Myostatin negatively regulates satellite cell activation and self-renewal (original) (raw)
Abstract
Satellite cells are quiescent muscle stem cells that promote postnatal muscle growth and repair. Here we show that myostatin, a TGF-β member, signals satellite cell quiescence and also negatively regulates satellite cell self-renewal. BrdU labeling in vivo revealed that, among the Myostatin-deficient satellite cells, higher numbers of satellite cells are activated as compared with wild type. In contrast, addition of Myostatin to myofiber explant cultures inhibits satellite cell activation. Cell cycle analysis confirms that Myostatin up-regulated p21, a Cdk inhibitor, and decreased the levels and activity of Cdk2 protein in satellite cells. Hence, Myostatin negatively regulates the G1 to S progression and thus maintains the quiescent status of satellite cells. Immunohistochemical analysis with CD34 antibodies indicates that there is an increased number of satellite cells per unit length of freshly isolated Mstn−/− muscle fibers. Determination of proliferation rate suggests that this elevation in satellite cell number could be due to increased self-renewal and delayed expression of the differentiation gene (myogenin) in Mstn−/− adult myoblasts. Taken together, these results suggest that Myostatin is a potent negative regulator of satellite cell activation and thus signals the quiescence of satellite cells.
Keywords: myostatin; GDF-8; satellite cell; quiescence; MyoD
Introduction
Muscle satellite cells are a distinct lineage of myogenic progenitors responsible for postnatal muscle growth. Satellite cells are mitotically quiescent and are known to express several myogenic markers. When stimulated by damage to the muscle, satellite cells are activated to reenter the cell cycle and express myogenic regulatory factors. The resulting myoblasts subsequently differentiate and fuse to form new replacement myofibers (Bischoff, 1989). The total number of quiescent satellite cells remains constant over repeated cycles of degeneration and regeneration, suggesting that the steady-state satellite cell population is maintained by self-renewal (Yoshida et al., 1998; Schmalbruch and Lewis, 2000; Heslop et al., 2001).
Several different growth factors, including HGFs (Maina et al., 1996) and FGFs (Flanagan-Steet et al., 2000), have been shown to affect the postnatal muscle growth by regulating satellite cell activation. Nowhere is the influence of growth factors on skeletal muscle development more dramatically demonstrated than by the remarkable widespread skeletal muscle mass of _myostatin_-deficient animals (Kambadur et al., 1997; McPherron and Lee, 1997; McPherron et al., 1997). Myostatin is a secreted growth and differentiation factor (GDF-8) that belongs to the TGF-β superfamily and shares several features with other members. The _myostatin_-null mice and cattle with mutation in myostatin showed increased muscling due to generalized hyperplasia and, to a lesser extent, hypertrophy of muscle. The phenotype of myostatin mutant cattle and mice has clearly established that Myostatin negatively regulates muscle growth. Consistent with this, work from our laboratory as well as other published results suggest that Myostatin inhibits myoblast proliferation (Thomas et al., 2000; Zhu et al., 2000; Taylor et al., 2001). Investigations into mechanisms revealed that one of the targets of Myostatin signaling is the cell cycle. In cell culture studies, Myostatin inhibited the progression of myoblasts at G1 and G2 phases of the cell cycle. This cell cycle arrest was shown to be mediated through p21, which was the only Cdk inhibitor to be up-regulated by Myostatin. In addition, Myostatin treatment also specifically reduced levels of Cdk2, the combination of which resulted in markedly reduced activity of Cdk2. This, in turn, led to the hypophosphorylation of retinoblastoma protein and thus inhibition of progression into S phase of the cell cycle (Thomas et al., 2000).
Several studies have implicated a role for Myostatin postnatally in muscle misuse and wasting. Serum concentrations of Myostatin have been reported to be increased in HIV-infected men with weight loss, suggesting that increased Myostatin levels may contribute to the pathophysiology of muscle wasting during HIV infection (Gonzalez-Cadavid et al., 1998). Similarly, a study by Wehling et al. (2000) and Carlson et al. (1999) indicated that atrophy-related muscle loss due to hind limb suspension in mice was associated with increased Myostatin levels in m. plantaris. One of the explanations for the elevated levels of Myostatin in hind limb unloading is that Myostatin may function as an inhibitor of satellite cell proliferation. Indeed, this is supported by the fact that _myostatin_-null mice display muscle hypertrophy and increased postnatal muscle growth, both of which have been related to increased satellite cell activity (Grounds and Yablonka-Reuveni, 1993; Mesires and Doumit, 2002). This circumstantial evidence led us to speculate that Myostatin may have a role in postnatal muscle growth by controlling the satellite cell quiescence. Here we report that Myostatin is expressed in satellite cells and that lack of myostatin in mice leads to increased number of satellite cells per unit length of muscle fiber. We also show that in _myostatin_-null mice, a higher percentage of the satellite cells are in an activated state as compared with wild-type mice. Therefore, the observed hypertrophy phenotype and increased postnatal muscle growth in _myostatin_-null mice could be due to increased number and activation of satellite cells.
Results
Myostatin is expressed in satellite cells and in the adult myoblasts
To assess if Myostatin is present in satellite cells, we performed immunocytochemistry and in situ hybridizations on serial cross sections of m. tibialis anterior. Satellite cells on fibers were identified by positive immunostaining with anti-Pax7 antibodies (Fig. 1 B). In the serial sections, the Pax7-positive satellite cells were also positive for Myostatin immunostaining (Fig. 1 A). Similarly, in situ hybridization studies with myostatin and Pax7 digoxigenin-labeled probes revealed that Pax7-positive satellite cells (Fig. 1 D) also express abundant levels of myostatin mRNA (Fig. 1 C). Expression of myostatin mRNA is not detected in the muscle sections derived from myostatin knockout mice, confirming the specificity of the myostatin probe (Fig. 1 E). Therefore, it is convincing that the satellite cells attached to the muscle fibers express Myostatin. To further prove that myostatin is present in satellite cells, RT-PCR and Western blot analysis were performed on the total RNA and protein extracts, respectively, of quiescent satellite cells isolated by Percoll gradient. The PCR primers designed to specifically amplify the processed portion of myostatin do indeed amplify the expected 515-bp product from the cDNA pool derived from satellite cell RNA (Fig. 1 F, i). Furthermore, anti-Myostatin antibodies specifically recognized the full-length Myostatin in the protein extracts, confirming that the satellite cells do express Myostatin (Fig. 1 F, ii). To demonstrate that the cells isolated by Percoll gradient techniques are indeed satellite cells, independent PCR reactions were performed with Pax7 and CD34 primers (satellite cell–specific markers). The results show that RT reactions indeed contained Pax7 and CD34 splice variant cDNAs (Fig. 1 F, i).
Figure 1.
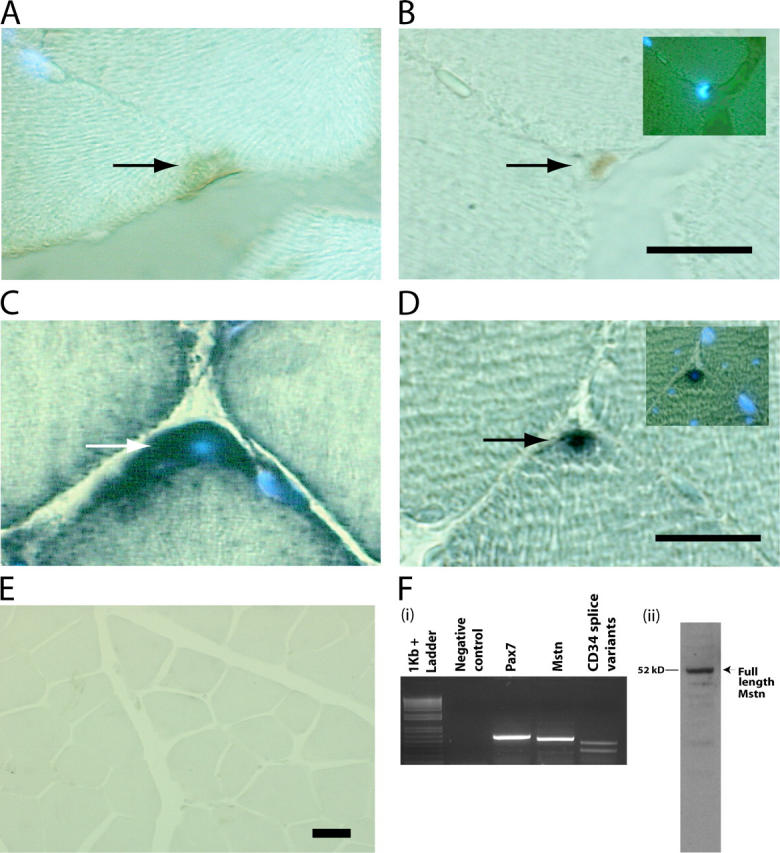
Myostatin is expressed in satellite cells. M. tibialis anterior was serially sectioned and immunostained with antibodies specific for (A) Myostatin and (B) Pax7. The myonuclei were stained with DAPI. The same muscle was used for in situ and probed for Myostatin (C) and Pax7 transcripts (D). Arrows indicate the stained satellite cells, and DAPI-stained myonuclei are also shown in the insets (B and D). (E) Micrograph showing in situ hybridization performed with myostatin antisense probe on muscle sections of _myostatin_-null mouse. (F, i) Agarose gel electrophoresis of RT-PCR products derived from satellite cell total RNA. Primers specific for 3′ region of myostatin amplify the expected 515-bp product in a combined RT-PCR reaction (Mstn). Amplicons are not detected in the absence of template (Negative control). Pax7 was amplified with primers designed to produce a 571-bp product (Pax7), and CD34 splice variants were also PCR amplified from the same RT reaction (CD34 splice variants). 1-kb plus DNA ladder is shown. (F, ii) Western blot showing the presence of the full-length 52-kD Myostatin protein in satellite cell protein extract. Bars, 10 μm.
To gain insight into the role of Myostatin in satellite cell activation and proliferation, low passage number primary cultures were isolated from hind limb muscles of 8-wk-old _myostatin_-null and wild-type mice to generate highly purified satellite cell–derived cultures and preclude the inclusion of neonatal myoblasts (see Materials and methods). Previous work has demonstrated that proliferating primary adult myoblasts derived from satellite cells express myoblast markers such as MyoD, Desmin, and adhesion molecules, but not terminal differentiation markers such as Myogenin or Myosin heavy chain. Therefore, to characterize the cultures used in this experiment, we immunostained the adult myoblast cultures with MyoD- and M-cadherin–specific antibodies. Over 95% of proliferating primary cells derived from the hind limb muscles of wild-type and myostatin knockout mice expressed M-cadherin and MyoD, indicating that the isolation procedure generated highly purified cultures of myogenic cells from both wild-type and myostatin knockout mice (Fig. 2, A and B). Proliferating myogenic precursor cells in vivo, and myoblasts in vitro, express the intermediate filament Desmin, whereas, satellite cells do not express Desmin (George-Weinstein et al., 1993). Hence, primary cultures were immunostained with antibodies reactive with Desmin to assess their developmental status. As shown in Fig. 2 C, uniform expression of Desmin is detected, indicating that the primary cultures are all activated myogenic precursor cells. It has been recently demonstrated that expression of CD34 protein is associated with skeletal muscle precursors but not with the differentiated fibers. To rule out any myogenic differentiation during the isolation procedure of primary cultures, the cells were immunostained with anti-CD34 antibodies. As shown in Fig. 2 D, almost all of the cultured cells were immunopositive for CD34, indicating that under the culturing conditions, the myogenic precursor cells are in active proliferation status (Beauchamp et al., 2000). The negative control did not show any nonspecific background (Fig. 2 E).
Figure 2.
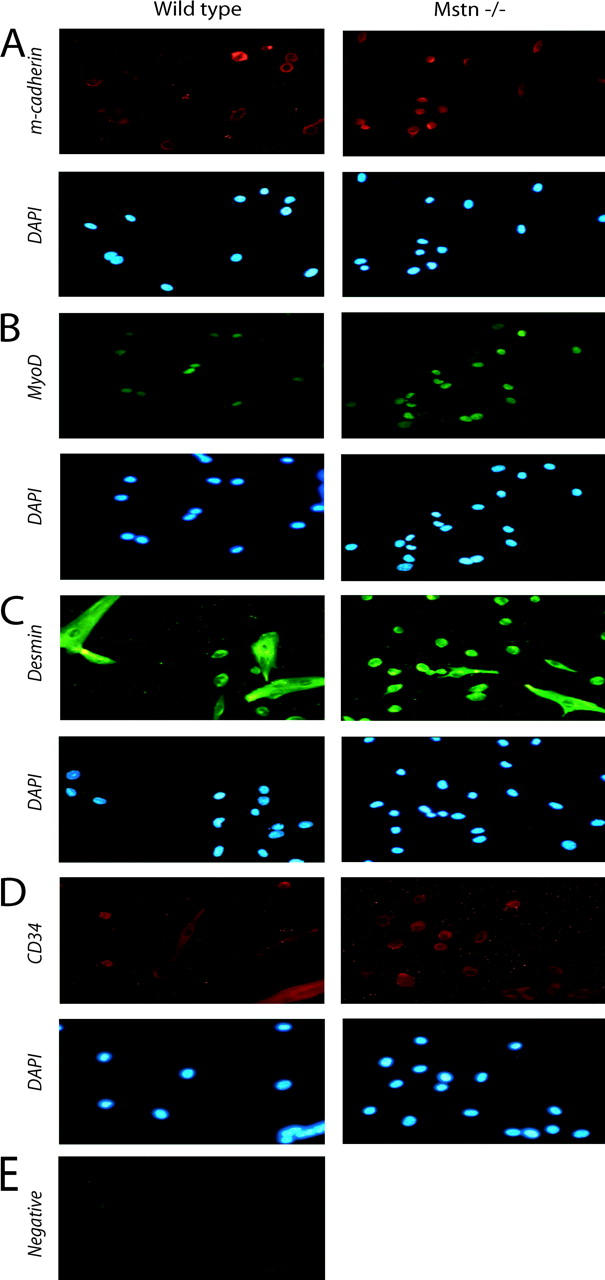
Immunocytochemistry of isolated satellite cells. Adult myoblasts were cultured from the hind leg muscles of either wild-type or Mstn−/−mice, fixed, and immunostained for M-cadherin (A), MyoD (B), Desmin (C), and CD34 (D). DAPI staining of the nuclei in the corresponding fields is also shown; panel E shows the background immunofluorescence when anti–mouse secondary antibody was used in the absence of primary antibody. Similar background was observed for all other secondary antibody negative controls. Greater than 95% of the isolated cultured cells were myogenic.
Three independent techniques, RT-PCR, Western blotting, and immunocytochemistry, were used to ascertain the expression of myostatin in adult myoblasts. As shown in Fig. 3 A, the PCR reaction amplified the expected size fragment of 1.2-kb myostatin ORF. In addition, anti-Myostatin antibodies specifically recognized Myostatin on the Western blot performed on the protein extracts derived from the adult myoblasts (Fig. 3 B). To further confirm the expression of Myostatin, primary cultures were immunostained with Myostatin antibodies. As shown in Fig. 3 C (i and ii), virtually all the cultured myoblasts showed staining with Myostatin antibodies. Thus, it is clear from these results that Myostatin is expressed in satellite cells and in the adult myoblasts derived from satellite cells.
Figure 3.
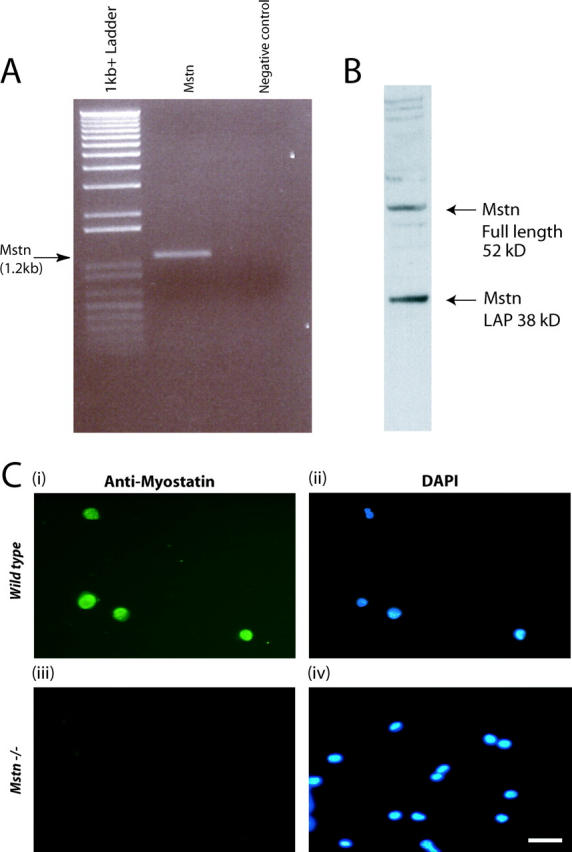
Myostatin is expressed in adult myoblasts derived from satellite cells. (A) Agarose gel electrophoresis of myostatin ORF amplified in a combined RT-PCR reaction using total RNA from adult myoblasts (lane 2). Amplicons are not detected in the absence of template (negative control) (lane 3). 1-kb plus DNA ladder is shown in lane 1. (B) Western analysis from proteins isolated from adult myoblasts cultured for 48 h showing both the full-length (52 kD) and LAP (38 kD) peptides of Myostatin. (C) Representative immunofluorescence showing the presence of Myostatin in wild-type adult myoblasts (i) and absence in _myostatin_-null (Mstn−/−) myoblasts (iii). DAPI staining of the nuclei is shown in the corresponding fields (ii and iv).
Increased activation of satellite cells in the absence of Myostatin in vivo
Using single-cell PCR analysis, Cornelison and Wold (1997) have previously speculated that Myostatin is an important regulator of the quiescent state of satellite cells. Thus, to determine the involvement of Myostatin in satellite cell quiescence, the cell cycle phase of the satellite cells attached to myofibers in vivo in wild-type and myostatin knockout mice was assessed. Both wild-type and myostatin knockout mice were pulsed with BrdU, and satellite cells from m. tibialis anterior muscle were isolated by Percoll gradient. Characterization of satellite cells with M-cadherin and CD34 immunostaining revealed that the cultures were >95% pure and that there is no difference in the yield or purity between wild-type and myostatin knockout mice (unpublished data). BrdU-positive satellite cells were identified by immunostaining. As shown in Fig. 4 A (i), ∼18% of satellite cells isolated from 4-wk-old wild-type mice were labeled with BrdU. However, in myostatin knockout mice, a significantly higher number (33%) of satellite cells was activated to enter S phase. In 8-wk-old wild-type muscle, 8% of the satellite cells isolated were BrdU positive, whereas in the _myostatin_-null muscle, 15% of the satellite cells were BrdU positive. When the same experiment was repeated on older mice (6 mo old), only 3.2% of the wild-type satellite cells had incorporated BrdU. A threefold higher number, ∼10% of Myostatin-deficient satellite cells, had incorporated BrdU (Fig. 4 A i), indicating that irrespective of the age, lack of functional Myostatin results in an increased number of activated satellite cells in uninjured muscle. To investigate the role of Myostatin in inhibiting satellite cell activation ex vivo, recombinant Myostatin was added to single isolated fibers in varying concentrations, and satellite cells were left to detach from the fiber and migrate to the well. Fig. 4 A (ii) graphically demonstrates that on average, 80% of the wells had identifiable satellite cells, which were subsequently differentiated in low serum to show they were myogenic. In comparison, in media containing 1 μg/ml of Myostatin, only 37% of fibers had migrated myogenic precursor cells in the well, while media with increased amounts of Myostatin, 2 and 5 μg/ml, had 12 and 0% migrated myogenic cells, respectively. These results confirm that a lack of myostatin expression leads to increased activation of satellite cells in vivo, and overexpression of myostatin inhibits activation.
Figure 4.
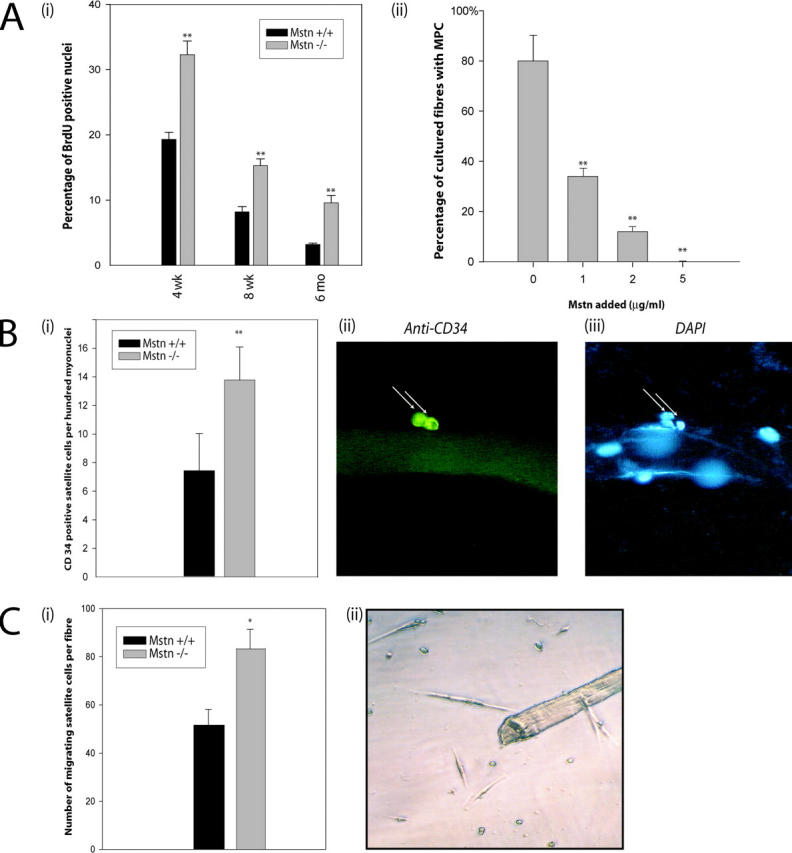
Lack of Myostatin increases the activation of satellite cells on myofibers. (A, i) In vivo quantification of activated satellite cells in the muscle of wild-type and _myostatin_-null mice. Activated satellite cells were labeled with BrdU in wild-type or _myostatin_-null (Mstn −/−) mice of 4 wk, 8 wk, or 6 mo and were isolated using Percoll gradient. 5,000–10,000 satellite cells were immunostained for BrdU, and percentages of nuclei that were positive for BrdU labeling are shown. **, P < 0.01 (as compared with wild type). At least a total of 1,000 cells were counted in each of three replicates. The data provided are an average of three animals each. (A, ii) Myostatin inhibits the migration of satellite cells from fibers. Single muscle fibers (n = 32) were isolated from the muscle and incubated in media conducive to the migration of myogenic precursor cells. The addition of Myostatin in increasing concentrations decreases the percentage of fibers with migrated satellite cells (**, P < 0.01). (B, i) The number of satellite cells (CD34 positive) per 100 myonuclei in wild-type and _myostatin_-null (Mstn−/−) myofibers is shown. **, P < 0.01 (as compared with wild type). More than 1,000 nuclei were counted in each of three replicates. The data presented are an average of three animals each. (B, ii) Micrograph showing typical CD34-immunostained satellite cell and (iii) myonuclei were visualized by counterstaining with DAPI. (C, i) An increased number of satellite cells migrate from _myostatin_-null fibers. Quantitative analysis demonstrates that an increased number of myogenic progenitor cells migrates from single myofibers isolated from _myostatin_-null mice as compared with wild-type myofibers (*, P < 0.05). (C, ii) An example of satellite cells surrounding an isolated myofiber after 72 h.
Steady-state satellite cell numbers are increased on Myostatin-deficient muscle fibers
A constant pool of satellite cells is maintained on muscle fibers during repeated cycles of degeneration and regeneration. A portion of myoblasts derived from the activated satellite cells returns to quiescent status, thus maintaining a constant number of satellite cells per unit length of muscle fiber by the self-renewal process. Adult myoblasts derived from the _myostatin_-null mice tend to proliferate faster (see the results below), suggesting the possibility of increased self-renewal of the satellite cells in these knockout mice. Thus, the steady-state number of satellite cells per fiber of the muscle fibers isolated from both wild-type and _myostatin_-null mice was measured. As shown in Fig. 4 B (i), there is a significantly higher number of steady-state satellite cells per unit of muscle fiber in myostatin knockout mice as compared with the wild-type mice. An example of CD34-positive satellite cells on the muscle fiber is shown in Fig. 4 B (ii).
To further demonstrate this increase in satellite cell number per fiber, myofiber explant culture (Fig. 4 C, ii) was used (Hawke et al., 2003). Results showed that a significantly higher number of myogenic precursor cells had migrated from the _myostatin_-null fiber (83 ± 8; n = 31) as compared with the wild type (51 ± 6; n = 31). The difference in total number of satellite cells between _myostatin_-null and wild-type fibers was significant (P < 0.05) (Fig. 4 C, i).
Primary and secondary myoblasts that lack Myostatin proliferate faster
Previously, we have shown that high concentrations of Myostatin inhibit the proliferation of myoblasts by controlling the G1 to S transition during cell cycle (Thomas et al., 2000). Thus, here, we determined the proliferation rate and the doubling time of adult myoblasts isolated from both wild-type and myostatin knockout mice. For this purpose, we plated secondary myoblasts cultured from embryonic day (E) 17 and adult myoblasts from postnatal muscle and determined their proliferation. Primary myoblasts isolated from wild-type and myostatin knockout mice underwent a lag period of 3 d before they became proliferative (unpublished data). Cultured secondary myoblasts, isolated from E17 embryos from myostatin knockout mice, proliferated significantly faster, resulting in an increase in the number of Mstn−/− myoblasts as compared with the wild-type myoblasts (Fig. 5 A). The increased proliferation rate of Mstn−/− myoblasts is also reflected by three times higher cumulative population doublings attained by the _myostatin_-null myoblasts, compared with the wild-type myoblasts (unpublished data). In addition, the proliferation rate of adult myoblasts derived from hind limbs of 4-wk-old _myostatin_-null and wild-type mice were also assayed using the methylene blue cell proliferation assay. As seen in Fig. 5 B, _myostatin_-null myoblasts proliferate significantly faster than wild-type myoblasts after 3 d in culture. The proliferation experiments described above were performed on myoblasts derived from satellite cells that were isolated from mixed muscles that contain both fast and slow twitch fibers. A recent report, however, suggests that the satellite cells isolated from fast and slow fibers are inherently different (Rosenblatt et al., 1996). Thus, to address the role of Myostatin in the proliferation of myoblasts from fast or slow fibers, we cultured adult myoblasts from m. tibialis anterior (fast twitch) or m. soleus (slow twitch) of both normal and myostatin knockout mice. The proliferation assay again indicates that both fast (Fig. 5 C) and slow (Fig. 5 D) fiber–specific adult myoblasts isolated from myostatin knockout mice proliferate much faster as compared with the wild-type adult myoblasts. Thus, regardless of the origin of satellite cells, lack of myostatin appears to increase the proliferation rate of both fast and slow fiber–specific satellite cells.
Figure 5.

_myostatin_-null adult myoblasts proliferate faster than the wild-type myoblasts. Adult myoblasts were isolated from hind limbs of C57BL/10 (Mstn+/+) or _myostatin_-null mice (Mstn−/−) and seeded at a low number. Proliferation rate determined for the myoblasts isolated from E17 (A) and 4-wk-old muscle (B). In addition, the proliferation rate determined for m. tibialis anterior (C) and m. soleus (D) muscle fiber–specific adult myoblasts is also shown. Experiments were done in triplicate. Data shown are an average of three animals. *, P < 0.05; **, P < 0.01 (as compared with wild type). To determine the direct effect of Myostatin on satellite cell proliferation, adult myoblasts were isolated from wild-type and _myostatin_-null mice. Exogenous Myostatin was added to the proliferating _myostatin_-null myoblast cultures. As the concentration increased, the enhanced proliferative potential of the _myostatin_-null myoblast decreased to that of the wild type (E).
Addition of exogenous Myostatin inhibits satellite cell activation and myoblast proliferation
To show the direct inhibitory effects of Myostatin on myoblast proliferation, tissue-dissociated satellite cells were isolated from both wild-type and _myostatin_-null mice and used in a methylene blue assay. The same number of wild-type and _myostatin_-null myoblasts were cultured in the media, and myostatin was added in increasing concentrations to only _myostatin-_null cultures. After 48 h of proliferation, the cells were fixed with 10% formal saline and stained with the methylene blue. When recombinant Myostatin was added to the media in increasing amounts, the enhanced proliferation rate seen in the _myostatin_-null myoblasts was reduced to that of the wild-type myoblasts (Fig. 5 E).
Cell cycle progression of wild-type and Myostatin-deficient satellite cells
Proliferation assays clearly demonstrate that adult myoblasts that lack Myostatin proliferate much faster than the wild-type myoblasts. Furthermore, our previous results with C2C12 cells indicate that addition of exogenous Myostatin to the growth medium (GM) arrests the proliferation of C2C12 cells in G1 phase of the cell cycle (Thomas et al., 2000). Thus, we next asked if the increased proliferation observed in the myostatin knockout myoblasts is due to deregulated G1 to S phase transition. FACS® analysis of the cell cycle distribution of asynchronous populations of wild-type and myostatin knockout adult myoblasts in proliferation media revealed that myostatin knockout myoblasts had a twofold increase in S phase cell population (Fig. 6 A). To confirm these results, we then synchronized the adult myoblasts in G0 phase by growing them in methionine-free media and initiated S phase entry by serum stimulation. To monitor the number of cells in S phase, the cells were pulsed with BrdU, and the percent of labeled cells was determined by immunostaining the fixed cells at the end of the assay. The results indicate that within the first hour after serum stimulation, 7% of Mstn−/− myoblasts entered S phase. In contrast, only 2% of wild-type myoblasts were found to be in S phase. Similarly, an enhanced number of BrdU-positive cells was observed up to 8 h after serum stimulation in Mstn−/− myoblasts (Fig. 6 B). These results indicate that lack of Myostatin increases the proliferation of myoblasts by deregulating the S phase entry.
Figure 6.
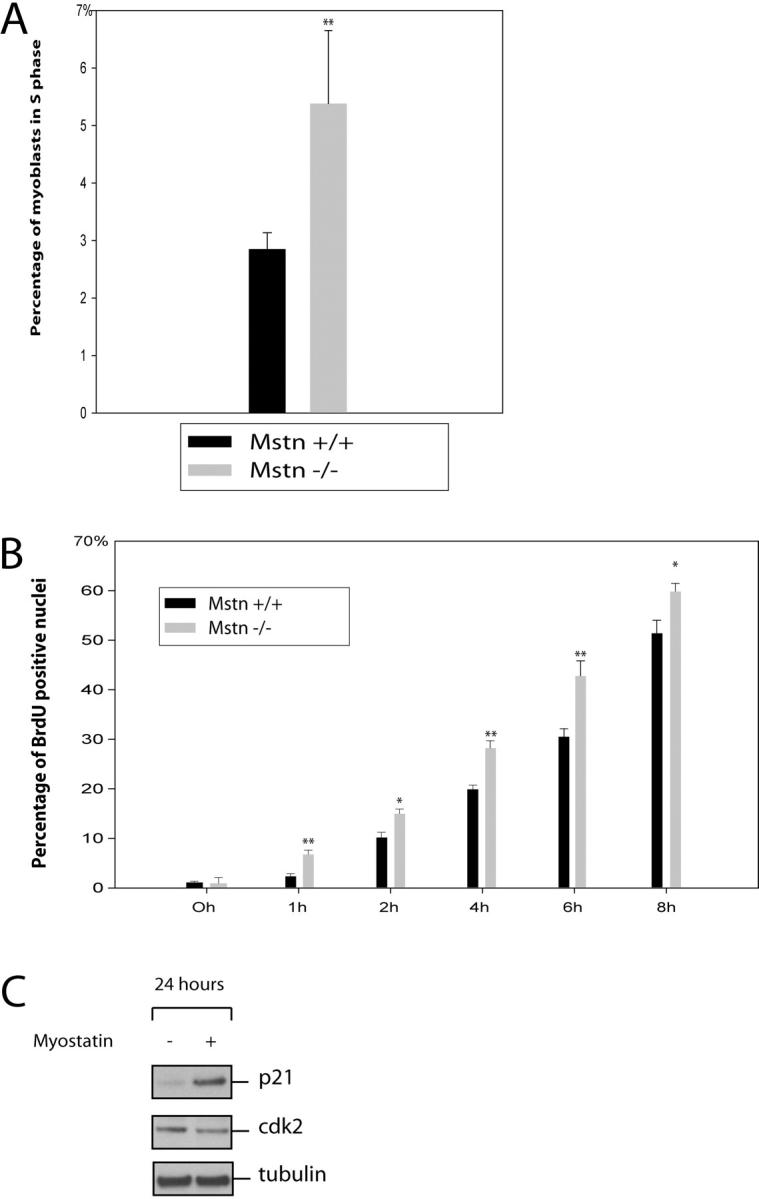
Lack of Myostatin results in deregulated S phase entry of adult myoblasts. (A) Freshly isolated adult myoblasts were stained with propidium iodide and analyzed on a flow cytometer. 10,000 cells of each genotype were analyzed on FACS® and were distributed into the phases of the cell cycle based on the DNA content. Percentage of _myostatin_-null (Mstn−/−) and wild-type (Mstn+/+) myoblasts in S phase are shown in panel A. (B) Isolated myoblasts were synchronized to G1 phase, and once released into S phase, they were BrdU labeled and the percentage of BrdU-positive cells was counted at various time points. _myostatin_-null cells progress into the S phase more rapidly as compared with wild-type myoblasts. **, P < 0.01. At least a total of 1,000 cells were counted in each of three replicates. Data presented are an average of three animals. (C) Western blot showing the levels of p21 and Cdk2 protein in primary myoblasts cultured with (+) or without (−) Myostatin for 24 h. p21 protein was detected using anti-p21 antibodies, and Cdk2 protein was detected using Cdk2 antibody. Tubulin protein levels are included to show equal loading.
Given that Myostatin inhibits the G1 to S phase progression of myoblasts, we speculated that Myostatin may affect one or more of the cell cycle control proteins. Of particular interest was the Cdk inhibitor p21, which has previously been shown to be involved in cell cycle arrest in the G1 phase. In addition, growth arrest by Myostatin signaling has been shown to influence p21 expression (Thomas et al., 2000; Joulia et al., 2003). To examine this possibility, actively growing adult myoblasts derived from satellite cells were cultured with or without Myostatin in the media for 24 h before harvesting. Western analysis with p21-specific antibodies revealed that Myostatin treatment up-regulated p21 expression (Fig. 6 C). To determine if the elevated p21 levels seen in Myostatin-treated myoblasts coincided with constant or down-regulated Cdk2, and therefore a change in the ratio of p21 to Cdk2, we performed Western blot analysis for Cdk2. Fig. 6 C shows that the level of Cdk2 was down-regulated in protein extracts from myoblasts treated with Myostatin.
Delayed differentiation potential of Myostatin-deficient myoblasts
Myogenic differentiation of myoblasts occurs upon withdrawal of actively growing myoblasts from cell cycle into G0 and subsequent expression of myogenic markers. As the adult myoblasts derived from the Mstn−/− muscle fibers have deregulated S phase entry, we next evaluated the ability of Mstn−/− myoblasts to withdraw from the cell cycle upon mitogen withdrawal. Adult myoblasts derived from both wild-type and myostatin knockout mice were switched to differentiation low serum medium and pulsed with BrdU to identify the total number of myoblasts that are still cycling through the cell cycle in differentiation medium (DM). As shown in Fig. 7 A, nearly 100% of cells incorporated BrdU when the myoblasts were in GM. The majority of wild-type myoblasts (67%) stopped proliferating and withdrew from the cell cycle at 6 h after switching to DM. However, at this time point, 50% of Mstn−/− myoblasts were still in S phase, indicating that these cells were still cycling through the S phase and thus had not withdrawn to the G0 stage to differentiate. Even at the 12-h time point, significantly more Mstn−/− myoblasts were in S phase as compared with wild-type myoblasts (Fig. 7 A). Although delayed, eventually, the majority of Mstn−/− myoblasts do withdraw from the cell cycle and differentiate within 48 h.
Figure 7.

Delayed cell cycle withdrawal and differentiation of _myostatin_-null adult myoblasts. Isolated myoblasts from C57BL/10 (Mstn+/+) or _myostatin_-null mice (Mstn−/−) were switched to DM, and the cells were pulsed with BrdU before fixing at 0-, 6-, 12-, 24-, or 48-h time points. Percentage of cells that incorporate BrdU are shown for every time point in panel A. *, P < 0.05; **, P < 0.01 (as compared with wild type). Over 1,000 cells were counted in each of three replicates. The data presented are an average of three animals (A). Freshly isolated myoblasts from C57BL/10 (WT) or _myostatin_-null mice (KO) were switched to DM, and proteins were extracted at indicated time points. Time 0 indicates freshly isolated quiescent satellite cells. Western analysis (n = 3) with MyoD (B) and Myogenin (C) antibodies on protein extracts from differentiating myoblasts is shown. Maximum expression was termed 100%, and relative expression at various time points was plotted. Anti–α-tubulin Western indicates equal loading.
The myoblast cell cycle withdrawal process is temporally regulated by the up-regulation of several cell cycle–specific genes and myogenic differentiation markers. To establish the molecular basis for this delayed differentiation of Mstn−/− cells, we performed expression analysis of differentiation-specific genes such as MyoD and Myogenin. For this purpose, adult myoblasts isolated from wild-type and myostatin knockout mice were switched to DM, and protein extracts were made from differentiating myoblasts at 0 (majority quiescent), 6, 12, 24, 48, and 72 h after switching to the low serum media. The levels of differentiation-specific genes were quantified by Western and densitometric analysis. As expected, MyoD protein levels peak immediately after switching to the DM (within 6 h) in both wild-type and myostatin knockout myoblasts. However, in the _myostatin_-null myoblasts, the peak levels of MyoD continue until 12 h and then slowly decline to basal levels at ∼48 h after switching to DM (Fig. 7 B). Whereas in the wild-type myoblasts, a sharp decline in the levels of MyoD was observed at 12 h of differentiation. Similar to MyoD, the levels of the early differentiation marker Myogenin peak within 6 h into differentiation in wild-type myoblasts. However, in the Myostatin-deficient myoblasts, the peak expression of Myogenin is delayed and is seen at 24 h into differentiation (Fig. 7 C).
Discussion
Investigations into the molecular basis of Myostatin function revealed that Myostatin functions by controlling the myoblast number during embryonic myogenesis (Thomas et al., 2000). Several lines of evidence, including the expression of Myostatin in postnatal muscle (Sharma et al., 1999), increased postnatal muscle growth in _myostatin_-null mice (McPherron et al., 1997), and expression of Myostatin in nascent myotubes after muscle regeneration (Kirk et al., 2000), compelled us to evaluate the role of Myostatin in adult myogenesis. The results presented here clearly demonstrate that Myostatin is expressed in satellite cells and that Myostatin regulates satellite cell quiescence and self-renewal.
Before delineating the role of Myostatin in adult myogenesis, we attempted to demonstrate the expression of myostatin mRNA and protein specifically in satellite cells, which are the precursors of adult muscle. Immunocytochemistry with anti-Myostatin antibodies on muscle transverse sections revealed intense Myostatin immunoreactivity in cells that are juxtaposed to the surface of muscle fibers. Further, immunostaining with antibodies against Pax7 (satellite cell marker) on serial sections established that the cells positive for Myostatin immunoreactivity are indeed the satellite cells (Seale et al., 2000) (Fig. 1, A and B). Myostatin expression was further confirmed in Pax7-positive satellite cells by in situ hybridization (Fig. 1, C and D). In addition, purified satellite cells appear to express myostatin mRNA and protein, confirming that Myostatin is present in satellite cells. These results thus are in agreement with an earlier report, where Cornelison et al. (2000) have shown the presence of myostatin mRNA in the total RNA isolated from the single satellite cell attached to the fiber. In the same study, they also showed that, on activation of satellite cells, the levels of Myostatin (GDF-8) were significantly down-regulated, suggesting that high levels of Myostatin might be involved in satellite cell quiescence.
Three independent techniques, RT-PCR, Western blotting, and immunocytochemistry, were used to ascertain the expression of myostatin in adult myoblasts. As shown in Fig. 3 A, the PCR reaction amplified the expected size fragment of 1.2-kb myostatin ORF. In addition, anti-Myostatin antibodies specifically recognized Myostatin on the Western blot performed on the protein extracts derived from the adult myoblasts (Fig. 3 B). To demonstrate the expression of myostatin in actively proliferating cells, primary cultures were immunostained with Myostatin antibodies. As shown in Fig. 3 C, virtually all the cultured myoblasts showed staining with Myostatin antibodies and did not detect Myostatin in myoblasts isolated from myostatin knockout mice. Thus, it is clear from these results that Myostatin is expressed in satellite cells and in the adult myoblasts derived from satellite cells.
If Myostatin negatively regulates the activation of satellite cells, we reasoned that in myostatin knockout mice there would be increased activation of satellite cells. Consistent with this hypothesis, a BrdU labeling experiment showed that in myostatin knockout animals, there is a significant increase in the pool of activated satellite cells as compared with wild-type mice. Although there is a significant reduction in the pool of activated satellite cells with age, _myostatin_-null muscle fibers exhibit considerably more activated satellite cells even at 6 mo (Fig. 4 A, i), which is consistent with the persistent postnatal muscle growth and lack of muscle wasting observed in _myostatin_-null mice (McPherron and Lee, 2002). Recently, the breeding of the _myostatin_-null mouse with the mdx mouse resulted in the amelioration of the symptoms (Wagner et al., 2002). Also, the injection of a specific antibody to Myostatin into the mdx mice alleviated the severity of the phenotype; in fact, the mice gained muscle mass and increased the production of the muscle protein creatine kinase, indicating improvement of muscle strength (Bogdanovich et al., 2002). Whittemore et al. (2003) also injected inhibitory Myostatin antibody into wild-type mice and demonstrated increased skeletal muscle mass and increased grip strength. These results further indicate that Myostatin acts postnatally as a negative regulator of satellite cell activation and skeletal muscle growth. In addition to the increased muscle growth through hyperplasia, _myostatin_-null mutant mice also display hypertrophy, which has been related to increased satellite cell activity. One explanation for the increased number of myonuclei, and thus hypertrophy of _myostatin_-null muscle fiber, is the observed spontaneous activation of satellite cells in the absence of Myostatin. Converse to this, the addition of Myostatin to isolated myofibers decreased the activation of satellite cells, when normally they would be induced to activate (Fig. 4 A, ii).
For activation, it is essential that the quiescent satellite cells either in G0 or G1 should reenter the S phase to undergo cell cycle progression. Hence, negative regulators of satellite cell activation, such as Myostatin, could possibly regulate satellite cell activation by controlling the G1 to S phase transition. Indeed, the results from this study (Fig. 6 C) suggest that high levels of Myostatin can inhibit G1 to S progression of the cell cycle by down-regulating Cdk2 and up-regulating p21, which would result in hypophosphorylation of pRB. Hence, it is quite possible that the higher levels of Myostatin observed in quiescent satellite cells (Cornelison et al., 2000) maintain the G0 status by regulating the G1/S check point. In the myostatin knockout mice, we also observed an increase in the number of CD34-positive satellite cells per unit length of fiber as compared with wild-type mice (Fig. 4 B, i). A recent report suggested that in order to meet the demand and maintain a constant pool of satellite cells on muscle fibers, satellite cells must self-renew like stem cells. The self-renewal process is accomplished by returning a pool of satellite cell–derived myoblasts to the quiescent status on muscle fibers (Seale and Rudnicki, 2000). An increased number of satellite cells per unit fiber thus means that in the absence of Myostatin, there is increased self-renewal of satellite cells, possibly by increased proliferation of satellite cell–derived myoblasts. This theory is supported by the observation that the adult myoblasts derived from the satellite cells of myostatin knockout mice appear to have a shorter doubling time and proliferate much faster as compared with the wild-type adult myoblasts (Fig. 5). This increased proliferation rate of Mstn−/− adult myoblasts could result in increased self-renewal and thus elevate the number of satellite cells per unit fiber. On the contrary, the addition of exogenous Myostatin to _myostatin_-null myoblasts decreases their proliferation rate to that of wild type (Fig. 5 E).
This increased proliferation of _myostatin_-null myoblasts can be attributed to a deregulated G1 to S transition. The use of FACS® and BrdU uptake demonstrated that this was indeed the case. Two to three times more _myostatin_-null myoblasts were in the S phase of the cell cycle at any one time (Fig. 6 A). Furthermore, after synchronizing _myostatin_-null or wild-type myoblasts, more _myostatin_-null cells entered into the S phase (Fig. 6 B).
Myoblast differentiation is a highly orchestrated step wherein the withdrawal of myoblasts from the cell cycle to G0 should occur before the committed myoblasts postmitotically differentiate into myotubes. Molecular analysis indicates that low mitogen medium triggers the up-regulation of MyoD, which in turn induces the expression of the p21 gene. Concomitant up-regulation of p21 and decrease of G1 cyclins and Cdks results in the withdrawal of myoblasts to G0 and terminal differentiation. Thus, the rate of withdrawal of myoblasts is an important step in the differentiation of myoblasts into myotubes. The studies with BrdU labeling clearly indicated that the wild-type myoblasts immediately withdraw from the cell cycle, whereas, the _myostatin_-null myoblasts appear to undergo the cell cycle longer, resulting in delayed withdrawal from the cell cycle (Fig. 7 A). The molecular basis for delayed withdrawal appears to be prolonged expression of MyoD and delayed peak expression of a differentiation-specific marker, such as Myogenin, in _myostatin_-null myoblasts (Fig. 7, B and C). These results certainly point out that Myostatin, by regulating MyoD expression, plays a key role in adult myoblast differentiation. Immunostaining with Myosin heavy chain antibody (unpublished data) and gene expression analysis revealed that loss of Myostatin results in delayed withdrawal from the cell cycle and differentiation of primary myoblasts derived from satellite cells. Consistent with this hypothesis, Rios et al. (2002) and Langley et al. (2002) very recently demonstrated that excess Myostatin in DM blocks the differentiation of C2C12 myoblasts by inhibiting MyoD expression. Eventually, however, the Myostatin-deficient myoblasts do differentiate, albeit delayed.
In conclusion, we propose that Myostatin blocks the activation of satellite cells and also negatively regulates self-renewal of satellite cells (Fig. 8). Increased activation of Mstn−/− satellite cells, and subsequent enhanced proliferation with delayed differentiation of _myostatin_-null myoblasts, could thus be the primary reason for the increased postnatal muscle growth and hypertrophy in _myostatin_-null animals. To our knowledge, this is the first report of a secreted growth factor that is expressed in satellite cells and maintains the satellite cell quiescence.
Figure 8.

A model for the role of Myostatin in postnatal muscle growth. Quiescent satellite cells on muscle fibers are activated in response to muscle injury to give rise to myoblasts. Proliferating myoblasts can either fuse with the existing fiber or differentiate into a nascent myotube. A portion of proliferating myoblasts, however, can revert to become quiescent satellite cells, thus resulting in self-renewal. As Myostatin is a negative regulator of cell cycle progression, high levels of Myostatin in satellite cells block the activation to maintain quiescence.
Materials and methods
Animals
_myostatin_-null mice (C57BL/10 background) were obtained from S.-J. Lee (The Johns Hopkins University, Baltimore, MD). Wild-type mouse strain C57BL/10 was bred at the Ruakura Small Animal Colony.
BrdU labeling of satellite cells in vivo
To determine the activated satellite cells in vivo at a given time point, BrdU labeling was used. Three C57BL/10 or Mstn−/− mice at ages 4 wk, 8 wk, or 6 mo were intraperitoneally injected with BrdU (30 mg/kg) as a single pulse 2 h before killing. Satellite cells from m. tibialis anterior were isolated using Percoll gradient as per the published protocol (Allen et al., 1997; Partridge, 1997; Yablonka-Reuveni et al., 1999). To detect activated satellite cells, 5,000–10,000 satellite cells were fixed using 0.25% glutaraldehyde and immunostained for BrdU using the cell proliferation kit (Amersham Biosciences). A minimum of 1,000 immunostained satellite cells were counted to calculate the percentage of BrdU-positive cells.
Determination of satellite cell number on muscle fiber
M. tibialis anterior was excised from six C57BL/10 or Mstn−/− mice, and individual muscle fibers were isolated by the method of Rosenblatt et al. (1995). Isolated fibers were fixed in an eight-well Lab-Tek chamber slide (Nunc) with 0.25% glutaraldehyde for 1 min and rinsed with PBS for immunocytochemistry. Satellite cell detection with CD34 antibodies (Santa Cruz Biotechnology, Inc.) and counting were performed according to the method of Beauchamp et al. (2000). The total number of migrated satellite cells was counted after 72 h incubation of the wild-type and _myostatin_-null tibialis anterior muscle fibers in fiber medium (FM) at 37°C, 5% CO2.
Effect of Myostatin on satellite cell migration
To determine the effect of Myostatin on the migration of satellite cells from the fiber, isolated muscle fibers were either placed in FM or in FM with titrating amounts of Myostatin for 96 h at 37°C, 5% CO2. Wells were analyzed for satellite cell migration, and satellite cells were counted. Replicates of at least 24 fibers were used for statistical analysis.
Cell cycle analysis of adult myoblasts
Adult myoblasts were cultured according to the published methods (Allen et al., 1997; Partridge, 1997; Yablonka-Reuveni et al., 1999). FACS® analysis of primary myoblasts was done according to the method of Thomas et al. (2000). To monitor the cell cycle progression, adult myoblasts were synchronized at the G1/S boundary and released to S phase by the method of Kitzmann et al. (1998). The myoblasts in S phase were pulsed with 0.1 mM BrdU for 15 min, fixed, and processed for immunocytochemistry. To determine the cell cycle withdrawal of adult myoblasts, proliferating adult myoblasts were switched to DM and pulsed with 0.1 mM BrdU at 0-, 6-, 12-, 24-, or 48-h time points. The myoblasts were fixed and immunostained for BrdU.
Myoblast proliferation assay
Primary myoblasts were cultured from hind limb muscles of three 4-wk-old mice or six to eight E17 mouse embryos according to the published protocols (Allen et al., 1997; Partridge, 1997; Yablonka-Reuveni et al., 1999). Embryonic primary myoblasts and satellite cells isolated from fast or slow fibers were seeded at low density of ∼100 cells/cm2 in GM, and cells were counted every 24 h. Adult myoblasts were cultured as described earlier and were seeded at a density of 6,097 cells/cm2 in GM on Matrigel-coated 96-well microtitre plates. Cell counting and methylene blue photometric endpoint assay were performed according to the published protocols (Oliver et al., 1989; Chakravarthy et al., 2000).
To demonstrate the direct effect of Myostatin on myoblast proliferation, myoblasts were isolated from _myostatin_-null and wild-type skeletal muscle. Isolated myoblasts were seeded at a density of 6,097 cells/cm2 in GM on Matrigel-coated 96-well microtitre plates. Exogenous Myostatin was added in increasing concentrations (n = 8 per concentration) to _myostatin_-null myoblasts, and 48 h later, cell number was assessed using the methylene blue assay according to the published protocols (Oliver et al., 1989; Chakravarthy et al., 2000).
In situ hybridization
M. tibialis anterior muscle tissue was fixed in 4% paraformaldehyde (wt/vol) in PBS for 16 h at room temperature, washed, dehydrated through an ascending ethanol series, and embedded in paraffin wax. Sections were then cut at 7 μm and mounted on microscope slides (SuperfrostPlus; Fisher Scientific). For the generation of digoxigenin probes, a 326-bp fragment of the mature myostatin cDNA (bp 905–1231; Genbank/EMBL/DDBJ accession no. NM_010834.1) or a 571-bp fragment of the Pax7 cDNA (bp 963–1534; Genbank/EMBL/DDBJ accession no. XM_131758.2) were cloned into the pGEM-T easy vector and transcribed according to the manufacturer's recommendation (DIG RNA Labeling Kit; SP6/T7; Boehringer). Paraffin-embedded sections were dewaxed, and in situ hybridizations were performed according to the method of Greiwe et al. (2001).
Immunocytochemistry
M. tibialis anterior muscle tissue was removed from C57BL/10 or Mstn−/− mice, fixed, and sectioned according to the method of Sharma et al. (1999). The sections were incubated with either 1:50 diluted Myostatin polyclonal antibody (Sharma et al., 1999) or with 1:100 diluted Pax7 mouse monoclonal antibody (DSHB). Subsequent steps of immunostaining were performed according to the published protocol (Thomas et al., 2000).
Primary cultures fixed with 0.25% glutaraldehyde on eight-well Lab-Tek chamber slides were permeabilized with 0.1% Triton X-100 in PBS at room temperature, and the immunocytochemistry was performed according to the method of Beauchamp et al. (2000). Detailed procedures are available on request. Photographs were taken using an Olympus BX50 microscope (Olympus) fitted with a DAGE-MTI DC-330 color camera (DAGE-MTI, Inc.).
Western blotting
Adult myoblasts were extracted from C57BL/10 or _myostatin_-null muscle tissue as described above. Percoll gradient–enriched quiescent satellite cells, proliferating myoblasts, or myoblasts changed to DM (DME, 2% [vol/vol] HS) for 0, 6, 12, 24 or 48 h were resuspended in 200 μl of protein lysis solution (50 mM Tris, pH 7.6, 250 mM NaCl, 5 mM EDTA, 0.1% Nonidet P-40; Complete™ protease inhibitor; Boehringer). The suspension was passed through a fine syringe needle to lyse the cells. Protein was measured using protein assay reagent (Bio-Rad Laboratories). Proteins (10 μg) were separated on SDS-PAGE (4–12% gradient; Novex) and then transferred to a nitrocellulose membrane. Western blots for MyoD and Myogenin were performed according to the protocol described previously (Thomas et al., 2000). Western blots for Myostatin were performed as described by Sharma et al. (1999). To detect the changes in the levels of p21 and Cdk2 upon Myostatin treatment, Western blotting was performed. Adult myoblasts were treated with 10 μg/ml Myostatin for 24 h, and the protein extracts from the myoblasts were made as described above. The Western analysis for p21 and Cdk2 was performed according to Thomas et al. (2000). Western blot of α-tubulin was performed to show equal protein loading.
Detection of Myostatin in fiber-dissociated satellite cells and adult myoblasts
Satellite cells and adult myoblasts were isolated from 6-wk-old mice using the Percoll gradient method (Goodell et al., 1996). Total RNA was isolated by using TRIZOL reagent (Invitrogen) according to the manufacturer's protocol, and RT-PCR was performed to amplify myostatin as described previously (Kambadur et al., 1997). Pax7 cDNA was PCR amplified using 5′-GCTGCCGGACTCTACCTACC-3′ and 5′-CCAGCACAGCGGAGTGTTCC-3′ primers and the following conditions: 95°C for 15 s, 55°C for 30 s, and 72°C for 30 s for 35 cycles. CD34 was PCR amplified using a published protocol (Beauchamp et al., 2000). Protein extracts from satellite cells were made as described above, and Western blot for Myostatin was performed according to the published method (Sharma et al., 1999).
Statistical analysis
To determine the significance between two groups, comparisons were made using a t test. The data are presented as means ± SEM.
Acknowledgments
The Pax 7 monoclonal antibody developed by Atsushi Kawakami was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa Department of Biological Sciences. We would like to thank Dr. S.J Lee for the donation of _myostatin_-null mice. The authors would like to thank Dr. John Bass and Brett Langley for critically reading the manuscript. We thank Alex Hennebry for technical help.
We are indebted to the Foundation for Research, Science, and Technology (New Zealand) and Royal Society of New Zealand (Marsden Fund) for financial support.
M. Sharma and R. Kambadur are co-senior authors.
Abbreviations used in this paper: DM, differentiation medium; E, embryonic day; FM, fiber medium; GM, growth medium.
References
- Allen, R.E., C.J. Temm-Grove, S.M. Sheehan, and G. Rice. 1997. Skeletal muscle satellite cell cultures. Methods Cell Biol. 52:155–176. [DOI] [PubMed] [Google Scholar]
- Beauchamp, J.R., L. Heslop, D.S. Yu, S. Tajbakhsh, R.G. Kelly, A. Wernig, M.E. Buckingham, T.A. Partridge, and P.S. Zammit. 2000. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J. Cell Biol. 151:1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff, R. 1989. Analysis of muscle regeneration using single myofibers in culture. Med. Sci. Sports Exerc. 21:S164–S172. [PubMed] [Google Scholar]
- Bogdanovich, S., T.O. Krag, E.R. Barton, L.D. Morris, L.A. Whittemore, R.S. Ahima, and T.S. Khurana. 2002. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 420:418–421. [DOI] [PubMed] [Google Scholar]
- Carlson, C.J., F.W. Booth, and S.E. Gordon. 1999. Skeletal muscle myostatin mRNA expression is fiber-type specific and increases during hindlimb unloading. Am. J. Physiol. 277:R601–R606. [DOI] [PubMed] [Google Scholar]
- Chakravarthy, M.V., T.W. Abraha, R.J. Schwartz, M.L. Fiorotto, and F.W. Booth. 2000. Insulin-like growth factor-I extends in vitro replicative life span of skeletal muscle satellite cells by enhancing G1/S cell cycle progression via the activation of phosphatidylinositol 3′-kinase/Akt signaling pathway. J. Biol. Chem. 275:35942–35952. [DOI] [PubMed] [Google Scholar]
- Cornelison, D.D., and B.J. Wold. 1997. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev. Biol. 191:270–283. [DOI] [PubMed] [Google Scholar]
- Cornelison, D.D., B.B. Olwin, M.A. Rudnicki, and B.J. Wold. 2000. MyoD−/− satellite cells in single-fiber culture are differentiation defective and MRF4 deficient. Dev. Biol. 224:122–137. [DOI] [PubMed] [Google Scholar]
- Flanagan-Steet, H., K. Hannon, M.J. McAvoy, R. Hullinger, and B.B. Olwin. 2000. Loss of FGF receptor 1 signaling reduces skeletal muscle mass and disrupts myofiber organization in the developing limb. Dev. Biol. 218:21–37. [DOI] [PubMed] [Google Scholar]
- George-Weinstein, M., R.F. Foster, J.V. Gerhart, and S.J. Kaufman. 1993. In vitro and in vivo expression of α7 integrin and desmin define the primary and secondary myogenic lineages. Dev. Biol. 156:209–229. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cadavid, N.F., W.E. Taylor, K. Yarasheski, I. Sinha-Hikim, K. Ma, S. Ezzat, R. Shen, R. Lalani, S. Asa, M. Mamita, et al. 1998. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc. Natl. Acad. Sci. USA. 95:14938–14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell, M.A., K. Brose, G. Paradis, A.S. Conner, and R.C. Mulligan. 1996. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 183:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiwe, J.S., B. Cheng, D.C. Rubin, K.E. Yarasheski, and C.F. Semenkovich. 2001. Resistance exercise decreases skeletal muscle tumor necrosis factor α in frail elderly humans. FASEB J. 15:475–482. [DOI] [PubMed] [Google Scholar]
- Grounds, M.D., and Z. Yablonka-Reuveni. 1993. Molecular and cell biology of skeletal muscle regeneration. Mol. Cell Biol. Hum. Dis. Ser. 3:210–256. [DOI] [PubMed] [Google Scholar]
- Hawke, T.J., N. Jiang, and D.J. Garry. 2003. Absence of p21CIP rescues myogenic progenitor cell proliferative and regenerative capacity in Foxk1 null mice. J. Biol. Chem. 278:4015–4020. [DOI] [PubMed] [Google Scholar]
- Heslop, L., J.R. Beauchamp, S. Tajbakhsh, M.E. Buckingham, T.A. Partridge, and P.S. Zammit. 2001. Transplanted primary neonatal myoblasts can give rise to functional satellite cells as identified using the Myf5nlacZl+ mouse. Gene Ther. 8:778–783. [DOI] [PubMed] [Google Scholar]
- Joulia, D., H. Bernardi, V. Garandel, F. Rabenoelina, B. Vernus, and G. Cabello. 2003. Mechanisms involved in the inhibition of myoblast proliferation and differentiation by myostatin. Exp. Cell Res. 286:263–275. [DOI] [PubMed] [Google Scholar]
- Kambadur, R., M. Sharma, T.P. Smith, and J.J. Bass. 1997. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 7:910–916. [DOI] [PubMed] [Google Scholar]
- Kirk, S., J. Oldham, R. Kambadur, M. Sharma, P. Dobbie, and J. Bass. 2000. Myostatin regulation during skeletal muscle regeneration. J. Cell. Physiol. 184:356–363. [DOI] [PubMed] [Google Scholar]
- Kitzmann, M., G. Carnac, M. Vandromme, M. Primig, N.J. Lamb, and A. Fernandez. 1998. The muscle regulatory factors MyoD and myf-5 undergo distinct cell cycle–specific expression in muscle cells. J. Cell Biol. 142:1447–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley, B., M. Thomas, A. Bishop, M. Sharma, S. Gilmour, and R. Kambadur. 2002. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 277:49831–49840. [DOI] [PubMed] [Google Scholar]
- Maina, F., F. Casagranda, E. Audero, A. Simeone, P.M. Comoglio, R. Klein, and C. Ponzetto. 1996. Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell. 87:531–542. [DOI] [PubMed] [Google Scholar]
- McPherron, A.C., and S.J. Lee. 1997. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA. 94:12457–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron, A.C., A.M. Lawler, and S.J. Lee. 1997. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature. 387:83–90. [DOI] [PubMed] [Google Scholar]
- McPherron, A.C., and S.J. Lee. 2002. Suppression of body fat accumulation in myostatin-deficient mice. J. Clin. Invest. 109:595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesires, N.T., and M.E. Doumit. 2002. Satellite cell proliferation and differentiation during postnatal growth of porcine skeletal muscle. Am. J. Physiol. Cell Physiol. 282:C899–C906. [DOI] [PubMed] [Google Scholar]
- Oliver, M.H., N.K. Harrison, J.E. Bishop, P.J. Cole, and G.J. Laurent. 1989. A rapid and convenient assay for counting cells cultured in microwell plates: application for assessment of growth factors. J. Cell Sci. 92:513–518. [DOI] [PubMed] [Google Scholar]
- Partridge, T.A. 1997. Tissue culture of skeletal muscle. Methods Mol. Biol. 75:131–144. [DOI] [PubMed] [Google Scholar]
- Rios, R., I. Carneiro, V.M. Arce, and J. Devesa. 2002. Myostatin is an inhibitor of myogenic differentiation. Am. J. Physiol. Cell Physiol. 282:C993–C999. [DOI] [PubMed] [Google Scholar]
- Rosenblatt, J.D., A.I. Lunt, D.J. Parry, and T.A. Partridge. 1995. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell. Dev. Biol. Anim. 31:773–779. [DOI] [PubMed] [Google Scholar]
- Rosenblatt, J.D., D.J. Parry, and T.A. Partridge. 1996. Phenotype of adult mouse muscle myoblasts reflects their fiber type of origin. Differentiation. 60:39–45. [DOI] [PubMed] [Google Scholar]
- Schmalbruch, H., and D.M. Lewis. 2000. Dynamics of nuclei of muscle fibers and connective tissue cells in normal and denervated rat muscles. Muscle Nerve. 23:617–626. [DOI] [PubMed] [Google Scholar]
- Seale, P., and M.A. Rudnicki. 2000. A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev. Biol. 218:115–124. [DOI] [PubMed] [Google Scholar]
- Seale, P., L.A. Sabourin, A. Girgis-Gabardo, A. Mansouri, P. Gruss, and M.A. Rudnicki. 2000. Pax7 is required for the specification of myogenic satellite cells. Cell. 102:777–786. [DOI] [PubMed] [Google Scholar]
- Sharma, M., R. Kambadur, K.G. Matthews, W.G. Somers, G.P. Devlin, J.V. Conaglen, P.J. Fowke, and J.J. Bass. 1999. Myostatin, a transforming growth factor-β superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J. Cell. Physiol. 180:1–9. [DOI] [PubMed] [Google Scholar]
- Taylor, W.E., S. Bhasin, J. Artaza, F. Byhower, M. Azam, D.H. Willard, Jr., F.C. Kull, Jr., and N. Gonzalez-Cadavid. 2001. Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am. J. Physiol. Endocrinol. Metab. 280:E221–E228. [DOI] [PubMed] [Google Scholar]
- Thomas, M., B. Langley, C. Berry, M. Sharma, S. Kirk, J. Bass, and R. Kambadur. 2000. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 275:40235–40243. [DOI] [PubMed] [Google Scholar]
- Wagner, K.R., A.C. McPherron, N. Winik, and S.J. Lee. 2002. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 52:832–836. [DOI] [PubMed] [Google Scholar]
- Wehling, M., B. Cai, and J.G. Tidball. 2000. Modulation of myostatin expression during modified muscle use. FASEB J. 14:103–110. [DOI] [PubMed] [Google Scholar]
- Whittemore, L.A., K. Song, X. Li, J. Aghajanian, M. Davies, S. Girgenrath, J.J. Hill, M. Jalenak, P. Kelley, A. Knight, et al. 2003. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem. Biophys. Res. Commun. 300:965–971. [DOI] [PubMed] [Google Scholar]
- Yablonka-Reuveni, Z., M.A. Rudnicki, A.J. Rivera, M. Primig, J.E. Anderson, and P. Natanson. 1999. The transition from proliferation to differentiation is delayed in satellite cells from mice lacking MyoD. Dev. Biol. 210:440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, N., S. Yoshida, K. Koishi, K. Masuda, and Y. Nabeshima. 1998. Cell heterogeneity upon myogenic differentiation: down-regulation of MyoD and Myf-5 generates “reserve cells.” J. Cell Sci. 111:769–779. [DOI] [PubMed] [Google Scholar]
- Zhu, X., M. Hadhazy, M. Wehling, J.G. Tidball, and E.M. McNally. 2000. Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett. 474:71–75. [DOI] [PubMed] [Google Scholar]