The uniformity of phagosome maturation in macrophages (original) (raw)
Abstract
Many studies of endocytosis and phagocytosis presume that organelles containing a single kind of internalized particle exhibit invariant patterns of protein and phospholipid association as they mature inside cells. To test this presumption, fluorescent protein chimeras were expressed in RAW 264.7 macrophages, and time-lapse ratiometric fluorescence microscopy was used to measure the maturation dynamics of individual phagosomes containing IgG-opsonized erythrocytes. Quantitative analysis revealed consistent patterns of association for YFP chimeras of β-actin, Rab5a, Rab7, and LAMP-1, and no association of YFP chimeras marking endoplasmic reticulum or Golgi. YFP-2xFYVE, recognizing phosphatidylinositol 3-phosphate (PI(3)P), showed two patterns of phagosome labeling. Some phagosomes increased labeling quickly after phagosome closure and then lost the label within 20 min, whereas others labeled more slowly and retained the label for several hours. The two patterns of PI(3)P on otherwise identical phagosomes indicated that organelle maturation does not necessarily follow a single path and that some features of phagosome maturation are integrated over the entire organelle.
Keywords: phagosomes; phagocytosis; macrophages; membrane proteins; phosphatidylinositol
Introduction
Phagocytosis of particles by macrophages provides an experimental model for analyzing the uniformity and integrity of organelle maturation inside cells. Fcγ receptor (FcR)–mediated phagocytosis occurs by the sequential engagement of particle-bound IgG by macrophage surface FcR. Ligated FcRs locally activate intracellular signaling that results in pseudopod extension over the particle and further engagement of particle-bound IgG by macrophage FcR. This zipper-like mechanism of pseudopod advance is followed by contractile activities that constrict the outer margin of these extensions to enclose the particle (Greenberg et al., 1991; Swanson et al., 1999). The newly formed individual organelle, the phagosome, matures by a series of interactions with endocytic compartments, eventually fusing with lysosomes (Desjardins et al., 1994). Phagosome maturation could be guided locally by continued FcR-associated signaling. Alternatively, mechanisms of maturation could be integrated over the phagosome. If maturation were strictly locally regulated, then individual FcR phagosomes should all mature alike. If maturation also entails integrative activities, however, one might expect instances in which a phagosome behaves as an integrated whole. It is therefore appropriate to measure the uniformity of maturation.
Phagocytosis and phagosome maturation are temporally coordinated through sequential activities of molecules that regulate the cytoskeleton and direct fusion with elements of the endocytic pathway. Rab GTPases and phosphoinositides regulate diverse functions associated with membrane trafficking, such as vesicle formation (McLauchlan et al., 1998), vesicle docking and fusion (Simonsen et al., 1998; Christoforidis et al., 1999; Gillooly et al., 2001), and organelle association with the cytoskeleton (Nielsen et al., 1999). Rab5a is associated with early endosomes and directs interaction of phagosomes with early endosomes, as well as homotypic endosome fusion (Bucci et al., 1992; Duclos et al., 2000). Rab5a recruits the type III phosphatidylinositol (PI) 3-kinase hVps34, which generates PI 3-phosphate (PI(3)P) (Christoforidis et al., 1999; Murray et al., 2002). The concomitant presence of Rab5a and PI(3)P is necessary for the recruitment of early endosomal antigen 1, which mediates vesicle docking and fusion (Gaullier et al., 2000; Lawe et al., 2000).
PI(3)P on phagosomes may facilitate assembly of the NADPH oxidase complex, which mediates oxidative destruction of internalized microorganisms (Babior, 1999). p40phox and p47phox are soluble proteins of the complex that contain PX domains. PI(3)P may coordinate recruitment of these proteins to forming complexes (Kanai et al., 2001; Zhan et al., 2002).
Rab7 is acquired by phagosomes during the progression from the early to late endosomal stages (Desjardins et al., 1994). Rab7 is localized to late endosomes and lysosomes and is an important regulator of membrane transport from early to late endosomes (Vitelli et al., 1997; Feng et al., 2001) and of lysosome biogenesis (Bucci et al., 2000). Lysosomes are distinguished from endosomes, in part, by the presence of lysosome-associated membrane protein (LAMP)-1 (Chang et al., 2002). Lysosomes are the terminal degradation compartment of the endocytic pathway (Kornfeld and Mellman, 1989) and play an important role in the degradation of phagocytosed material (Funato et al., 1997).
Recent work has indicated that phagosomes interact not only with components of the endocytic pathway but with other intracellular compartments as well. Proteins of the ER have been localized by immunoelectron microscopy to phagosomes containing latex beads or erythrocytes (Gagnon et al., 2002). Phagosomes containing the obligate intracellular parasite Chlamydia trachomatis communicate with the Golgi apparatus and do not acquire endocytic or lysosomal markers (Scidmore et al., 2003).
Progression through the endocytic pathway can be monitored in live cells with fluorescent protein chimeras of intracellular markers (Sonnichsen et al., 2000; Feng et al., 2001; Jaiswal et al., 2002). Rab5a, Rab7, and LAMP-1 have been localized with GFP-tagged proteins. ER and Golgi membranes have been marked with GFP containing calreticulin ER-targeting and retrieval sequences (Kendall et al., 1994; Roderick et al., 1997), or the Golgi-targeting sequence of human β 1,4-galactosyltransferase, respectively (Gleeson et al., 1994; Llopis et al., 1998). PI(3)P has been localized using fluorescently tagged PX and FYVE domains that bind PI(3)P with high affinity and specificity (Gaullier et al., 2000; Sato et al., 2001). Although chimeric proteins containing a single FYVE domain do not localize significantly to PI(3)P-rich membranes (Gillooly et al., 2000), a chimeric fluorescent protein encoding two tandem FYVE domains (GFP-2xFYVE) from hepatocyte growth factor–regulated tyrosine kinase (Hrs) binds PI(3)P specifically (Gillooly et al., 2000; Stenmark et al., 2002).
Maturation patterns determined from phagosomes isolated or fixed during synchronized phagocytic responses presume that all phagosomes of a given class mature alike. Although there is abundant evidence that different particles can follow different maturation pathways in cells (Oh and Swanson, 1996; Aderem and Underhill, 1999), the assumption that all particles of a single class exhibit identical patterns of maturation has not been tested. Here, we followed the maturation in live cells of individual phagosomes containing IgG-opsonized erythrocytes (E-IgG). We provide evidence that maturation is largely uniform but that patterns of maturation vary with respect to PI(3)P. This indicates that signaling for maturation is not regulated strictly by local interactions but is integrated over the entire organelle.
Results
The interactions of proteins and organelles with newly formed phagosomes were measured in RAW 264.7 macrophages expressing soluble CFP plus YFP chimeras. The movements of β-actin, Rab5a, Rab7, and LAMP-1 were inferred using YFP chimeras of those proteins. PI(3)P was localized with a YFP chimera of tandem Hrs FYVE domains. ER was localized with YFP containing calreticulin ER targeting sequences, and the Golgi apparatus was localized using YFP with Golgi targeting sequence of human β 1,4-galactosyltransferase. Ratiometric imaging of YFP chimeras and CFP allowed mapping of the distributions of the maturation markers during FcR-mediated phagocytosis of E-IgG. Three images were collected at regular intervals during a phagocytic response: phase-contrast, YFP, and CFP. These were then compiled to generate three time-series movies and a processed time-series movie representing ratios of YFP to CFP fluorescence (Fig. 1, a–d). YFP signals were normalized to the distribution of the cytosol, using CFP. The YFP/CFP ratio images represented two-dimensional projections of the relative intracellular concentrations of the YFP chimeras. In cells expressing nonchimeric YFP and CFP, the ratio images appeared uniformly gray despite cell movement and considerable variation in cytoplasmic thickness. This indicated that the ratiometric image processing of YFP chimeras reliably localized distributions of the tagged proteins (Fig. 1, e and f).
Figure 1.

Distribution of YFP-actin during phagocytosis. (a) Time series showing phase-contrast, the component fluorescence images of (b) CFP, (c) YFP-actin, and (d) ratio images representing YFP-actin/CFP. Each column contains component images of one time point, separated from the adjacent columns by the time, in minutes, indicated in the phase-contrast image panel. The YFP-actin/CFP ratios were high in the forming phagocytic cup but declined quickly after closure of the cup. (e) Time series of phase-contrast images from a single phagocytic event and (f) the corresponding ratio images in a cell expressing YFP and CFP. The ratio was uniform at each time point.
Dynamics of YFP chimeras during phagocytosis
The temporal dynamics of YFP chimeras during phagocytosis were analyzed quantitatively by automated image analysis. A morphometric routine tracked the phagosome through a time series and measured at each time point the ratio of YFP/CFP on the phagosome (Rp) and the fraction of the phagosomal region of interest that contained CFP signal above a set threshold (Fig. 2). Only cells that completed phagocytosis by lateral extension of pseudopodia (i.e., side view) were selected for processing. Rp was then divided by the ratio of YFP/CFP for the entire cell (Rc) to obtain a normalized ratio of YFP/CFP in the phagosome (Rp/Rc). When Rp/Rc = 1.0, the concentration of YFP relative to CFP in the phagosome was identical to that of the cytoplasm, that is, YFP was not localized to the phagosome. To compare Rp/Rc from multiple phagocytic events, time series were aligned temporally using the rate of pseudopod extension (Fig. 3 a). For each series, the time point of complete pseudopod extension around the erythrocyte was set at 5 min (Fig. 3 a). For macrophages expressing different YFP chimeras, the rates at which the binary mask filled phagosomal regions of interest were all similar, indicating that the probes did not measurably inhibit phagocytosis. Increases in Rp/Rc above 1.0 indicated increased concentration of the YFP chimera in the phagosome (Fig. 3 b). Phagocytosis by macrophages expressing YFP and CFP gave a constant Rp/Rc value of 1 at all times (Fig. 3 c).
Figure 2.

The particle-tracking image analysis routine. The phagosome was marked in the phase-contrast image by two squares. The inner square defined the center of the object, and the outer square defined the search region. Regions of interest were positioned in the corresponding YFP, CFP, and CFP-thresholded images from the centroid of the phagosome determined above. From time 1 to time 2, the tracking routine followed the phagosome and adjusted the positions of the region of interest accordingly. For each frame, the mean fluorescence intensities, standard deviation, or fractional thresholded area were recorded for the phagosomal region of interest.
Figure 3.

Quantitative analysis of YFP-actin dynamics during phagocytosis. (a) Rate of phagosome extension in cells expressing YFP-actin and CFP was measured as the rate at which the binary mask filled the region of interest. The time point for maximal threshold was set to 5 min, and all the image stacks and corresponding data were realigned accordingly; this synchronized the measurements (e.g., the tracings shown in panel a were used to align the ratios for panel b). (b) Synchronized data from the tracking routine outputs for phagocytic events in cells expressing YFP-actin and CFP. The YFP-actin/CFP ratio in the phagosome (Rp) was normalized with respect to the YFP-actin/CFP ratio for the entire cell, yielding Rp/Rc, (diamonds). Rp/Rc and fraction thresholded area of the region of interest (circles) were plotted relative to time during pseudopod extension around the particle. Before and after extension, Rp/Rc was near 1, but this ratio increased sharply during pseudopod extension (n = 11). (c) Rp/Rc for cells expressing nonchimeric YFP and CFP (n = 5). The bars indicate SEM.
Actin, Rab5a, Rab7, and LAMP-1 dynamics were consistent for all phagosomes
Phagocytosis of E-IgG was accompanied by the rapid accumulation of YFP-actin in the forming phagocytic cup (Fig. 1 d). Actin moved as a circumferential band over the erythrocyte, consistent with sequential activation of Fc receptors along the extending pseudopod (Griffin and Silverstein, 1974) (Fig. 1 c). Fig. 3 b shows measurements from the time series of YFP-actin/CFP. Prior to internalization of the opsonized erythrocyte (0 min), the ratio Rp/Rc was slightly greater than 1 due to the presence of cortical actin. Rp/Rc then increased on the phagosome (0.25–4 min) and decreased (4.0–7 min), indicating the transient localization of F-actin in the forming phagosome. These results were similar to those obtained with rhodamine-labeled actin (Swanson et al., 1999) and GFP-actin (Araki et al., 2003).
The early endosome marker YFP-Rab5a was absent from the forming phagocytic cup but present during phagosome closure (Fig. 4 a). The YFP-Rab5a/CFP ratio increased after internalization (5–7 min). The labeling of early phagosomes with YFP-Rab5a was transient and persisted for up to 10 min, after which the ratio returned to 1 (Fig. 5 a). These dynamics were evident in every phagocytic event studied.
Figure 4.

Distributions of YFP-Rab5a, -Rab7, –LAMP-1, -Golgi, and -ER during phagocytosis. Each column contains component images of one time point, separated from the adjacent columns by the time, in minutes, indicated in panel a, with the exception of panel e. (a) Time series of YFP-Rab5a/CFP ratio images. The ratio increased after phagosome formation and began to decrease after 10 min. (b) Time series of YFP-Rab7/CFP ratio images. The ratio began to rise 5 min after phagosome closure and remained high throughout the observation. (c) Time series of YFP-Golgi/CFP and (d) YFP-ER/CFP ratio images. The ratios were flat at all time points. (e) Time series of YFP–LAMP-1/CFP ratio images. The ratio increased 20 min after internalization and remained high throughout the observation.
Figure 5.
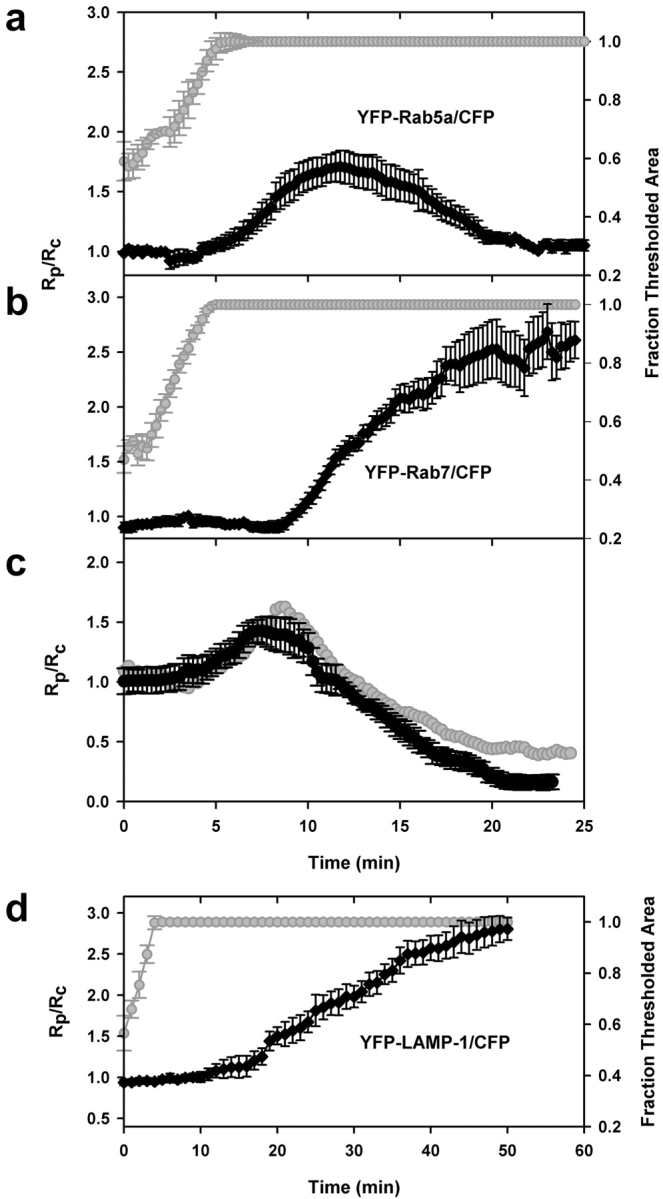
Rab5a, Rab7, and LAMP-1 dynamics were consistent for all phagosomes. Synchronized data from the tracking routine outputs for phagocytic events in cells expressing (a) YFP-Rab5a and CFP (n = 10), (b) YFP-Rab7 and CFP (n = 14), (c) YFP-Rab5a and CFP-Rab7 (n = 11), and (d) YFP–LAMP-1 and CFP (n = 4). The ratio data for each set were aligned as described in Fig. 3. The Rp/Rc values for each marker were similar at all time points. The gray curves in a, b, and d indicate the rates of pseudopod extension. The gray curve in panel c was obtained by dividing the Rp/Rc values for Rab5a (a) by the Rp/Rc values for Rab7 (b).
E-IgG phagosomes acquired YFP-Rab7 subsequent to YFP-actin dissociation (Fig. 4 b). The ratio of YFP-Rab7 to CFP increased beginning at 9 min after phagocytosis, continued for 10 min, and reached a plateau by 20 min (Fig. 5 b). This concentration was maintained for at least 6 h (not depicted), indicating that YFP-Rab7 was abundant on both late endosomal and lysosomal membranes. To verify this result, cells transfected with YFP-Rab7 were pulsed with Texas red dextran followed by an overnight chase to label lysosomes. YFP-Rab7 labeling partially colocalized with Texas red dextran–labeled lysosomes (unpublished data). These Rab7-positive organelles may represent exchanges of contents and/or membrane proteins between late endosomes and lysosomes (Mullock et al., 1998), or fusion of late endosomes with lysosomes to form hybrid compartments (Griffiths, 1996).
Quantitative analysis indicated that phagosomes should contain both Rab5a and Rab7 for a brief period during their maturation. To determine the extent of Rab5a and Rab7 overlap on phagosomes, ratiometric imaging was applied to RAW macrophages cotransfected with CFP-Rab7 and YFP-Rab5a. The ratio of YFP-Rab5a to CFP-Rab7 was high for ∼5 min after particle engulfment, corresponding to the period when phagosomes contained Rab5a but not Rab7. During the next 2.5 min, phagosomes were labeled with both markers; this was indicated by Rp/Rc values of 1 between 10 and 12.5 min (Fig. 5 c). The phagosomal YFP-Rab5a/CFP-Rab7 ratio decreased due to both dissociation of YFP-Rab5a and association of CFP-Rab7. Then the ratio dropped below 1 as Rab5a left the phagosome and Rab7 remained. Importantly, the probe dynamics measured in cells expressing YFP-Rab5a plus CFP-Rab7 (Fig. 5 c) were nearly identical to the dynamics inferred from cells expressing YFP-Rab5a plus CFP or YFP-Rab7 plus CFP. This indicated that sample sizes were sufficient to measure the dynamics of the chimeras on phagosomes.
The progression of phagosomes to late endosomes or lysosomes was indicated by their acquisition of YFP–LAMP-1 (Fig. 4 e). The ratio of YFP–LAMP-1 to CFP was near 1 until 20 min, after which Rp/Rc slowly increased (Fig. 5 d). Thus, two markers of late endosomes, Rab7 and LAMP-1, labeled phagosomes with different kinetics.
ER and Golgi markers were absent from erythrocyte phagosomes
The fluorescent chimera used to mark ER membrane consisted of YFP-tagged ER targeting sequence and retrieval sequences of calreticulin, which have been shown to localize proteins to the ER lumen (Kendall et al., 1994; Roderick et al., 1997). YFP-ER fluorescence in RAW macrophages exhibited a typical ER network throughout the cell, with no fluorescence visible in the nucleus (Fig. 4 d). YFP-Golgi, consisting of YFP-tagged Golgi targeting sequence of human β 1,4-galactosyltransferase, brightly labeled a small area of membrane juxtaposed to the nucleus, which is typical of trans-medial Golgi staining (Fig. 4 c). During phagocytosis, the ratio of YFP-ER or YFP-Golgi to CFP in the phagosome did not change, but remained at 1, indicating that neither ER nor Golgi membranes were associated with E-IgG phagosomes (Fig. 6, a and b). Absence of YFP-ER from E-IgG phagosomes was inconsistent with results of Gagnon et al. (2002). For a more direct comparison, we measured YFP-ER localization to phagosomes containing latex beads. YFP-ER was observed on latex bead phagosomes up to 30 min after internalization (Fig. 6 c).
Figure 6.

ER and Golgi markers were absent from erythrocyte phagosomes. Synchronized data from the tracking routine outputs for phagocytic events in cells expressing (a) YFP-Golgi and CFP (n = 6) and (b) YFP-ER and CFP (n = 9). The unchanging Rp/Rc values in cells indicated absence of these markers on phagosomes. (c) Compiled YFP-ER/CFP ratios for opsonized erythrocytes (n = 9) as compared with YFP-ER/CFP ratios for 3.5-μm latex beads (n = 15). P < 0.0001.
PI(3)P displayed heterogeneous association and dissociation profiles
YFP-2xFYVE localized to intracellular vesicular structures when expressed in macrophages. Incubation of cells with wortmannin, an inhibitor of PI 3-kinase, abolished the localization of YFP-2xFYVE on intracellular vesicular structures (unpublished data). YFP-2xFYVE was observed on E-IgG phagosomes after closure, indicating that PI(3)P was generated early (Fig. 7 a). The association of YFP-2xFYVE with phagosomes slightly preceded that of YFP-Rab5a, indicating possible generation of PI(3)P by enzymes other than hVps34.
Figure 7.


PI(3)P exhibited two distinct kinetic profiles on phagosomes. (a) Time series of ratio images in a cell expressing YFP-2xFYVE and CFP. YFP-2xFYVE was removed from the phagosome by 10 min. (b) Time series of ratio images in a cell expressing YFP-2xFYVE and CFP. YFP-2xFYVE remained on the phagosome for the length of the observation (20 min). (c) Temporally aligned data from the tracking routine outputs for phagocytic events in cells expressing YFP-2xFYVE and CFP. Rp/Rc increased immediately after internalization for all phagosomes, but after 9 min, Rp/Rc varied (n = 10). (d) Rp/Rc values for phagosomes that lost YFP-2xFYVE at early time points (filled diamonds) (n = 5) as compared with Rp/Rc values for phagosomes that retained YFP-2xFYVE (open diamonds) (n = 5). (e) Quantitative analysis of dynamics in macrophages cotransfected with YFP-2xFYVE and CFP-Rab7 (black circles) (n = 15) as compared with Rp/Rc values for YFP-2xFYVE plus CFP (a) divided by the Rp/Rc values for YFP-Rab7 plus CFP (Fig. 5 b) (gray circles). Rp/Rc was high initially due to high concentrations of PI(3)P on phagosomes and decreased as CFP-Rab7 accumulated on phagosomes. The higher ratio of PI(3)P to Rab7 in the cotransfected group reflects a higher proportion of phagosomes that retained YFP-2xFYVE. 12 of 15 phagosomes retained the marker.
The timing of PI(3)P association and dissociation was variable. Some phagosomes displayed high YFP-2xFYVE/CFP ratios immediately after sealing from the plasma membrane and lost the marker after ∼10 min, while for other phagosomes, the ratio increased slowly and remained high for the duration of the experiments (Fig. 7 b). This differential 2xFYVE localization could be observed on two phagosomes within the same macrophage. Overall, 32 of 68 recorded phagocytic events displayed early loss of PI(3)P, and the remainder retained the marker.
The duration of PI(3)P localization to phagosomes did not correlate with the number of phagosomes per macrophage, nor with the relative timing of phagocytic events in a cell. To determine whether variation in opsonization of the erythrocytes could account for variation in the maturation patterns exhibited by E-IgG phagosomes, the density of IgG bound to erythrocytes was quantified by flow cytometry. The mean fluorescence of erythrocytes labeled with various concentrations of antibody was compared to determine relative density. As the concentration of antibody was increased, the mean fluorescence of the erythrocytes increased accordingly until it reached a maximum. The maximum density was achieved at antibody concentrations below those used for phagocytosis experiments, indicating that the erythrocytes used were maximally coated with antibody. The variation in opsonization was determined by the coefficient of variance. At maximum antibody density, the coefficient of variance was 30%, indicating that the variation in antibody density was minimal (unpublished data).
Quantitative analysis of YFP-2xFYVE/CFP ratios revealed two distinct profiles of PI(3)P localization during phagocytosis. Comparing these phagocytic events with those that occurred in cells expressing YFP-Rab5a or YFP-Rab7 and CFP, it was apparent that the first PI(3)P localization profile preceded YFP-Rab5a accumulation, and the second PI(3)P profile coincided with YFP-Rab5a accumulation. The quantitative data for PI(3)P kinetics was collected from the 10 side-view phagocytic events that could be aligned using binary masks. 5 of the 10 exhibited early loss of PI(3)P, and 5 exhibited late loss (Fig. 7 d).
Heterogeneous localization of YFP-2xFYVE was also observed on latex bead phagosomes. The localization of YFP-2xFYVE to phagosomes containing opsonized 5-μm latex beads was observed at fixed time points. At 15 min, half of latex bead phagosomes were labeled with YFP-2xFYVE, while half were unlabeled. At 30 min, >90% of latex bead phagosomes lacked YFP-2xFYVE labeling. A small percentage of phagosomes still retained the marker at 60 min. No latex bead phagosomes were labeled at 75 min.
Although PI(3)P is known as a marker of early endosomes, the persistence of YFP-2xFYVE on some phagosomes suggested that it may also be present on late endosomes and lysosomes. To investigate this possibility, RAW macrophages were cotransfected with YFP-2xFYVE and CFP-Rab7. The YFP/CFP ratio was initially high, due to high concentrations of PI(3)P on E-IgG phagosomes, and then decreased as CFP-Rab7 accumulated on phagosomes. The slow decrease in ratio indicated that PI(3)P and Rab7 colocalized on phagosomes for varying time periods (Fig. 7 e). Thus, PI(3)P was present on late endosomal membranes but was not a reliable late endosomal marker. To ascertain PI(3)P expression on lysosomes, cells transfected with YFP-2xFYVE were pulsed with Texas red dextran followed by an overnight chase to label lysosomes. YFP-2xFYVE labeling did not correspond to Texas red–labeled lysosomes, indicating that PI(3)P was not present on lysosomes (unpublished data).
Discussion
The high sensitivity and temporal resolution provided by ratiometric imaging of individual phagosomes indicated that some aspects of maturation were integrated over the entire organelle. Maturation was largely similar from one event to the next: actin was present briefly during phagosome formation, Rab5a and Rab7 associated sequentially, with a brief period of overlap on the nascent phagosome, and LAMP-1 associated later than Rab7. Yet despite the constant patterns of maturation for these markers, the timing of PI(3)P formation and removal from phagosomes fell into two very different patterns. The underlying mechanism for the divergence remains unknown, but its existence indicates integrative activities in organelle maturation.
Quantitative analysis of single phagocytic events in live cells revealed subtleties in membrane marker dynamics that would be missed by analyses of fixed cells or isolated phagosomes. The sensitivity of ratiometric fluorescence microscopy to low concentrations of chimeric molecules on phagosomes detected the gradual association and dissociation of markers on phagosomes, creating complete temporal profiles of marker dynamics. By normalizing YFP chimera distributions with CFP, the localization and relative concentrations of the membrane markers could be measured throughout the phagocytic response, independent of path length variations or cell movement (Swanson, 2002). Ratiometric fluorescence microscopy was also used to quantify the relative dynamics of two markers.
The use of fluorescently tagged probes may be complicated by competition between those probes and nonfluorescent endogenous molecules. For example, the loss of fluorescence from a phagosome could reflect either the normal removal kinetics of the endogenous protein or the displacement of the fluorescent probe by endogenous protein. Competitive displacement seems unlikely, however, as the dynamics reported here are consistent with those inferred by other methods.
Invariant features of maturation
The maturation pathway followed by phagosomes containing opsonized erythrocytes was defined by the sequential appearance of actin, Rab5a, Rab7, and LAMP-1 on phagosomal membranes. The timing of association and dissociation of these markers on phagosomes defined a pattern displayed by all E-IgG phagosomes measured and was strikingly similar from one phagosome to the next. The coordinated activity of these markers may be necessary for phagosome trafficking to lysosomes, as disruption of this pathway through inhibition of Rab5a or Rab7 alters phagosome progression to lysosomes (Duclos et al., 2000; Roberts et al., 2000; Feng et al., 2001).
The sensitivity of ratiometric fluorescence microscopy to slight increases in levels of markers on phagosomes relative to the cytoplasm confirmed the presence of an ER marker on latex bead phagosomes and the absence of the ER marker on E-IgG phagosomes. The differential recruitment of markers between the two types of phagosomes may be due to differences in the initial binding of particles to macrophage surface receptors, which initiate signaling for particle engulfment (Greenberg et al., 1991). The surface receptors ligated by latex beads and opsonized erythrocytes may not be the same. It was not surprising then to find differences in markers between E-IgG phagosomes and latex bead phagosomes.
This differential association of ER with E-IgG phagosomes and latex bead phagosomes may underlie differences in antigen delivery to major histocompatibility complex (MHC) class I molecules via phagocytosis. Antigen delivery from latex bead phagosomes into the cytoplasm for MHC class I–restricted antigen presentation has been demonstrated (Rock et al., 1994). The ER present on latex bead phagosomes contains components of the MHC class I antigen presentation pathway, which facilitate loading of antigens delivered on latex bead phagosomes onto MHC class I molecules of those same phagosomes (Guermonprez et al., 2003; Houde et al., 2003). Interestingly, antigen delivered via E-IgG phagosomes was shown to be less available for MHC class I presentation than antigen delivered via latex beads (Oh et al., 1997). The different efficiencies of loading may be due to differences in the extent of ER recruitment between latex bead phagosomes and E-IgG phagosomes.
Variable features of maturation
Amidst this tight coordination of maturation molecules, the dynamics of PI(3)P on phagosomes were not uniform. It is unlikely that these results reflect occasional deregulation of the maturation pathway, as the acquisition of Rab7 and LAMP-1 remained constant in all cases. Rather, these results point to the existence of two pathways of maturation. Both pathways maintain a constant timing of interactions between phagosomes, early endosomes, and late endosomes, yet phagosomal PI(3)P levels rise and fall in two patterns. The patterns may be determined stochastically, by random fluctuations in the activities of lipid kinases or phosphatases that then become amplified to one of two extremes. Or they may be determined by some still-unmeasured functional variation in phagosomal maturation.
Heterogeneous PI(3)P dynamics have also been observed for _Salmonella typhimurium_–containing vacuoles. PI(3)P appeared and reappeared on _Salmonella_-containing vacuoles for varying time periods, producing asynchronous “flashes” of 2xFYVE-GFP labeling (Pattni et al., 2001). It was suggested that competing activities of bacterial and cellular enzymes caused this phenomenon. The production and degradation of PI(3)P are affected by type III PI 3-kinase, the phosphatase myotubularin, and probably other lipid kinases and phosphatases. The competing activities of these enzymes, together with positive or negative feedback effects, could produce the two different labeling patterns observed on 20-min-old phagosomes.
What functional differences could explain the different patterns of PI(3)P labeling? Imperceptible differences in the combination of phagosomal proteins or lipids at 9 min could create a small bias that becomes amplified for further PI(3)P generation or its removal. The timing of the labeling indicates a possible role for Rab5a and Rab7. YFP-2xFYVE association began similarly in all phagosomes, increasing rapidly 5 min after the initiation of phagocytosis. At 9 min, however, when half of the phagosomes continued to increase PI(3)P and the other half lost it, Rab5a accumulation slowed and Rab7 levels began to increase. Small differences in the levels or activities of these GTPases could bias the feedback loops affecting lipid kinases or phosphatases.
Another possible functional difference defining the two patterns could be the levels of activity of the NADPH oxidase. PI(3)P can recruit effector proteins containing FYVE or PX domains. There are >40 FYVE domain–containing proteins and >150 proteins with PX domains, including many regulators of vesicular trafficking (Stenmark and Aasland, 1999; Ellson et al., 2002). Of particular interest are the p40phox and p47phox subunits of NADPH oxidase, which bind PI(3)P. Heterogeneous PI(3)P association with phagosomes may allow for variation in NADPH oxidase activity on phagosomes. An early and transient oxidative response may be appropriate for some phagosomes, while a sustained oxidative response may be necessary for others. Different levels of reactive oxygen intermediates generated inside the phagosome could bias the chemistries affecting PI(3)P levels.
The dynamic levels of phagosomal PI(3)P lead to either of two steady states on the 20-min-old phagosome: maintenance of a high level of PI(3)P or complete absence of PI(3)P. The complete conversion to one of these two states indicates that the signals governing phagosome membrane chemistry are integrated over the entire organelle. A signal above a certain threshold precipitates an all-or-none response. This contrasts with the mechanism of particle engulfment. Internalization of IgG-opsonized particles by FcR-mediated phagocytosis occurs through the local activation of receptors along the extending pseudopod, a hallmark of “zippering” phagocytosis (Griffin and Silverstein, 1974). FcR signaling is spatially confined to the region of cytoplasm near the receptor. If the levels of PI(3)P were also determined solely by localized signaling from ligated receptors, then all IgG-erythrocyte–containing phagosomes would have to mature similarly, with PI(3)P levels reflecting the average of the many receptor-proximal signals. Instead, each organelle commits entirely to one path or the other.
Materials and methods
Cell culture of RAW 264.7 cells
RAW 264.7 cells were grown in DME (Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen) and 100 U/ml of penicillin/streptomycin mixture (Sigma-Aldrich) at 37°C with 5% CO2. During microscopic observation, the cells were maintained at 37°C on a heated stage in Ringer's buffer (155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM NaH2PO4, 10 mM Hepes, and 10 mM glucose, pH 7.2).
CFP and YFP constructs
GFP–LAMP-1 was provided by N. Andrews (Yale University School of Medicine, New Haven, CT). PCR-amplified LAMP-1 sequences were inserted into pEYFP-N1 (CLONTECH Laboratories, Inc.) to create YFP–LAMP-1. GFP-Rab5a was provided by P. Stahl (Washington University, St. Louis, MO). GFP-2xFYVE, containing two linked FYVE finger domains from hepatocyte growth factor–regulated tyrosine kinase substrate (Hrs), was supplied by H. Stenmark (Norwegian Radium Hospital, Oslo, Norway). Rab7 cDNA was a gift from A. Wandinger-Ness (University of New Mexico, Albuquerque, NM). The PCR-amplified coding sequences of Rab5a, 2xFYVE, and Rab7 were subcloned into pEYFP-C1 and pECFP-C1 vectors (CLONTECH Laboratories, Inc.) to create YFP-Rab5a, CFP-Rab5a, YFP-2xFYVE, YFP-Rab7, and CFP-Rab7. All constructs were verified by sequencing. Mammalian expression vectors for pECFP-N1 (CFP), pEYFP-actin, pEYFP-ER, and pEYFP-Golgi were purchased from CLONTECH Laboratories, Inc.
Transient transfection and fluorescent labeling
All DNA used for transfection was prepared with a MaxiPREP kit (QIAGEN). For cells transfected by electroporation, RAW cells were suspended at ∼3 × 106 cells/ml in DME. 400 μl of the cell suspension was mixed with 2 μg of plasmid DNA in a 4 mM gap electroporation cuvette (BTX). Electroporation was performed in an ECM 630 (BTX) at 300 V, 1,000 μf, and 25 Ω, to yield a time constant of ∼13 ms. Cells were then plated on 25-mm coverslips and grown in DME with FBS for at least 18 h before observation. For some experiments, RAW cells were transfected with Fugene 6 (Roche), according to the manufacturer's instructions. For pulse-chase experiments, RAW macrophages transfected with YFP chimeras were pulsed with 0.5 mg/ml Texas red dextran, M r 10,000 (Molecular Probes) in DME for 30 min, washed four times with warm Ringer's buffer, and then incubated overnight in DME to allow Texas red dextran redistribution to lysosomes. The specificity of YFP-2xFYVE for PI(3)P was tested by incubating transfected RAW cells for 30 min with 100 nM wortmannin (Sigma-Aldrich) and measuring redistribution of the chimera.
Phagocytic assays
Sheep erythrocytes were opsonized with IgG rabbit anti–sheep erythrocyte (E-IgG) (Knapp and Swanson, 1990). Phagocytosis was initiated by adding of 108 E-IgG to the RAW cells at 37°C. Opsonized erythrocytes were labeled with a FITC-conjugated goat anti–rabbit IgG antibody (Sigma-Aldrich) and analyzed using a FACScan® instrument and CellQuest software (BD Biosciences). 3.5 or 5-μm latex beads were incubated with 10 mg/ml BSA (Sigma-Aldrich) in Ringer's buffer for 1 h at 37°C and washed by centrifugation. BSA-coated beads were opsonized with IgG mouse anti-BSA (Sigma-Aldrich) for 30 min at 37°C. Latex beads were added to RAW cells at 4°C, after which the coverslips were centrifuged at 700 g for 2 min. The coverslips were warmed to 37°C before image acquisition.
Microscopy
An inverted fluorescence microscope (Nikon TE-300) was equipped with a temperature-controlled stage, shutters for trans- and epifluorescence illumination, filter wheels for both excitation and emission filters, dichroic mirrors that allowed simultaneous detection of multiple fluorophores, a 60x 1.4 oil Planapo objective, and a cooled digital CCD camera (Quantix; Photometrics). Electronics were controlled by Metamorph image processing software (Universal Imaging Corp.). Imaging employed the two filter wheels and selective dichroic mirrors for viewing both YFP and CFP (Omega Optical). Time-lapse movies of phagocytosis were obtained by collecting three 12-bit images: a phase-contrast image and then two fluorescence images, YFP and CFP. Such series were collected every 15 s for 15–30 min, unless indicated otherwise. Light exposure and image collection were adjusted to obtain maximal pixel values between 200 and 400 (12-bit range of 0–4096; background + offset intensities were 100–125).
Image analysis
Methods for image analysis and contrast enhancement were applied uniformly using an automated processing routine developed in Metamorph. Primary fluorescence images were background corrected for each frame by subtracting the mean value from a cell-free region. A ratio image was obtained by dividing the YFP image by the corresponding CFP image and multiplying by 1,000. A binary mask was produced from the addition of the YFP and CFP images and a manual threshold. The binary and divided images were combined in a logical AND to produce ratio images that excluded noncellular signals. Processed images were prepared for presentation using Adobe Photoshop and Adobe Premiere (Adobe Systems Inc.).
The dynamics of YFP chimeras relative to CFP were analyzed quantitatively using a novel particle-tracking routine developed using Metamorph software. The routine used a cross-correlation centroid tracking algorithm to locate the center of the phagosome in each phase-contrast image of a time series. The centroid was then used to position regions of interest in the thresholded fluorescence images (YFP and CFP). These regions were circles large enough to include the entire sheep erythrocyte and its surrounding cytoplasm, generally with a diameter of 5 μm. The output of this routine included the position of the phagosome, the mean YFP and CFP intensities inside the threshold, and the fractional area of the region included in the binary mask defining the cell profile. A second region drawn around the entire cell was used to measure the average fluorescence intensities of YFP and CFP over the entire cell. Relative ratios of YFP/CFP in the phagosome (Rp) were then divided by the YFP/CFP for the entire cell (Rc) to obtain a cell-normalized phagosome ratio (Rp/Rc). In all cases, Rc changed very little during the course of a time series, indicating that changes in Rp/Rc primarily indicated changes in Rp.
Acknowledgments
The authors wish to thank Norma Andrews, Philip Stahl, Harald Stenmark, and Angela Wandinger-Ness for their gifts of cDNA.
This work was supported by National Institutes of Health grant AI35950.
Abbreviations used in this paper: E-IgG, IgG-opsonized erythrocyte; FcR, Fcγ receptor; LAMP, lysosome-associated membrane protein; MHC, major histocompatibility complex; PI, phosphatidylinositol; PI(3)P, PI 3-phosphate;
References
- Aderem, A., and D.M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593–623. [DOI] [PubMed] [Google Scholar]
- Araki, N., T. Hatae, A. Furukawa, and J.A. Swanson. 2003. Phosphoinositide-3-kinase-independent contractile activities associated with Fcγ-receptor-mediated phagocytosis and macropinocytosis in macrophages. J. Cell Sci. 116:247–257. [DOI] [PubMed] [Google Scholar]
- Babior, B.M. 1999. NADPH oxidase: an update. Blood. 93:1464–1476. [PubMed] [Google Scholar]
- Bucci, C., R.G. Parton, I.H. Mather, H. Stunnenberg, K. Simons, B. Hoflack, and M. Zerial. 1992. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 70:715–728. [DOI] [PubMed] [Google Scholar]
- Bucci, C., P. Thomsen, P. Nicoziani, J. McCarthy, and B. van Deurs. 2000. Rab7: a key to lysosome biogenesis. Mol. Biol. Cell. 11:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, M.H., L.E. Karageorgos, and P.J. Meikle. 2002. CD107a (LAMP-1) and CD107b (LAMP-2). J. Biol. Regul. Homeost. Agents. 16:147–151. [PubMed] [Google Scholar]
- Christoforidis, S., H.M. McBride, R.D. Burgoyne, and M. Zerial. 1999. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 397:621–625. [DOI] [PubMed] [Google Scholar]
- Desjardins, M., L.A. Huber, R.G. Parton, and G. Griffiths. 1994. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 124:677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclos, S., R. Diez, J. Garin, B. Papadopoulou, A. Descoteaux, H. Stenmark, and M. Desjardins. 2000. Rab5 regulates the kiss and run fusion between phagosomes and endosomes and the acquisition of phagosome leishmanicidal properties in RAW 264.7 macrophages. J. Cell Sci. 113:3531–3541. [DOI] [PubMed] [Google Scholar]
- Ellson, C.D., S. Andrews, L.R. Stephens, and P.T. Hawkins. 2002. The PX domain: a new phosphoinositide-binding module. J. Cell Sci. 115:1099–1105. [DOI] [PubMed] [Google Scholar]
- Feng, Y., B. Press, W. Chen, J. Zimmerman, and A. Wandinger-Ness. 2001. Expression and properties of Rab7 in endosome function. Methods Enzymol. 329:175–187. [DOI] [PubMed] [Google Scholar]
- Funato, K., W. Beron, C.Z. Yang, A. Mukhopadhyay, and P.D. Stahl. 1997. Reconstitution of phagosome-lysosome fusion in streptolysin O-permeabilized cells. J. Biol. Chem. 272:16147–16151. [DOI] [PubMed] [Google Scholar]
- Gagnon, E., S. Duclos, C. Rondeau, E. Chevet, P.H. Cameron, O. Steele-Mortimer, J. Paiement, J.J. Bergeron, and M. Desjardins. 2002. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 110:119–131. [DOI] [PubMed] [Google Scholar]
- Gaullier, J.M., E. Ronning, D.J. Gillooly, and H. Stenmark. 2000. Interaction of the EEA1 FYVE finger with phosphatidylinositol 3-phosphate and early endosomes. Role of conserved residues. J. Biol. Chem. 275:24595–24600. [DOI] [PubMed] [Google Scholar]
- Gillooly, D.J., I.C. Morrow, M. Lindsay, R. Gould, N.J. Bryant, J.M. Gaullier, R.G. Parton, and H. Stenmark. 2000. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19:4577–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillooly, D.J., A. Simonsen, and H. Stenmark. 2001. Phosphoinositides and phagocytosis. J. Cell Biol. 155:15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson, P.A., R.D. Teasdale, and J. Burke. 1994. Targeting of proteins to the Golgi apparatus. Glycoconj. J. 11:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg, S., J. el Khoury, F. di Virgilio, E.M. Kaplan, and S.C. Silverstein. 1991. Ca2+-independent F-actin assembly and disassembly during Fc receptor-mediated phagocytosis in mouse macrophages. J. Cell Biol. 113:757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, F.M., and S.C. Silverstein. 1974. Segmental response of the macrophage plasma membrane to a phagocytic stimulus. J. Exp. Med. 139:323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, G. 1996. On vesicles and membrane compartments. Protoplasma. 195:37–58. [Google Scholar]
- Guermonprez, P., L. Saveanu, M. Kleijmeer, J. Davoust, P. Van Endert, and S. Amigorena. 2003. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 425:397–402. [DOI] [PubMed] [Google Scholar]
- Houde, M., S. Bertholet, E. Gagnon, S. Brunet, G. Goyette, A. Laplante, M.F. Princiotta, P. Thibault, D. Sacks, and M. Desjardins. 2003. Phagosomes are competent organelles for antigen cross-presentation. Nature. 425:402–406. [DOI] [PubMed] [Google Scholar]
- Jaiswal, J.K., N.W. Andrews, and S.M. Simon. 2002. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 159:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai, F., H. Liu, S.J. Field, H. Akbary, T. Matsuo, G.E. Brown, L.C. Cantley, and M.B. Yaffe. 2001. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat. Cell Biol. 3:675–678. [DOI] [PubMed] [Google Scholar]
- Kendall, J.M., M.N. Badminton, R.L. Dormer, and A.K. Campbell. 1994. Changes in free calcium in the endoplasmic reticulum of living cells detected using targeted aequorin. Anal. Biochem. 221:173–181. [DOI] [PubMed] [Google Scholar]
- Knapp, P.E., and J.A. Swanson. 1990. Plasticity of the tubular lysosomal compartment in macrophages. J. Cell Sci. 95:433–439. [DOI] [PubMed] [Google Scholar]
- Kornfeld, S., and I. Mellman. 1989. The biogenesis of lysosomes. Annu. Rev. Cell Biol. 5:483–525. [DOI] [PubMed] [Google Scholar]
- Lawe, D.C., V. Patki, R. Heller-Harrison, D. Lambright, and S. Corvera. 2000. The FYVE domain of early endosome antigen 1 is required for both phosphatidylinositol 3-phosphate and Rab5 binding. Critical role of this dual interaction for endosomal localization. J. Biol. Chem. 275:3699–3705. [DOI] [PubMed] [Google Scholar]
- Llopis, J., J.M. McCaffery, A. Miyawaki, M.G. Farquhar, and R.Y. Tsien. 1998. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA. 95:6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLauchlan, H., J. Newell, N. Morrice, A. Osborne, M. West, and E. Smythe. 1998. A novel role for Rab5-GDI in ligand sequestration into clathrin-coated pits. Curr. Biol. 8:34–45. [DOI] [PubMed] [Google Scholar]
- Mullock, B.M., N.A. Bright, C.W. Fearon, S.R. Gray, and J.P. Luzio. 1998. Fusion of lysosomes with late endosomes produces a hybrid organelle of intermediate density and is NSF dependent. J. Cell Biol. 140:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, J.T., C. Panaretou, H. Stenmark, M. Miaczynska, and J.M. Backer. 2002. Role of Rab5 in the recruitment of hVps34/p150 to the early endosome. Traffic. 3:416–427. [DOI] [PubMed] [Google Scholar]
- Nielsen, E., F. Severin, J.M. Backer, A.A. Hyman, and M. Zerial. 1999. Rab5 regulates motility of early endosomes on microtubules. Nat. Cell Biol. 1:376–382. [DOI] [PubMed] [Google Scholar]
- Oh, Y.K., and J.A. Swanson. 1996. Different fates of phagocytosed particles after delivery into macrophage lysosomes. J. Cell Biol. 132:585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, Y.K., C.V. Harding, and J.A. Swanson. 1997. The efficiency of antigen delivery from macrophage phagosomes into cytoplasm for MHC class I-restricted antigen presentation. Vaccine. 15:511–518. [DOI] [PubMed] [Google Scholar]
- Pattni, K., M. Jepson, H. Stenmark, and G. Banting. 2001. A PtdIns(3)P-specific probe cycles on and off host cell membranes during Salmonella invasion of mammalian cells. Curr. Biol. 11:1636–1642. [DOI] [PubMed] [Google Scholar]
- Roberts, R.L., M.A. Barbieri, J. Ullrich, and P.D. Stahl. 2000. Dynamics of rab5 activation in endocytosis and phagocytosis. J. Leukoc. Biol. 68:627–632. [PubMed] [Google Scholar]
- Rock, K.L., C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, D. Hwang, and A.L. Goldberg. 1994. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 78:761–771. [DOI] [PubMed] [Google Scholar]
- Roderick, H.L., A.K. Campbell, and D.H. Llewellyn. 1997. Nuclear localisation of calreticulin in vivo is enhanced by its interaction with glucocorticoid receptors. FEBS Lett. 405:181–185. [DOI] [PubMed] [Google Scholar]
- Sato, T.K., M. Overduin, and S.D. Emr. 2001. Location, location, location: membrane targeting directed by PX domains. Science. 294:1881–1885. [DOI] [PubMed] [Google Scholar]
- Scidmore, M.A., E.R. Fischer, and T. Hackstadt. 2003. Restricted fusion of Chlamydia trachomatis vesicles with endocytic compartments during the initial stages of infection. Infect. Immun. 71:973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.M. Gaullier, A. Brech, J. Callaghan, B.H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Sonnichsen, B., S. De Renzis, E. Nielsen, J. Rietdorf, and M. Zerial. 2000. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark, H., and R. Aasland. 1999. FYVE-finger proteins–effectors of an inositol lipid. J. Cell Sci. 112:4175–4183. [DOI] [PubMed] [Google Scholar]
- Stenmark, H., R. Aasland, and P.C. Driscoll. 2002. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett. 513:77–84. [DOI] [PubMed] [Google Scholar]
- Swanson, J.A. 2002. Ratiometric fluorescence microscopy. Methods in Microbiology. Vol. 31. P. Sansonetti and A. Zychlinsky, editors. Academic Press Inc., London. 1–18.
- Swanson, J.A., M.T. Johnson, K. Beningo, P. Post, M. Mooseker, and N. Araki. 1999. A contractile activity that closes phagosomes in macrophages. J. Cell Sci. 112:307–316. [DOI] [PubMed] [Google Scholar]
- Vitelli, R., M. Santillo, D. Lattero, M. Chiariello, M. Bifulco, C.B. Bruni, and C. Bucci. 1997. Role of the small GTPase Rab7 in the late endocytic pathway. J. Biol. Chem. 272:4391–4397. [DOI] [PubMed] [Google Scholar]
- Zhan, Y., J.V. Virbasius, X. Song, D.P. Pomerleau, and G.W. Zhou. 2002. The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J. Biol. Chem. 277:4512–4518. [DOI] [PubMed] [Google Scholar]