Clustering of neuronal potassium channels is independent of their interaction with PSD-95 (original) (raw)
Abstract
Voltage-dependent potassium channels regulate membrane excitability and cell–cell communication in the mammalian nervous system, and are found highly localized at distinct neuronal subcellular sites. Kv1 (mammalian Shaker family) potassium channels and the neurexin Caspr2, both of which contain COOH-terminal PDZ domain binding peptide motifs, are found colocalized at high density at juxtaparanodes flanking nodes of Ranvier of myelinated axons. The PDZ domain–containing protein PSD-95, which clusters Kv1 potassium channels in heterologous cells, has been proposed to play a major role in potassium channel clustering in mammalian neurons. Here, we show that PSD-95 colocalizes precisely with Kv1 potassium channels and Caspr2 at juxtaparanodes, and that a macromolecular complex of Kv1 channels and PSD-95 can be immunopurified from mammalian brain and spinal cord. Surprisingly, we find that the high density clustering of Kv1 channels and Caspr2 at juxtaparanodes is normal in a mutant mouse lacking juxtaparanodal PSD-95, and that the indirect interaction between Kv1 channels and Caspr2 is maintained in these mutant mice. These data suggest that the primary function of PSD-95 at juxtaparanodes lies outside of its accepted role in mediating the high density clustering of Kv1 potassium channels at these sites.
Keywords: juxtaparanode; myelin; ion channel; MAGUK; node of Ranvier
Introduction
The localization of ion channels, receptors, cell adhesion molecules, and other signaling proteins in discrete plasma membrane domains is critical for neuronal function. Especially pronounced examples of this are excitatory chemical synapses of central neurons and nodes of Ranvier of myelinated axons. Although a great deal is known about the repertoire of synaptic proteins, their molecular interactions and the dynamic mechanisms of their trafficking and clustering (Scannevin and Huganir, 2000; Husi and Grant, 2001), much less is known about the molecular mechanisms that define the structure and function of the node of Ranvier (for reviews see Arroyo and Scherer, 2000; Peles and Salzer, 2000; Rasband and Trimmer, 2001a). The node of Ranvier is defined by the high density clustering of voltage-dependent sodium (Nav)* channels responsible for the rapid inward sodium currents that support saltatory conduction (Hille, 2001). On each side of the node are paranodes; regions where sequential layers of the myelin sheath are anchored and terminate in septate-like axoglial junctions (Rosenbluth, 1976; Einheber et al., 1997). Finally, just inside the innermost axoglial junction is a region called the juxtaparanode, where voltage-dependent potassium (Kv) channels of the Kv1 (mammalian Shaker) subfamily are clustered at high densities in both peripheral and central myelinated nerve fibers (Wang et al., 1993, 1994). These juxtaparanodal Kv1 channels modulate axonal excitability (David et al., 1995; Vabnick et al., 1999) and consist of various heteromultimeric combinations of pore-forming Kv1 α and cytoplasmic Kvβ subunits, including Kv1.1, Kv1.2, Kv1.4, Kv1.6, and Kvβ2 (Wang et al., 1993; Rhodes et al., 1997; Rasband et al., 1999, Rasband and Trimmer, 2001a).
A number of candidate proteins have been identified that may be involved in the aggregation of sodium channels and in the formation of paranodal axoglial junctions (Kordeli et al., 1995; Lambert et al., 1997; Menegoz et al., 1997; Peles et al., 1997; Berghs et al., 2000; Tait et al., 2000; Boyle et al., 2001). However, much less is known of proteins that interact with and/or cluster Kv1 channels. At juxtaparanodes, one likely contributor is Caspr2, a member of the neurexin superfamily that colocalizes with juxtaparanodal Kv1 channels throughout development and is found associated with Kv1.2 and Kvβ2 subunits immunopurified from rat brain (Poliak et al., 1999, 2001). Although Caspr2 and Kv1.2 are found associated in vivo, the interaction is indirect (Poliak et al., 1999), and thought to require a multiple PDZ domain–containing protein that binds the COOH-terminal class I PDZ-binding peptide motifs (TDV) of Kv1.1 and Kv1.2, and the COOH-terminal class II PDZ-binding peptide motif (EWLI) of Caspr2 (for a discussion of PDZ domain classification see Sheng and Sala, 2001). Thus, it is attractive to speculate that Kv1 channels and Caspr2 interact indirectly via a multiple PDZ domain scaffolding protein present at juxtaparanodes. Importantly, membrane-associated guanylate kinases (MAGUKs), typified by PSD-95, contain three such PDZ domains. PSD-95 and other MAGUKs exhibit a high affinity interaction with the class I PDZ-binding peptide motifs at the COOH termini of Kv1.1, Kv1.2, and Kv1.4 (Kim et al., 1995). Although PSD-95 and Kv1 channels expressed separately in transfected cells have uniform plasma membrane expression, when coexpressed PSD-95 and Kv1 channels are reciprocally co-clustered in high density plasma membrane complexes (Kim and Sheng, 1996; Tiffany et al., 2000). In mammalian neurons, PSD-95 is present at high levels at the postsynaptic density of excitatory chemical synapses, where it contributes to synaptic function through interaction with NMDA receptors and other synaptic proteins (Sheng and Sala, 2001). In mammalian brain, PSD-95 is also found colocalized with Kv1.1, Kv1.2, and Kvβ2 in presynaptic basket cell terminals in the cerebellum (Kim et al., 1995; Laube et al., 1996), but not at many other sites containing high density clusters of Kv1 channels (Rhodes et al., 1997). However, immunostaining for PSD-95 or another highly related MAGUK at juxtaparanodes has been reported previously, suggesting a potential role for MAGUKs in the juxtaparanodal localization of Kv1 channels (Baba et al., 1999; Rasband and Trimmer, 2001a).
Here, we provide a definitive molecular characterization that PSD-95, but not other MAGUKs, is present at juxtaparanodes of mammalian myelinated axons where it colocalizes with Kv1 channels and Caspr2. Moreover, we demonstrate that an in vivo interaction exists between Kv1 channel subunits and PSD-95. However, we find that the association and high density clustering of Kv1 channels and Caspr2 is normal in mutant mice lacking juxtaparanodal PSD-95. Together, these results suggest that although PSD-95 is found at juxtaparanodes of mammalian myelinated axons, its function at these sites lies outside of potassium channel clustering.
Results
PSD-95 is present at juxtaparanodes and colocalizes with Kv1 potassium channels
To address if PSD-95 itself or another highly related MAGUK was present at juxtaparanodes, we used a recombinant fragment of PSD-95 to generate a large panel of mAbs (Tiffany et al., 2000). These mAbs were assayed for specificity against COS-1 cells transfected with cDNAs for each of the known mammalian MAGUKs (PSD-95, Chapsyn-110, SAP-97, and SAP-102). Although certain mAbs (such as K28/86) were found to be ‘pan-MAGUK,’ exhibiting robust cross reactivity to all known MAGUKs, others (such as mAb K28/43) recognize only PSD-95. Consistent with previous reports of the localization of PSD-95 in mammalian brain, K28/43 robustly labels excitatory synapses (unpublished data), as well as cerebellar basket cell terminals (Kim et al., 1995; Laube et al., 1996), where the staining colocalizes precisely with that for Kv1.2 (Fig. 1A, arrows). Juxtaparanodes of myelinated nerve fibers in cerebellar white matter are labeled with both the pan-MAGUK K28/86 mAb (unpublished data) and the PSD-95-specific mAb K28/43 (Fig. 1 B, arrowheads). Double staining using a Kv1.2-specific rabbit polyclonal antibody shows that Kv1.2 immunoreactivity colocalizes precisely with PSD-95 immunostaining at these cerebellar juxtaparanodes (Fig. 1 B, arrowheads). PSD-95 immunoreactivity is also present and colocalizes with Kv1 channels at juxtaparanodes of mammalian sciatic and optic nerves, and spinal cord (unpublished data).
Figure 1.
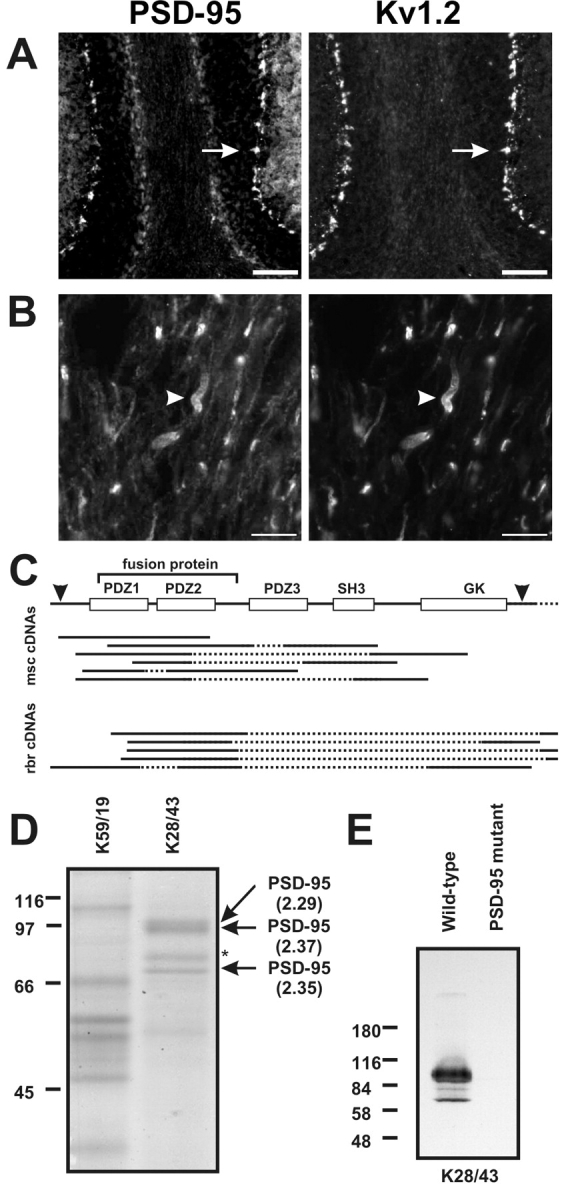
PSD-95 is present at juxtaparanodes and colocalizes with Kv1.2. (A and B) Immunoreactivity for PSD-95 (K28/43 mAb) and Kv1.2 colocalize at cerebellar basket cell terminals (A, arrows) and juxtaparanodes (B, arrowheads). (C) Schematic showing the full-length cDNA for dlg4 (PSD-95), including translation start and end sites (arrowheads), and the region corresponding to the fusion protein used to generate the K28 mAbs. Expression cloning from mouse spinal cord (msc) and rat brain (rbr) libraries resulted in the depicted cDNAs, all of which corresponded to PSD-95. Sequenced regions for each clone are shown as solid lines. (D) Purification of PSD-95 using a K28/43 immunoaffinity column. The major proteins purified on the K28/43 column were compared with an unrelated IgG precolumn (K59/19), excised, digested with trypsin, and the masses of the resulting peptides were determined by mass spectrometry. Search of the protein databases using ProFound yielded high probability matches (Z values given in parentheses) to PSD-95 for three of the four high molecular mass bands. The unidentified band (asterisk) did not significantly match any protein in the databases. (E) mAb K28/43 immunoblot of brain homogenate from wild-type and mutant PSD-95 mice; 20 μg of protein were loaded in each lane. PDZ, PSD-95, discs-large, zona occludens; SH3, Src homology 3; GK, guanylate kinase. Bars: (A) 100 μm, (B) 10 μm.
PSD-95 is the only MAGUK antigen for mAb K28/43
Although among known MAGUKs mAb K28/43 recognizes only PSD-95, there remains the possibility it also reacts with a highly related but as yet undescribed MAGUK. Therefore, we performed several experiments providing evidence that the only antigen recognized by K28/43 is bona fide PSD-95. We first performed expression cloning on both mouse spinal cord and rat brain cDNA libraries using mAb K28/43. A total of 2.1 × 105 and 9.6 × 105 plaque forming units were screened from the brain and spinal cord libraries, respectively. A total of 13 and 6 clones, respectively, were isolated and sequenced from the brain and spinal cord libraries. All 19 clones identified from the spinal cord or brain libraries using this strategy corresponded to PSD-95 itself (Fig. 1 C). Importantly, each clone that was isolated during the screening included the region corresponding to the fragment of PSD-95 used to generate the K28 mAbs (Fig. 1 C).
In parallel with the expression cloning approach, we used a K28/43 mAb immunoaffinity column for chromatographic purification of the K28/43 antigen from spinal cord, as this tissue is highly enriched in nodes of Ranvier (Arroyo et al., 2001; Rasband and Trimmer, 2001b). Detergent extracts of rat spinal cord membranes (RSCM) were initially chromatographed on a mAb affinity precolumn made using an unrelated IgG mAb (K59/19) to eliminate and/or identify spinal cord proteins that bind nonspecifically to mouse IgG affinity columns. The flow-through from the precolumn was then applied to the K28/43 mAb affinity column, and peak fractions from the precolumn and K28/43 column were pooled and the component proteins fractionated by SDS-PAGE. Coomassie blue staining revealed four distinct high M r bands unique to the K28/43 column (Fig. 1 D). These were excised from the gel, and subjected to trypsin proteolysis and mass spectrometric analysis for protein identification. By comparing the measured masses of the peptides against databases, three of the four bands (Fig. 1 D, arrows) were found to correspond to PSD-95 with very high probability matches (Z ≥ 2.29). The fourth protein band (Fig. 1 D, asterisk), did not yield a match to any protein in the database.
Finally, we performed immunoblots on brain membranes from wild-type and mutant mice with a targeted mutation in PSD-95 that introduces a stop codon within the third PDZ domain (Migaud et al., 1998). The robust K28/43 immunoreactivity observed on immunoblots of wild-type mouse brain membranes is completely absent in brain membranes from the PSD-95 mutant animals (Fig. 1 E; note that other immunoblots reveal the presence of K28/43 immunoreactivity to the expected M r = 40 kD truncated PSD-95 band in brain membranes prepared from mutant mice). Together, the expression cloning, mass spectrometry, and immunoblotting data show that the only antigen detected by mAb K28/43 is PSD-95 itself.
PSD-95 and Kv1 channels form a macromolecular protein complex in vivo
Because mAb K28/43 robustly labels juxtaparanodes and colocalizes precisely with Kv1 channels (Fig. 1 B), we sought to demonstrate an in vivo interaction between Kv1 channels and PSD-95. We made detergent extracts from both rat brain membranes (RBMs) and RSCM, in either lysis buffer containing 1% Triton X-100, known to preserve Kv channel intersubunit interactions (Trimmer, 1991; Rhodes et al., 1995), or by extracting first in buffer containing 2% SDS, followed by a fivefold dilution in Triton X-100 lysis buffer; the latter conditions have been shown to preserve the interaction between NMDA receptors and the MAGUK SAP102 (Lau et al., 1996). In each case, the cognate antigens were efficiently immunoprecipitated in both detergent buffers; however, only the extraction in Triton X-100 alone preserves the PSD-95–Kv1.2 interaction (Fig. 2A). It is important to note that coimmunoprecipitation of Kv1.2 and PSD-95 was observed in reactions performed on both RBM and RSCM extracts. Antibodies against Kvβ2, a cytoplasmic component subunit of juxtaparanodal Kv1 channels, are also effective at coimmunoprecipitating PSD-95 from both brain and spinal cord, but immunoprecipitation using antibodies against Kv2.1, a potassium channel subunit that lacks a PDZ-binding motif and does not coassociate with Kv1 channels, failed to coimmunoprecipitate PSD-95 (unpublished data). Overall, we found that the yield of coimmunoprecipitated protein was small compared with the yield of cognate antigen obtained by direct immunoprecipitation. Thus, although it is possible to demonstrate an association between Kv1 channels and PSD-95 in native tissue, these results suggest that either only a small subset of the total Kv1.2 and PSD-95 protein pools associate in vivo_,_ that the majority of Kv1.2–PSD-95 complexes are detergent-insoluble, and/or that the bulk of the in situ complexes are disrupted by the detergent extractions and incubations required for these experiments.
Figure 2.
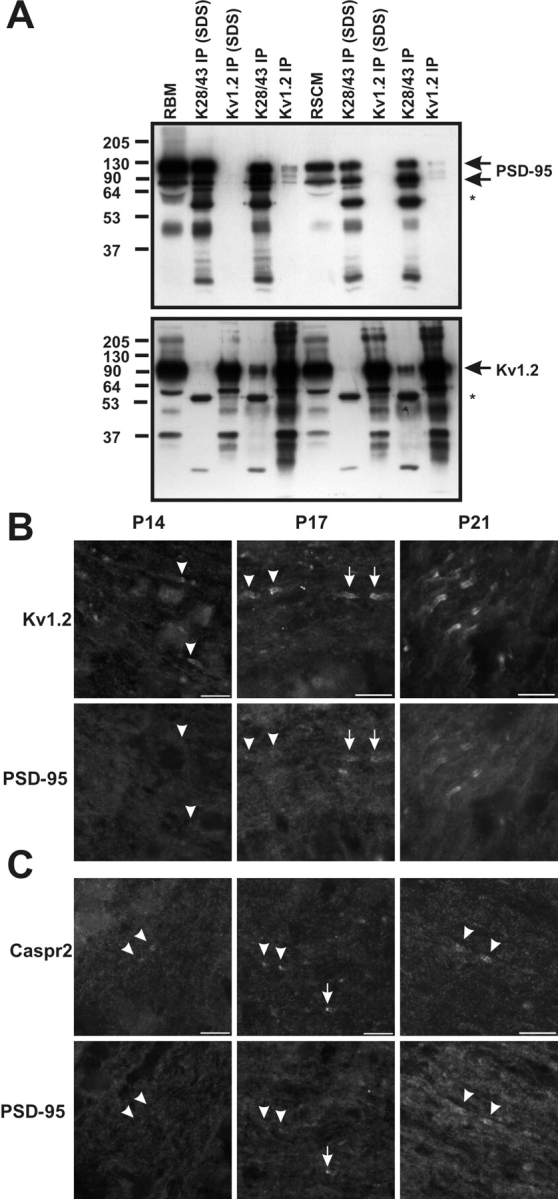
Kv1.2 and PSD-95 form a macromolecular protein complex in vivo. (A) Immunoprecipitation reactions performed on RBM and RSCM, using a pAb against Kv1.2 and an mAb against PSD-95 (K28/43). Immunoblotting for PSD-95 (top) and Kv1.2 (bottom) show that they are coimmunoprecipitated, and that this interaction is sensitive to the presence of SDS in the extraction buffer. The mouse IgG band is indicated by an asterisk. 50 μg of crude membranes was loaded in the RBM and RSCM lanes, and immunoprecipitated proteins from the equivalent of 100 μg of membranes were loaded for each IP lane. (B and C) Developmental coexpression of Kv1.2, PSD-95, and Caspr2 at rat optic nerve juxtaparanodes using indirect immunofluorescence. Kv1.2 (B) and Caspr2 (C) were detected as early as P14 (arrowheads), but PSD-95 was not seen to colocalize with this staining until P17 and later at P21. At P17, both more mature (arrows) and immature (arrowheads) juxtaparanodes were detected.
PSD-95 is developmentally expressed at juxtaparanodes after Kv1 channels and Caspr2
If PSD-95 initiates recruitment and clustering of Kv1 channels and Caspr2 to juxtaparanodes in myelinated nerve fibers, then it should be detectable at these sites concomitant with, or before the appearance of clustered Kv1.2 and Caspr2. We have previously characterized the developmental expression of Kv1 channels at juxtaparanodes in central myelinated axons of the optic nerve. We found that Kv1 channel subunits are first detected immediately adjacent to paranodal axoglial junctions after the onset of myelination and several days after the expression and clustering of nodal Nav channels (Rasband et al., 1999). When we analyzed the time course of expression of PSD-95 in optic nerve sections double-labeled with antibodies against Kv1.2 (Fig. 2 B) or Caspr2 (Fig. 2 C), we found no detectable immunoreactivity for Kv1.2, PSD-95, or Caspr2 at postnatal day 7 (P7; unpublished data). Consistent with our earlier study (Rasband et al., 1999), clustered Kv1.2 and Caspr2 were first detected at P14; however, PSD-95 immunoreactivity was still not observed (Fig. 2, B and C, P14, arrowheads). The earliest detectable expression of PSD-95 at juxtaparanodes was at P17, when PSD-95 staining was found to colocalize with both Kv1.2 and Caspr2 (Fig. 2, B and C, P17, arrows). In these same fields of view, examples of Kv1.2- and Caspr2-labeled juxtaparanodes devoid of PSD-95 immunoreactivity are also evident (Fig. 3, B and C; P17, arrowheads); these sites presumably represent a more immature state of juxtaparanode formation. By P21, many juxtaparanodes had colocalized PSD-95, Kv1.2, and Caspr2 (Fig. 3, B and C, P21). Although not conclusive, these observations suggest that Kv1 channels and Caspr2 can cluster at juxtaparanodes before the expression of PSD-95 at these sites.
Figure 3.
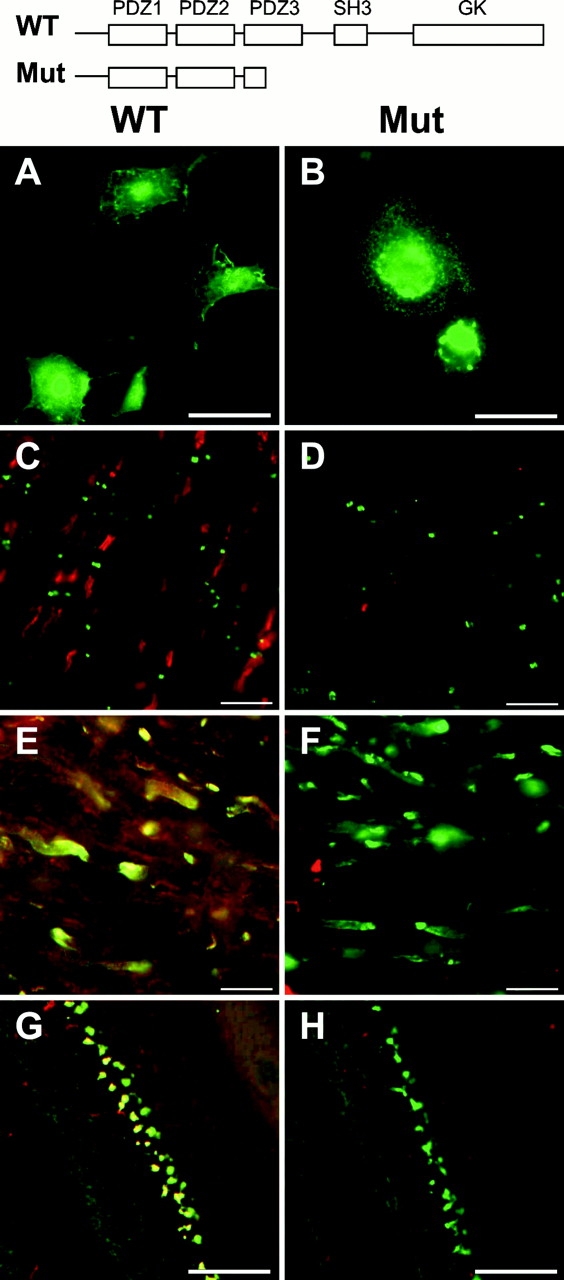
Kv1.2 is clustered in PSD-95 mutant animals. (A and B) K28/43 immunostaining of COS-1 fibroblasts expressing wild-type PSD-95 (WT; A) or the truncated form of PSD-95 (Mut; B), corresponding to that present in the PSD-95 mutant mouse (cartoon at top of this figure). (C and D) Immunolabeling of WT (C) and mutant (D) mouse optic nerves with mAb K28/43 against PSD-95 (red) and Nav1.6 (green). (E and F) PSD-95 (red) and Kv1.2 (green) colocalize in optic nerves from WT mice, but in mutant mice only clustered Kv1.2 is detected (green). (G and H) At basket cell terminals, PSD-95 (red) and Kv1.2 (green) colocalize in WT mice, but in mutant mice only clustered Kv1.2 is detected (green). Bars: (A and B) 50 μm; (C–F) 10 μm; (G and H) 100 μm. PDZ, PSD-95, discs-large, zona occludens; SH3, Src homology 3; GK, guanylate kinase.
PSD-95 is not required for juxtaparanodal localization of Kv1.2 or Caspr2
Because mutation of the Drosophila discs-large (dlg) gene, a homologue of PSD-95, results in failure to correctly localize Shaker potassium channels to the neuromuscular junction of flies (Tejedor et al., 1997), we reasoned that mutating the murine PSD-95 protein might result in loss of clustered Kv1 channels at juxtaparanodes and at basket cell terminals. To test this hypothesis, we compared the expression and localization of Kv1 α and Kvβ subunits, and Caspr2 between wild-type mice, and PSD-95 mutant mice (Migaud et al., 1998) expressing truncated PSD-95 (Fig. 3, top). First, we verified in transfected COS-1 cells that mAb K28/43 recognizes both full-length PSD-95 (Fig. 3 A) and the truncated form of the protein expressed in the mutant mouse (Fig. 3 B). In wild-type mice, as in rats, PSD-95 staining (red) was restricted to juxtaparanodes where it did not overlap with staining for Nav1.6 (Fig. 3 C; green), the predominant nodal Nav channel in adult myelinated nerve fibers (Caldwell et al., 2000; Boiko et al., 2001). Moreover, in wild-type mice PSD-95 immunoreactivity colocalized precisely with juxtaparanodal Kv1.2 (Fig. 3 E; overlap is yellow). In contrast, when we examined the optic nerves of PSD-95 mutant mice, we were unable to detect any juxtaparanodal PSD-95 staining (red), but Nav1.6 channels (green) were still clustered at nodes of Ranvier (Fig. 3 D), indicating that nodes formed normally in the mutant mice. Surprisingly, when we examined the expression of potassium channel Kv1 α and Kvβ subunits in the PSD-95 mutant mice they were also found clustered at high density in their normal juxtaparanodal localization, at levels indistinguishable from those observed in wild-type mice (Fig. 3 F, green; data for Kv1.1, Kv1.4, and Kvβ2 staining are not depicted). Moreover, the localization of Kv1 channels at basket cell terminals was also unaffected by the mutation in PSD-95 (Fig. 3, G and H). Finally, we observed no difference between wild-type and mutant animals in the expression of Caspr2 at optic nerve juxtaparanodes (Fig. 4A, arrowheads).
Figure 4.
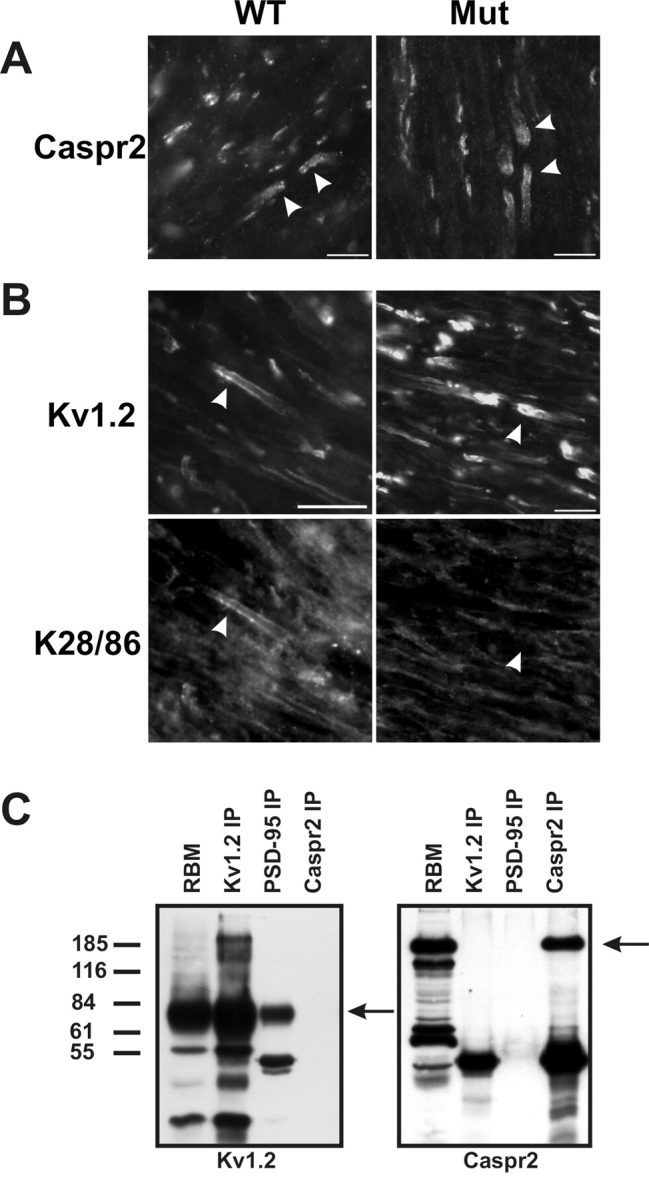
Immunostaining of WT and PSD-95 mutant mice, and immunoprecipitation (IP) of Kv1.2, PSD-95, and Caspr2 from RBM. (A) Caspr2 is clustered in both WT and mutant mice (arrowheads). (B) Although Kv1.2 is clustered in both WT and PSD-95 mutant mice (B_,_ top), the pan-MAGUK antibody K28/86 only stains juxtaparanodes in WT mice (arrowheads). (C) Coimmunoprecipitation from RBM using pAbs against Kv1.2 and Caspr2, and mAb K28/43 that recognizes PSD-95. Arrows indicate the positions of the respective antigens. The RBM lanes were loaded with 50 μg of crude membrane protein, and each immunoprecipitation lane was loaded with precipitated proteins from 500 μg of starting material. Bars, 10 μm.
We considered the possibility that other MAGUKs may be up-regulated and compensate for PSD-95 at juxtaparanodes, facilitating the observed clustering of Kv1 channels and Caspr2. Therefore, we used the pan-MAGUK mAb K28/86 to label juxtaparanodes, but found that whereas the K28/86 staining colocalized with that for Kv1.2 in wild-type animals (Fig. 4 B, arrowheads), no immunoreactivity was observed at juxtaparanodes (Fig. 4 B) or at basket cell terminals (unpublished data) in the PSD-95 mutant animals. We also immunolabeled myelinated nerve fibers using antibodies specific for each of the other known MAGUKs SAP-97, Chapsyn-110, and SAP-102. Although each of these antibodies labeled the cognate antigen in transfected cells, no juxtaparanodal or basket cell terminal immunoreactivity for these MAGUKs was observed in either wild-type or mutant animals (unpublished data). These results show that whereas PSD-95 is normally expressed at juxtaparanodes and at basket cell terminals in both rats and mice, the truncated PSD-95 protein expressed in these mutant mice is not found at these sites. Moreover, because compensatory up-regulation of the other known MAGUKs does not occur, yet Kv1 channels and Caspr2 are still found clustered at these sites in mutant animals, these MAGUKs are not essential for clustering of either Kv1 channels or Caspr2.
PSD-95 is not required for Kv1 channel–Caspr2 interactions
To further define the molecular interactions between the juxtaparanodal proteins PSD-95, Kv1.2, and Caspr2, we performed coimmunoprecipitation experiments from Triton X-100 solubilized RBM using antibodies against each juxtaparanodal component (Fig. 4 C). Under these conditions, we were able to consistently coimmunoprecipitate Kv1.2 and PSD-95, but we were unable to demonstrate a robust interaction between Caspr2 and these proteins. Similarly, we performed coimmunoprecipitation experiments using antibodies against Kv1.2, Caspr2, and PSD-95 using WT and PSD-95 mutant mouse brain membranes. These experiments were performed under two different extraction conditions: (1) using Triton X-100 extraction buffer as above; or (2) using the detergent sucrose monolaurate (SML), as this has been used previously to demonstrate an in vivo interaction between Caspr2 and Kv1.2 (Poliak et al., 1999). Fig. 5A demonstrates no obvious change in the expression levels of either Kv1.2 or Caspr2 in the mutant mice, and that we were able to directly immunoprecipitate Caspr2 using either detergent buffer. Further, although Kv1.2 showed poor solubility in the SML detergent buffer, small amounts of Kv1.2 were found in Caspr2 immunoprecipitation reactions from brain membranes performed in SML buffer (it should be noted that we have loaded ∼1/200 of the amount of starting material as compared with previous reports describing Kv1.2–Caspr2 interactions; Poliak et al., 2001). Importantly, Kv1.2 was coimmunoprecipitated by antibodies against Caspr2 from both the wild-type and the PSD-95 mutant mouse brains. We were also able to detect a very small amount of Caspr2 in the Kv1.2 immunoprecipitation reactions. On the other hand, PSD-95 coimmunoprecipitated Kv1.2 only in Triton X-100, and not in the SML buffer. Together, these results demonstrate that PSD-95 is not required for an interaction between Kv1 channels and Caspr2. Further, these results imply that Kv1.2–PSD-95 interactions and protein complexes are biochemically distinct from Kv1.2–Caspr2 complexes because extraction in the various detergents differentially disrupts the ability to coimmunoprecipitate the three demonstrated juxtaparanodal protein components (Kv1 channels, Caspr2, and PSD-95).
Figure 5.

Immunoprecipitation reactions and immunoblotting from wild-type and PSD-95 mutant mouse brain membranes show that Kv1 channels and Caspr2 interact independently of PSD-95. (A) Coimmunoprecipitation reactions in Triton X-100 or SML detergent containing buffers. WT and Mut brain lanes were loaded with 50 μg of crude brain membranes (1-min exposure shown), and each immunoprecipitation lane was loaded with precipitated proteins from 250 μg of starting brain membranes (1-h exposure shown). Immunoblots were probed with monoclonal anti-Kv1.2 or anti-Caspr2 rabbit serum. (B) Detergent-insoluble membrane pellets from WT and Mut mice obtained after extraction in buffers containing the indicated concentrations of Triton X-100 (% vol/vol) were assayed by immunoblots probed with antibodies against Kv1.2 or PSD-95. Each lane represents the detergent-insoluble material from 100 μg of starting brain membrane.
PSD-95 does not recruit Kv1 channels to detergent-insoluble complexes
We considered the possibility that rather than clustering Kv1 channels directly, PSD-95 may recruit channels into detergent-insoluble protein complexes similar to those derived from the postsynaptic density. We extracted Kv1 channels from brain membranes prepared from wild-type and mutant mice using varying concentrations of Triton X-100 (0.01–0.25%), and analyzed the detergent-insoluble fractions for PSD-95 and Kv1.2 by immunoblot. Fig. 5 B (bottom) shows that whereas the bulk of wild-type PSD-95 protein was detergent insoluble (a faint, low M r band of immunoreactivity, not depicted, was observed in brain membranes from mutant animals), there was no difference in the detergent extractability of Kv1.2 between brain membranes prepared from wild-type or PSD-95 mutant mice (Fig. 5 B, top). Similarly, much higher detergent concentrations (1–4% Triton X-100) did not lead to any difference in Kv1.2 extractability (unpublished data). Thus, PSD-95 is not required for recruitment of Kv1 channels into detergent-insoluble protein complexes.
Discussion
A number of previous studies aimed at elucidating the mechanisms underlying the highly restricted subcellular localization of potassium channels in mammalian neurons have focused on Kv1 channels clustered at juxtaparanodes of myelinated axons. These include studies of Kv1 channel localization during remyelination (Rasband et al., 1998), in dys- and hypomyelinating mutant animals (Wang et al., 1995; Baba et al., 1999), during developmental myelination (Rasband et al., 1999; Vabnick et al., 1999), and in transgenic animals with defects in paranodal structures (Dupree et al., 1999; Bhat et al., 2001; Boyle et al., 2001; Poliak et al., 2001). These previous studies have all pointed to the formation of compact myelin, and of axoglial junctions (Rosenbluth, 1976), as critical to establishing the highly restricted high density clustering of Kv1 channels in juxtaparanodal axonal membrane domains. In contrast, there remains a paucity of information on the specific molecular components of Kv1 channel-containing protein complexes at juxtaparanodes. One molecule that is found colocalized with Kv1 channels at juxtaparanodes is the neurexin Caspr2. It has been suggested that the large extracellular domain of Caspr2 may be important in mediating specific neuroglial interactions that serve to establish and maintain the unique characteristics of the juxtaparanodal axonal membrane domain. This presumably occurs through the formation of a juxtaparanodal-specific protein complex that includes Kv1 channel subunits and other juxtaparanode-specific axonal and glial proteins (Fig. 6) . Recently, a cell adhesion molecule called TAG-1 was found localized to the juxtaparanode and present on the surface of the myelinating glia (Traka et al., 2002); such a protein could serve as a binding partner for Caspr2. However, the precise roles of Caspr2 and TAG-1 in organizing the juxtaparanodal domain remain to be determined.
Figure 6.

Model of the molecular components of the juxtaparanode. The juxtaparanodal protein complex includes the known components PSD-95, Caspr2, and Kv1 channels (α and β subunits). Additional components likely include a putative glial binding partner for Caspr2 (possibly TAG-1, a PDZ-domain containing scaffolding protein linking Caspr2 and Kv1 channels) and cytoskeletal proteins. The paranodal axoglial junctions are also shown because these structures are essential for the localization of juxtaparanodal proteins.
The MAGUK PSD-95 is an attractive candidate for an axonal Kv1 channel binding partner. Robust PSD-95 immunoreactivity is found at juxtaparanodes of myelinated axons in optic nerve (Baba et al., 1999; Rasband and Trimmer, 2001a), cerebellar white matter, and in sciatic nerve. PSD-95 is also found at one other site of high density clustering of Kv1 channels in the mammalian nervous system, in cerebellar basket cell terminals (Kim et al., 1995; Laube et al., 1996). Moreover, PSD-95 interacts with and clusters plasma membrane Kv1 channels in heterologous expression systems (Manganas and Trimmer, 2000; Tiffany et al., 2000). As such, PSD-95 represents an especially attractive candidate for a Kv1 channel clustering protein, given the cross-linking that could occur between the three class I PDZ domains of PSD-95, and the four class I PDZ-binding peptide motifs present (one per Kv1 α subunit) in a tetrameric Kv1 potassium channel. PSD-95 could also play a role in linking Kv1 channels to Caspr2, as both axonal membrane proteins contain PDZ-binding motifs.
PSD-95 is not essential for Kv1 channel and Caspr2 clustering
Does PSD-95 in fact function in myelinated nerve fibers to induce a Caspr2-independent clustering of Kv1 channels at juxtaparanodes, similar to that seen for heterologous expression systems? Alternatively, does PSD-95 provide a molecular bridge between Kv1 channels and Caspr2, implicating Caspr2 as the principal clustering protein, potentially through extracellular interactions with glial binding partners like TAG-1? Because PSD-95 is not expressed during development until well after the initial detection of Kv1.2 and Caspr2 at juxtaparanodes, it is unlikely that PSD-95 recruits and/or concentrates channels to these subcellular domains. More convincingly, Kv1.2 and Caspr2 remain clustered at juxtaparanodes in the absence of PSD-95 and other MAGUKs in the PSD-95 mutant mice. Moreover, Kv1.2 and Caspr2 still associate in mice lacking juxtaparanodal PSD-95. These results do not exclude the possibility that Caspr2 may be the organizing protein responsible for Kv1 channel aggregation and/or maintenance at juxtaparanodes, perhaps through a scaffolding protein that binds to the class II PDZ-binding motif at the Caspr2 COOH terminus, and the class I motif on Kv1 channels. Any function for Caspr2 in the clustering of juxtaparanodal potassium channels awaits the identification of an as yet unknown bridging molecule linking Kv1 channels and Caspr2 (Fig. 6).
Although colocalized with Kv1 channels at juxtaparanodes, it is clear that PSD-95 is not essential for their recruitment or anchoring at these sites, and it is not the mediator of the observed Kv1 channel–Caspr2 interaction. It is important to emphasize that our results cannot completely rule out the possibility that Kv1 channels do interact with both Caspr2 and PSD-95, or that PSD-95 plays a role in localizing Kv1 channels at juxtaparanodes; this function of PSD-95 might be unmasked by a more in-depth analysis of Kv1 channel clustering at juxtaparanodes in developing or remyelinating PSD-95 mutant mice. Because Kv1 channels are tetrameric and harbor four distinct PDZ-binding motifs, individual Kv1 channels might interact with both PSD-95 and another multi-domain PDZ protein through interactions that are differentially sensitive to detergent extraction (Fig. 6). It is still reasonable to postulate that an as yet unknown protein containing multiple PDZ-binding domains links Caspr2 and Kv1 channels. Such a protein could account for the normal recruitment and anchoring of Kv1 channels in high density clusters in both wild-type and PSD-95 mutant mice, and could provide a link to the cytoskeleton, perhaps through interactions with CRIPT-like proteins (Niethammer et al., 1998).
Other mechanisms of Kv1 channel localization in myelinated nerve fibers
Some evidence suggests that the aggregation of Kv1 channels and Caspr2 at juxtaparanodes may occur passively, via an exclusion of these proteins from axoglial junctions (Rosenbluth, 1976), followed by ‘tethering’ to PDZ domain-containing proteins and glial components. This sequence of events may be consistent with the detection of PSD-95 at juxtaparanodes after the initial clustering of Kv1 channels and Caspr2. Further, recent experiments using mutant mice with disrupted axoglial junctions have shown that loss of the normal paranodal structure results in Kv1 channels being mislocalized into paranodal zones (Dupree et al., 1999; Poliak et al., 2001). Interestingly, these same channels are still excluded from the node itself and do not colocalize with Nav channels. One might suppose that the high density of Nav channels at the node can exclude Kv1 channels from this membrane domain, but experiments during remyelination have shown that Kv1 channels and Nav channels can coexist at the node (Rasband et al., 1998). Thus, it is more likely that glial interactions are responsible for restricting Kv1 channels from the node.
What is the function of PSD-95?
Rather than clustering, anchoring, or linking Kv1 channels and Caspr2, perhaps PSD-95 plays a more dynamic role as a signaling, versus structural, scaffolding protein. PSD-95, through its multiple protein–protein interaction motifs, could link Kv1 channels and Caspr2 to other juxtaparanodal signaling proteins important in regulating the function of this axonal domain. Recent evidence shows that myelination and specific neuroglial interactions may regulate not only the localization of nodal ion channels, but also their expression levels (Boiko et al., 2001), suggesting the existence of a signaling pathway linking axoglial interaction at nodes of Ranvier and the neuronal nucleus. The idea that PSD-95 may participate in such a signaling complex is particularly attractive and intriguing when considering the node of Ranvier as an analogue to the chemical synapse in terms of subcellular specialization, molecular organization, and certain aspects of function. Although at present no evidence for this function of PSD-95 at the juxtaparanode exists, there is substantial support for such a role for PSD-95 at the excitatory chemical synapse. Migaud et al. (1998) showed that in this same PSD-95 mutant mouse strain that whereas NMDA receptors were still normally targeted and clustered at the postsynaptic density, long-term potentiation, as well as learning and memory, were substantially altered. These results suggested that PSD-95 is critical for subcellular signaling underlying synaptic plasticity in a much broader sense than clustering of NMDA receptors, perhaps through a role in mediating diverse intermolecular interactions between signaling proteins. Indeed, interactions between PSD-95, kinases, phosphatases, and other signaling proteins at post-synaptic sites have been well described (Fanning and Anderson, 1999; Kawachi et al., 1999; Colledge et al., 2000; Husi et al., 2000; Sheng, 2001). To determine if PSD-95 plays a similar role in myelinated nerve fibers, it will be necessary to both identify additional PSD-95 binding partners at juxtaparanodes and assay in more detail node of Ranvier function in the PSD-95 mutant mice. However, whatever the role of the high density of PSD-95 protein at juxtaparanodes of myelinated mammalian axons, based on the results presented here, the traditional view that PSD-95 functions principally as a channel clustering molecule must be challenged.
Materials and methods
Antibodies
The antigen used for production of the K28 mAbs (Tiffany et al., 2000) corresponds to amino acids 77–299 of PSD-95 (clone 2; Kim et al., 1995). The general strategy and methods used to generate these antibodies have been described previously in detail (Bekele-Arcuri et al., 1996). Other MAGUK-specific antibodies used here include a rabbit polyclonal anti-SAP102 antibody (Affinity BioReagents, Inc.), a rabbit anti–Chapsyn-110 antibody made against amino acids 347–416 from mouse Chapsyn-110 (a gift from Dr. M. Watanabe, Hokkaido University School of Medicine, Sapporo, Japan; Fukaya and Watanabe, 2000), and a monoclonal anti–SAP-97 antibody made against a SAP-97 GST fusion protein. The polyclonal and monoclonal anti-Kv1.2 and -Kvβ2 antibodies have been described previously (Rhodes et al., 1995; Bekele-Arcuri et al., 1996). The polyclonal Caspr2 antibody has also been described previously (Poliak et al., 1999).
Transient transfection and immunostaining of COS-1 cells
Cells were transfected with mammalian expression vectors for PSD-95, SAP97, Chapsyn-110, and SAP102 (Kim et al., 1995; Kim and Sheng, 1996 [the SAP102 cDNA was a gift from Dr. Craig Garner, University of Alabama, Birmingham, AL]) by calcium phosphate precipitation (Trimmer, 1998). COS-1 cells were seeded at 10% confluence on 25 μg/ml poly-l-lysine–coated coverslips and grown at 37°C in DME containing 10% (vol/vol) calf serum. The calcium phosphate DNA mixture was added within 24 h of seeding, when cells were approximately twice the original plating density, and left for 18 h. The transfection media was then removed, and after the addition of fresh media, the cells were incubated at 37°C for an additional 24 h. Cells were then washed three times in ice-cold PBS (10 mM phosphate buffer, pH 7.4, and 0.15 M NaCl) containing 1 mM MgCl2 and 1 mM CaCl2, and then fixed in the same buffer containing 3% (wt/vol) PFA and 0.1% Triton X-100 for 30 min at 4°C. After three washes with TBS (10 mM Tris-HCl, pH 8.0, and 0.15 M NaCl) containing 0.1% Triton X-100 (TBS + T), nonspecific protein-binding sites were blocked with Blotto (4% nonfat dry milk powder in TBS) containing 0.1% Triton X-100 (Blotto + T) for 1 h at RT. This was followed by incubation with the K28/43 mAb for 1 h at RT. Cells were washed three times with Blotto + T, incubated with Alexa 488 goat anti–mouse diluted in Blotto + T for 1 h, washed three times with TBS + T, mounted in 0.1 mg/ml _p_-phenylenediamine in 10% PBS, pH 9.0, 90% glycerol, and viewed under indirect immunofluorescence on an Axioscope microscope (Carl Zeiss MicroImaging, Inc.).
Expression cloning
A rat brain cDNA library, made from RNA isolated from P14–17 rat (insert size ranging from 2 to 4 kb, a gift of Dr. Terry Snutch, University of British Columbia, Vancouver, BC, Canada), and a mouse spinal cord cDNA library (insert size 0.3–4.5 kb) purchased from CLONTECH Laboratories, Inc., were immunoscreened using mAb K28/43. Each library was titrated to give ∼12,000 pfu per 15-cm plate. λ phage was combined with XL-1 Blue MRF' bacterial cultures for 15 min at 37°C, combined with top agar, and distributed across the surface of agar plates. The plates were then incubated for 3–5 h at 42°C until small plaques became visible. Nitrocellulose membranes were then prewet with 10 mM IPTG, and gently applied to the top of the agar plates. The agar plates were then incubated for 3.5 h at 37°C. The nitrocellulose filters were then removed and immunoblotted as previously described (Rhodes et al., 1997). A second prewet duplicate filter was then applied to each plate, incubated for 4–5 h at 37°C, and immunoblotted. Positive clones were picked based on the duplicate immunoblotting results and were placed in SM buffer (100 mM NaCl, 50 mM Tris-Hcl, pH 7.5, and 10 mM MgSO4) containing 4% chloroform. λ phage were retitered and rescreened as described above. A total of three screens were performed to obtain pure λ phage clones. Excision of the Bluescript phagemid was accomplished using the ExAssist® interference-resistant helper phage with SOLR strain (Stratagene) according to the manufacturer's protocol. Recovered plasmids were sequenced using a fluorescent DNA sequencer (ABI Prism; Applied Biosystems).
Affinity chromatography and mass spectrometry
Purified mAbs (K28/43 and K59/19) were cross-linked to Affi-Gel 10 (Bio-Rad Laboratories) per the supplier's protocol. RSCMs were prepared as described previously (Rasband and Trimmer, 2001b). 50 mg RSCM was extracted in a lysis buffer (TBS, pH 8.0, 5 mM EDTA, 1% Triton X-100, 1 mM iodoacetamide, and a protease inhibitor cocktail [2 μg/ml aprotinin, 1 μg/ml leupeptin, 2 μg/ml antipain, 10 μg/ml benzamidine, and 0.5 mM PMSF]) for 1 h, followed by clarification of the extract by centrifugation at 16,000 g for 30 min. Affinity columns were prewashed with ≥10 column volumes of ‘column buffer’ (TBS, pH 8.0, 5 mM EDTA, and 0.1% Triton X-100). The RSCM extract was passed over the K59/19 mAb column three times, after which the flow-through was passed over the K28/43 column three times. The columns were then washed with ≥10 volumes of column buffer to remove any unbound proteins. Bound proteins were then eluted with 0.1 M diethylamine, pH 11.5, and collected in 1-ml fractions. The fractions with peak immunoreactivity (determined by immunoblotting using the K28/43 mAb) were precipitated in 10% TCA for 30 min on ice, followed by centrifugation at 20,000 g for 10 min. The pellets were washed in 90% acetone (−20°C), and then solubilized in a reducing sample buffer for size fractionation by 9% polyacrylamide SDS-PAGE. Proteins were visualized by Coomassie blue staining, and distinct individual protein bands were excised, washed in H2O, and dried. Gel pieces were subjected to in situ trypsin digestion and subsequent mass determination of component peptides by mass spectrometry at the Stanford Protein and Nucleic Acid Facility. Obtained masses were used to identify proteins using ProFound software.
Immunoprecipitation
Immunoprecipitation reactions were performed essentially as described previously (Rhodes et al., 1995). 1 mg RBM or RSCM was solubilized in 1 ml of lysis buffer for 1 h at 4°C. Detergent extracts were collected by centrifugation at 16,000 g for 30 min, and then incubated with 1–5 μg of purified antibody for 2 h to overnight. 50 μl protein A or G conjugated to agarose or sepharose (50% vol/vol slurry) was added to the antibody–membrane extract lysate and then incubated for an additional 45 min. The agarose beads were then washed seven times with lysis buffer, pelleted by centrifugation, and the immunoprecipitation products eluted from the beads by addition of reducing sample buffer and boiling. For some experiments, membranes were first extracted in 200 μl of 2% SDS in immunoprecipitation buffer (137 mM NaCl, 2.7 mM KCl, 4.3 mM, Na2HPO4, 1.4 mM KH2PO4, 5 mM EDTA, 5 mM NaF, and 0.5 mM PMSF) and then diluted with 5 volumes of cold 1% Triton X-100 in lysis buffer for extract preparation (Lau et al., 1996). For some immunoprecipitation reactions involving mutant mouse brains (Migaud et al., 1998), membranes were solubilized in 100 μl SML buffer (PBS, pH 7.4, 2% SML, 2 mM MgCl2 and the protease inhibitor cocktail) and subsequently diluted 1:10 in PBS containing 2 mM MgCl2, followed by centrifugation for 30 min at 16,000 g (Poliak et al., 1999).
Immunoblotting and SDS-PAGE
Membranes, membrane extracts, insoluble membrane pellets, fractions from affinity columns, and/or products of coimmunoprecipitation reactions were separated on 7.5% or 9% SDS–polyacrylamide gels. For RBM and RSCM, 20–50 μg membrane protein was loaded per lane. Membrane extractions and affinity column fractions were added to an equal volume of 2× concentrated reducing sample buffer, boiled, and then loaded on gels. For coimmunoprecipitation analysis, 20 μl of the suspended beads (equivalent yield from 50 to 500 μg of starting membrane proteins) were applied per lane. Lauryl sulfate (Sigma-Aldrich) was the SDS source used in all SDS-PAGE experiments (Shi et al., 1994). Size-fractionated proteins were transferred electrophoretically to nitrocellulose paper, and the resultant immunoblots stained with mAb tissue culture supernatants (diluted 1:2–1:4) or a Caspr2 rabbit serum (diluted 1:250), as described previously (Rhodes et al., 1997). The blots were then incubated in enhanced chemiluminescence substrate (NEN Life Science Products) for 1 min and autoradiographed on Kodak XLS-1 film.
Immunofluorescence
Brains and optic nerves from adult and neonatal rats and adult mice were dissected and immediately fixed in ice-cold 4% PFA in 0.1 M phosphate buffer, pH 7.2, for 30 min (optic nerves) or 2 h (brains). Fixed tissue was transferred to ice-cold 20% sucrose (wt/vol) in 0.1 M phosphate buffer until equilibrated, then transferred again to 30% sucrose. The tissue was then frozen in Tissue-Tek OCT mounting medium. Sections were cut, placed in 0.1 M phosphate buffer, spread on gelatin coated coverslips, and allowed to air dry. Immunofluorescence staining was performed as described previously (Rasband et al., 2001). Secondary antibodies were Alexa 488– or Alexa 594–conjugated to goat anti–rabbit or goat anti–mouse antibodies (Molecular Probes, Inc.). Digital images were collected on a fluorescence microscope (Axioskop 2; Carl Zeiss MicroImaging, Inc.) fitted with an Axiocam color CCD camera. All digital images (both immunofluorescence and immunoblots) were prepared using Adobe Photoshop® and CorelDRAW®.
Acknowledgments
This work was supported by National Institutes of Health grants R37NS34383 and F32NS10906, and by the Spinal Cord Research Foundation grant 2040. E. Peles is an incumbent of the Madeleine Haas Russell Career Development Chair.
M.N. Rasband's present address is Dept. of Neuroscience, University of Connecticut Health Center, Farmington, CT 06030.
Footnotes
*
Abbreviations used in this paper: Kv, voltage-dependent potassium; MAGUK, membrane-associated guanylate kinase; Nav, voltage-dependent sodium; RBM, rat brain membrane; RSCM, rat spinal cord membrane; SML; sucrose monolaurate.
References
- Arroyo, E.J., and S.S. Scherer. 2000. On the molecular architecture of myelinated fibers. Histochem. Cell Biol. 113:1–18. [DOI] [PubMed] [Google Scholar]
- Arroyo, E.J., T. Xu, S. Poliak, M. Watson, E. Peles, and S.S. Scherer. 2001. Internodal specializations of myelinated axons in the central nervous system. Cell Tissue Res. 305:53–66. [DOI] [PubMed] [Google Scholar]
- Baba, H., H. Akita, T. Ishibashi, Y. Inoue, K. Nakahira, and K. Ikenaka. 1999. Completion of myelin compaction, but not the attachment of oligodendroglial processes triggers K+ channel clustering. J. Neurosci. Res. 58:752–764. [PubMed] [Google Scholar]
- Bekele-Arcuri, Z., M.F. Matos, L. Manganas, B.W. Strassle, M.M. Monaghan, K.J. Rhodes, and J.S. Trimmer. 1996. Generation and characterization of subtype-specific monoclonal antibodies to K+ channel α- and β-subunit polypeptides. Neuropharmacology. 35:851–865. [DOI] [PubMed] [Google Scholar]
- Berghs, S., D. Aggujaro, R. Dirkx, E. Maksimova, P. Stabach, J.M. Hermel, J.P. Zhang, W. Philbrick, V. Slepnev, T. Ort, and M. Solimena. 2000. βIV spectrin, a new spectrin localized at axon initial segments and nodes of Ranvier in the central and peripheral nervous system. J. Cell Biol. 151:985–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat, M.A., J.C. Rios, Y. Lu, G.P. Garcia-Fresco, W. Ching, M. St Martin, J. Li, S. Einheber, M. Chesler, J. Rosenbluth, et al. 2001. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 30:369–383. [DOI] [PubMed] [Google Scholar]
- Boiko, T., M.N. Rasband, S.R. Levinson, J.H. Caldwell, G. Mandel, J.S. Trimmer, and G. Matthews. 2001. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 30:91–104. [DOI] [PubMed] [Google Scholar]
- Boyle, M.E., E.O. Berglund, K.K. Murai, L. Weber, E. Peles, and B. Ranscht. 2001. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 30:385–397. [DOI] [PubMed] [Google Scholar]
- Caldwell, J.H., K.L. Schaller, R.S. Lasher, E. Peles, and S.R. Levinson. 2000. Sodium channel Na(v)1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA. 97:5616–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge, M., R.A. Dean, G.K. Scott, L.K. Langeberg, R.L. Huganir, and J.D. Scott. 2000. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 27:107–119. [DOI] [PubMed] [Google Scholar]
- David, G., B. Modney, K.A. Scappaticci, J.N. Barrett, and E.F. Barrett. 1995. Electrical and morphological factors influencing the depolarizing after-potential in rat and lizard myelinated axons. J. Physiol. 489:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree, J.L., J.-A. Girault, and B. Popko. 1999. Axoglial interactions regulate the localization of axonal paranodal proteins. J. Cell Biol. 147:1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber, S., G. Zanazzi, W. Ching, S. Scherer, T.A. Milner, E. Peles, and J.L. Salzer. 1997. The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J. Cell Biol. 139:1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning, A.S., and J.M. Anderson. 1999. Protein modules as organizers of membrane structure. Curr. Opin. Cell Biol. 11:432–439. [DOI] [PubMed] [Google Scholar]
- Fukaya, M., and M. Watanabe. 2000. Improved immunohistochemical detection of postsynaptically located PSD-95/SAP90 protein family by protease section pretreatment: a study in the adult mouse brain. J. Comp. Neurol. 426:572–586. [PubMed] [Google Scholar]
- Hille, B. 2001. Ionic Channels of Excitable Membranes. Sinauer Associates, Inc., Sunderland, MA. 814 pp.
- Husi, H., and S.G. Grant. 2001. Proteomics of the nervous system. Trends Neurosci. 24:259–266. [DOI] [PubMed] [Google Scholar]
- Husi, H., M.A. Ward, J.S. Choudhary, W.P. Blackstock, and S.G. Grant. 2000. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3:661–669. [DOI] [PubMed] [Google Scholar]
- Kawachi, H., H. Tamura, I. Watakabe, T. Shintani, N. Maeda, and M. Noda. 1999. Protein tyrosine phosphatase zeta/RPTPβ interacts with PSD-95/SAP90 family. Brain Res. Mol. Brain Res. 72:47–54. [DOI] [PubMed] [Google Scholar]
- Kim, E., and M. Sheng. 1996. Differential K+ channel clustering activity of PSD-95 and SAP97, two related membrane-associated putative guanylate kinases. Neuropharmacology. 35:993–1000. [DOI] [PubMed] [Google Scholar]
- Kim, E., M. Niethammer, A. Rothschild, Y.N. Jan, and M. Sheng. 1995. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 378:85–88. [DOI] [PubMed] [Google Scholar]
- Kordeli, E., S. Lambert, and V. Bennett. 1995. AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J. Biol. Chem. 270:2352–2359. [DOI] [PubMed] [Google Scholar]
- Lambert, S., J.Q. Davis, and V. Bennett. 1997. Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J. Neurosci. 17:7025–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, L.F., A. Mammen, M.D. Ehlers, S. Kindler, W.J. Chung, C.C. Garner, and R.L. Huganir. 1996. Interaction of the N-methyl-D-aspartate receptor complex with a novel synapse-associated protein, SAP102. J. Biol. Chem. 271:21622–21628. [DOI] [PubMed] [Google Scholar]
- Laube, G., J. Roper, J.C. Pitt, S. Sewing, U. Kistner, C.C. Garner, O. Pongs, and R.W. Veh. 1996. Ultrastructural localization of Shaker-related potassium channel subunits and synapse-associated protein 90 to septate-like junctions in rat cerebellar Pinceaux. Brain Res. Mol. Brain Res. 42:51–61. [DOI] [PubMed] [Google Scholar]
- Manganas, L.N., and J.S. Trimmer. 2000. Subunit composition determines Kv1 potassium channel surface expression. J. Biol. Chem. 275:29685–29693. [DOI] [PubMed] [Google Scholar]
- Menegoz, M., P. Gaspar, M. Le Bert, T. Galvez, F. Burgaya, C. Palfrey, P. Ezan, F. Arnos, and J.A. Girault. 1997. Paranodin, a glycoprotein of neuronal paranodal membranes. Neuron. 19:319–331. [DOI] [PubMed] [Google Scholar]
- Migaud, M., P. Charlesworth, M. Dempster, L.C. Webster, A.M. Watabe, M. Makhinson, Y. He, M.F. Ramsay, R.G. Morris, J.H. Morrison, et al. 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 396:433–439. [DOI] [PubMed] [Google Scholar]
- Niethammer, M., J.G. Valtschanoff, T.M. Kapoor, D.W. Allison, T.M. Weinberg, A.M. Craig, and M. Sheng. 1998. CRIPT, a novel postsynaptic protein that binds to the third PDZ domain of PSD-95/SAP90. Neuron. 20:693–707. [DOI] [PubMed] [Google Scholar]
- Peles, E., and J.L. Salzer. 2000. Molecular domains of myelinated axons. Curr. Opin. Neurobiol. 10:558–565. [DOI] [PubMed] [Google Scholar]
- Peles, E., M. Nativ, M. Lustig, M. Grumet, J. Schilling, R. Martinez, G.D. Plowman, and J. Schlessinger. 1997. Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J. 16:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak, S., L. Gollan, R. Martinez, A. Custer, S. Einheber, J.L. Salzer, J.S. Trimmer, P. Shrager, and E. Peles. 1999. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 24:1037–1047. [DOI] [PubMed] [Google Scholar]
- Poliak, S., L. Gollan, D. Salomon, E.O. Berglund, R. Ohara, B. Ranscht, and E. Peles. 2001. Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J. Neurosci. 21:7568–7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband, M.N., and J.S. Trimmer. 2001. a. Developmental clustering of ion channels at and near the node of Ranvier. Dev. Biol. 236:5–16. [DOI] [PubMed] [Google Scholar]
- Rasband, M.N., and J.S. Trimmer. 2001. b. Subunit composition and novel localization of K+ channels in spinal cord. J. Comp. Neurol. 429:166–176. [DOI] [PubMed] [Google Scholar]
- Rasband, M.N., J.S. Trimmer, T.L. Schwarz, S.R. Levinson, M.H. Ellisman, M. Schachner, and P. Shrager. 1998. Potassium channel distribution, clustering, and function in remyelinating rat axons. J. Neurosci. 18:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband, M.N., J.S. Trimmer, E. Peles, S.R. Levinson, and P. Shrager. 1999. K+ channel distribution and clustering in developing and hypomyelinated axons of the optic nerve. J. Neurocytol. 28:319–331. [DOI] [PubMed] [Google Scholar]
- Rasband, M.N., E.W. Park, T. Vanderah, J. Lai, F. Porreca, and J.S. Trimmer. 2001. Distinct K+ channels on pain sensing neurons. Proc. Natl. Acad. Sci. USA. 98:13373–13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes, K.J., S.A. Keilbaugh, N.X. Barrezueta, K.L. Lopez, and J.S. Trimmer. 1995. Association and colocalization of K+ channel α- and β-subunit polypeptides in rat brain. J. Neurosci. 15:5360–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes, K.J., B.W. Strassle, M.M. Monaghan, Z. Bekele-Arcuri, M.F. Matos, and J.S. Trimmer. 1997. Association and colocalization of the Kvβ1 and Kvβ2 β-subunits with Kv1 α-subunits in mammalian brain K+ channel complexes. J. Neurosci. 17:8246–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth, J. 1976. Intramembranous particle distribution at the node of Ranvier and adjacent axolemma in myelinated axons of the frog brain. J. Neurocytol. 5:731–745. [DOI] [PubMed] [Google Scholar]
- Scannevin, R.H., and R.L. Huganir. 2000. Postsynaptic organization and regulation of excitatory synapses. Nat. Rev. Neurosci. 1:133–141. [DOI] [PubMed] [Google Scholar]
- Sheng, M. 2001. Molecular organization of the postsynaptic specialization. Proc. Natl. Acad. Sci. USA. 98:7058–7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, M., and C. Sala. 2001. PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 24:1–29. [DOI] [PubMed] [Google Scholar]
- Shi, G., A.K. Kleinklaus, N.V. Marrion, and J.S. Trimmer. 1994. Properties of Kv2.1 K+ channels expressed in transfected mammalian cells. J. Biol. Chem. 269:23204–23211. [PubMed] [Google Scholar]
- Tait, S., F. Gunn-Moore, J.M. Collinson, J. Huang, C. Lubetzki, L. Pedraza, D.L. Sherman, D.R. Colman, and P.J. Brophy. 2000. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axoglial junction. J. Cell Biol. 150:657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor, F.J., A. Bokhari, O. Rogero, M. Gorczyca, J. Zhang, E. Kim, M. Sheng, and V. Budnik. 1997. Essential role for dlg in synaptic clustering of Shaker K+ channels in vivo. J. Neurosci. 17:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffany, A.M., L.N. Manganas, E. Kim, Y.P. Hsueh, M. Sheng, and J.S. Trimmer. 2000. PSD-95 and SAP97 exhibit distinct mechanisms for regulating K+ channel surface expression and clustering. J. Cell Biol. 148:147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka, M., J.L. Dupree, B. Popko, and D. Karagogeos. 2002. The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J. Neurosci. 22:3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer, J.S. 1991. Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc. Natl. Acad. Sci. USA. 88:10764–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer, J.S. 1998. Analysis of K+ channel biosynthesis and assembly in transfected mammalian cells. Methods Enzymol. 293:32–49. [DOI] [PubMed] [Google Scholar]
- Vabnick, I., J.S. Trimmer, T.L. Schwarz, S.R. Levinson, D. Risal, and P. Shrager. 1999. Dynamic potassium channel distributions during axonal development prevent aberrant firing patterns. J. Neurosci. 19:747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., D.D. Kunkel, T.M. Martin, P.A. Schwartzkroin, and B.L. Tempel. 1993. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature. 365:75–79. [DOI] [PubMed] [Google Scholar]
- Wang, H., D.D. Kunkel, P.A. Schwartzkroin, and B.L. Tempel. 1994. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J. Neurosci. 14:4588–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., M.L. Allen, J.J. Grigg, J.L. Noebels, and B.L. Tempel. 1995. Hypomyelination alters K+ channel expression in mouse mutants shiverer and Trembler. Neuron. 15:1337–1347. [DOI] [PubMed] [Google Scholar]