Palmitoylation regulates plasma membrane–nuclear shuttling of R7BP, a novel membrane anchor for the RGS7 family (original) (raw)
Abstract
The RGS7 (R7) family of RGS proteins bound to the divergent Gβ subunit Gβ5 is a crucial regulator of G protein–coupled receptor (GPCR) signaling in the visual and nervous systems. Here, we identify R7BP, a novel neuronally expressed protein that binds R7–Gβ5 complexes and shuttles them between the plasma membrane and nucleus. Regional expression of R7BP, Gβ5, and R7 isoforms in brain is highly coincident. R7BP is palmitoylated near its COOH terminus, which targets the protein to the plasma membrane. Depalmitoylation of R7BP translocates R7BP–R7–Gβ5 complexes from the plasma membrane to the nucleus. Compared with nonpalmitoylated R7BP, palmitoylated R7BP greatly augments the ability of RGS7 to attenuate GPCR-mediated G protein–regulated inward rectifying potassium channel activation. Thus, by controlling plasma membrane nuclear–shuttling of R7BP–R7–Gβ5 complexes, reversible palmitoylation of R7BP provides a novel mechanism that regulates GPCR signaling and potentially transduces signals directly from the plasma membrane to the nucleus.
Introduction
Hundreds of neurotransmitters regulate neuronal development and function by signaling through G protein–coupled receptors (GPCRs; for review see Gainetdinov et al., 2004). GPCRs exert their effects by regulating ion channels, second messenger production, and protein kinase cascades, which in turn control neuronal activity, gene expression, plasticity, differentiation, morphogenesis, and migration.
GPCR signaling is tightly regulated to determine the sensitivity, kinetics, and fidelity of neuronal activity. Disruption of GPCR regulatory mechanisms, such as the GPCR phosphorylation–arrestin pathway, dramatically affects processes such as nociception and addiction (for reviews see Chao and Nestler, 2004; Gainetdinov et al., 2004). GPCR signaling also is potently regulated by RGS proteins (regulators of G protein signaling; for review see Hollinger and Hepler, 2002). RGS proteins attenuate signaling by functioning as GTPase-activating proteins (GAPs) for Gα subunits (Berman et al., 1996; Hunt et al., 1996; Watson et al., 1996). Certain RGS proteins, such as the RGS protein p115RhoGEF (Hart et al., 1998), also function as Gα effectors.
Among more than 20 RGS family members in vertebrates, the R7 subfamily consisting of RGS6, RGS7, RGS9-1, RGS9-2, and RGS11 is emerging as an important set of neuronal GPCR-signaling regulators. The R7 family is highly expressed in the central and peripheral nervous systems (Gold et al., 1997; Zhang et al., 2000; Witherow et al., 2003; Larminie et al., 2004). R7 proteins selectively deactivate the Gi/o-class of Gα subunits that mediate the action of GPCRs for many modulatory neurotransmitters (Posner et al., 1999; Rose et al., 2000; Hooks et al., 2003). RGS9 is the best understood R7 family member (Cowan et al., 2001; Witherow and Slepak, 2003; Jones et al., 2004). RGS9 potently regulates GPCR-mediated Ca2+ channel inhibition in striatal neurons (Cabrera-Vera et al., 2004). RGS9 knockout mice exhibit augmentation of the antinociceptive and rewarding effects of μ-opioid receptors and enhancement of the locomotor and rewarding effects mediated by D2 dopamine receptors (Rahman et al., 2003; Zachariou et al., 2003). Human or mouse RGS9 mutants exhibit bradyopsia, a visual defect caused by prolonged activation of the Gα subunit transducin, resulting in impaired light adaptation and contrast detection (Chen et al., 2000; Nishiguchi et al., 2004).
R7 proteins function as complexes with Gβ5, a divergent member of the Gβ family (Watson et al., 1994; Jones et al., 2004). Mice lacking Gβ5 exhibit visual defects indistinguishable from RGS9 knockout mice (Krispel et al., 2003) and high mortality and slow growth due to destabilization of the entire R7 family (Chen et al., 2003). In the retina, RGS9-1–Gβ5L complexes are anchored to disk membranes by binding the retina-specific membrane protein R9AP (RGS9-anchoring protein; Hu and Wensel, 2002). R9AP also stimulates the GAP activity of RGS9-1 (Lishko et al., 2002; Hu et al., 2003). R9AP knockout mice recapitulate the visual phenotypes of RGS9 or Gβ5 knockout mice (Keresztes et al., 2004). Likewise, humans with R9AP or RGS9 mutations exhibit similar defects in visual perception (Nishiguchi et al., 2004).
Because R9AP is expressed insignificantly in nonretinal tissues (Hu and Wensel, 2002), the mechanisms that control subcellular localization and function of R7–Gβ5 complexes in the central and peripheral nervous systems are unknown. However, R7–Gβ5 localization and function probably are regulated tightly because these proteins localize both to membranes and nuclei in neurons where they regulate GPCR signaling at the plasma membrane and gene expression in the nucleus (Zhang and Simonds, 2000; Bouhamdan et al., 2004; Cabrera-Vera et al., 2004; Krumins et al., 2004; Liu and Fisher, 2004). Mechanisms that regulate R7–Gβ5 localization and function may be neuron-specific because R7 isoforms associate poorly with the plasma membrane when expressed in nonneuronal cells (Posner et al., 1999; Chatterjee et al., 2003; Witherow et al., 2003; Bouhamdan et al., 2004; Liu and Fisher, 2004; Takida et al., 2005).
Here, we report the identification and characterization of R7BP, a novel palmitoylated R9AP-related protein that is highly expressed in the nervous system. We present evidence indicating that reversible palmitoylation of R7BP controls the shuttling of R7–Gβ5 complexes between the plasma membrane and nucleus and the ability of an R7 protein to regulate GPCR signaling.
Results
Identification, cloning, and expression of R7BP
We identified R9AP-like proteins by performing PSI-BLAST searches of the mouse genome. This identified a novel 257-residue protein that for reasons presented in subsequent paragraphs was named R7BP (RGS7 family binding protein; Fig. 1, A and B). Analysis of several genomic and EST databases indicated that R7BP and R9AP are the only closely related members of this family (Fig. 1 C; accession numbers listed in Materials and methods). Both proteins also display weak similarity to SNARE proteins that mediate vesicular trafficking (unpublished data). The predicted R7BP gene contains six exons and is localized to chromosome 5q12.3 in humans, 13D1 in mouse, and 2q13 in rat (unpublished data). R7BP or R9AP homologues were not detected in Caenorhabditis elegans or Drosophila melanogaster, which do possess Gβ5 and R7 homologues. Therefore, R7BP and R9AP evolved after Gβ5 and the R7 family.
Figure 1.
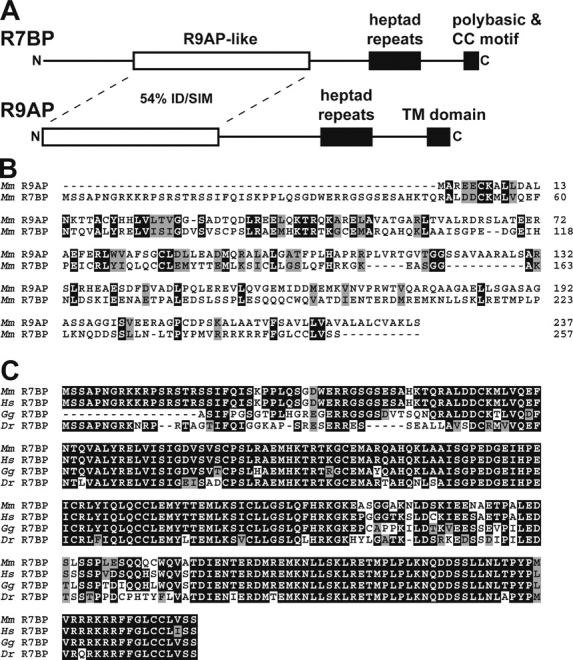
Identification of R7BP. (A) Schematic diagram of R7BP. Mouse R7BP (RGS7 family binding protein) was identified in PSI-BLAST searches as the protein most closely related to retinal R9AP (RGS9-anchoring protein). The deduced R7BP protein is 257 aa in length and consists of a nonconserved NH2-terminal region of ∼50 aa, a central domain (residues 48–151) exhibiting 28% identity and 54% similarity to R9AP, a putative coiled-coil domain (residues 194–221), and a COOH-terminal domain containing a polybasic stretch followed by a double-cysteine motif. (B) Protein sequence alignments of mouse R9AP and R7BP. Identical (black) and similar (gray) residues are indicated. (C) Multiple sequence alignment of full or partial R7BP protein sequences deduced from Mus musculus (mouse), Homo sapiens (human), Gallus gallus (chicken), and Danio rerio (zebrafish) databases. Identical (black) and similar (gray) residues are indicated.
We cloned and sequenced a cDNA encoding the R7BP ORF from mouse brain, which confirmed database predictions. A mouse R7BP cDNA sequence assembled from EST databases predicted an mRNA of ∼6.4 kb. In the deduced mRNA, the ORF encoding R7BP apparently is preceded by a GC-rich (∼66%) stretch of ∼720 nucleotides containing four short open reading frames, which could regulate R7BP translation (for review see Meijer and Thomas, 2002).
R7BP and R9AP exhibit similar domain organizations (Fig. 1 A). Within their NH2-terminal regions these proteins are 28% identical and 54% similar. Distal to this region is a predicted coiled-coil domain followed by a putative membrane localization domain. The membrane localization domain of R9AP is a transmembrane segment, whereas R7BP contains a COOH-terminal polybasic sequence and a double cysteine motif that is an excellent candidate for lipid modification (Zhang and Casey, 1996; Smotrys and Linder, 2004).
Results of Northern blotting experiments indicated that R7BP is encoded by an ∼6.4-kb mRNA expressed highly in brain and at much lower levels in other tissues examined (Fig. 2 A), similar to the R7 family and Gβ5 (Gold et al., 1997; Zhang et al., 2000; Larminie et al., 2004). There was no indication of alternative splicing suggested by Northern blotting or assembly of ESTs (Fig. 2 A and not depicted). Results of in situ hybridization experiments revealed that R7BP mRNA is highly expressed in the cerebellum, neocortex, thalamus, hippocampus, striatum, and piriform cortex and is readily detectable in olfactory bulb, several midbrain nuclei, and hindbrain (Fig. 2, B and C). This regional expression pattern is largely coincident with the aggregate expression pattern of the R7 family (Gold et al., 1997). However, our results do not exclude that some brain nuclei expressing certain R7 family members at relatively high levels may poorly express R7BP. R7BP mRNA is also detectable in mouse retina, organ of Corti, pituitary, aorta, vein, colon, hematopoetic stem cells, spinal cord, and embryonic heart, as indicated by EST database searches.
Figure 2.

R7BP mRNA is highly and broadly expressed in brain. (A) Northern blot analysis of R7BP mRNA expression. A probe consisting of the R7BP open reading frame was used to detect R7BP mRNA in a mouse multiple tissue Northern blot. The same blot was stripped and probed for β-actin mRNA. (B) In situ hybridization analysis of R7BP mRNA expression in mouse brain. An antisense RNA probe for the R7BP open reading frame was hybridized to coronal sections of mouse brain followed by autoradiography. A labeled RNA sense probe was hybridized to half-brain coronal sections as a specificity control. (C) In situ analysis performed as in B, except that sagittal sections were analyzed. A representative medial (left) and a lateral (right) section are shown. ctx, neocortex; cpu, caudate/putamen (striatum); th, thalamus; hp, hippocampus; pf, piriform cortex; ob, olfactory bulb; cbl, cerebellum.
R7BP binds R7–Gβ5 complexes
To determine whether R7BP binds R7 proteins, we performed coimmunoprecipitation experiments using HEK293 cells that coexpressed FLAG-tagged R7BP (FLAG-R7BP), MYC-tagged Gβ5, and HA-tagged forms of RGS6, RGS7, RGS9-1, RGS9-2, or RGS11. Immunoblotting of anti-FLAG immunoprecipitates indicated that FLAG-R7BP bound Gβ5 and each R7 family member (Fig. 3 A). Expression of R7BP moderately increased the total levels of Gβ5 and R7 proteins, suggesting that R7BP stabilizes Gβ5–R7 complexes. In the absence of an R7 protein, Gβ5 did not associate with FLAG-R7BP (Fig. 3 B). When Gβ5 was not coexpressed, an R7 family member (RGS9-2) could bind FLAG-R7BP (Fig. 3 B), albeit less efficiently. Therefore, R7BP binds the R7 subunit of the R7–Gβ5 complex; Gβ5 may promote this interaction by stabilizing or changing the conformation of the R7 subunit. R7BP–R7 interaction was specific because tagged forms of two RGS proteins of a different subfamily (RGS2 and RGS4 of the R4 family) that do not bind Gβ5 were undetectable in FLAG-R7BP immunoprecipitates (Fig. 3 C).
Figure 3.

R7BP interacts with R7 RGS proteins. (A) R7BP binds each of the five R7 RGS proteins. HEK293 cells were transfected with plasmids encoding an HA-tagged R7 RGS protein (RGS6, RGS7, RGS9-1, RGS9-2, or RGS11) and MYC-tagged versions of Gβ5 with or without a FLAG-tagged version of R7BP. FLAG antibody immunoprecipitates were assayed for the presence of the RGS and Gβ5 proteins by blotting with the indicated antibodies. Western blots of total cell lysates indicated the levels of the expressed proteins. (B) R7BP forms a complex with R7–Gβ5 proteins. HEK293 cells were transfected with different combinations of plasmids expressing the indicated tagged forms of R7BP, RGS9-2, and Gβ5. FLAG-R7BP was immunoprecipitated, and the precipitated material and total cell lysates were analyzed as in A. (C) R7BP does not interact with R4 family RGS proteins. Plasmids encoding a GFP-tagged form of RGS4 or a 3MYC-tagged version of RGS2 were transfected into HEK293 cells along with FLAG-R7BP. The presence of RGS2 or RGS4 in the FLAG-immunoprecipitate and total lysate was assayed by Western blotting.
R7BP recruits R7 RGS proteins to the plasma membrane
To determine whether R7BP associates with cell membranes and recruits R7 family members, we performed confocal microscopy experiments with cells expressing R7BP tagged at its NH2 terminus with GFP or FLAG. The results showed that GFP- or FLAG-R7BP localized primarily to the plasma membrane as well as the cytoplasm and nucleus (Fig. 4 A). Quantification of confocal images indicated that ∼40% of GFP-R7BP (SEM 15%; n = 30 cells) was nuclear, whereas the remainder localized to the plasma membrane or cytoplasm.
Figure 4.
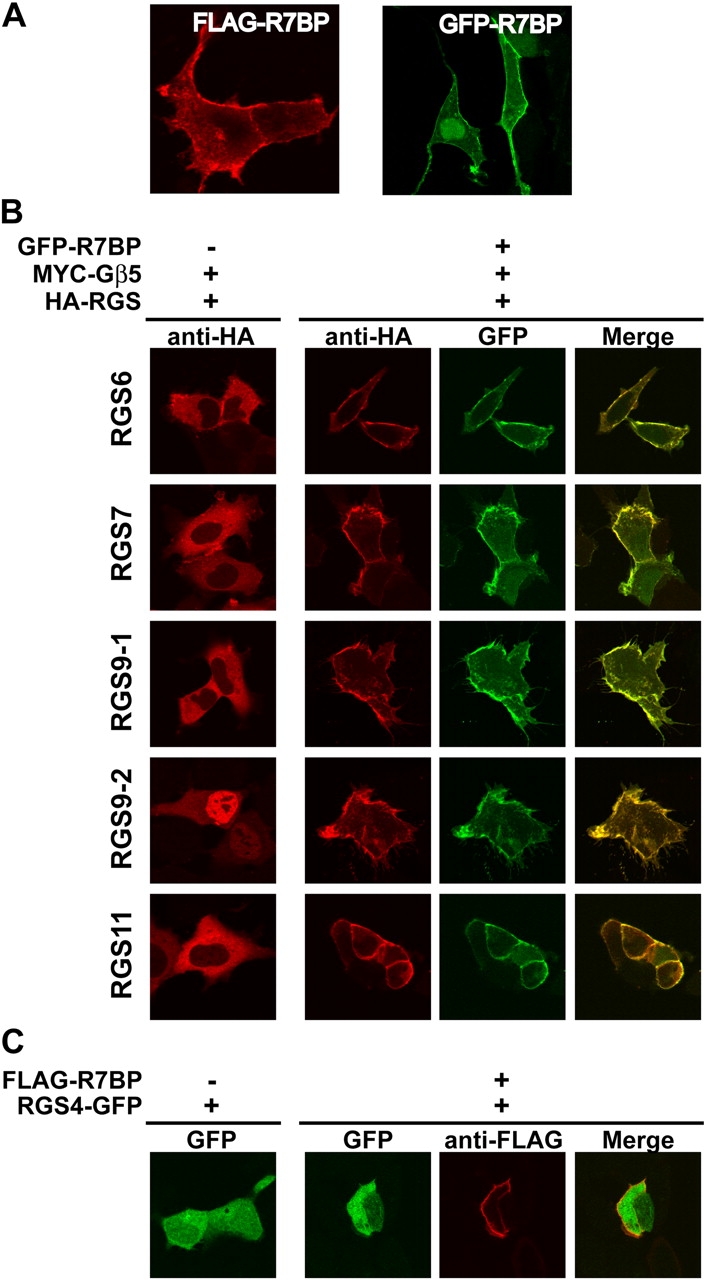
R7BP specifically recruits R7 RGS proteins to the plasma membrane. (A) R7BP is a plasma membrane protein. HEK293 cells expressing GFP-R7BP or FLAG-R7BP were analyzed by laser scanning confocal microscopy. (B) R7BP recruits R7 family RGS proteins to the plasma membrane. Cells expressing HA-tagged R7 RGS proteins (RGS6, RGS7, RGS9-1, RGS9-2, or RGS11) and MYC-Gβ5 were assayed for localization of the R7 family member by immunofluorescence confocal microscopy in the presence or absence of coexpressed GFP-R7BP. Anti-HA indicates the location of the RGS protein, whereas GFP-fluorescence indicates the localization of R7BP. Colocalization of R7BP and the RGS protein was determined by merging the data from the two channels; colocalization is indicated by yellow. (C) RGS4 is not recruited to the plasma membrane by R7BP. RGS4-GFP was coexpressed with FLAG-R7BP and the localization of these proteins was determined by confocal microscopy.
Results of similar experiments indicated that R7BP recruits each R7 family member to the plasma membrane. We expressed MYC-tagged Gβ5 to stabilize expression of HA-tagged R7 subunits. In the absence of R7BP, R7 proteins failed to associate detectably with the plasma membrane (Fig. 4 B) and were cytoplasmic (RGS6, 7, 9–1 and 11) or nuclear (RGS9-2), which is in agreement with previous studies. In contrast, coexpression of GFP-R7BP resulted in efficient recruitment of each HA-R7 protein to the plasma membrane coincident with the localization of GFP-R7BP (Fig. 4 B). This effect was specific for the R7 family because FLAG-R7BP failed to recruit GFP-RGS4 to the plasma membrane (Fig. 4 C). Therefore, R7BP is a plasma membrane–anchoring protein specific for the R7 family of RGS proteins.
Palmitoylation of R7BP
R7BP possesses a dicysteine motif near its COOH terminus (CCLVSS-COOH) that is a potential lipid modification site (Fig. 5 A). This sequence resembles the COOH terminus of H-Ras (CCAAX-COOH), which is palmitoylated and prenylated (for reviews see Zhang and Casey, 1996; Smotrys and Linder, 2004). However, the COOH-terminal region of R7BP contains four rather than three amino acids after the last cysteine, which should preclude prenylation by displacing this residue from the prenyltransferase active site (Long et al., 2002).
Figure 5.
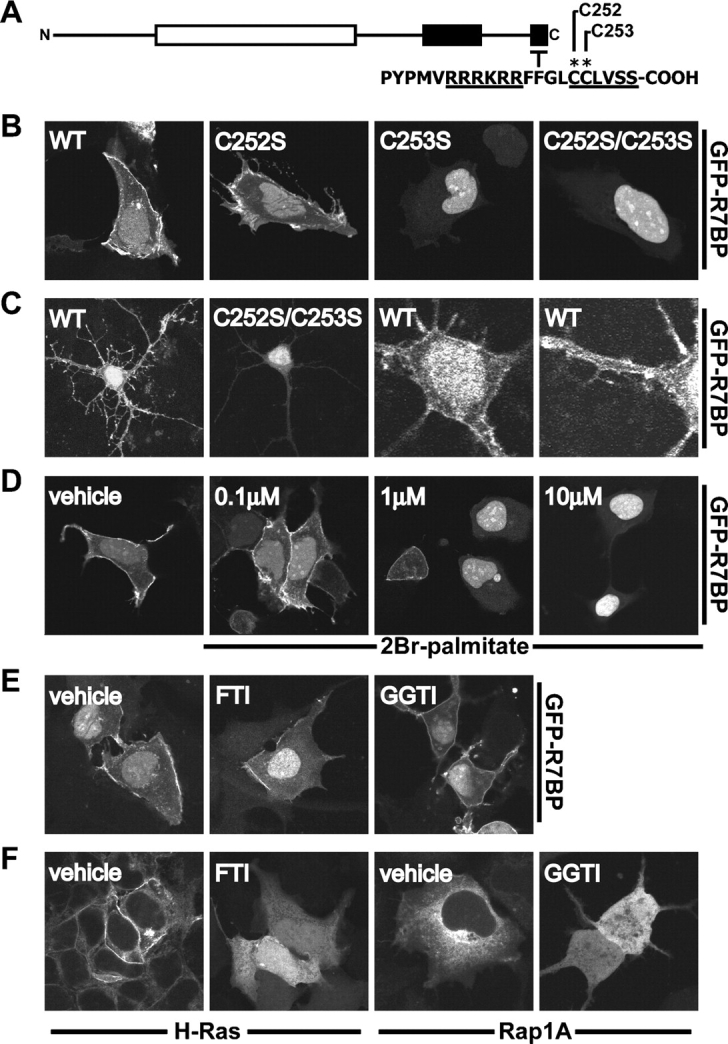
R7BP localization is palmitoylation dependent. (A) Schematic diagram of the R7BP COOH terminus. Cysteine residues that were mutated are indicated with an asterisk. (B) The R7BP double-cysteine motif is necessary for plasma membrane localization. Plasmids encoding GFP-tagged wild type and mutant forms of R7BP were transfected into HEK293 cells and localized by confocal microscopy. The following mutants were used: C252S, C253S, and a C252S/C253S double mutant. (C) R7BP localization in neurons. Neonatal rat hippocampal neurons were transfected with wild type or the C252S/C253S mutant form of GFP-R7BP and analyzed by confocal microscopy. High magnification images of a cell transfected with wild-type GFP-R7BP show localization in the cell soma and dendrites. (D) A palmitoylation inhibitor (2Br-palmitate) disrupts R7BP membrane localization. HEK293 cells expressing wild-type GFP-R7BP were treated with the indicated dose of 2Br-palmitate for 16 h, followed by fixation and analysis by confocal microscopy. (E) Prenylation inhibitors do not block R7BP plasma membrane localization. HEK293 cells expressing GFP-R7BP were treated 16 h with vehicle (DMSO), a farnesyltransferase inhibitor (FTI-277; 30 μM), or a geranylgeranyltransferase inhibitor (GGTI-298; 20 μM). Fixed cells were analyzed by confocal microscopy. (F) FTI and GGTI controls. HEK293 cells expressing GFP-H-Ras (farnesylated protein) or GFP-Rap1A (geranylgeranylated protein) were treated with vehicle or the indicated compounds as in E. Cells were fixed and analyzed by confocal microscopy.
We investigated the roles of COOH-terminal cysteine residues in GFP-tagged R7BP by changing either or both to serine. We then analyzed the localization of wild-type and mutant proteins expressed in HEK293 cells or primary rat hippocampal neurons. Wild-type GFP-R7BP localized to the plasma membrane and nuclei of HEK293 cells and punctate dendritic structures and nuclei of hippocampal neurons (Fig. 5, B and C). In contrast, the C253S single mutant or the C252S/C253S double mutant form of GFP-R7BP did not concentrate on the plasma membrane of HEK293 cells or dendritic foci in neurons but instead was cytoplasmic and/or nuclear localized (Fig. 5, B and C). The C252S mutant exhibited a localization phenotype intermediate between the wild-type protein and other mutants (Fig. 5 B). Quantification of confocal images indicated that whereas ∼40% of wild-type GFP-R7BP was nuclear in HEK293 cells (SEM 15%; n = 30 cells), ∼80% of the GFP-R7BP-C252S/C353S double mutant was nuclear (SEM 8%; n = 30 cells). Similar results were obtained using wild type and mutant forms of FLAG-tagged R7BP (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200502007/DC1), indicating that nuclear accumulation was not due to the presence of the GFP tag. Thus, the COOH-terminal cysteine residues profoundly regulate the distribution of R7BP between the plasma membrane, cytoplasm, and nucleus. The R7BP cysteine mutants migrated identically with wild-type R7BP upon SDS-PAGE (unpublished data), suggesting that R7BP is not subject to proteolytic processing characteristic of many prenylated proteins (Zhang and Casey, 1996).
To determine whether R7BP is palmitoylated, we analyzed the localization of GFP-R7BP when cells were treated with agents that block palmitoylation or prenylation. Doses of 2-bromopalmitate (2Br-palmitate) that block the modification of known palmitoylated proteins strikingly decreased plasma membrane localization of GFP-R7BP and increased nuclear localization of the protein (Fig. 5 D). Quantification of confocal images indicated that ∼35% of GFP-R7BP in vehicle-treated cells was nuclear (SEM 8%; n = 30 cells), whereas ∼67% of the protein in 2Br-palmitate–treated cells was nuclear (SEM 11%; n = 30). In contrast, neither a selective farnesyltransferase inhibitor (FTI-277) nor a geranylgeranyltransferase inhibitor (GTI-298) resulted in loss of GFP-R7BP from the plasma membrane (Fig. 5 E). However, these inhibitors did disrupt the localization of farnesylated and geranylgeranylated proteins (GFP–H-Ras and -Rap1A, respectively; Fig. 5 F; Fukasawa et al., 2004).
Second, we determined whether FLAG-R7BP expressed in HEK293 cells could be labeled with [3H]palmitate. Fluorography of anti-FLAG immunoprecipitates resolved by SDS-PAGE revealed that wild-type R7BP but not a control protein (FLAG-ERK2) was labeled with [3H]palmitate (Fig. 6 A). Palmitate labeling of R7BP was reduced when cells expressed the C252S or the C253S single mutants (Fig. 6 A), suggesting that both cysteines are palmitoylated. Labeling was undetectable when both cysteines were changed to serine (Fig. 6 A). Palmitate labeling of wild-type R7BP was reversed upon treating gels with neutral hydroxylamine (Fig. 6 B). Reverse-phase thin layer chromatography indicated that the labeled lipid released from R7BP comigrated with C16:0 palmitate (Fig. 6 C) rather than a palmitate metabolite. Together these results indicated that labeled palmitate was attached to R7BP via a base-labile thioester linkage characteristic of palmitoylated proteins.
Figure 6.

R7BP is palmitoylated at its COOH-terminal cysteine residues. (A) Palmitate labeling of R7BP. HEK293 cells expressing wild type or mutant (C252S, C253S, or C252S/C253S) FLAG-tagged forms of R7BP were metabolically labeled with [3H]palmitic acid or [35S]methionine as a control. FLAG-R7BP proteins were immunoprecipitated and resolved by SDS-PAGE. Radiolabeled R7BP was detected by fluorography ([3H]palmitate; 7-d exposure) or autoradiography ([35S]methionine; 4-h exposure) of dried gels (left). Expression and labeling controls for FLAG-R7BP and the control protein FLAG-ERK2 are shown in the right panel (13-d exposure for 3H-labeled samples). (B) Palmitate is attached to R7BP by a thioester linkage. HEK293 cells expressing FLAG-R7BP were labeled with [3H]palmitic acid or [35S]methionine followed by immunoprecipitation with FLAG-M2 agarose. Immunoprecipitates resolved on duplicate gels were treated with 0.5 M Tris, pH 7.0, or 0.5 M hydroxylamine, pH 7.0, and analyzed by fluorography (12-d exposure) and autoradiography (16-h exposure). (C) R7BP is labeled with palmitate. HEK293 cells expressing FLAG-R7BP or FLAG-ERK2 proteins were labeled with [3H]palmitic acid, immunoprecipitated, resolved by SDS-PAGE, and excised from the gel. Fatty acids were released from samples by base treatment and analyzed by fractionation on reverse-phase TLC plates relative to the indicated standards (18-d exposure).
R7BP binds R7 proteins independently of its palmitoylation state
Because R7 family members in brain localize both to the plasma membrane and nucleus, plasma membrane–nuclear shuttling of R7–Gβ5 complexes may be controlled by the palmitoylation state of R7BP. This hypothesis requires that R7 proteins bind palmitoylated or unpalmitoylated R7BP. Accordingly, we compared the ability of RGS7–Gβ5 and RGS9-2–Gβ5 complexes to bind FLAG-tagged forms of wild-type or nonpalmitoylated (C252S/C253S; “SS” mutant) R7BP in coimmunoprecipitation experiments. The results indicated that RGS7–Gβ5 and RGS9-2–Gβ5 complexes bind wild-type or nonpalmitoylated R7BP (Fig. 7, A and B). Likewise, the nonpalmitoylated R7BP mutant recruited RGS7 or RGS9-1 to the nucleus (Fig. 7 C; RGS9-2 was used rather than RGS9-1 because the latter molecule localizes to the nucleus without R7BP). These results suggested that palmitoylation/depalmitoylation of R7BP provides a mechanism that controls shuttling of R7–Gβ5 complexes between the plasma membrane and nucleus.
Figure 7.
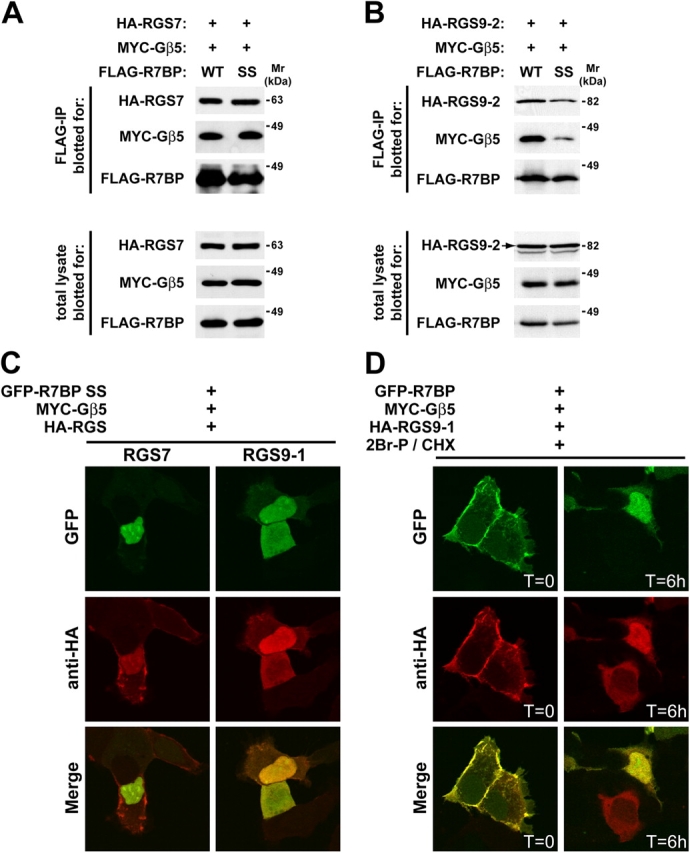
R7BP binds to R7 proteins independently of its palmitoylation state. (A) RGS7 binds wild type or the C252S/C253S mutant form of R7BP. Wild-type or mutant (SS) FLAG-R7BP was expressed in HEK293 cells with HA-RGS7 and MYC-Gβ5. FLAG immunoprecipitates and total lysates were resolved on SDS–polyacrylamide gels and blotted for the expressed proteins. (B) RGS9-2 binds wild type or the C252S/C253S mutant form of R7BP. Methods were identical to A except that HA-tagged RGS9-2 was expressed. (C) Unpalmitoylated R7BP recruits R7 proteins to the nucleus. Experiments were performed identically to those shown in Fig. 4 B except that the C252S/C253S double mutant form of R7BP was used. (D) Depalmitoylation of R7BP results in translocation of RGS9-1 to the nucleus. HEK293 cells were transfected with plasmids expressing wild-type GFP-R7BP, HA-RGS9-1, and MYC-Gβ5. Cells were treated at t = 0 with cycloheximide (30 μg/ml) and 2Br-palmitate (10 μM) to block, respectively, new protein synthesis and repalmitoylation of R7BP that had been depalmitoylated by endogenous thioesterase activity. Confocal microscopy was used to analyze cells fixed at the indicated times after drug treatment.
To test this hypothesis, we determined whether or not depalmitoylation of wild-type R7BP by endogenous protein-thioesterase activity results in shuttling of R7BP–RGS9-1–Gβ5 complexes to the nucleus. We treated cells coexpressing GFP-R7BP, HA-RGS9-1, and MYC-Gβ5 with cycloheximide to block new protein synthesis. As R7BP was depalmitoylated, it was trapped in the unmodified state by blocking repalmitoylation with 2Br-palmitate. Confocal imaging at various times after drug treatment was used to detect translocation of GFP-R7BP and HA-RGS9-1 from the plasma membrane to the nucleus (Fig. 7 D). Quantification of the images indicated that immediately after drug treatment ∼90% of cells showed colocalization of GFP-R7BP and HA-RGS9-1 at the plasma membrane. In striking contrast, within 6 h after drug treatment ∼65% of cells exhibited colocalization of R7BP and RGS9-1 in the nucleus. The time course of R7BP translocation to the nucleus was similar to the basal rate with which signaling proteins are depalmitoylated in cells (for review see Smotrys and Linder, 2004). Therefore, these results indicated that R7BP palmitoylation/depalmitoylation regulates plasma membrane–nuclear shuttling of R7BP–R7–Gβ5 complexes.
R7BP augments RGS7-mediated regulation of G protein–regulated inward rectifying potassium (GIRK) channel activity
To determine whether or not R7BP affects R7 protein function, we performed experiments that used G protein–mediated activation of GIRK channels expressed in Xenopus laevis oocytes as an assay (Doupnik et al., 2004). Because R7 proteins are GAPs for Gi/oα subunits (Posner et al., 1999; Rose et al., 2000; Hooks et al., 2003), we examined the effects of R7BP on GIRK channels activated by m2 muscarinic acetylcholine (ACh) receptors selectively coupled to Goα using pertussis toxin (PTX)-insensitive GoαA(C351A) (Zhang et al., 2002). As shown in Fig. 8 A, in this system GFP-R7BP dramatically accelerated the kinetics of ACh-activated GIRK currents; this robust effect depended on coexpression with RGS7–Gβ5 complexes. Coexpression of RGS7–Gβ5 and GFP-R7BP accelerated the kinetics of GIRK channel deactivation, activation, and acute desensitization, which are the hallmarks of GIRK channel modulation by the GAP activity of RGS proteins (Doupnik et al., 1997; Saitoh et al., 1997; Chuang et al., 1998).
Figure 8.

R7BP augments RGS7–Gβ5–modulated GIRK currents activated by Goα-coupled m2 muscarinic receptors expressed in X. laevis oocytes by a mechanism facilitated by its palmitoylation sites. (A) Superimposed GIRK currents evoked by a range of ACh concentrations (25-s application) from individual oocytes under three different expression conditions. Left traces, control conditions (no R7BP expression); center traces, GFP-R7BP expression (5 ng cRNA/oocyte); right trace, palmitoylation-deficient GFP-R7BP-C252S/C253S mutant expression (GFP-R7BP-SS, 5 ng cRNA/oocyte). All oocytes were injected with cRNAs for RGS7 and Gβ5 (1 ng each/oocyte), and equivalent amounts of Kir3.1, Kir3.2a, muscarinic m2 receptor, GoαA (C351G), and pertussis toxin (PTX-S1; see Materials and methods for details). Bars indicate 1 μA and 10 s. (B) R7BP accelerates GIRK current deactivation kinetics by a mechanism dependent on RGS7–Gβ5 concentration and R7BP palmitoylation sites (C252 and C253). Derived time constants for GIRK deactivation after application of 10 μM ACh at three levels of RGS7–Gβ5 expression, with coexpression of either GFP-R7BP (5 ng cRNA/oocyte) or GFP-R7BP-SS (5 ng cRNA/oocyte). (C) R7BP accelerates GIRK current activation kinetics. Activation time constants derived from the time course for GIRK current activation with different ACh concentrations. (D) R7BP does not significantly affect the ACh dose–response relation for steady-state GIRK current activation or the maximal GIRK current amplitude. Error bars show SEM.
To quantify the effects of GFP-R7BP, we varied the expression levels of RGS7–Gβ5 by injecting increasing amounts of cRNA. In the absence of GFP-R7BP, increasing the expression of RGS7–Gβ5 moderately accelerated the rate of GIRK current deactivation (Fig. 8 B), as previously reported (Kovoor et al., 2000). Further increases in RGS7–Gβ5 expression (or RGS7 alone) resulted in marked suppression of GIRK current amplitude (unpublished data), as demonstrated previously (Zhang et al., 2002). At the lowest level of RGS7–Gβ5 expression (0.1 ng each cRNA/oocyte), GFP-R7BP (5 ng cRNA/oocyte) had an insignificant effect on GIRK current deactivation rate (Fig. 8 B). However, at moderate (1 ng each cRNA/oocyte) or higher (5 ng each cRNA/oocyte) RGS7–Gβ5 expression, GFP-R7BP significantly increased the rates of agonist-independent GIRK deactivation (Fig. 8 B) and agonist-dependent activation (Fig. 8 C) four- to fivefold. Coexpression of GFP-R7BP and RGS7–Gβ5 at these levels did not significantly change the ACh dose–response relation (Fig. 8 D). Together, these functional effects are consistent with previous studies implicating the existence of GPCR-GIRK channel signaling complexes that include RGS proteins (Sadja et al., 2003; Benians et al., 2005). Therefore, our findings indicate R7BP strikingly facilitates the formation of GIRK channel signaling complexes containing R7 RGS proteins.
Finally, to determine whether or not palmitoylation of R7BP affects its ability to augment RGS7–Gβ5 activity, we examined the effects of palmitoylation-deficient GFP-R7BP (C252S/C253S; “SS” mutant). By varying RGS7–Gβ5 expression level, we found that GFP-R7BP-SS had an insignificant effect on GIRK channel deactivation rates at low level of RGS7–Gβ5 (1 ng cRNA/oocyte) and diminished effect relative to wild-type GFP-R7BP at higher level of RGS7–Gβ5 expression (5 ng cRNA/oocyte; Fig. 8 B). The effects of R7BP palmitoylation on GIRK channel activation kinetics were examined at a fixed RGS7–Gβ5 expression level (1 ng each cRNA/oocyte) by varying the ACh dose (Fig. 8 C). These results indicated that nonpalmitoylated GFP-R7BP-SS exhibited significantly reduced activity relative to wild-type GFP-R7BP. The palmitoylation-independent effects of mutant R7BP may occur by stabilizing RGS7–Gβ5 complexes and/or augmenting the GAP activity of RGS7–Gβ5 complexes, similar to what has been shown with R9AP and RGS9-1 (Lishko et al., 2002; Hu et al., 2003). Nevertheless, because wild-type R7BP exhibited significantly greater activity relative to nonpalmitoylated R7BP, we conclude that palmitoylation of R7BP is required for efficient targeting of R7BP–RGS7–Gβ5 complexes to the plasma membrane, thereby facilitating modulation of receptor-coupled GIRK channels.
Discussion
We identify R7BP as a novel palmitoylated protein that regulates plasma membrane–nuclear shuttling of any member of the RGS7 (R7) family of G protein regulators bound to Gβ5. We suggest that R7BP is the principal plasma membrane–anchoring protein for R7–Gβ5 complexes in the nervous system because: (a) R7BP, Gβ5, and R7 proteins are expressed highly only in the nervous system (Gold et al., 1997; Zhang et al., 2000; Larminie et al., 2004), in contrast to the R7 anchor R9AP, which is expressed highly only in retina (Hu and Wensel, 2002); (b) R7BP and R9AP are the only closely related members of this protein family of membrane anchors; and (c) the spatial expression of R7BP, Gβ5, and the R7 family in brain are highly coincident (results presented herein; Gold et al., 1997; Zhang et al., 2000).
In parallel with our studies, Martemyanov et al. (2005) recently identified R7BP as a protein that copurifies nearly stoichiometrically with RGS9-2–Gβ5 complexes isolated from brain extracts. They showed that recombinant R7BP interacts in vitro with each R7 isoform, that endogenously expressed R7BP and each R7 isoform can be coprecipitated from brain membrane extracts, and that R7BP protein is expressed highly in various brain regions and in retina but not in several other tissues. These findings complement our results showing that R7BP is as a palmitoylation-regulated plasma membrane–nuclear shuttling protein that augments R7 protein function by targeting them to the plasma membrane. Thus, there is strong evidence that R7BP is the primary membrane anchoring protein for R7 family members in the nervous system.
Why do vertebrate genomes encode two distinct membrane-anchoring proteins for the R7 family: R9AP in retinal photoreceptor cells and R7BP throughout the nervous system? The nonoverlapping expression patterns and different membrane association mechanisms of R7BP and R9AP indicate that these proteins have distinct physiologic and mechanistic functions. R9AP may function constitutively as a membrane-anchoring protein because it possesses a transmembrane domain. R9AP also may possesses unique structural or sequence motifs that target R9AP–RGS9-1–Gβ5L complexes to outer segments of photoreceptor cells, analogous to the outer segment-targeting domain at the COOH terminus of rhodopsin (Tam et al., 2000).
R7BP, in contrast, apparently functions as a regulated plasma membrane–nuclear shuttling protein (Fig. 9). R7BP is palmitoylated, a reversible lipid modification that profoundly affects the localization and function of R7BP–R7–Gβ5 complexes. Palmitoylation may target R7BP–R7–Gβ5 complexes to lipid rafts or other specialized domains of the plasma membrane, similar to other palmitoylated proteins (El-Husseini and Bredt, 2002). Indeed, in hippocampal neurons GFP-R7BP concentrates in dendritic foci, as do the neuronal palmitoylated proteins PSD-95 and GAP-43 (El-Husseini and Bredt, 2002). More strikingly, palmitoylation regulates shuttling of R7BP–R7–Gβ5 complexes between the plasma membrane and nucleus. Accordingly, we hypothesize that reversible palmitoylation of R7BP provides a novel plasticity or “tuning” mechanism that modulates GPCR signaling. Depalmitoylation of R7BP and consequent delocalization of R7BP–R7–Gβ5 complexes from the plasma membrane may sensitize signaling mediated by Gi/o-coupled GPCRs. Palmitoylation of R7BP could have the opposite effect by attenuating or desensitizing signaling via Gi/o-coupled receptors or it could enable R7–Gβ5 complexes transduce signals to plasma membrane effectors.
Figure 9.

Regulation of the RGS7 family and Gβ5 by R7BP palmitoylation. R7BP, Gβ5, and an R7 family member form a trimolecular complex. R7BP is palmitoylated and recruits R7BP–R7–Gβ5 complexes to the plasma membrane where they regulate GPCR signal transduction. Upon depalmitoylation of R7BP, either unregulated or signal-induced R7BP–R7–Gβ5 complexes release from the plasma membrane and translocate to the nucleus where they potentially regulate gene expression or other processes. Unpalmitoylated R7BP–R7–Gβ5 complexes may shuttle between the nucleus and cytoplasm, allowing them to become palmitoylated and targeted to the plasma membrane. RGS7 can be palmitoylated (Rose et al., 2000; Takida et al., 2005), which may influence trafficking and function of R7BP–RGS7–Gβ5 complexes.
If R7BP depalmitoylation occurs after GPCR activation, R7BP–R7–Gβ5 complexes would be targeted to the nucleus. Because the R7 protein RGS6 can regulate gene expression (Liu and Fisher, 2004), signal-triggered depalmitoylation of R7BP could provide a novel mechanism for transmitting GPCR signals directly from the plasma membrane to the nucleus. Conversely, signal-regulated palmitoylation of R7BP could recruit quiescent nuclear R7–Gβ5 complexes to the plasma membrane to regulate GPCR signaling. These hypotheses are supported by evidence indicating that endogenously expressed R7 proteins and Gβ5 in neurons localize to membranes and nuclei (Zhang and Simonds, 2000; Bouhamdan et al., 2004; Krumins et al., 2004) and that GPCR activation can regulate the palmitoylation status of signaling proteins (for review see Smotrys and Linder, 2004).
Palmitoylation-regulated translocation of R7BP–R7–Gβ5 complexes to and from the plasma membrane would have profound consequences for GPCR-mediated regulation of ion channels and neuronal excitability. The concentration-dependent kinetic effects of RGS7 toward m2 receptor/Goα–coupled GIRK channel activity and the augmented effects caused by R7BP coexpression indicate that palmitoylation-regulated R7BP–R7–Gβ5 translocation controls both the amplitude and kinetics of ion channels regulated by Gi/o-coupled receptors, especially GIRK channels and voltage-gated calcium channels (Wickman and Clapham, 1995). Palmitoylated R7BP also may promote GPCR-selective ion channel regulation by targeting specific GPCR–R7–ion channel complexes to lipid rafts where Go, selected GPCRs, and ion channels colocalize (Cabrera-Vera et al., 2004).
R7BP may prove to be crucial for several functions of the central and peripheral nervous systems. This hypothesis is based on evidence indicating that the R7 substrate Goα is required in the central nervous system for regulation of motor control, motor behavior, and pain sensation (Jiang et al., 1998), and in retinal “ON” bipolar cells for light response (Dhingra et al., 2002). It also is suggested by evidence indicating that RGS9 is required to regulate antinociception via μ-opioid receptors and locomotor activity via D2 dopamine receptors (Rahman et al., 2003; Zachariou et al., 2003).
In conclusion, many neuronal signaling proteins including β-adrenergic receptors, G protein α subunits, R7BP, several RGS isoforms, PSD-95, nonreceptor tyrosine kinases, and H-ras are palmitoylated (for reviews see El-Husseini and Bredt, 2002; Smotrys and Linder, 2004). The diversity of palmitoylated proteins and the large family enzymes that palmitoylate them (Fukata et al., 2004) raise intriguing questions about how palmitoylation occurs in a specific and regulated manner to modulate nervous and sensory system function. Further analysis of R7BP palmitoylation may reveal novel mechanistic principles applicable to a variety of neuronal signaling proteins.
Materials and methods
Reagents and antibodies
For Western blots, anti-FLAG-M2 HRP conjugate (Sigma-Aldrich) was used at a dilution of 1:5,000, anti-MYC 9E10 HRP conjugate (Roche) at 1:1,000, anti-HA 12CA5 HRP conjugate (Roche) at 1:2,000, and HRP-conjugated goat anti–mouse secondary antibodies (Pierce Chemical Co.) at 1:10,000. For immunofluorescence, the anti-HA-11 antibody (BAbco) was used at a dilution of 1:500, anti-FLAG M2 (Sigma-Aldrich) at 1:1,000, and goat anti–mouse Alexa 568 secondary (Molecular Probes) at 1:100. 2Br-palmitate was obtained from Sigma-Aldrich. Hydroxylamine and sodium salicylate were purchased from Fisher Scientific. FTI-277 and GGTI-298 were obtained from Calbiochem. Unless noted otherwise, all chemicals were obtained from Sigma-Aldrich.
Cell culture and transfection
HEK293 cells (American Type Culture Collection) were maintained and transfected in DME/F12 with 10% FBS (Atlanta Biologicals) plus penicillin/streptomycin. Cells were grown at 37°C and 5% CO2. Transfection of HEK293 cells was performed using Effectene (QIAGEN) according to the manufacturer's instructions. Neonatal rat hippocampal neurons were isolated and cultured on glass coverslips coated with laminin and poly-D,L-ornithine as described previously (Wilding and Huettner, 2001). Neurons were transfected with R7BP plasmids via calcium phosphate precipitation.
R7BP cloning
RNA was prepared from adult male mouse brain using TRIZOL (Invitrogen) according to the manufacturer's instructions. 1 μg of total RNA was used in a reverse transcriptase reaction using oligo-dT primers and Superscript II (Invitrogen) according to manufacturer's instructions. cDNA from reverse transcriptase reactions was used as template for PCR to amplify the coding region of R7BP using the following primers that included engineered BamHI and EcoRI sites: 5′-gtggatccgtatgcatgagttctg-3′; 5′-aagaattcagcatagaaaacatgg-3′. A 904-bp PCR product was generated, which subsequently was cloned into the BamHI–EcoRI sites of pcDNA3.1. The entire insert was sequenced and found to be identical to a consensus cDNA sequence for R7BP that was generated by assembling all sequence information from publicly available cDNA and EST databases. Database searches identified putative R7BP homologues in the following species: human (XP_376386), orangutan (CAH92694), chimpanzee (XP_518191), mouse (BAC32849), rat (XP_215473), African clawed frog (AAH73094), spotted green pufferfish (CAF93443), pig (CO949728), chicken (BI390910), dog (CO656749), zebra finch (CK317390), zebrafish (CK363290), Salmo salar (CB513384), Oncorhynchus mykiss (CA386277).
Plasmids
R7BP was fused to the COOH terminus of GFP by cloning a BamHI–EcoRI fragment containing R7BP into the BglII–EcoRI sites of pEGFP-C1 (CLONTECH Laboratories, Inc.). 3FLAG-R7BP was constructed by cloning a HindIII–EcoRI fragment containing the R7BP coding region into p3FLAG-CMV10 (Sigma-Aldrich). pBlueScript-R7BP was constructed by cloning a BamHI–EcoRI fragment containing the R7BP coding region into pBlueScript II SK(+). Plasmids expressing HA-tagged versions of RGS6, RGS7, RGS9-1, RGS9-2, and RGS11 along with a FLAG-tagged version of Gβ5 were gifts of T.K. Harden (University of North Carolina, Chapel Hill, NC). A MYC-tagged version of Gβ5 was constructed by amplifying the Gβ5 coding region (flanked with BamHI and XhoI sites) from FLAG-Gβ5. This PCR product was digested and cloned into the BamHI–XhoI sites of pCMV-Tag 3A (Stratagene) to create a MYC-tagged fusion protein. RGS4-GFP was generated by cloning a fragment containing the complete RGS4-GFP fusion from pVT-RGS4-GFP (Srinivasa et al., 1998) into pCR3 for expression in mammalian cells. GFP-tagged versions of Rap1A and H-Ras were gifts of M. Phillips (New York University School of Medicine, New York, NY). A FLAG-tagged ERK2 plasmid was a gift of M. Weber (University of Virginia Health Sciences Center, Charlottesville, VA). R7BP point mutants were generated using Quickchange (Stratagene) according to the manufacturer's instructions. All constructs and point mutants were verified by DNA sequencing.
Cell lysis, immunoblotting, and immunoprecipitation
Cells were washed once with ice cold PBS and lysed in lysis buffer (MCLB: 50 mM Tris, pH 8.0, 5 mM EDTA, 0.5% NP-40, 100 mM NaCl, 1 mM sodium orthovanadate, and 1 mM PMSF) supplemented with protease inhibitor tablets (Roche). Cells were incubated at 4°C for 5 min with MCLB followed by further lysis by end-over-end rotation at 4°C for 15 min. Cell debris and unbroken cells were pelleted by centrifugation (15,000 g) for 10 min at 4°C. Supernatant fractions were used directly for Western blot analysis or for immunoprecipitation. For Western blots, 50–100 μg of total protein was electrophoresed through 10% SDS–polyacrylamide gels followed by transfer to PVDF (Millipore) membranes. Membranes were blocked for 1 h in 5% milk/TBST followed by overnight incubation with primary antibodies. HRP-conjugated primary antibodies were removed, membranes were washed with TBST, and signal was detected with ECL reagent (Amersham Biosciences). For some Westerns, primary antibody was washed and membranes were incubated with HRP-conjugated secondary antibodies for 1 h followed by processing. For immunoprecipitation, cleared cell lysates were incubated with 50 μl of FLAG M2-agarose affinity gel (Sigma-Aldrich) for 90 min followed by 3× 5-min washes in MCLB. Immunoprecipitates were denatured with 1× SDS-sample buffer for 5 min at 100°C.
Northern blotting
The BamHI–EcoRI fragment encoding R7BP in pcDNA3.1 was used as a template for random priming reactions (RediPrime II labeling system; Amersham Biosciences) to generate a radiolabeled probe with Redivue stabilized α-[32P]dCTP (Amersham Biosciences). This probe and one for β-actin were used to detect their cognate mRNAs in a mouse multiple tissue Northern blot containing poly(A) RNA (BD Biosciences) according to the manufacturer's instructions.
In situ hybridization
Antisense and sense R7BP cRNA probes labeled with α-[33P]UTP were generated by run-off transcription using T7 RNA polymerase. C57/B6 adult male mice were transcardially perfused with 20 ml D-PBS (DEPC treated) followed by fixation with 4% PFA in D-PBS (DEPC treated). Brains were removed and immersion fixed for an additional 24 h at 4°C in 4% PFA in D-PBS, and transferred to 10% sucrose in DEPC D-PBS for 36 h. Brains were frozen in OCT in a dry ice-ethanol bath and cut into 20-μm sections (coronal or sagittal). Sections were fixed at RT under vacuum, followed by in situ analysis as described previously (Muglia et al., 1999), with the following modifications. Slides were digested with 0.001% proteinase K, rinsed in 0.1 M TEA, and followed by blocking in 0.25% acetic anhydride. Slides were washed in 2× SSC followed by dehydration in a graded ethanol series (50%, 75%, 95%, 100%). Slides were dried under vacuum followed by hybridization at 58°C for 20 h in a humidified chamber. Slides were washed with 4× SSC, RNase-treated, rinsed, and de-salted in a graded SSC series (2×, 1×, 0.5×), and then dehydrated in a graded ethanol series. Slides were dried under a vacuum and exposed to film for 20–40 h.
Confocal immunofluorescence microscopy
Cells were grown and transfected on glass coverslips before preparation for microscopy as follows. Cells were washed with PBS, fixed for 10 min in fixative (3% PFA and 2% sucrose in PBS, pH 7.0) at 37°C, washed in PBS, and, if immunofluorescence was performed, permeabilized in ice-cold buffer (20 mM Hepes, pH 7.4, 0.5% Triton X-100, 50 mM NaCl, 3 mM MgCl2, and 300 mM sucrose). Cells were washed with PBS, blocked with 0.1% BSA (in PBS) for 10 min at 4°C, and washed again with PBS. Cells were incubated with primary antibodies (diluted in 5% goat serum/TBST) for 20 min at 37°C in a humidified chamber followed by 10 consecutive washes in TBST. Secondary antibodies (diluted in 5% goat serum/TBST) were incubated with cells for 15 min at RT followed by 10 washes in TBST. Cells were mounted on slides for confocal microscopy. Confocal microscopy was performed with a laser scanning confocal microscope (model LSM-510; Carl Zeiss MicroImaging, Inc.). GFP-R7BP localization in medial confocal sections of z-axis stacks was quantified by using Adobe Photoshop to determine integrated pixel intensities in the nucleus versus the entire cell and expressing the data as a nuclear/total cell ratio.
Metabolic labeling
HEK293 cells were grown and transfected in 6-well plates for labeling experiments. For [35S]methionine labeling, cells were starved 60 min of methionine (methionine-free DME/F12 with sodium pyruvate and penicillin/streptomycin) followed by labeling for 90 min in labeling media (starvation media plus 10% dialyzed FBS and [35S]methionine [Amersham Biosciences] at 100 μCi per well). After labeling, cells were lysed and processed for immunoprecipitation. After SDS-PAGE, gels were dried and exposed to film at −70°C. For [3H]palmitate labeling, cells were washed with media (DME/F12 with sodium pyruvate, nonessential amino acids, and penicillin/streptomycin) and incubated with labeling media for 90 min (media plus 10% dialyzed FBS and [3H]palmitic acid [PerkinElmer] at 1 mCi per well). Cell lysis, immunoprecipitation, and SDS-PAGE were performed as described for [35S]methionine labeling. Gels containing [3H]palmitate were treated with fluor solution (1 M sodium salicylate in 15% methanol) for 30 min before drying and fluorography at −70°C. For hydroxylamine treatment, gels containing either [35S]methionine- or [3H]palmitate-labeled R7BP were treated overnight with 0.5 M NH2OH, pH 7.0, or 0.5 M Tris, pH 7.0, in 50% 2-propanol. Gels were washed several times over 2 d with 50% 2-propanol before fluorography.
Lipid analysis
Cells transfected with either FLAG-R7BP or FLAG-ERK2 were labeled with [3H]palmitic acid. Cells were lysed, immunoprecipitated with anti-FLAG antibodies, and resolved on SDS-polyacrylamide gels. FLAG-tagged proteins were excised from the gel and subjected to treatment with NaOH, as described previously (Linder et al., 1995). Lipids were extracted from base hydrolysates with chloroform/methanol (1:2), dissolved in chloroform, spotted on reverse-phase TLC plates (Whatman) along with 3H-labeled C14:0, C16:0, and C18:0 standards. Plates were developed in acetonitrile/acetic acid (9:1), dried, sprayed with EN3HANCE (PerkinElmer), and exposed to film at −70°C.
Electrophysiology
All procedures for the use and handling of X. laevis (Xenopus One) were approved by the University of South Florida Institutional Animal Care and Use Committee in accordance with National Institutes of Health guidelines. Oocytes were enzymatically isolated from ovarian tissue as described previously (Zhang et al., 2002). Stage V–VI oocytes were selected and maintained in oocyte culture medium (OCM) at 19°C in 35-mm dishes on an orbital shaker. OCM was changed one to two times daily and was composed of 82.5 mM NaCl, 2.5 mM KCl, 1.0 mM CaCl2, 1.0 mM MgCl2, 1.0 mM NaHPO4, 2.5 mM Na-pyruvate, and 5.0 mM Hepes, pH 7.5, containing 2% heat-inactivated horse serum.
The effects of R7BP on GIRK channels activated by Gαo-coupled muscarinic m2 receptors were examined with and without RGS7 coexpression in experimental groups of oocytes injected with different mixtures of cRNAs synthesized in vitro from linearized cDNA vectors (mMessage mMachine; Ambion). Stock cRNAs for each cDNA were dissolved in DEPC-treated water and concentrations quantified by spectrophotometric absorbance at 260 nm. Absorbance profiles from 220 to 320 nm were performed to assess the quality of the stock cRNAs. Mixtures of different cRNAs were prepared in DEPC-treated H2O so that amounts of each cRNA injected per oocyte reflected that from a 50-nl injection of the cRNA mixture using a positive-displacement nanoliter injector (Nanoliter2000; World Precision Instruments). All experimental groups were injected with cRNAs for rat Kir3.1 (0.5 ng/oocyte), mouse Kir3.2a (0.5 ng/oocyte), human m2 muscarinic receptor (0.5 ng/oocyte), PTX-insensitive mouse GαoA(C352G) mutant subunit (5 ng cRNA/oocyte), and PTX-S1 cRNA (1 ng/oocyte) to inactivate endogenous Gαi/o subunits (Zhang et al., 2002).
Agonist-activated GIRK currents (IK,ACh) and receptor-independent basal GIRK channel activity (IK,basal) were measured for each of the various experimental groups by two-electrode voltage clamp recording (GeneClamp 500; Axon Instruments, Inc.). Electrodes were constructed from borosilicate glass capillary tubes (1.5-mm outside diameter, 0.86-mm inside diameter; GC150F-10; Warner Instruments) using a programmable microelectrode puller (P-97; Sutter Instrument Co.). The electrodes had tip resistances of 0.8–1.0 MΩ after filling with 3 M KCl. Membrane currents from voltage clamped oocytes were digitized (Digidata 1200 acquisition system; Axon Instruments, Inc.) and stored on a PC computer running pCLAMP 8.0 software (Axon Instruments, Inc.).
Oocytes were initially superfused with a minimal salt solution composed of 98 mM NaCl, 1 mM MgCl2, and 5 mM Hepes, pH 7.5 (NaOH). After electrode impalement and clamping the membrane potential to −80 mV, the solution was changed to a high K+ solution composed of 20 mM KCl, 78 mM NaCl, 1 mM MgCl2, and 5 mM Hepes, pH 7.5 (NaOH). High K+ induced an inward current (IK,basal) that is comprised primarily of receptor-independent GIRK channel activity (Dascal et al., 1993). Rapid application and washout of a range of ACh concentrations (Sigma-Aldrich) was performed with a computer controlled superfusion system (SF-77B; Warner Instruments; Doupnik et al., 2004). Voltage ramps from −80 to +20 mV and 1 s in duration were evoked before and during agonist application to monitor inward rectification of the ACh-evoked current. All recordings were performed at RT (21–23°C). GIRK current deactivation kinetics were analyzed using pCLAMP software to derive deactivation time constants (τdeact) associated with agonist washout. Statistical comparisons between the various experimental groups were performed by one-way ANOVA where P < 0.05 was considered significant. Experiments were each replicated in oocytes from two separate batches (dissections) of oocytes.
Online supplemental material
Fig. S1 shows localization of FLAG-tagged wild type and palmitoylation site mutant (C252S/C253S) R7BP expressed in HEK293 cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.20502007/DC1.
Acknowledgments
We thank Ben Kolber, Wendy Greentree, Monica Croke, and Alec Dickson for technical assistance. We are grateful to Drs. T. Kendall Harden, Mark Phillips, and Michael J. Weber for providing plasmids. We thank members of the Blumer laboratory for helpful advice and discussion.
This work was supported by grants from the National Institutes of Health (AG18876 and AA12957 to L.J. Muglia; NS30888 to J.E. Huettner; GM51466 to M.E. Linder; and GM44592 and HL075632 to K.J. Blumer) and the American Heart Association, Florida and Puerto Rico Affiliate (C.A. Doupnik). M.E. Linder is an Established Investigator of the American Heart Association. R.M. Drenan was supported by a predoctoral fellowship from the American Heart Association (0415310Z).
Abbreviations used in this paper: 2Br-palmitate, 2-bromopalmitate; ACh, acetylcholine; GAP, GTPase-activating protein; GIRK, G protein–regulated inward rectifying potassium; GPCR, G protein–coupled receptor; PTX, pertussis toxin.
References
- Benians, A., M. Nobles, S. Hosny, and A. Tinker. 2005. Regulators of G-protein signaling form a quaternary complex with the agonist, receptor, and G-protein. A novel explanation for the acceleration of signaling activation kinetics. J. Biol. Chem. 280:13383–13394. [DOI] [PubMed] [Google Scholar]
- Berman, D.M., T.M. Wilkie, and A.G. Gilman. 1996. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell. 86:445–452. [DOI] [PubMed] [Google Scholar]
- Bouhamdan, M., S.K. Michelhaugh, I. Calin-Jageman, S. Ahern-Djamali, and M.J. Bannon. 2004. Brain-specific RGS9-2 is localized to the nucleus via its unique proline-rich domain. Biochim. Biophys. Acta. 1691:141–150. [DOI] [PubMed] [Google Scholar]
- Cabrera-Vera, T.M., S. Hernandez, L.R. Earls, M. Medkova, A.K. Sundgren-Andersson, D.J. Surmeier, and H.E. Hamm. 2004. RGS9-2 modulates D2 dopamine receptor-mediated Ca2+ channel inhibition in rat striatal cholinergic interneurons. Proc. Natl. Acad. Sci. USA. 101:16339–16344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, J., and E.J. Nestler. 2004. Molecular neurobiology of drug addiction. Annu. Rev. Med. 55:113–132. [DOI] [PubMed] [Google Scholar]
- Chatterjee, T.K., Z. Liu, and R.A. Fisher. 2003. Human RGS6 gene structure, complex alternative splicing, and role of N terminus and G protein gamma-subunit-like (GGL) domain in subcellular localization of RGS6 splice variants. J. Biol. Chem. 278:30261–30271. [DOI] [PubMed] [Google Scholar]
- Chen, C.K., M.E. Burns, W. He, T.G. Wensel, D.A. Baylor, and M.I. Simon. 2000. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature. 403:557–560. [DOI] [PubMed] [Google Scholar]
- Chen, C.K., P. Eversole-Cire, H. Zhang, V. Mancino, Y.J. Chen, W. He, T.G. Wensel, and M.I. Simon. 2003. Instability of GGL domain-containing RGS proteins in mice lacking the G protein beta-subunit Gbeta5. Proc. Natl. Acad. Sci. USA. 100:6604–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang, H.H., M. Yu, Y.N. Jan, and L.Y. Jan. 1998. Evidence that the nucleotide exchange and hydrolysis cycle of G proteins causes acute desensitization of G-protein gated inward rectifier K+ channels. Proc. Natl. Acad. Sci. USA. 95:11727–11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan, C.W., W. He, and T.G. Wensel. 2001. RGS proteins: lessons from the RGS9 subfamily. Prog. Nucleic Acid Res. Mol. Biol. 65:341–359. [DOI] [PubMed] [Google Scholar]
- Dascal, N., W. Schreibmayer, N.F. Lim, W. Wang, C. Chavkin, L. DiMagno, C. Labarca, B.L. Kieffer, C. Gaveriaux-Ruff, D. Trollinger, et al. 1993. Atrial G protein-activated K+ channel: expression cloning and molecular properties. Proc. Natl. Acad. Sci. USA. 90:10235–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra, A., M. Jiang, T.L. Wang, A. Lyubarsky, A. Savchenko, T. Bar-Yehuda, P. Sterling, L. Birnbaumer, and N. Vardi. 2002. Light response of retinal ON bipolar cells requires a specific splice variant of Galpha(o). J. Neurosci. 22:4878–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik, C.A., N. Davidson, H.A. Lester, and P. Kofuji. 1997. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. USA. 94:10461–10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik, C.A., C. Jaen, and Q. Zhang. 2004. Measuring the modulatory effects of RGS proteins on GIRK channels. Methods Enzymol. 389:131–154. [DOI] [PubMed] [Google Scholar]
- El-Husseini, A.E., and D.S. Bredt. 2002. Protein palmitoylation: a regulator of neuronal development and function. Nat. Rev. Neurosci. 3:791–802. [DOI] [PubMed] [Google Scholar]
- Fukasawa, M., O. Varlamov, W.S. Eng, T.H. Sollner, and J.E. Rothman. 2004. Localization and activity of the SNARE Ykt6 determined by its regulatory domain and palmitoylation. Proc. Natl. Acad. Sci. USA. 101:4815–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata, M., Y. Fukata, H. Adesnik, R.A. Nicoll, and D.S. Bredt. 2004. Identification of PSD-95 palmitoylating enzymes. Neuron. 44:987–996. [DOI] [PubMed] [Google Scholar]
- Gainetdinov, R.R., R.T. Premont, L.M. Bohn, R.J. Lefkowitz, and M.G. Caron. 2004. Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27:107–144. [DOI] [PubMed] [Google Scholar]
- Gold, S.J., Y.G. Ni, H.G. Dohlman, and E.J. Nestler. 1997. Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J. Neurosci. 17:8024–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart, M.J., X. Jiang, T. Kozasa, W. Roscoe, W.D. Singer, A.G. Gilman, P.C. Sternweis, and G. Bollag. 1998. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science. 280:2112–2114. [DOI] [PubMed] [Google Scholar]
- Hollinger, S., and J.R. Hepler. 2002. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol. Rev. 54:527–559. [DOI] [PubMed] [Google Scholar]
- Hooks, S.B., G.L. Waldo, J. Corbitt, E.T. Bodor, A.M. Krumins, and T.K. Harden. 2003. RGS6, RGS7, RGS9, and RGS11 stimulate GTPase activity of Gi family G-proteins with differential selectivity and maximal activity. J. Biol. Chem. 278:10087–10093. [DOI] [PubMed] [Google Scholar]
- Hu, G., and T. Wensel. 2002. R9AP, a membrane anchor for the photoreceptor GTPase accelerating protein, RGS9-1. Proc. Natl. Acad. Sci. USA. 99:9755–9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, G., Z. Zhang, and T. Wensel. 2003. Activation of RGS9-1 GTPase acceleration by its membrane anchor, R9AP. J. Biol. Chem. 278:14550–14554. [DOI] [PubMed] [Google Scholar]
- Hunt, T.W., T.A. Fields, P.J. Casey, and E.G. Peralta. 1996. RGS10 is a selective activator of G alpha i GTPase activity. Nature. 383:175–177. [DOI] [PubMed] [Google Scholar]
- Jiang, M., M.S. Gold, G. Boulay, K. Spicher, M. Peyton, P. Brabet, Y. Srinivasan, U. Rudolph, G. Ellison, and L. Birnbaumer. 1998. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc. Natl. Acad. Sci. USA. 95:3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, M.B., D.P. Siderovski, and S.B. Hooks. 2004. The Gbetagamma dimer as a novel source of selectivity in G-protein signaling: GGL-ing at convention. Mol. Interv. 4:200–214. [DOI] [PubMed] [Google Scholar]
- Keresztes, G., K.A. Martemyanov, C.M. Krispel, H. Mutai, P.J. Yoo, S.F. Maison, M.E. Burns, V.Y. Arshavsky, and S. Heller. 2004. Absence of the RGS9.Gbeta5 GTPase-activating complex in photoreceptors of the R9AP knockout mouse. J. Biol. Chem. 279:1581–1584. [DOI] [PubMed] [Google Scholar]
- Kovoor, A., C.K. Chen, W. He, T.G. Wensel, M.I. Simon, and H.A. Lester. 2000. Co-expression of Gbeta5 enhances the function of two Ggamma subunit-like domain-containing regulators of G protein signaling proteins. J. Biol. Chem. 275:3397–3402. [DOI] [PubMed] [Google Scholar]
- Krispel, C.M., C.K. Chen, M.I. Simon, and M.E. Burns. 2003. Prolonged photoresponses and defective adaptation in rods of Gbeta5−/− mice. J. Neurosci. 23:6965–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumins, A.M., S.A. Barker, C. Huang, R.K. Sunahara, K. Yu, T.M. Wilkie, S.J. Gold, and S.M. Mumby. 2004. Differentially regulated expression of endogenous RGS4 and RGS7. J. Biol. Chem. 279:2593–2599. [DOI] [PubMed] [Google Scholar]
- Larminie, C., P. Murdock, J.P. Walhin, M. Duckworth, K.J. Blumer, M.A. Scheideler, and M. Garnier. 2004. Selective expression of regulators of G-protein signaling (RGS) in the human central nervous system. Brain Res. Mol. Brain Res. 122:24–34. [DOI] [PubMed] [Google Scholar]
- Linder, M.E., C. Kleuss, and S. Mumby. 1995. Palmitoylation of G-protein alpha subunits. Methods Enzymol. 250:314–330. [DOI] [PubMed] [Google Scholar]
- Lishko, P.V., K.A. Martemyanov, J.A. Hopp, and V.Y. Arshavsky. 2002. Specific binding of RGS9-Gbeta 5L to protein anchor in photoreceptor membranes greatly enhances its catalytic activity. J. Biol. Chem. 277:24376–24381. [DOI] [PubMed] [Google Scholar]
- Liu, Z., and R.A. Fisher. 2004. RGS6 interacts with DMAP1 and DNMT1 and inhibits DMAP1 transcriptional repressor activity. J. Biol. Chem. 279:14120–14128. [DOI] [PubMed] [Google Scholar]
- Long, S.B., P.J. Casey, and L.S. Beese. 2002. Reaction path of protein farnesyltransferase at atomic resolution. Nature. 419:645–650. [DOI] [PubMed] [Google Scholar]
- Martemyanov, K.A., P.J. Yoo, N.P. Skiba, and V.Y. Arshavsky. 2005. R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. J. Biol. Chem. 280:5133–5136. [DOI] [PubMed] [Google Scholar]
- Meijer, H.A., and A.A.M. Thomas. 2002. Control of eukaryotic protein synthesis by upstream open reading frames in the 5′-untranslated region of an mRNA. Biochem. J. 367:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muglia, L.M., M.L. Schaefer, S.K. Vogt, G. Gurtner, A. Imamura, and L.J. Muglia. 1999. The 5′-flanking region of the mouse adenylyl cyclase type VIII gene imparts tissue-specific expression in transgenic mice. J. Neurosci. 19:2051–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi, K.M., M.A. Sandberg, A.C. Kooijman, K.A. Martemyanov, J.W. Pott, S.A. Hagstrom, V.Y. Arshavsky, E.L. Berson, and T.P. Dryja. 2004. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature. 427:75–78. [DOI] [PubMed] [Google Scholar]
- Posner, B.A., A.G. Gilman, and B.A. Harris. 1999. Regulators of G protein signaling 6 and 7. Purification of complexes with gbeta5 and assessment of their effects on g protein-mediated signaling pathways. J. Biol. Chem. 274:31087–31093. [DOI] [PubMed] [Google Scholar]
- Rahman, Z., J. Schwarz, S.J. Gold, V. Zachariou, M.N. Wein, K.H. Choi, A. Kovoor, C.K. Chen, R.J. DiLeone, S.C. Schwarz, et al. 2003. RGS9 modulates dopamine signaling in the basal ganglia. Neuron. 38:941–952. [DOI] [PubMed] [Google Scholar]
- Rose, J.J., J.B. Taylor, J. Shi, M.I. Cockett, P.G. Jones, and J.R. Hepler. 2000. RGS7 is palmitoylated and exists as biochemically distinct forms. J. Neurochem. 75:2103–2112. [DOI] [PubMed] [Google Scholar]
- Sadja, R., N. Alagem, and E. Reuveny. 2003. Gating of GIRK channels: details of an intricate, membrane-delimited signaling complex. Neuron. 39:9–12. [DOI] [PubMed] [Google Scholar]
- Saitoh, O., Y. Kubo, Y. Miyatani, T. Asano, and H. Nakata. 1997. RGS8 accelerates G-protein-mediated modulation of K+ currents. Nature. 390:525–529. [DOI] [PubMed] [Google Scholar]
- Smotrys, J.E., and M.E. Linder. 2004. Palmitoylation of intracellular signaling proteins: regulation and function. Annu. Rev. Biochem. 73:559–587. [DOI] [PubMed] [Google Scholar]
- Srinivasa, S.P., L.S. Bernstein, K.J. Blumer, and M.E. Linder. 1998. Plasma membrane localization is required for RGS4 function in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 95:5584–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takida, S., C.C. Fischer, and P.B. Wedegaertner. 2005. Palmitoylation and plasma membrane targeting of RGS7 are promoted by alpha o. Mol. Pharmacol. 67:132–139. [DOI] [PubMed] [Google Scholar]
- Tam, B.M., O.L. Moritz, L.B. Hurd, and D.S. Papermaster. 2000. Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xenopus laevis. J. Cell Biol. 151:1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, A.J., A. Katz, and M.I. Simon. 1994. A fifth member of the mammalian G-protein beta-subunit family. Expression in brain and activation of the beta 2 isotype of phospholipase C. J. Biol. Chem. 269:22150–22156. [PubMed] [Google Scholar]
- Watson, N., M.E. Linder, K.M. Druey, J.H. Kehrl, and K.J. Blumer. 1996. RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha subunits. Nature. 383:172–175. [DOI] [PubMed] [Google Scholar]
- Wickman, K., and D.E. Clapham. 1995. Ion channel regulation by G proteins. Physiol. Rev. 75:865–885. [DOI] [PubMed] [Google Scholar]
- Wilding, T.J., and J.E. Huettner. 2001. Functional diversity and developmental changes in rat neuronal kainate receptors. J. Physiology. 532.2:411-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherow, D.S., and V.Z. Slepak. 2003. A novel king of G protein heterodimer: the Gbetagamma-RGS complex. Receptors Channels. 9:205–212. [PubMed] [Google Scholar]
- Witherow, D.S., S.C. Tovey, Q. Wang, G.B. Willars, and V.Z. Slepak. 2003. G beta 5.RGS7 inhibits G alpha q-mediated signaling via a direct protein-protein interaction. J. Biol. Chem. 278:21307–21313. [DOI] [PubMed] [Google Scholar]
- Zachariou, V., D. Georgescu, N. Sanchez, Z. Rahman, R. DiLeone, O. Berton, R.L. Neve, L.J. Sim-Selley, D.E. Selley, S.J. Gold, and E.J. Nestler. 2003. Essential role for RGS9 in opiate action. Proc. Natl. Acad. Sci. USA. 100:13656–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F.L., and P.J. Casey. 1996. Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem. 65:241–269. [DOI] [PubMed] [Google Scholar]
- Zhang, J.-H., and W.F. Simonds. 2000. Copurification of brain G-protein beta5 with RGS6 and RGS7. J. Neurosci. 20:RC59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J.-H., Z. Lai, and W.F. Simonds. 2000. Differential expression of the G protein beta(5) gene: analysis of mouse brain, peripheral tissues, and cultured cell lines. J. Neurochem. 75:393–403. [DOI] [PubMed] [Google Scholar]
- Zhang, Q., M.A. Pacheco, and C.A. Doupnik. 2002. Gating properties of GIRK channels activated by Galpha(o)- and Galpha(i)-coupled muscarinic m2 receptors in Xenopus oocytes: the role of receptor precoupling in RGS modulation. J. Physiol. 545:355–373. [DOI] [PMC free article] [PubMed] [Google Scholar]