridized first with a randomly primed MVP probe (bases 1–330) and then with a human β-actin cDNA probe supplied by Origene.
Subcellular Fractionation and Analysis of p193 and MVP
Preparation of HeLa cell extracts (S100 and P100) and discontinuous sucrose gradient fractionation of the P100 extracts were carried out as described (Kickhoefer et al. 1998). The 100,000 g pellet was resuspended by dounce homogenization with a Teflon pestle. Both S100 and P100 extracts and sucrose gradient fractions were resolved by SDS-PAGE and transferred to Hybond membrane (Amersham Pharmacia Biotech). The equivalent of 1.2 × 106 cells were represented in each lane of the S100 and P100 extracts. Equivalent aliquots of each of the sucrose gradient fractions were represented. The membrane was incubated with affinity-purified anti-p193 antibody (1:500), followed by an HRP-conjugated secondary antibody and visualized by ECL (Amersham).
Antibody Production
Two fragments of the p193-containing aa 408–611 (p193rbd) or 1471–1724 (p193int) were expressed in the pET expression system (Novagen) or as GST fusion proteins (Amersham Pharmacia Biotech). The p193int (pET) protein was purified on a His-bind column (Novagen) and injected into a rabbit. Conversely, the p193rbd (pET) protein was present in the insoluble fraction and was purified on an SDS-polyacrylamide gel; the appropriate fragment was excised, minced, and injected into the same rabbit. A p193 (408–611 or 1471–1724) containing GST fusion protein was coupled to Affi-Gel 15 resin (Bio-Rad Laboratories) to make an affinity column. Antiserum was initially purified on protein A as described (Sambrook et al. 1989). After affinity purification on Affi-Gel 15 columns, the antibody protein concentrations were too low to be measured by protein assay and appropriate dilutions were determined by immunoblot analysis. Affinity-purified Ig was used at 1:500 for immunoblot analysis.
Catalytic Activity Assays
The catalytic domain (aa 255–611) of p193 was amplified by PCR and inserted into the EcoR1 and XhoI sites of the pET28b expression vector. His tagged p193cat (aa 255–611) protein was purified on a His-bind affinity column. Poly(ADP-ribose) activity assays were carried out as described previously (Simonin et al. 1993a,Simonin et al. 1993b; Smith et al. 1998). Reactions contained 1 μg of purified p193 cat or 3 μg of purified rat liver vaults. Vaults were purified from rat liver as described (Kedersha and Rome 1986; Kong et al. 1999). Reactions were incubated at 25°C for 30 min in assay buffer (0.1 ml) containing 50 mM Tris-HCl, pH 8.0, 4 mM MgCl2, 0.2 mM DTT, 1.3 μM [32P]NAD+ (4 μCi; New England Nuclear), and 1 mM of unlabeled NAD+. Some assays contained the PARP inhibitor 3-aminobenzamide (3ABA) at 1 mM final concentration. Reactions were stopped by the addition of TCA containing deoxycholate (as a carrier) to a final concentration of 20% and 0.8 mg/ml, respectively. Precipitated proteins were suspended in SDS loading buffer, and fractionated by SDS-PAGE. Proteins were visualized by Coomassie blue stain and exposed to PhosphorImager screens (Molecular Dynamics).
Immunofluorescence
HeLa cells or human foreskin fibroblasts were grown on coverslips until ∼50–70% confluent, then cells were fixed in 4% paraformaldehyde in PHEM buffer (60 mM Pipes, 25 mM Hepes, 10 mM EGTA, 2 mM MgCl2, pH 6.9), permeabilized and postfixed in methanol (Kedersha and Rome 1990). The DNA damage experiments were carried out on human foreskin fibroblasts that were exposed to a UV Stratalinker 1800 (Stratagene) using energy settings 50, 100, 150, and 200 μJ. UV-treated cells were allowed to recover for 24 h before immunostaining. Cells were stained with DAPI (Sigma Chemical Co.) at 0.5 μg/ml. The following antibodies were used for our immunofluorescence studies: affinity-purified anti-p193 polyclonal IgG was used at 1:100; affinity-purified anti–rat vault polyclonal IgG was used at 1:100; a monoclonal anti-MVP (LRP56; kindly provided by Dr. Rik Scheper, Academic Hospital, Vrije Universiteit, Amsterdam, The Netherlands) (Scheper et al. 1993) IgG was used at 10 μg/ml; a monoclonal anti-p53 (BP53; Sigma Chemical Co.) was used at 1:200; a polyclonal anti-PARP (Boehringer Mannheim) antibody was used at 1:2,000; and a monoclonal anti-tubulin (T4026; Sigma Chemical Co.) was used at 1:200. Cells were incubated for 1 h in primary antibody followed by a 30-min incubation with goat anti–rabbit Cy3 (1:250 dilution; Jackson ImmunoResearch Laboratories), goat anti–mouse Cy3 (1:250 dilution; Jackson ImmunoResearch Laboratories), or goat anti–mouse FITC (1:200 dilution; Jackson ImmunoResearch Laboratories). COS cells were transfected with the VSVG-tagged p193 cDNA using the lipid reagent DMRIE (Life Technologies, Inc.). Transiently transfected cells were fixed as described above and immunostained with a monoclonal antiserum to the 14 aa VSVG epitope tag, anti-VSVG (P5D4; Sigma Chemical Co.) antiserum at 1:500, followed by incubation with a goat anti–mouse Cy3 (1:250 dilution; Jackson ImmunoResearch Laboratories). Fixed cells were mounted in polyvinyl alcohol–based mounting medium and viewed with a Nikon FXA epifluorescence microscope.
Results
p193 Interacts with the MVP in a Yeast Two-Hybrid Screen
To identify cellular proteins which interact with the MVP, we pursued a yeast two-hybrid strategy (Fields and Song 1989; Durfee et al. 1993; Gyuris et al. 1993). A cDNA sequence encoding the rat MVP (sequence data available from EMBL/GenBank/DDBJ under accession no. U09870), missing the NH2-terminal 67 aa, was inserted into the expression vector, pEG202. The resultant plasmid plex-MVP encodes a hybrid protein containing the DNA-binding domain of lexA fused to MVP residues 68–885. We then transformed the yeast strain EGY48 containing the lacZ reporter (pSH18-34) and lex-MVP along with a galactose-inducible HeLa acid fusion cDNA library. About 1 million library transformants were screened, and 6 clones were isolated that coactivated the lexA-responsive LEU2 and lacZ reporter genes of EGY48. Three of the isolates interacted specifically with lex-MVP in a yeast two-hybrid retransformation assay where an irrelevant protein (lexA-bicoid) was used as a negative control (Table ). Nucleotide sequence analysis of the three isolates identified a previously determined nucleotide sequence of unknown function (KIAA0177; sequence data available from EMBL/GenBank/DDBJ under accession no. D79999). The three overlapping clones encoded the COOH terminus, beginning at aa 1471, 1510, and 1562, respectively (Fig. 1). The region encoding aa 1562–1724 was designated the MVP interaction domain (Fig. 1, IV), since it is the smallest domain that we have tested that interacts with MVP.
Table 1.
Specificity of Interaction of p193int Domain Using Two-Hybrid Analysis
| p193int | Bait proteins | |
|---|---|---|
| lex-MVP | lex-bicoid | |
| 8 | + | − |
| 15 | + | − |
| 21 | + | − |
Figure 1.
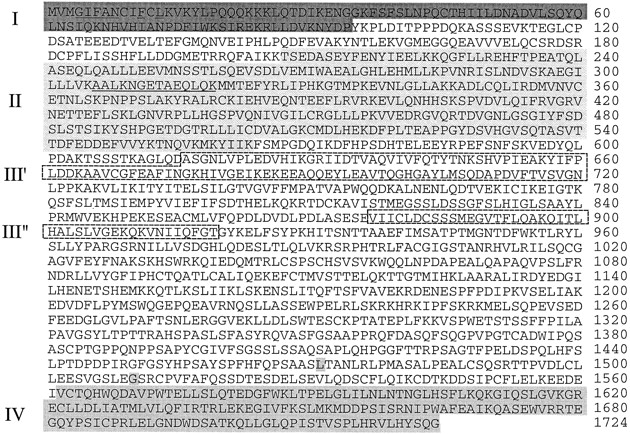
p193 is 1724 aa in length and can be divided into four domains (I–IV). A PROSITE sequence motif search identified the NH2 terminus (aa 1–94) as a BRCT domain (dark grey, I). aa 209–563 share 28% sequence identity with the catalytic subunit of PARP (light grey, II). aa 616–706 and 877–919 share 30 and 29% identity, respectively, with the inter–α-trypsin inhibitor heavy chain (broken boxes, III), although the significance of this homology is not clear at present. The COOH terminus (aa 1562–1724) defines the region necessary for interaction with the MVP (medium gray, IV). The nucleotide sequence data are available from GenBank/EMBL/DDBJ under accession no. AF158255.
The results of the two-hybrid assay were consistent with the results from an in vitro binding assay using a GST fusion protein. In this experiment, the p193 MVP interaction domain (p193int) was expressed in Escherichia coli as a GST fusion protein and was then bound to glutathione beads. The beads were incubated with reticulocyte lysate containing in vitro–translated 35S-labeled MVP, and washed. Binding was assessed by fractionation on SDS-PAGE, followed by PhosphorImager analysis (Fig. 2).
Figure 2.

In vitro binding assay of p193–MVP interaction. In vitro–translated [35S]methionine-labeled MVP was incubated with GST fusion proteins, GST only (lane 1), or GST-p193int (lane 2) and glutathione-agarose beads. After extensive washing, proteins extracted from the beads were analyzed by SDS-PAGE followed by PhosphorImager analysis.
Concurrently, highly purified vaults from monkey liver were fractionated by SDS-PAGE, stained, and the appropriate gel fragments (p193) were excised and sent to William S. Lane (Harvard Microchemistry Facility) for peptide sequence analysis. One peptide sequence was obtained (AALKNGETAEQLQK) and was determined to correspond to nucleotides 639–680 of the KIAA0177 by a TBLASTN search of the nonredundant nucleotide sequence database (Fig. 1). These results confirmed the identity of the KIAA0177 sequence to be a truncated form of the 193-kD vault protein. The 5′ end of the p193 cDNA clone was obtained using 5′ RACE. The predicted NH2 terminus was confirmed by earlier NH2-terminal aa sequence analysis of p193 protein purified from bovine spleen vaults carried out by Audree Fowler (UCLA Protein Microsequencing Facility). The bovine NH2-terminal sequence MTV(L/G)IFAN(S/L)(T/P)F(Q/V)L verifies the NH2 terminus of the p193 protein (Fig. 1). The sequence differences between the human and bovine p193 proteins probably represent species-specific variation. Although the degenerate sequence was not useful for cDNA cloning, it allows us to conclude that we have identified the authentic NH2 terminus.
Structural Analysis of p193
The composite p193 cDNA is 5490 bases, with a short untranslated 5′ end; the coding region encompasses bases 107–5281 and encodes a protein of 1724 aa. Fig. 1 shows the 1724 aa sequence encoded by the p193 cDNA. The size of the predicted protein was calculated to be 192.7 kD. A PROSITE protein sequence analysis of the aa sequence revealed several interesting features, thus allowing the sequence to be separated into four domains (Fig. 1, I–IV). First, aa 1–94 were identified as a BRCT domain (Fig. 1I). BRCT domains were first identified in the BRCA1 gene and later were determined to define a superfamily of cell cycle checkpoint DNA damage response proteins (Wu et al. 1996; Bork et al. 1997; Callebaut and Mornon 1997). The BRCT domain is thought to be important for protein–protein interactions. Domain II is discussed below. Domain III is formed by aa 616–706 and 877–919. This domain shares 30 and 27% identity, respectively, with the inter–α-trypsin inhibitor heavy chain–related protein, a novel human plasma glycoprotein (Fig. 1, III) (Choi-Miura et al. 1995). Domain IV is the MVP interacting domain (discussed above; Fig. 1, IV).
The second domain, aa 209–563, shares 29% identity with the catalytic subunit of PARP (PARP, sequence data available from EMBL/GenBank/DDBJ under accession no. G130781) (Fig. 3). PARP is a nuclear protein that can be divided into three domains: the NH2-terminal DNA binding domain (containing two zinc fingers), a central automodification domain, and a COOH-terminal catalytic domain (for review see de Murcia et al. 1991). The catalytic subunit binds to NAD+, hydrolyzes the nicotine moiety, and polymerizes the ADP-ribose group in response to DNA damage. Poly(ADP-ribose) is attached mainly to PARP, but also to other substrates including histones H1 and H2B (Simonin et al. 1993b). A number of drugs have been shown to bind to the active site of the catalytic subunit, thus blocking NAD binding (including PD128763 and 3ABA; Ruf et al. 1996, Ruf et al. 1998). The minimum region necessary for PARP to retain catalytic activity is a 40-kD fragment (aa 654–1014; Simonin et al. 1990). The crystal structure of the catalytic fragment of PARP has been determined (Ruf et al. 1996, Ruf et al. 1998). Based on the crystal structure, the residues that form the NAD binding pocket are conserved between PARP and p193 (Fig. 3, shaded residues). These data suggest that this region of the p193 will form a similar binding pocket, which could have catalytic activity.
Figure 3.

Alignment of p193 and PARP catalytic domains. Colons indicate conserved aa. Periods indicate semi-conserved aa. Highlighted aa form the binding pocket of the catalytic domain in the crystal structure of the catalytic fragment of PARP (Ruf et al. 1996, Ruf et al. 1998).
p193 Catalytic Activity
To determine whether p193 has PARP activity, the catalytic domain of p193, aa 255–611, were expressed in E. coli as a His-tagged fusion protein and purified. An in vitro PARP activity assay, which measures the addition of radiolabeled ADP-ribose to protein acceptors with [32P]NAD+ used as a substrate, was carried out. A Coomassie stain of the gel before exposure to a PhosphorImager screen shows that equal amounts of proteins were used in all of the assays (Fig. 4, left panel). Like PARP, the catalytic domain of p193 contains ADP-ribosylation activity, and it ADP-ribosylates itself (Fig. 4, right panel). This activity is heat inactivatable (Fig. 4, right panel). The addition of unlabeled NAD+ (1 mM) decreased the level of labeled ADP-ribose polymers added to p193 (255–611) about threefold (Fig. 4, right panel). To confirm that the labeling reaction with p193 was analogous to PARP-catalyzed poly(ADP-ribosyl)ation, the PARP-specific inhibitor 3ABA was included in a reaction. Modification of p193 (255–611) was decreased about twofold in the presence of the inhibitor (Fig. 4, right panel). Furthermore, modified p193 (255–611) reacted with a monoclonal anti–poly (ADP-ribose) antibody (data not shown), consistent with it carrying ADP-ribose polymers. These data indicate that p193 (255–611) is a PARP.
Figure 4.

The catalytic subunit of p193 is a PARP that ADP-ribosylates itself. The catalytic domain of p193 (255–611) was expressed and purified from E. coli. In vitro assays containing p193 (255–611) were incubated in the presence of [32P]NAD+ and the products separated by SDS-PAGE followed by Coomassie blue staining (left panel) and by autoradiography (right panel). Reactions contained 1 μg of p193 (255–611) and 1.3 μM [32P]NAD+. Preincubation of p193 (255–611) at 65°C for 10 min before the addition of labeled NAD+ inactivated the activity (heat). Reactions were supplemented with either 1 mM unlabeled NAD+ (cold chase) or with 1 mM 3ABA, the PARP inhibitor (inhibitor).
Next, we wanted to investigate whether full-length endogenous p193 within the vault particle would possess enzymatic activity. Highly purified vault particles were incubated with [32P]NAD+ in the presence and absence of inhibitor or unlabeled NAD+. The most prominently modified protein in purified vaults was the MVP. However, there was some labeling in the vicinity of the p193 and a high molecular weight smear was also detected (Fig. 5). Modification of all of these products was competed for by the addition of unlabeled NAD+ and partially competed by the addition of the inhibitor 3ABA. These data indicate that full-length p193 is a PARP that is active in the vault particle with at least one specific substrate, MVP.
Figure 5.

In purified vaults, p193 is a PARP that ADP-ribosylates itself and the MVP. Vaults (3 μg) purified from rat liver were incubated in the presence of [32P]NAD+ (as indicated) and the products separated by SDS-PAGE followed by Coomassie blue staining (left panel), and by autoradiography (right panel). Reactions were supplemented with either unlabeled NAD+ (Cold Chase) or with the PARP inhibitor 3ABA (Inhibitor). Arrows indicate the specific ADP-ribosylated proteins. The bracket indicates the smear of higher molecular weight ADP-ribosylated products.
Heterogenous Expression of p193 in Human Tissues
We determined the expression of p193 by Northern blot analysis of human tissues, including brain, heart, kidney, spleen, liver, and leukocytes (Fig. 6). In all tissues, except brain, a 5.4-kb mRNA was readily detectable in 2 μg of poly(A)+ RNA. The highest level of expression was seen in kidney, with about equal levels detectable in spleen and liver. The p193 mRNA tissue expression pattern is similar to that of MVP; however, the level of expression in individual tissues is variable, as there is a higher level of MVP mRNA in spleen compared with liver (Fig. 6).
Figure 6.

Tissue-specific expression patterns of p193 and MVP are similar. A Northern blot (Origene) containing poly(A)+ RNA from human tissues was hybridized with either a 32P-labeled probe specific for p193 (top), MVP (middle), or β-actin (bottom) as described in Materials and Methods. The tissue sources are indicated at the top of the blot.
Subcellular Fractionation of p193
A polyclonal anti-p193 antibody was generated from bacterially expressed fragments of p193 (aa 408–611 and 1471–1724; see Materials and Methods). The anti-p193 antibody recognizes a single protein species of 193 kD by immunoblot analysis (Fig. 7 A). To compare the subcellular distribution of p193 with MVP, extracts from tissue culture cells were isolated and fractionated on a discontinuous sucrose gradient followed by immunoblot analysis (Fig. 7). Vaults are cytoplasmic particles that typically pellet with the microsomes at 100,000 g (Kedersha and Rome 1986). Detergent-lysed HeLa cells were centrifuged at 20,000 g, resulting in a nuclear (N) pellet. The supernatant was further fractionated by centrifugation at 100,000 g and the supernatant (S100) and pellet (P100) fractions were analyzed by immunoblotting with anti-p193 antibody. Interestingly, unlike MVP, which primarily fractionates with the P100, all of the fractions contained the p193 protein (Fig. 7 A). We should note that the N fraction does not represent purified nuclei, and a certain portion of the cells are in mitosis at any given time, so the amount of p193 detected by immunoblotting may not be comparable to that seen by immunofluorescence (see below). Further fractionation of the P100 extract on a discontinuous sucrose gradient revealed that the majority of p193 sedimented to the 45/50% sucrose layer, coinciding with the pattern observed for the MVP (Fig. 7 B). These results suggest that all of the p193 protein in the P100 fraction is associated with the vault particle. Immunoblot analysis of vaults purified from rat liver revealed that the p193 vault protein is recognized by the anti-p193 antibody, further confirming its association with vaults (Fig. 7 B). The shifted mobility of p193 in vaults purified from rat liver is probably due to species-specific differences.
Figure 7.

p193 exists in cells associated with vaults and in other subcellular fractions. Comparison of levels of p193 during biochemical fractionation of HeLa cells. (A) Cellular protein fractions, nuclear (N), supernatant S100 (S), and microsomal pellet P100 (P) were generated from HeLa extracts and resolved by SDS-PAGE (see Materials and Methods). Immunoblot analysis was carried out using affinity-purified polyclonal antibody prepared against recombinant p193 fragment. (B) The P100 fraction was further fractionated on a discontinuous sucrose gradient (20/30/40/45/50/60%), and the proteins from the gradient fractions were resolved by SDS-PAGE. Immunoblot analysis was carried out using either affinity-purified p193 polyclonal antibody (top) or affinity-purified anti-vault polyclonal antibody (bottom). Sucrose gradient fraction sources are indicated at the top. P100 represents the starting material, and the vault sample was purified from rat liver (a lighter exposure of the detected MVP in purified vaults is shown). Arrows indicate the positions of p193 and MVP.
Immunolocalization of p193
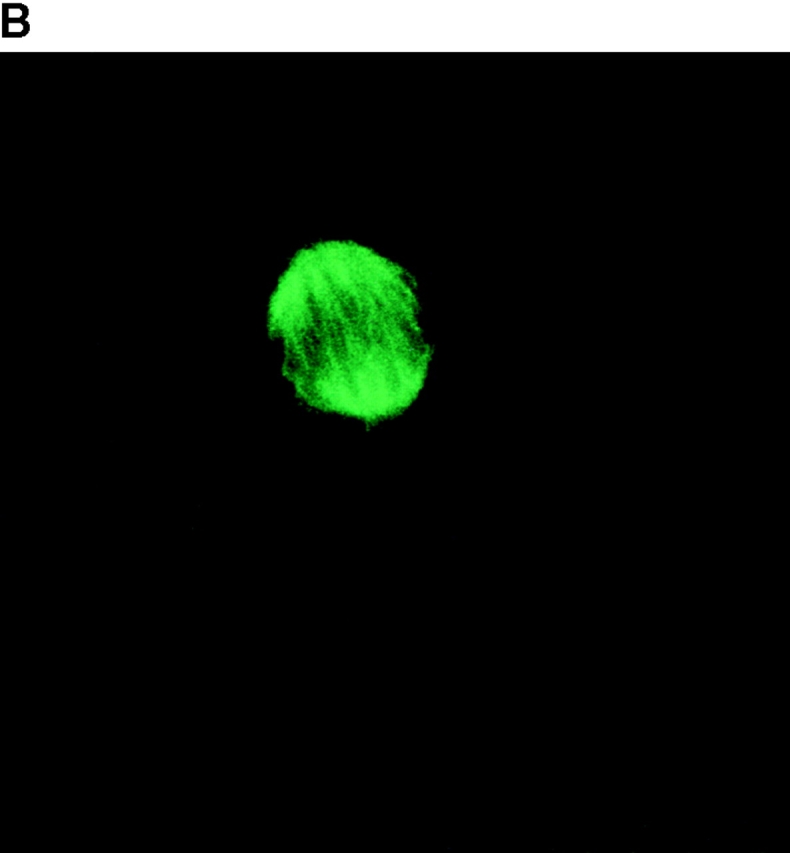
To determine the intracellular distribution of p193 in tissue culture cells, we carried out immunostaining using affinity-purified anti-p193 antibody. Vaults have a punctate cytoplasmic distribution (Fig. 8 and see Fig. 10; Kedersha and Rome 1990). They have also been localized to lamellipodia and at the tips of actin filaments at or near adhesion plaques (Kedersha and Rome 1990). Immunofluorescence patterns observed for p193 have a similar punctate cytoplasmic distribution. However, a variable number of nuclear speckles are also detectable with anti-p193 antibody that is not seen with anti-vault antibody (Fig. 8 and see Fig. 10). Double-immunofluorescence images revealed coincident staining of p193 and MVP in the cytoplasm, but no coincident staining is detected in the nucleus. The lack of completely coincident staining of p193 and MVP in the cytoplasm is not surprising, as our fractionation studies showed that not all of the p193 is associated with vaults. Transfection of COS cells with a cDNA encoding p193 containing a VSVG epitope tag (VSVG-193) revealed that the recombinant protein is distributed similarly to endogenous p193 (Fig. 8 F). The p193 localization pattern is very different from that seen for anti-PARP antiserum, which showed a mostly nuclear staining pattern (Fig. 8 G). Interestingly, in mitotic cells a portion of the p193 immunoreactivity localizes to the mitotic spindle (Fig. 9 A), like β-tubulin (Fig. 9 B), with merged images depicting their colocalization (Fig. 9c and Fig. d). Vault staining of mitotic cells is diffuse and punctate throughout the cytoplasm (data not shown). Only a portion of the p193 is localized to the mitotic spindle, as another exposure also shows the diffuse punctate p193 staining in the cytoplasm (Fig. 9 D), presumably representing the p193 in vaults. Next, we determined whether the distribution of p193 or vaults varied in response to DNA damage. Human foreskin fibroblasts were UV irradiated (100 μJ), and the distribution of p53, p193, and vaults was monitored by immunofluorescence (Fig. 10). p53 is known to be activated in response to DNA damage and is upregulated and localizes to the nucleus (for review see Levine 1997). No change in either the distribution of p193 or vaults was detectable in the UV-treated cells (Fig. 10).
Figure 8.
Subcellular localization of p193. Indirect immunofluorescence of endogenous p193 (A) and vaults (B) in HeLa cells reveals a punctate cytoplasmic staining pattern with some nuclear speckle staining with affinity-purified p193 antibody but not with the vault mAb (LRP56). By merging the images of A and B, coincident staining is seen as yellow (C), revealing a partial overlap in the cytoplasm and highlighting the nuclear staining by p193. The nucleus is stained with DAPI (D). Preimmune p193 antiserum reveals background staining (E). COS cells transiently expressing VSVG-tagged p193 revealed that the recombinant protein has an expression pattern similar to endogenous p193 (F). PARP is predominantly localized to the nucleus (G).







Figure 10.
Vaults and p193 are not relocalized in response to DNA damage by UV light. Human foreskin fibroblasts were either not treated (A–C) or treated with UV light (D–F). After 24 h, cells were examined by indirect immunofluorescence with a monoclonal p53 antibody (A and D), affinity-purified p193 antibody (B and E), or affinity-purified vault antibody (C and F).
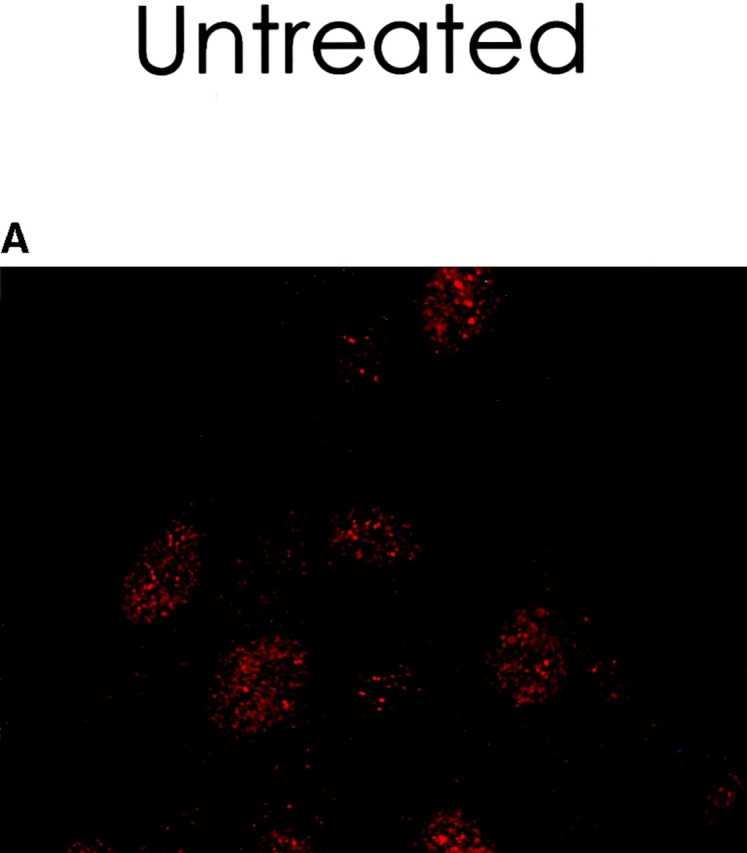





Figure 9.
A portion of the p193 colocalizes with β-tubulin to the mitotic spindle. Mitotic HeLa cells were stained with anti-p193 (red, A) and anti-β tubulin (green, B). A merged image of A and B where coincident staining is yellow (C). Another coincident staining is shown in D where the anti-p193 cytoplasmic staining is also evident (red punctate staining).



Discussion
Identification of the 193-kD vault protein completes the molecular characterization of the repertoire of proteins that form the basic vault particle. The identities of both the 193- and 240-kD vault proteins have led to unexpected but tantalizing findings. The determination that the 240-kD protein is identical to the telomerase-associated protein, TEP1, suggests that this protein may have a more general role in RNP structure, function, or assembly (Kickhoefer et al. 1999). The data presented here demonstrates that the p193 provides vaults with an enzymatic activity. We show that p193 is a PARP that ADP-ribosylates itself and the major vault protein in purified vaults. Based on this data we propose that p193 be named VPARP for vault poly(ADP-ribose) polymerase. To date there are only two other proteins that contain poly(ADP-ribosyl)ation activity: PARP and tankyrase. PARP is a nuclear protein that has been studied for nearly 20 yr (for reviews see de Murcia et al. 1991; Jeggo 1998). It is activated in response to DNA damage, where it binds to DNA at single or double-strand breaks and covalently attaches ADP-ribose moieties derived from NAD+ to itself and other nuclear proteins (de Murcia et al. 1991; Jeggo 1998). PARP has also been shown to be a target of caspases during apoptosis, where it is cleaved near the DNA binding domain (Kaufmann et al. 1993; Lazebnik et al. 1994). Although PARP knockout mice are viable, they are more sensitive to gamma irradiation and treatment with the alkylating agent _N_-methyl-_N_-nitrosourea (de Murcia et al. 1997). Mice lacking PARP have recently been shown to be resistant to pancreatic β-cell destruction and development of type I diabetes induced by streptozocin (Burkart et al. 1999; Masutani et al. 1999).
Recently, another PARP, tankyrase, was identified through its ability to interact with TRF1 (Smith et al. 1998). TRF1 is a mammalian telomeric protein that binds to double-stranded telomere repeat containing DNA at chromosome ends (for review see Smith and de Lange 1997). Overexpression of TRF1 induces telomere shortening, whereas expression of a dominant negative TRF1 results in longer telomeres (van Steensel and de Lange 1997). These data indicate that TRF1 is a negative regulator of telomere length. TRF1 is not a component of telomerase; therefore, it must interact with other proteins that mediate the interaction of telomerase with telomeres. One such protein may be tankyrase. Tankyrase was shown to ADP-ribosylate itself and TRF1, and this modification abolishes the ability of TRF1 to bind to telomeric DNA sequences in an in vitro gel shift assay. These results suggest that poly(ADP-ribosyl)ation may negatively regulate telomere length. Since the precise function of vaults is unknown, it is difficult to assess the impact of poly(ADP-ribosyl)ation on its function. However, it may enhance or negate vault interaction with other proteins in the cell, or it may allow for changes in vault conformation, i.e., opening and closing of vaults.
A BRCT domain has been identified at the NH2 terminus of VPARP. More than 50 distinct proteins have been identified that contain a BRCT domain, and many of these proteins have defined roles in the cellular response to DNA damage (Wu et al. 1996; Bork et al. 1997; Callebaut and Mornon 1997). BRCT domains are thought to mediate protein–protein interactions and are usually found at either the NH2 or COOH termini of proteins. Some proteins contain multiple copies of the BRCT domain. Interestingly, PARP also contains a BRCT domain in the central automodification domain upstream of the catalytic domain (Bork et al. 1997). The BRCT domain in PARP is separated from the catalytic domain by ∼145 aa, similar to the distance that separates these two domains in VPARP (115 aa). In some respects, multidrug resistance could be considered a response to DNA damage. Many chemotherapeutic drugs are DNA-damaging agents (e.g., doxorubicin and mitoxanthrone). The upregulation of vaults in some types of multidrug-resistant cancers (Kickhoefer et al. 1998; Scheffer et al. 1995), along with p193's homology to PARP, suggested that vaults may have a role in DNA damage response. However, we have shown that when cellular DNA is damaged by exposure to UV light sufficient to activate p53, the distribution of vaults and VPARP remains unchanged. In addition, we have determined that VPARP activity is not activated by damaged DNA in extracts using an in vitro ADP-ribosylation assay (data not shown). It seems reasonable to propose that like PARP, VPARP activity will be activated by some as yet undetermined signal. Other functional PARPs must exist, as PARP-deficient mouse cells have recently been shown to synthesize ADP-ribose polymers in response to the DNA-damaging agent, _N_-methyl-nitro-nitrosoguanidine (Shieh et al. 1998).
Northern blot analysis revealed a single VPARP mRNA that is heterogenously expressed in human tissues, with the highest amounts detectable in kidney. MVP showed a similar pattern of expression, although levels varied depending on the tissue being examined. Subcellular fractionation of tissue culture cells revealed that ∼90% of the MVP is present in the 100,000 g pellet (P100, crude vault fraction) (Kickhoefer et al. 1998). However, only a portion of VPARP, TEP1, and vRNA are associated with the 100,000 g vault particle fraction (Kickhoefer et al. 1999). Factors governing vault particle formation, function, or assembly have not been determined. It is possible that like TEP1, VPARP may be a shared protein interacting with other cellular proteins. TEP1 is associated with telomerase activity (Harrington et al. 1997; Nakayama et al. 1997), and may have a more general role in RNP structure, function, or assembly (Kickhoefer et al. 1999). The role of vRNA in vault particle function has not yet been defined. However, previous studies have demonstrated that the vRNA is not a structural component of the vault particle (Kedersha et al. 1991). Here we show that VPARP provides vaults with an enzymatic activity, and that this activity will likely be important in VPARP's vault-independent function. There are three potential nuclear localization signals in VPARP (aa 19 [PQQQKKK], aa 1237 [KRKHRK], and aa 1244 [PFSKRF]) and a portion of the VPARP protein is localized to the nucleus by subcellular fractionation and in a variable number of nuclear speckles by immunofluorescence. VPARP is probably associated with other cellular proteins and substrates that have not yet been identified.
The localization of VPARP, but not of vaults, to the mitotic spindle is particularly intriguing, inasmuch as neither vaults nor VPARP appears to associate with interphase microtubules in HeLa cells. Vaults have been reported to associate with microtubules in neurite extensions of differentiated PC-12 cells (Herrmann et al. 1999), and sea urchin vaults were originally discovered because of their ability to copurify in vitro with egg microtubules through several cycles of polymerization and depolymerization (Hamill and Suprenant 1997), although in adult sea urchin coelomocytes, sea urchin vaults do not appear to associate with microtubules by double immunofluorescence. It is possible that a putative sea urchin VPARP could mediate the vault association with egg microtubules in vitro, and that the assembly and/or disassembly of egg microtubules copurify selected microtubule-associated proteins specifically associated with the mitotic spindle. Examples of other proteins that selectively associate with mitotic spindle microtubules, but not interphase microtubules, include human Eg5 and protein phosphatase γ1 (Blangy et al. 1995; Andreassen et al. 1998). The association of human Eg5 with the mitotic spindle requires phosphorylation of a specific threonine residue by p34cdc2. Protein phosphatase γ1 is an isoform of protein phosphatase 1 (PP1), a family of serine/threonine phosphatases that has many important regulatory functions in mammalian cells (Shenolikar 1994). Recently it was recognized that the three isoforms (α1, δ1, and γ1) are each localized to distinct sites in both mitotic and interphase cells (Andreassen et al. 1998). These findings suggest that these distinct localizations may allow the various isoforms to control multiple cellular processes. Precisely how VPARP is recruited to the mitotic spindle is unknown. Posttranslational modifications to VPARP such as phosphorylation, self-modification by conjugation of poly(ADP-ribose) moieties, or interactions with other protein(s) during mitosis are all candidate mechanisms that could mediate interaction with the mitotic spindle. The key question that remains to be addressed in future studies is whether the spindle-associated VPARP is enzymatically active, and if so, what is the function of such localized PARP activity. It is clear that vaults, like Pandora's box, contain surprises in addition to their enigmatic contents.
Acknowledgments
We are grateful to Dion Baybridge for purification of the vaults from monkey liver; Hedi Roseboro for excellent technical assistance; Dr. Roger Brent for providing us with the yeast two-hybrid system; Carles Serra-Pages and Stephen O'Brien for assistance with the yeast two-hybrid screen; Dr. Rik Scheper for providing us with the monoclonal anti-MVP antibody (LRP56); and Dr. Owen Witte for the human cDNA library. We thank Drs. Andrew Stephen and Sujna Raval for critical comments and helpful discussions of the manuscript.
This research was supported by U.S. Public Health Service grants from the National Institutes of Health (GM38097 to L.H. Rome and CA55547 to M. Streuli) and a grant from the Margaret E. Early Foundation to L.H. Rome.
Note Added in Proof. While this manuscript was being reviewed, another PARP (PARP2) was identified by Gilbert de Murcia and colleagues. Ame, J.-C., V. Rolli, V. Schreiber, C. Niedergang, F. Apiou, P. Decker, S. Muller, T. Hoger, J. Mennissie-de Murcia, and G. de Murcia. 1999. PARP2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 274:17860–17868.
Footnotes
1.used in this paper: 3ABA, 3-aminobenzamide; aa, amino acid(s); BRCT, BRCA1 COOH terminus; GST, glutathione S-transferase; MVP, major vault protein; PARP, poly(ADP-ribose) polymerase; RACE, rapid amplification of cDNA ends; TEP1, telomerase-associated protein 1; vRNA, vault-associated ribonucleic acid; VPARP, vault PARP; VSVG, vesicular stomatitis virus glycoprotein
References
- Abbondanza C., Rossi V., Roscigno A., Gallo L., Belsito A., Piluso G., Medici N., Nigro V., Molinari A.M., Moncharmont B., Puca G.A. Interaction of vault particles with estrogen receptor in the MCF-7 breast cancer cell. J. Cell Biol. 1998;141:1301–1310. doi: 10.1083/jcb.141.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen P.R., Lacroix F.B., Villa-Moruzzi E., Margolis R.L. Differential subcellular localization of protein phosphatase-1 α, γ1, and δ isoforms during both interphase and mitosis in mammalian cells. J. Cell Biol. 1998;141:1207–1215. doi: 10.1083/jcb.141.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl, editors. 1995. Current Protocols in Molecular Biology. John Wiley & Sons, Inc., New York.
- Blangy A., Lane H.A., d'Herin P., Harper M., Kress M., Nigg E.A. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- Bork P., Hofmann K., Bucher P., Neuwald A.F., Altschul S.F., Koonin E.V. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- Burkart V., Wang Z.Q., Radons J., Heller B., Herceg Z., Stingl L., Wagner E.F., Kolb H. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat. Med. 1999;5:314–319. doi: 10.1038/6535. [DOI] [PubMed] [Google Scholar]
- Callebaut I., Mornon J.P. From BRCA1 to RAP1a widespread BRCT module closely associated with DNA repair. FEBS Lett. 1997;400:25–30. doi: 10.1016/s0014-5793(96)01312-9. [DOI] [PubMed] [Google Scholar]
- Choi-Miura N.H., Sano Y., Oda E., Nakano Y., Tobe T., Yanagishita T., Taniyama M., Katagiri T., Tomita M. Purification and characterization of a novel glycoprotein which has significant homology to heavy chains of inter-alpha-trypsin inhibitor family from human plasma. J. Biochem. 1995;117:400–407. doi: 10.1093/jb/117.2.400. [DOI] [PubMed] [Google Scholar]
- Chugani D.C., Kedersha N.L., Rome L.H. Vault immunofluorescence in the brainnew insights regarding the origin of microglia. J. Neurosci. 1991;11:256–268. doi: 10.1523/JNEUROSCI.11-01-00256.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugani D.C., Rome L.H., Kedersha N.L. Evidence that vault ribonucleoprotein particles localize to the nuclear pore complex. J. Cell Sci. 1993;106:23–29. doi: 10.1242/jcs.106.1.23. [DOI] [PubMed] [Google Scholar]
- de Murcia G., Menissier-de Murcia J., Schreiber V. Poly(ADP-ribose) polymerasemolecular biological aspects. Bioessays. 1991;13:455–462. doi: 10.1002/bies.950130905. [DOI] [PubMed] [Google Scholar]
- de Murcia J.M., Niedergang C., Trucco C., Ricoul M., Dutrillaux B., Mark M., Oliver F.J., Masson M., Dierich A., LeMeur M. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee T., Becherer K., Chen P.L., Yeh S.H., Yang Y., Kilburn A.E., Lee W.H., Elledge S.J. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev. 1993;7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- Fields S., Song O. A novel genetic system to detect protein–protein interaction. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Gyuris J., Golemis E.A., Chertkov H., Brent R. Cdi1, a human G1- and S-phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Hamill D.R., Suprenant K.A. Characterization of the sea urchin major vault proteina possible role for vault ribonucleoprotein particles in nucleocytoplasmic transport. Dev. Biol. 1997;190:117–128. doi: 10.1006/dbio.1997.8676. [DOI] [PubMed] [Google Scholar]
- Harrington L., McPhail T., Mar V., Zhou W., Oulton R., Bass M.B., Arruda I., Robinson M.O. A mammalian telomerase-associated protein. Science. 1997;275:973–977. doi: 10.1126/science.275.5302.973. [DOI] [PubMed] [Google Scholar]
- Herrmann C., Volknandt W., Wittich B., Kellner R., Zimmermann H. The major vault protein (MVP100) is contained in cholinergic nerve terminals of electric ray electric organ. J. Biol. Chem. 1996;271:13908–13915. doi: 10.1074/jbc.271.23.13908. [DOI] [PubMed] [Google Scholar]
- Herrmann C., Zimmermann H., Volknandt W. Analysis of a cDNA encoding the major vault protein from the electric ray Discopyge ommata . Gene. 1997;188:85–90. doi: 10.1016/s0378-1119(96)00781-0. [DOI] [PubMed] [Google Scholar]
- Herrmann C., Golkaramnay E., Inman E., Rome L., Volknandt W. Recombinant major vault protein is targeted to neuritic tips of PC12 cells. J. Cell Biol. 1999;144:1163–1172. doi: 10.1083/jcb.144.6.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeggo P.A. DNA repairPARP - another guardian angel? Curr. Biol. 1998;8:R49–R51. doi: 10.1016/s0960-9822(98)70032-6. [DOI] [PubMed] [Google Scholar]
- Kaufmann S.H., Desnoyers S., Ottaviano Y., Davidson N.E., Poirier G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerasean early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–3985. [PubMed] [Google Scholar]
- Kedersha N.L., Rome L.H. Isolation and characterization of a novel ribonucleoprotein particlelarge structures contain a single species of small RNA. J. Cell Biol. 1986;103:699–709. doi: 10.1083/jcb.103.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N.L., Rome L.H. Vaultslarge cytoplasmic RNPs that associate with cytoskeletal elements. Mol. Biol. Rep. 1990;14:121–122. doi: 10.1007/BF00360441. [DOI] [PubMed] [Google Scholar]
- Kedersha N.L., Miquel M.C., Bittner D., Rome L.H. Vaults. II. Ribonucleoprotein structures are highly conserved among higher and lower eukaryotes. J. Cell Biol. 1990;110:895–901. doi: 10.1083/jcb.110.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N.L., Heuser J.E., Chugani D.C., Rome L.H. Vaults. III. Vault ribonucleoprotein particles open into flower-like structures with octagonal symmetry. J. Cell Biol. 1991;112:225–235. doi: 10.1083/jcb.112.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kickhoefer V.A., Rome L.H. The sequence of a cDNA encoding the major vault protein from Rattus norvegicus . Gene. 1994;151:257–260. doi: 10.1016/0378-1119(94)90667-x. [DOI] [PubMed] [Google Scholar]
- Kickhoefer V.A., Searles R.P., Kedersha N.L., Garber M.E., Johnson D.L., Rome L.H. Vault ribonucleoprotein particles from rat and bullfrog contain a related small RNA that is transcribed by RNA polymerase III. J. Biol. Chem. 1993;268:7868–7873. [PubMed] [Google Scholar]
- Kickhoefer V.A., Vasu S.K., Rome L.H. Vaults are the answer, what is the question? Trends Cell Biol. 1996;6:174–178. doi: 10.1016/0962-8924(96)10014-3. [DOI] [PubMed] [Google Scholar]
- Kickhoefer V.A., Rajavel K.S., Scheffer G.L., Dalton W.S., Scheper R.J., Rome L.H. Vaults are up-regulated in multidrug-resistant cancer cell lines. J. Biol. Chem. 1998;273:8971–8974. doi: 10.1074/jbc.273.15.8971. [DOI] [PubMed] [Google Scholar]
- Kickhoefer V.A., Stephen A.J., Harrington L., Robinson M.O., Rome L.H. Vaults and telomerase share a common subunit TEP1. J. Biol. Chem. 1999;In press doi: 10.1074/jbc.274.46.32712. [DOI] [PubMed] [Google Scholar]
- Kong L.B., Siva A.C., Rome L.H., Stewart P.L. Structure of the vault, a ubiquitous cellular component. Structure. 1999;7:371–379. doi: 10.1016/s0969-2126(99)80050-1. [DOI] [PubMed] [Google Scholar]
- Lazebnik Y.A., Kaufmann S.H., Desnoyers S., Poirier G.G., Earnshaw W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Levine A.J. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Masutani M., Suzuki H., Kamada N., Watanabe M., Ueda O., Nozaki T., Jishage K., Watanabe T., Sugimoto T., Nakagama H. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA. 1999;96:2301–2304. doi: 10.1073/pnas.96.5.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J., Saito M., Nakamura H., Matsuura A., Ishikawa F. TLP1a gene encoding a protein component of mammalian telomerase is a novel member of WD repeats family. Cell. 1997;88:875–884. doi: 10.1016/s0092-8674(00)81933-9. [DOI] [PubMed] [Google Scholar]
- Rome L., Kedersha N., Chugani D. Unlocking vaultsorganelles in search of a function. Trends Cell Biol. 1991;1:47–50. doi: 10.1016/0962-8924(91)90088-q. [DOI] [PubMed] [Google Scholar]
- Ruf A., Mennissier-de Murcia J., de Murcia G., Schulz G.E. Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc. Natl. Acad. Sci. USA. 1996;93:7481–7485. doi: 10.1073/pnas.93.15.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf A., de Murcia G., Schulz G.E. Inhibitor and NAD+ binding to poly(ADP-ribose) polymerase as derived from crystal structures and homology modeling. Biochemistry. 1998;37:3893–3900. doi: 10.1021/bi972383s. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular CloningA Laboratory Manual. 2nd ed. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Scheffer G.L., Wijngaard P.L., Flens M.J., Izquierdo M.A., Slovak M.L., Pinedo H.M., Meijer C.J., Clevers H.C., Scheper R.J. The drug resistance-related protein LRP is the human major vault protein. Nat. Med. 1995;1:578–582. doi: 10.1038/nm0695-578. [DOI] [PubMed] [Google Scholar]
- Scheper R.J., Broxterman H.J., Scheffer G.L., Kaaijk P., Dalton W.S., van Heijningen T.H.M., van Kalken C.K., Slovak M.L., de Vries E.G.E., van der Valk P. Overexpression of a M(r) 110,000 vesicular protein in non-P-glycoprotein-mediated multidrug resistance. Cancer Res. 1993;53:1475–1479. [PubMed] [Google Scholar]
- Shenolikar S. Protein serine/threonine phosphatases—new avenues for cell regulation. Annu. Rev. Cell Biol. 1994;10:55–86. doi: 10.1146/annurev.cb.10.110194.000415. [DOI] [PubMed] [Google Scholar]
- Shieh W.M., Ame J.C., Wilson M.V., Wang Z.Q., Koh D.W., Jacobson M.K., Jacobson E.L. Poly(ADP-ribose) polymerase–null mouse cells synthesize ADP-ribose polymers. J. Biol. Chem. 1998;273:30069–30072. doi: 10.1074/jbc.273.46.30069. [DOI] [PubMed] [Google Scholar]
- Simonin F., Menissier-de Murcia J., Poch O., Muller S., Gradwohl G., Molinete M., Penning C., Keith G., de Murcia G. Expression and site-directed mutagenesis of the catalytic domain of human poly(ADP-ribose) polymerase in Escherichia coli. Lysine 893 is critical for activity. J. Biol. Chem. 1990;265:19249–19256. [PubMed] [Google Scholar]
- Simonin F., Poch O., Delarue M., de Murcia G. Identification of potential active-site residues in the human poly(ADP-ribose) polymerase J. Biol. Chem. 268 1993. 8529 8535a [PubMed] [Google Scholar]
- Simonin F., Hofferer L., Panzeter P.L., Muller S., de Murcia G., Althaus F.R. The carboxyl-terminal domain of human poly(ADP-ribose) polymerase. Overproduction in Escherichia coli, large scale purification, and characterization J. Biol. Chem. 268 1993. 13454 13461b [PubMed] [Google Scholar]
- Smith S., de Lange T. TRF1, a mammalian telomeric protein. Trends Genet. 1997;13:21–26. doi: 10.1016/s0168-9525(96)10052-4. [DOI] [PubMed] [Google Scholar]
- Smith S., Giriat I., Schmitt A., de Lange T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science. 1998;282:1484–1487. doi: 10.1126/science.282.5393.1484. [DOI] [PubMed] [Google Scholar]
- van Steensel B., de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385:740–743. doi: 10.1038/385740a0. [DOI] [PubMed] [Google Scholar]
- Vasu S.K., Rome L.H. Dictyostelium vaultsdisruption of the major proteins reveals growth and morphological defects and uncovers a new associated protein. J. Biol. Chem. 1995;270:16588–16594. doi: 10.1074/jbc.270.28.16588. [DOI] [PubMed] [Google Scholar]
- Vasu S.K., Kedersha N.L., Rome L.H. cDNA cloning and disruption of the major vault protein alpha gene (mvpA) in Dictyostelium discoideum . J. Biol. Chem. 1993;268:15356–15360. [PubMed] [Google Scholar]
- Wu L.C., Wang Z.W., Tsan J.T., Spillman M.A., Phung A., Xu X.L., Yang M.C., Hwang L.Y., Bocock A.M., Baer R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]