Tyrosine phosphorylation of type Iγ phosphatidylinositol phosphate kinase by Src regulates an integrin–talin switch (original) (raw)
Abstract
Engagement of integrin receptors with the extracellular matrix induces the formation of focal adhesions (FAs). Dynamic regulation of FAs is necessary for cells to polarize and migrate. Key interactions between FA scaffolding and signaling proteins are dependent on tyrosine phosphorylation. However, the precise role of tyrosine phosphorylation in FA development and maturation is poorly defined. Here, we show that phosphorylation of type Iγ phosphatidylinositol phosphate kinase (PIPKIγ661) on tyrosine 644 (Y644) is critical for its interaction with talin, and consequently, localization to FAs. PIPKIγ661 is specifically phosphorylated on Y644 by Src. Phosphorylation is regulated by focal adhesion kinase, which enhances the association between PIPKIγ661 and Src. The phosphorylation of Y644 results in an ∼15-fold increase in binding affinity to the talin head domain and blocks β-integrin binding to talin. This defines a novel phosphotyrosine-binding site on the talin F3 domain and a “molecular switch” for talin binding between PIPKIγ661 and β-integrin that may regulate dynamic FA turnover.
Keywords: PIPKIγ661; focal adhesion; phosphotyrosine-binding domain; FAK; β-integrin
Introduction
Integrin binding to the ECM stimulates the formation of cell–matrix adhesions and mediates the linkage between the ECM and actin cytoskeleton (for review see Geiger et al., 2001; Zamir and Geiger, 2001). This linkage is generated by a submembrane interconnecting complex that consists of structural proteins such as talin, vinculin, paxillin, α-actinin, and tensin, signaling molecules including tyrosine kinases such as FAK and Src, and serine/threonine kinases, as well as various adaptor proteins. The molecular complexity of cell–matrix adhesions enables them to modulate both cell anchorage and transmembrane signaling (Sastry and Burridge, 2000; Geiger and Bershadsky, 2001). The most common forms of integrin-mediated cell–matrix adhesions in cultured cells are termed focal adhesions (FAs), fibrillar adhesions, or focal complexes, based on their size, shape, and location within the cell. Signal transduction through FAs has been implicated in the regulation of a number of key cellular processes, including growth factor–induced mitogenic signals, cell survival, cell locomotion and morphogenesis, and tissue assembly (Burridge and Chrzanowska-Wodnicka, 1996).
The dynamic regulation of FA assembly and disassembly is the central process for cell motility in response to extracellular stimuli (for review see Parsons et al., 2000). In addition to regulation by Rho family small GTPases, another key mechanism for modulation of FA organization and stability is tyrosine phosphorylation. Nonreceptor tyrosine kinases, FAK, and Src family kinases are involved in the phosphorylation of several FA proteins including paxillin, p130Cas, tensin, and FAK itself (for review see Cary and Guan, 1999; Frame et al., 2002). Tyrosine phosphorylation provides docking sites for additional molecules that contain phosphotyrosine (pY)-binding (PTB) domains (for review see Zamir and Geiger, 2001). Inhibition of tyrosine kinases decreases the level of intracellular tyrosine phosphorylation and blocks the development of FAs (for review see Frame et al., 2002). Consistently, activation of protein tyrosine phosphatases disrupts FAs, whereas their inhibition stimulates FA assembly. On the other hand, overactivated Src results in high levels of tyrosine phosphorylation but disruption of FAs (Kellie et al., 1986), and Src−/− cells form FAs that fail to turn over into fibrillar adhesions (Volberg et al., 2001). In addition, Src family kinases were implicated in regulation of integrin–cytoskeleton interactions (Felsenfeld et al., 1999). These data suggest that tyrosine phosphorylation plays an essential role in the formation and turnover of FAs. However, the exact mechanism underlying tyrosine phosphorylation–mediated modification of the integrin–cytoskeleton linkage and FA dynamics are still poorly defined.
Phosphatidylinositol 4,5-bisphosphate (PI4,5P2), a lipid second messenger, modulates FA formation by binding to vinculin, α-actinin, and talin facilitating protein–protein interactions and by regulating Rho family small G proteins (for review see Sechi and Wehland, 2000). The concentration of PI4,5P2 appears to be regulated locally, enabling spatial and temporal changes in its availability and signaling. Type Iγ661 phosphatidylinositol phosphate kinase (PIPKIγ661) targets to FAs and regulates FA assembly by generating PI4,5P2 and targeting talin to FAs (Di Paolo et al., 2002; Ling et al., 2002). Our previous data showed that PIPKIγ661 was tyrosine phosphorylated by FAK signaling, and this enhances PIP kinase activity and correlated with increased talin association (Ling et al., 2002). Here, we demonstrate that PIPKIγ661 is tyrosine phosphorylated on Y644 by Src family kinases and is potentially regulated by FAK signaling. Tyrosine phosphorylation of PIPKIγ661 is required for its strong talin interaction and stable FA targeting. Interestingly, PIPKIγ661 tyrosine phosphorylation strengthens its interaction with talin, effectively competing with β1-integrin for binding to talin. The combined data support a novel mechanism regulating FA dynamics by tyrosine phosphorylation and PI4,5P2 signaling.
Results
FAK signaling mediates the tyrosine phosphorylation of PIPKIγ661 at Y644
Previously, we observed that tyrosine phosphorylation of PIPKIγ661 was dependent on adhesion and FAK signaling (Ling et al., 2002). Here, we show that PIPKIγ661 tyrosine phosphorylation is dependent on both FAK phosphorylation on Y397 and FAK activity because neither coexpressed Y397F nor kinase-dead mutants of FAK induced phosphorylation of PIPKIγ661 (Fig. 1 A). To map the phosphorylation sites, we used the following PIPKIα/γ chimeras: PIPKIα/γ635, PIPKIα/γ661, and PIPKIα/γ636–661 as defined in the Materials and methods. We coexpressed the chimeras with wild-type FAK, and then analyzed their tyrosine phosphorylation by immunoprecipitation and immunoblotting. Both PIPKIα/γ661 and PIPKIα/γ636–661 (but not PIPKIα/γ635) were phosphorylated after FAK coexpression (Fig. 1 B). The COOH-terminal truncation mutants of PIPKIγ661 showed consistent results; PIPKIγ661Δ20 (1–641) was not phosphorylated, PIPKIγ661Δ13 (1–648) was phosphorylated but to a lesser extent, and PIPKIγ661Δ3 (1–657) was phosphorylated similar to the wild-type kinase (unpublished data). These data suggest that the major FAK-mediated phosphorylation sites are in the last 26 amino acids of PIPKIγ661. To define the phosphorylated residues, two Y to F point mutants were made, Y644F and Y649F (Fig. 1 C). The wild-type or Y to F mutant PIPKIγ661 was coexpressed in HEK293T cells with wild-type FAK or vector control, and tyrosine phosphorylation of PIPKIγ661 was determined after immunoprecipitation using anti-PIPKIγ antibody. As shown in Fig. 1 C, when FAK was overexpressed, PIPKIγ661Y649F was phosphorylated similar to the wild type. However, PIPKIγ661Y644F and PIPKIγ661Y644/649F showed dramatically reduced phosphorylation (Fig. 1 C), suggesting that Y644 is the phosphorylation site downstream of FAK signaling.
Figure 1.
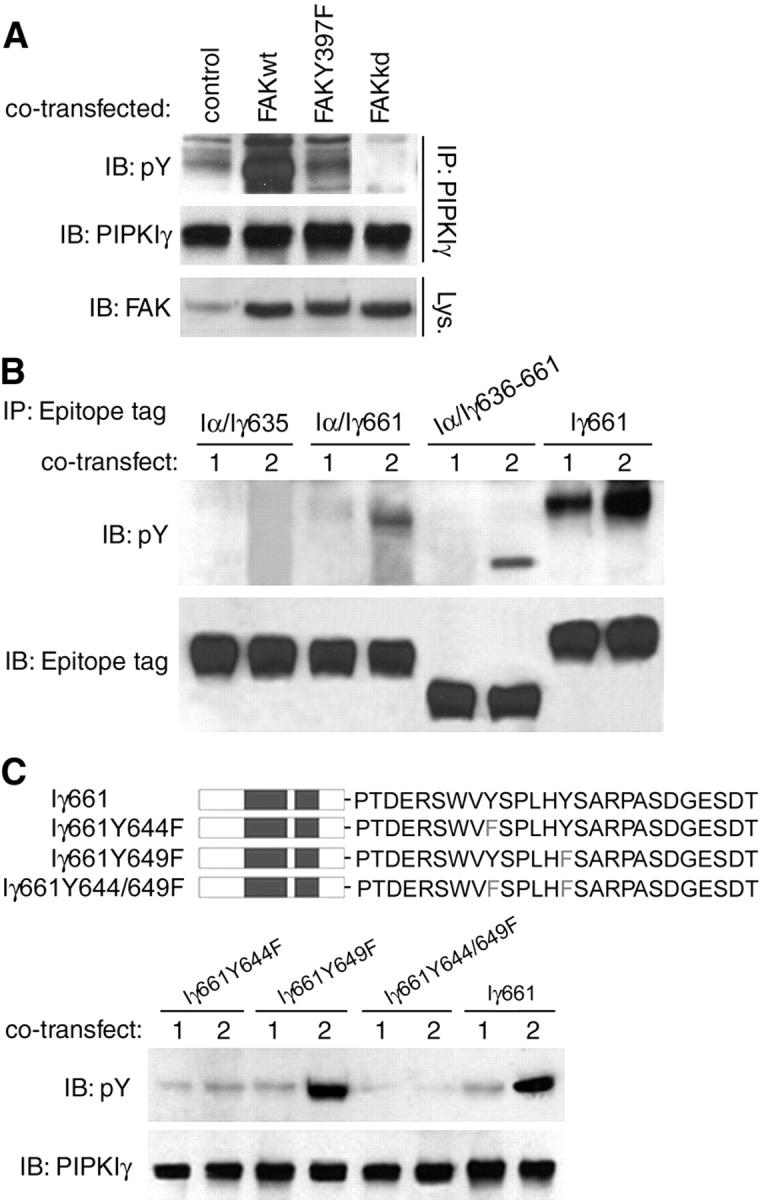
FAK signaling results in phosphorylation of PIPKIγ661 at Y644. (A) PIPKIγ661 was coexpressed with pcDNA3 (control), wild-type FAK (FAKwt), Y397F FAK (FAKY397F), or kinase-dead FAK (FAKkd) in HEK293T cells as indicated. After 48 h, tyrosine phosphorylation of PIPKIγ661 was analyzed by immunoprecipitation and Western blot as indicated. pY, phosphotyrosine. (B) The epitope-tagged PIPKIα/γ chimeras c-Myc-PIPKIα/γ635 (Iα/Iγ635), c-Myc-PIPKIα/γ661 (Iα/Iγ661), c-Myc-PIPKIα/γ636–661 (Iα/Iγ636–661), and wild-type HA-tagged PIPKIγ661 (Iγ661) were cotransfected with pcDNA3 (1) or wild-type FAK (2) in HEK293 cells for 48 h. Tyrosine phosphorylation of the PIPKI constructs was examined by immunoprecipitation and Western blot as indicated. (C) The wild-type (Iγ661) and Y to F mutant (Iγ661Y644F, Iγ661Y649F, Iγ661Y644/649F) PIPKIγ661 constructs were co-overexpressed in HEK293T cells with pcDNA3 (1) or wild-type FAK (2) for 48 h, and tyrosine phosphorylation of the PIPKIγ661 constructs was examined using immunoprecipitation and Western blot as indicated.
c-Src directly phosphorylates PIPKIγ661 at Y644
Src family tyrosine kinases are key factors that associate with FAK, thereby playing a fundamental role in downstream signaling and FAK-regulated processes (Parsons et al., 2000). Y397 is the autophosphorylation site in FAK and serves as a Src-binding site. Previous results have shown that either the Src-binding site (Y397) mutant or a kinase-dead mutant abrogated FAK's ability to promote PIPKIγ661 phosphorylation (Fig. 1 A). This suggests that phosphorylation of PIPKIγ661 is mediated by Src or FAK. To address the role of Src family kinases in PIPKIγ661 phosphorylation, Src activity was examined. The Src-specific inhibitor PP2 (but not the inactive analogue PP3) inhibited both basal and FAK-induced tyrosine phosphorylation of PIPKIγ661 (Fig. 2 A), showing that Src family kinases play an important role in tyrosine phosphorylation of PIPKIγ661. To further explore this possibility, we used the c-Src, Fyn, and Yes triple knockout cells (SFY−/−) and c-Src–reintroduced SFY−/− cells (Src+/+) to analyze the tyrosine phosphorylation of endogenous PIPKIγ661. As shown in Fig. 2 B, endogenous PIPKIγ661 showed very low phosphorylation in SFY−/− cells, whereas reintroduced c-Src restored PIPKIγ661 phosphorylation. Our results indicate that c-Src is directly involved in the tyrosine phosphorylation of PIPKIγ661, whereas the role of other Src family kinases members Fyn and Yes need further clarification. When the FAK knockout (FAK−/−) or their parent (FAK+/+) cells were examined, the endogenous PIPKIγ661 showed similar tyrosine phosphorylation in both cell lines (Fig. 2 B), indicating that FAK did not phosphorylate PIPKIγ661 directly, but enhanced its phosphorylation via regulation of Src.
Figure 2.

c-Src phosphorylates PIPKIγ661 at Y644. (A) HEK293T cells, transfected with PIPKIγ661 and the indicated constructs, were treated with 1 μM PP2 or PP3 for 30 min at 37°C. Then, cells were lysed and tyrosine phosphorylation of immunoprecipitated PIPKIγ661 was examined. (B) 2 mg lysate of c-Src/Fyn/Yes triple-knockout (SFY−/−), SFY−/− cells reintroduced with c-Src (Src+/+), FAK knockout (FAK−/−), or the FAK−/− parent (FAK+/+) cells were used for immunoprecipitation using purified anti-PIPKIγ antibody. Then, tyrosine phosphorylation of immunoprecipitated PIPKIγ was analyzed by Western blot as indicated. (C) In vitro Src kinase assay with His-tagged wild-type (wt-T) or mutant (Y644F-T, Y649F-T, or Y644/649F-T) PIPKIγ661 tails as substrates. Phosphorylation data were analyzed using SigmaPlot 4.0 from at least three independent experiments. (D) Analysis of tyrosine phosphorylation by tryptic phosphopeptide mapping. In vitro Src kinase assays using the substrates described in C were resolved by SDS-PAGE, and the tryptic digests of the tyrosine-phosphorylated substrates were analyzed on two-dimensional maps (exposure for 7 d at −70°C). Arrows indicate the spots corresponding to the loss of phosphorylation in the Y644F mutant. The large horizontal arrow points to the cathode and the large vertical arrow indicates the direction of chromatographic resolution.
These whole-cell assays were confirmed by in vitro kinase assays, which demonstrated that c-Src directly phosphorylated the His-tagged recombinant PIPKIγ661 tail (439–661; Fig. 2 C), whereas FAK only phosphorylated it very weakly (unpublished data). Both His-tagged recombinant PIPKIγ661Y644F and PIPKIγ661Y644/649F tails were not efficiently phosphorylated by c-Src in vitro compared with the wild-type PIPKIγ661, whereas the PIPKIγY649F tail was efficiently phosphorylated (Fig. 2 C). Furthermore, the phosphopeptide-mapping results confirmed that Y644 is the major substrate of c-Src because the Y644F mutant failed to yield two major phosphorylation spots compared with the wild-type PIPKIγ661 map (Fig. 2 D). These results demonstrate that Y644 is the major tyrosine phosphorylation site of PIPKIγ661 and that Src is the enzyme responsible for PIPKIγ661 phosphorylation.
c-Src associates with PIPKIγ661, and this is enhanced by FAK
To decipher the mechanism of PIPKIγ661 phosphorylation by Src, we examined whether there was an interaction between Src and PIPKIγ661. HEK293T cells were cotransfected with c-Src and PIPKIγ661 for 48 h, harvested, and PIPKIγ661 was immunoprecipitated with purified anti-PIPKIγ antibody. As shown in Fig. 3 A, c-Src was coimmunoprecipitated with PIPKIγ661. To further explore FAK's role in PIPKIγ661 phosphorylation, we coexpressed FAK with c-Src and PIPKIγ661. Coexpression of the wild-type FAK enhanced both tyrosine phosphorylation and Src association with PIPKIγ661 (Fig. 3 B). FAKY397F, which lacks the docking site for Src, inhibited the PIPKIγ661–c-Src association and failed to increase PIPKIγ661 phosphorylation to the same extant as FAK (Fig. 3 B). This demonstrates that the interaction of Src with FAK is required for FAK signaling to enhance Src binding and phosphorylation of PIPKIγ661.
Figure 3.
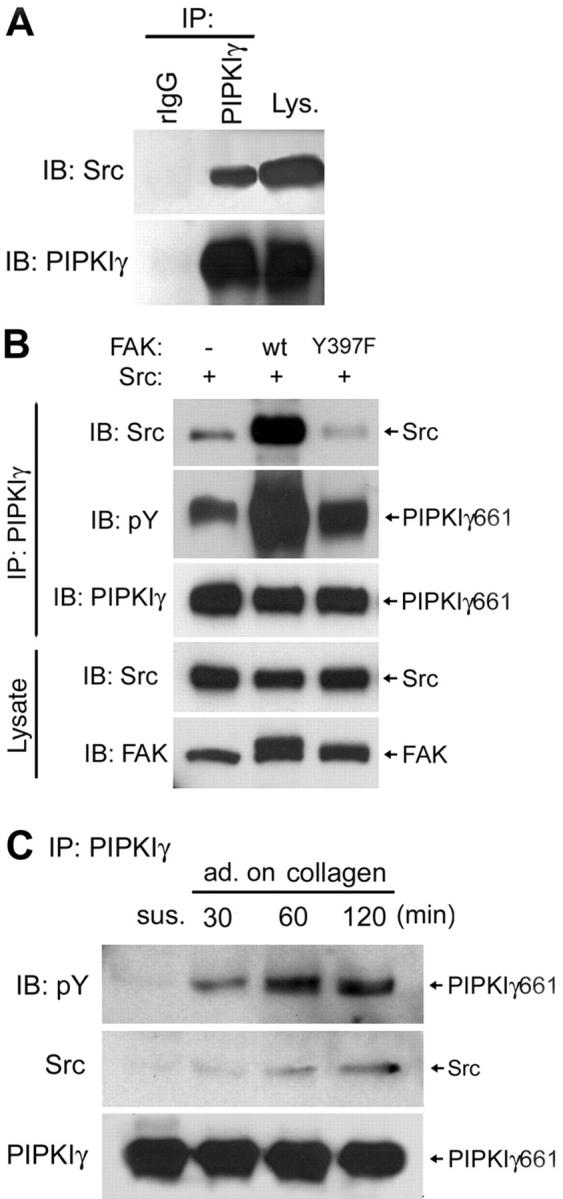
Src association with PIPKIγ661 is enhanced by FAK in vivo. (A) 48 h after cotransfection with c-Src and PIPKIγ661 constructs, HEK293T cells were immunoprecipitated using normal rabbit IgG (rIgG) or purified anti-PIPKIγ antibody. The immunoprecipitates and cell lysate were analyzed by Western blot as indicated. (B) HEK293T cells were transfected with PIPKIγ661, FAK, and/or Src constructs as indicated for 48 h, and then immunoprecipitated using the indicated antibodies. Immunoprecipitates were analyzed by Western blot as indicated. (C) A431 cells were lifted by trypsin, washed three times, and kept in suspension for 1 h at 37°C in serum-free DME. Cells were lysed (sus.) or plated on type I collagen-coated (10 μg/ml) plates in serum-free DME for the indicated time and then lysed, immunoprecipitated, and analyzed by Western blot as indicated.
Nevertheless, the PIPKIγ661 phosphorylation by Src is not absolutely dependent on FAK because PIPKIγ661 can be phosphorylated in FAK-null cells (Fig. 2 B). In addition, PIPKIγ635 and the PIPKIγ661 Y to F mutants coimmunoprecipitated with c-Src (unpublished data), suggesting that c-Src association with PIPKIγ requires a site distinct from the last 26 amino acids of PIPKIγ661 and independent of phosphorylation at Y644 or Y649. In A431 cells, endogenous PIPKIγ661 was tyrosine phosphorylated and associated with endogenous Src upon cell adhesion to type I collagen (Fig. 3 C). These results indicate that signals initiated from integrin binding to ligand regulate the Src–PIPKIγ661 association, and consequently, tyrosine phosphorylation of PIPKIγ661. The requirement of integrin-mediated cell adhesion suggests that FAK regulation is one potential signaling pathway that modulates the Src–PIPKIγ interaction. A431 cells highly express EGF receptors and, as a result, Src activity could be greatly enhanced. In other cell lines assayed, including HEK293T, NRK, and NIH3T3, the interaction between endogenous PIPKIγ661 and Src was more difficult to detect, although endogenous PIPKIγ661 is tyrosine phosphorylated in these cell lines and is dependent on adhesion on type I collagen (unpublished data; Ling et al., 2002).
Tyrosine phosphorylation regulates the PIPKIγ661–talin association and FA targeting of PIPKIγ661
Tyrosine phosphorylation of FA proteins regulates cell spreading and migration by generating protein–protein interaction sites for SH2 and PTB domains (for review see Panetti, 2002). To further explore the physiological significance of PIPKIγ661 tyrosine phosphorylation, we determined if the endogenous PIPKIγ661 bound to talin is tyrosine phosphorylated. Compared with endogenous PIPKIγ661 immunoprecipitated by anti-PIPKIγ antibody, endogenous PIPKIγ661 coimmunoprecipitated by anti-talin antibody had equivalent tyrosine phosphorylation but less PIPKIγ661 (Fig. 4 A), suggesting that PIPKIγ661 bound to talin is tyrosine phosphorylated to a greater extent than free PIPKIγ661 in vivo.
Figure 4.

Y644 of PIPKIγ661 is required for talin association. (A) Immunoprecipitations were performed with HEK293T cell lysate with normal rabbit IgG (rIgG), purified anti-PIPKIγ antibody, normal mouse IgG (mIgG), or monoclonal anti-talin antibody. The immunoprecipitates were analyzed by Western blot as indicated. (B) Top, HEK293T cells were cotransfected with PIPKIγ661 and indicated FAK construct, and then used for immunoprecipitation with purified anti-PIPKIγ antibody after 48 h. The immunoprecipitates were analyzed by Western blot as indicated. Bottom, quantification of talin association. (C) Top, HEK293 cells were transfected with wild-type (Iγ661), last 20 amino acids truncated (Iγ661Δ20), last 13 amino acids truncated (Iγ661Δ13), last 3 amino acids truncated (Iγ661Δ3), or Y to F mutant (Iγ661Y644F, Iγ661Y649F, Iγ661Y644/649F) PIPKIγ constructs for 48 h. Talin association with wild-type or mutant PIPKIγ661 was examined by immunoprecipitation and Western blot as indicated. Bottom, quantification of talin association. Data were quantified using NIH image 1.62 and plotted using SigmaPlot 4.0 from at least three independent experiments.
Cotransfection experiments also indicated that increased PIPKIγ661 tyrosine phosphorylation, induced by FAK, correlates with an increase in PIPKIγ661 bound to talin (Fig. 4 B; Ling et al., 2002). To characterize the role of PIPKIγ661 tyrosine phosphorylation, we examined the talin interaction with PIPKIγ661 phosphorylation-defective mutants by coimmunoprecipitation using HEK293T cells. Compared with the wild-type PIPKIγ661, PIPKIγ661Δ20 did not interact with talin, PIPKIγ661Δ13 had decreased talin association, and PIPKIγ661Δ3 maintained identical talin association (Fig. 4 C). The tyrosine point mutants PIPKIγ661Y644F and PIPKIγ661Y644/649F both showed substantially diminished talin association, whereas PIPKIγY649F retained talin association comparable with wild-type PIPKIγ661 (Fig. 4 C). These data demonstrate that tyrosine phosphorylation of the PIPKIγ661 COOH terminus regulates its interaction with talin.
The PIPKIγ661–talin association is important for targeting PIPKIγ661 to FAs (Di Paolo et al., 2002; Ling et al., 2002). To further explore targeting requirements, FA localization of PIPKIγ661 mutants were examined in NRK cells. Consistent with the ability to interact with talin in coimmunoprecipitation experiments, PIPKIγ661Δ20 lost FA targeting, PIPKIγ661Δ3 targeted to FAs and colocalized with talin similar to wild-type PIPKIγ661, and PIPKIγ661Δ13 partially retained FA targeting (Fig. 5 A). These data indicate that in addition to the minimum region 642WVYSPLH648 for in vitro talin binding (Di Paolo et al., 2002), in vivo talin association, FA targeting, and phosphorylation of PIPKIγ661 also require other residues in the COOH-terminal 26 amino acids. Consistently, the Y to F mutants lacking phosphorylation showed reduced talin association (Fig. 4 C) and lost FA targeting, whereas PIPKIγY649F localized to FAs similar to its wild-type counterpart (Fig. 5 A). Analysis of 600 cells under each condition in three independent experiments revealed a correlation with talin association (Fig. 4 C), FA targeting (Fig. 5 B), and tyrosine phosphorylation of PIPKIγ661 (Fig. 1 B). Additionally, PIPKIγ661 overexpressed in Src+/+ cells showed significantly greater FA targeting and colocalization with the pY staining compared with those overexpressed in SFY−/− cells (Fig. 5 C). In the FAK−/− cells, overexpressed PIPKIγ661 showed comparable FA targeting as in the parent cells (unpublished data), consistent with results demonstrating that FAK does not directly phosphorylate PIPKIγ661 in vivo. Together, these data show that c-Src–induced tyrosine phosphorylation of PIPKIγ661 enhances its FA targeting.
Figure 5.

Tyrosine phosphorylation of PIPKIγ661 is required for focal adhesion targeting. (A) NRK cells were transfected with wild-type or mutant PIPKIγ constructs using FuGENE™ 6 for 24 h. Overexpressed PIPKIγ constructs and endogenous talin in NRK cells were visualized by anti-PIPKIγ antibody (green) and anti-talin antibody (red). Bars, 10 μm. (B) Quantification of A. 200 transfected NRK cells were counted per slide. Percentage of FA targeting positive cells on each slide were calculated and plotted using SigmaPlot 4.0 from at least three independent experiments. (C) Src, Fyn, and Yes triple-knockout cells (SFY−/−) or c-Src–reintroduced SFY−/− cells (Src+/+) were transfected with wild-type PIPKIγ661 using FuGENE™ 6 for 24 h. Overexpressed PIPKIγ661 and pY were visualized by anti-PIPKIγ antibody (red) and anti-pY (PY99) antibody (green). Bars, 10 μm.
Tyrosine phosphorylation of PIPKIγ661 enhances talin interaction
To determine whether phenylalanine substitution or loss of the phosphorylated tyrosine is responsible for the reduced talin binding of PIPKIγ661 mutants, talin interactions were assessed by GST pull-down using recombinant nonphosphorylated His-PIPKIγ661 wild-type or Y to F mutant COOH terminus and GST-talin head domain. His-PIPKIγ661 Y to F mutants showed identical binding to talin compared with the wild-type protein (Fig. 6 A), suggesting that the decreased in vivo talin association resulted from the lack of tyrosine phosphorylation and not the phenylalanine substitution.
Figure 6.
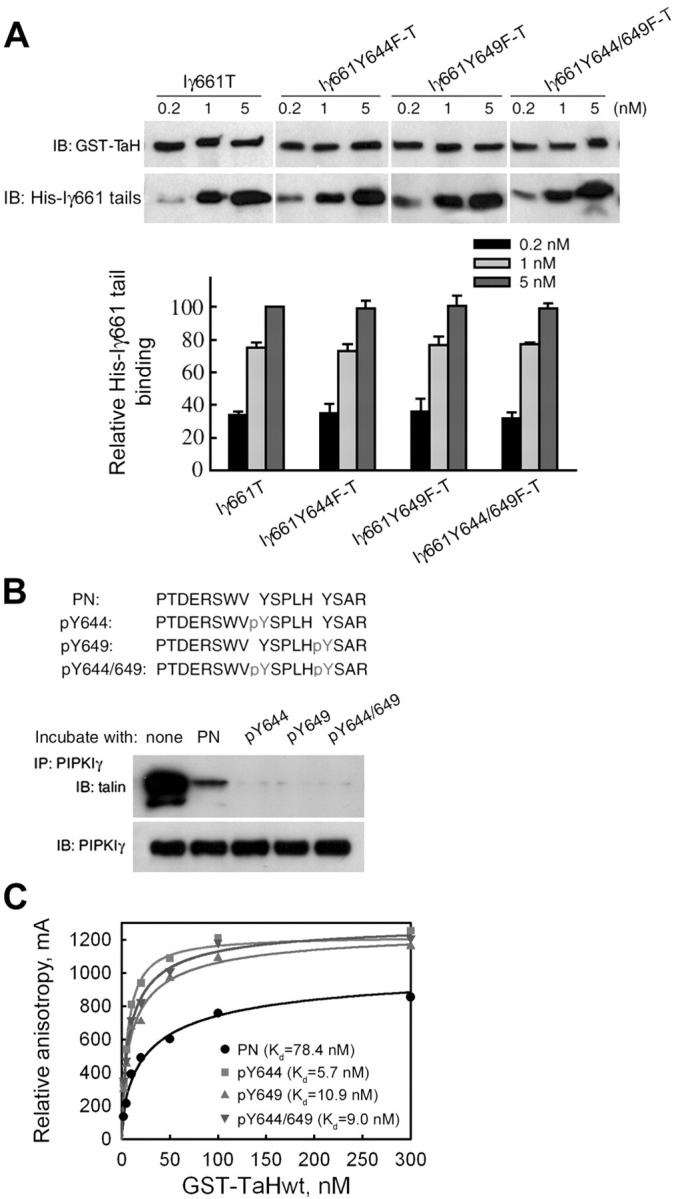
Tyrosine phosphorylation of PIPKIγ661 at Y644 and Y649 enhances its interaction with talin. (A) Top, effect of the Y to F mutation on PIPKIγ661 binding to talin. 50 nM GST talin head (GST-TaH) was incubated with indicated amount of His-tagged wild-type (Iγ661T) or mutant PIPKIγ661 tails (Iγ661Y644F-T, Iγ661Y649F-T, or Iγ661Y644/649F-T). Bottom, bound PIPKIγ661 tail bands were scanned, and the gray scales were quantified by NIH image 1.62 and then plotted using SigmaPlot 4.0 from at least three independent experiments. (B) Inhibition of PIPKIγ661–talin interaction by phosphorylated PIPKIγ661 peptides. Top, sequences and nomenclature of synthesized PIPKIγ661 peptides. Bottom, immunoprecipitation was performed using PIPKIγ661 overexpressing HEK293T lysates supplemented with 5 μM indicated PIPKIγ661 peptide. The talin–PIPKIγ661 association was analyzed from the immunoprecipitates by Western blot as indicated. (C) Direct binding curves of synthesized PIPKIγ661 peptides with wild-type GST fusion talin head domain. 20 nM fluorescein-labeled PIPKIγ661 peptides were incubated respectively with increased concentration of talin head in 0.1% BSA-PBS for 30 min at RT, and then anisotropy values were measured and analyzed using SigmaPlot 4.0 from three independent experiments. Kd values of wild-type talin head binding to each PIPKIγ661 peptide are shown as insets.
To address the role of tyrosine phosphorylation, we designed PIPKIγ661 peptides containing the talin-binding sequence 642WVYSPLH648 and flanking sequences, including normal peptide (PN), Y644-phosphorylated (pY644), Y649-phosphorylated (pY649), or Y644/Y649 dual-phosphorylated (pY644/649) peptides (Fig. 6 B). HEK293T cell lysates were supplemented with these peptides and subsequently used for immunoprecipitation. The phosphorylated peptides inhibited PIPKIγ661–talin association much more efficiently than the nonphosphorylated peptide (Fig. 6 B). To further explore this, we labeled the PIPKIγ661 peptides with fluorescein at the NH2 terminus and examined their direct binding with purified GST-talin head using fluorescence polarization anisotropy. As shown in Fig. 6 C, all of the phosphorylated peptides showed higher binding affinity for GST-talin head compared with the nonphosphorylated peptide. These results demonstrate that PIPKIγ661 tyrosine phosphorylation enhances talin binding.
Phosphorylated PIPKIγ661 competes with β1-integrin for talin binding
Talin plays a key role in integrin-mediated signaling by linking integrins to the actin cytoskeleton and regulating integrin activation (Liddington and Ginsberg, 2002). The F3 lobe of the band 4.1, ezrin, radixin, moesin homology (FERM) domain of talin, structurally homologous to a PTB domain, binds to the β-integrin cytoplasmic tail (Calderwood et al., 2002; Garcia-Alvarez et al., 2003) and the last 26 amino acids of PIPKIγ661 (Di Paolo et al., 2002; Ling et al., 2002). Because tyrosine phosphorylation of PIPKIγ661 enhanced its interaction with talin, it is important to explore whether PIPKIγ661 would displace β-integrin from talin in a phosphorylation-dependent manner. We examined the ability of recombinant PIPKIγ proteins or peptides to compete with His-β1-integrin tail for binding to talin in vitro. His-PIPKIγ661 tail, but not His-PIPKIγ635 tail, competed with His-β1-integrin tail in a dose-dependent manner (Fig. 7 A), consistent with the recent report by Barsukov et al. (2003). In addition, we found that the phosphorylated PIPKIγ661 peptides were ∼20-fold more effective in displacement of His-β1-integrin tail from GST-talin head than the nonphosphorylated peptide (Fig. 7 B). The analysis of the integrin competition data revealed that Y644 or Y649 single- or double-phosphorylated PIPKIγ661 peptides shared similar efficiencies for displacing His-β1-integrin tail from GST-talin head that is ∼20-fold higher than the nonphosphorylated PIPKIγ661 peptide (Fig. 7 C). Our current data suggest that tyrosine-phosphorylated PIPKIγ661 could be a key element in Src-mediated regulation of integrin–cytoskeleton linkage by competing with β-integrin binding to the talin FERM domain.
Figure 7.
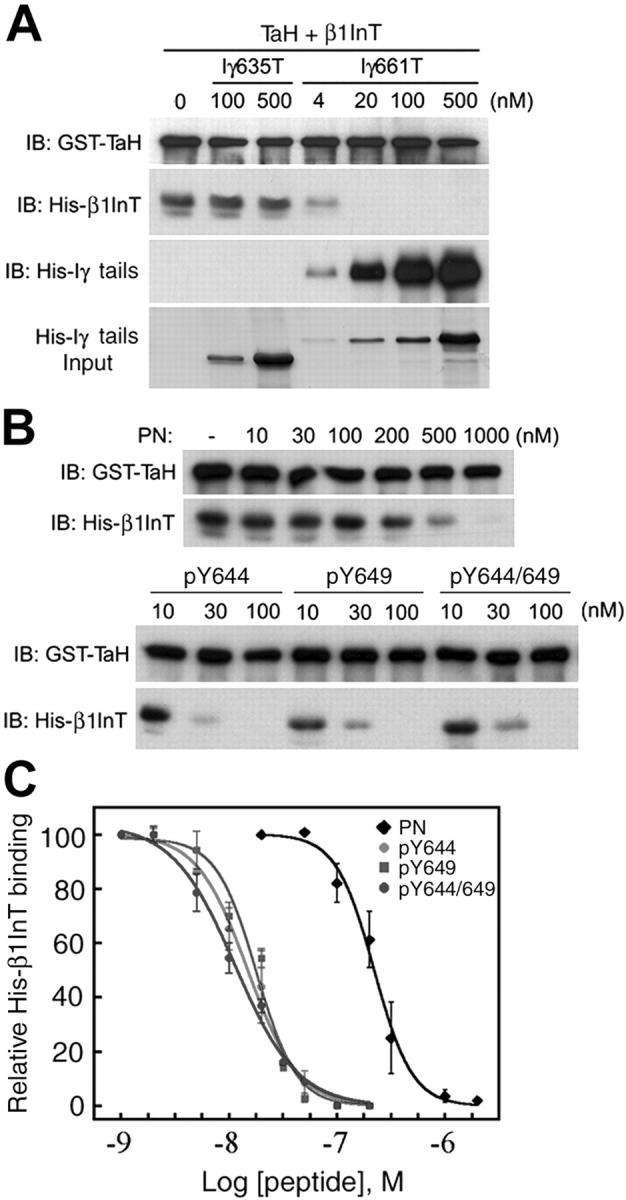
Phosphorylation of PIPKIγ661 disrupts integrin binding to talin. (A) 100 nM GST-TaH and 100 nM His-β1-integrin tail (His-β1InT) were used for GST pull-down assays with indicated amount of His-PIPKIγ635 tail (Iγ635T) or His-Iγ661T (Iγ661T). (B) The interactions between 100 nM GST-TaH and 100 nM His-β1InT, with indicated amounts of synthesized PIPKIγ661 peptides, were examined by GST pull-down assay and analyzed by Western blot as indicated. (C) The displacement curves of synthesized PIPKIγ661 peptides binding to GST-TaH in competition with His-β1InT were obtained by GST pull-down and Western blot. Both talin head bands and integrin tail bands were quantified by NIH image 1.62, and data were plotted by SigmaPlot 4.0 from at least three independent experiments.
PIPKIγ661 binds to talin on an overlapping yet distinct site from β-integrin, and K357 and R358 of talin are responsible for the enhanced binding of phosphorylated PIPKIγ661
As a key residue for binding to talin (Di Paolo et al., 2002), W642 of PIPKIγ661 aligns with the conserved tryptophan in the β-integrin tail (Garcia-Alvarez et al., 2003). PIPKIγ661 binding to talin through an interaction with W642 would position Y644 of PIPKIγ661 adjacent to K357 and R358 of talin. To determine if the increased binding affinity of the phosphorylated PIPKIγ661 sequences was due to these two basic residues, each residue was respectively mutated to glutamine, which is the most conservative mutation that eliminates charge but maintains side chain size. Although both K357Q and R358Q mutants abolished His-β1-integrin tail binding, neither mutation had a detectable effect on His-PIPKIγ661 tail binding (Fig. 8 A). The binding curves of His-PIPKIγ661 tail to the wild-type or mutant GST-talin head showed comparable properties (Fig. 8 B), showing that both K357Q and R358Q mutants have no effect on talin binding to PIPKIγ661. These data indicate that PIPKIγ661 binds to talin on an overlapping yet distinct site compared with β1-integrin, as concluded by Barsukov et al. (2003).
Figure 8.

PIPKIγ661 and β1-integrin bind to distinct sites on the talin head region. (A) The interactions between 50 nM wild-type His-Iγ661T or His-β1InT and 50 nM wild-type (GST-TaH), K357Q mutant (GST-TaHK357Q), R358Q mutant (GST-TaHR358Q) talin head region, or GST alone. (B) Binding curves of His-Iγ661T with 20 nM GST-TaH, GST-TaHK357Q, or GST-TaHR358Q were obtained by GST pull-down, Western blot, and quantification using NIH image 1.62, and were plotted using SigmaPlot 4.0 from at least three independent experiments. (C) Interactions between 20 nM His-Iγ661T and 20 nM GST-TaH, GST-TaHK357Q, or GST-TaHR358Q, with or without 500 nM synthesized PIPKIγ661 peptide as indicated, were analyzed by GST pull-down and Western blot as indicated. (D) Direct binding curves of synthesized PIPKIγ661 peptides with mutant GST fusion talin head domains were determined by fluorescence anisotropy. Kd values of wild-type or mutant talin head binding to each PIPKIγ661 peptide are shown as insets.
To assess the impact of PIPKIγ661 phosphorylation, the phosphorylated peptides were used to compete with His-PIPKIγ661 binding to the wild-type or mutant GST-talin head. As shown in Fig. 8 C, the K357Q mutant retained high affinity binding for the Y644-phosphorylated peptide, but lower affinity for the Y649-phosphorylated peptide. R358Q showed a loss of increased affinity for both Y644- and Y649-phosphorylated peptides (Fig. 8 C). These results indicate that both phosphorylated Y644 and Y649 require R358 for high affinity binding, but phosphorylated Y649 appeared to also require K357. To further examine this hypothesis, we examined the direct binding of fluorescein-labeled PIPKIγ661 peptides with the mutant GST-talin head. To the K357Q mutated talin head, the binding affinity of Y649-phosphorylated peptide dropped to the comparable level with the nonphosphorylated peptide, whereas the Y644- and double-phosphorylated peptides maintained their higher binding affinity (Fig. 8 D). All of the phosphorylated peptides showed decreased binding affinity to the R358Q mutant compared with the wild-type talin head (Fig. 8 D), suggesting that R358 is critical for phosphorylated PIPKIγ661 peptide binding. The anisotropy data revealed that phosphorylation at Y644 or Y649 enhanced the binding affinity to talin head ∼15-fold. These results support the GST pull-down displacement data that K357 is responsible for tighter binding induced by phosphorylation of Y649, whereas R358 is critical for tighter binding to talin induced by phosphorylation of both Y644 and Y649. Both nonphosphorylated and phosphorylated PIPKIγ661 peptides showed 3–4 times lower maximum change of anisotropy with the R358Q talin head mutant, consistent with the recent work by Barsukov et al. (2003). Previous data demonstrate that there is no detectable effect on full-length PIPKIγ661 tail binding to R358Q talin head (Fig. 8, A and B). This gives rise to the possibility that residues in the last 26 amino acids other than the 15 amino acids in the synthesized PIPKIγ661 peptide are involved in the interaction with talin head domain. In addition, the Kd value of nonphosphorylated PIPKIγ661 peptide for the full talin head is eightfold lower than the reported value for F2F3 domain of talin (Barsukov et al., 2003), suggesting additional contribution to the binding energy.
Because dimerization of GST may introduce artifactual results, we also performed experiments using His-tagged PIPKIγ661 tail and S-tagged recombinant talin head proteins to repeat the representative results. These experiment showed consistent results with those obtained from the GST pull-downs (unpublished data).
Tyrosine phosphorylation of β1-integrin tail decreases its interaction with talin
The enhanced association of the phosphorylated PIPKIγ661 peptides with talin raised the question of whether the β1-integrin tail may bind to talin with increased affinity when phosphorylated at the 780WDT782 and 785NPIY788 motif because the WDT motif aligns with 642WVY644 of PIPKIγ661. To address this question, β1-integrin peptides phosphorylated at T783 and/or Y788 were synthesized and used to compete integrin tail binding to talin. The T783-phosphorylated peptide competed with the integrin tail for talin to a similar extent as the nonphosphorylated peptide, whereas the Y788-phosphorylated peptide showed lower binding affinity (Fig. 9). These data indicate that phosphorylation of the Y788 residue diminished the interaction with GST-talin, consistent with previous reports (Tapley et al., 1989; Garcia-Alvarez et al., 2003). These results identify a region on the F3 lobe of talin as a PTB domain that can bind both phosphorylated PIPKIγ661 or unphosphorylated integrin. The loss of talin binding to phosphorylated integrin and the enhanced binding to phosphorylated PIPKIγ661 defines a mechanism by which tyrosine phosphorylation of PIPKIγ661 tightly regulates FA dynamics via an integrin–talin switch.
Figure 9.

Phosphorylation of β1-integrin tail does not increase interaction with talin. Top, sequences and nomenclature of the synthesized β1-integrin peptides. Bottom, the interactions between 100 nM GST-TaH and 100 nM His-β1InT with indicated amount of indicated β1-integrin peptides.
Discussion
PIPKIγ661 is a newly recognized FA-targeting phosphatidylinositol phosphate kinase isoform, and was identified as an important regulator for FA assembly through recruiting talin to and generating PI4,5P2 at FAs (Di Paolo et al., 2002; Ling et al., 2002). Previously, the tyrosine phosphorylation of PIPKIγ661 was shown to regulate its PIP kinase activity (Ling et al., 2002). The present work demonstrates that the PIPKIγ661–talin interaction and the PIPKIγ661 FA targeting require Src-mediated tyrosine phosphorylation of PIPKIγ661 at Y644. This enables PIPKIγ661 to produce PI4,5P2 at FAs, thereby regulating FA assembly. Tyrosine phosphorylation has been shown to regulate FA turnover; however, the underlying mechanism is poorly defined. We speculate that tyrosine phosphorylation of PIPKIγ661 provides a plausible mechanism for the dynamic regulation of FA organization.
PIPKIγ661 binds talin via the talin FERM domain. The binding region has been narrowed to the F3 lobe of the FERM domain, and this lobe has structural homology to PTB domains (Calderwood et al., 2002; Garcia-Alvarez et al., 2003). Here, we have shown that phosphorylated PIPKIγ661 peptides bind with high affinity to the F3 lobe, and K357 and R358 are key residues that bestow increased affinity to tyrosine-phosphorylated PIPKIγ661. To understand the interaction between talin and phosphorylated PIPKIγ661, we structurally modeled the talin F3 lobe with the dual phosphorylated PIPKIγ661 peptide (Fig. 10 A). This model shows that Y644 is located close to K357 and R358 of talin and forms a charge–charge interaction with these residues when phosphorylated. This model is supported by the observation that mutation of K357 and R358 to glutamine ablated the enhanced binding of the Y644-phosphorylated sequences. Unlike the integrin sequence, P646 of PIPKIγ661 creates a turn that also positions Y649 directly adjacent to K357 of talin. This model is consistent with the result that the K357Q mutant lost the enhanced binding to the Y649-phosphorylated PIPKIγ661 peptide. Y649 of PIPKIγ661 is in a similar position to Y788 of β1A-integrin, which was reported to be phosphorylated in Src-transformed cells (Sakai et al., 2001). However, phosphorylation of Y788 resulted in the loss of integrin binding to the talin head (Fig. 9; Tapley et al., 1989; Garcia-Alvarez et al., 2003). This indicates that P786 of integrin locates integrin Y788 to a position where the tyrosine-phosphorylated residue may be sterically hindered from interacting with K357 or R358 of talin.
Figure 10.

Models of talin–PIPKIγ661 interaction and PIPKIγ661-mediated regulation of focal adhesion organization. (A) Structure model of talin F3 lobe with dual phosphorylated PIPKIγ661 peptide (green) bound (phosphates are highlighted as red). Structure model was generated with the Sybyl molecular modeling program using the crystal structure as guide to dock the phosphorylated peptides, which were then energy minimized with the talin F3 lobe. (B) Signaling mechanism depicting PIPKIγ661 regulation of FAs and the interplay with β-integrin.
Our data demonstrate that the FERM domain of talin acts as a PTB domain by coordinating binding of two different tyrosine-phosphorylated sequences in PIPKIγ661. This may be a general function of FERM domains because the K357 and R358 positions are conserved in other FERM domains including moesin, radixin, ezrin, band 4.1. This suggests that there is structurally conserved binding between FERM domains and phosphorylated tyrosine residues in sequences similar to the PIPKIγ661 tail. In addition, the F3 lobe of talin does bind tyrosine-phosphorylated residues in the context of Y649 in the sequence PLHY, which is similar to the NPXY sequences that PTB domains normally bind. This demonstrates that the F3 lobe of talin is a remarkably flexible PTB domain that can bind two distinct phosphorylated tyrosine residues in different sequence contexts.
Current results suggest a mechanism as illustrated in Fig. 10 B, where talin switches between an association with integrin or PIPKIγ661 in a dynamic and highly regulated fashion. It is demonstrated here that both FAK and Src are involved in PIPKIγ661 tyrosine phosphorylation. Endogenous PIPKIγ661 tyrosine phosphorylation was dependent on adhesion to type I collagen, an α2β1 integrin-dependent signaling process. As a component of this pathway, FAK activates Src, which then phosphorylates PIPKIγ661. In addition, FAK stimulates an association between Src and PIPKIγ661. This association was also stimulated by adhesion to type I collagen, indicating that the Src association is physiologically relevant. The Src–PIPKIγ661 association is likely important for efficient phosphorylation of PIPKIγ661. These combined data suggest that α2β1 integrin signaling is one mechanism that leads to phosphorylation of PIPKIγ661.
The PIPKIγ661-binding site on talin overlaps with the integrin-binding site, and this competitive interaction is likely the key for modulation of PIPKIγ661 function at FAs. Current results demonstrate that tyrosine phosphorylation of PIPKIγ661 dramatically enhances talin interaction and FA targeting, which enable the generation of PI4,5P2 at FAs to improve the integrin–talin interaction. The enhancement of the integrin–talin association by PI4,5P2 may ultimately displace PIPKIγ661 from talin, resulting in a reduction of PI4,5P2. Via this mechanism, PI4,5P2 production is self limiting, and in the context of this model would be highly dynamic. The tyrosine phosphorylation of PIPKIγ661 may also be regulated by PI4,5P2 production because the kinase-dead enzyme is 20-fold less tyrosine phosphorylated (Ling et al., 2002). This results in a spatial and temporal generation of PI4,5P2 that is regulated by integrin binding to ECM, and by Src, FAK, or other signals. Although we have not yet identified a kinase that phosphorylates Y649, this potential phosphorylation site may be a link to other signaling pathways for the modulation of PIPKIγ661–talin association and FA dynamics.
FAK and Src family members are important regulators of FA dynamics (for review see Cary and Guan, 1999; Frame et al., 2002). Src is required in directed migration stimulated by growth factors. There is also evidence that adhesion to the ECM is required for cells to become responsive to growth factors, and this responsiveness correlates with stimulation of PI4,5P2 production by ECM adhesion (McNamee et al., 1993). As shown in Fig. 10 B, Src activation by integrin leads to phosphorylation of PIPKIγ661, enhancing both talin binding and PIP kinase activity (Ling et al., 2002). Because growth factor receptor and integrin signaling regulate Src activity, both pathways may lead to phosphorylation of PIPKIγ661 and regulation of migration.
Recent work has revealed that talin provides the initial connections between the ECM, integrin, and the cytoskeleton (Jiang et al., 2003). Moreover, talin binding to integrin has been identified as the final step leading to integrin activation (Tadokoro et al., 2003). This “inside-out” integrin signaling modulates both integrin-binding affinity to the ECM and internal connection to the cytoskeleton via the regulated association with talin (Calderwood and Ginsberg, 2003; Calderwood et al., 1999, 2002; Ulmer et al., 2003). Currently, the only known enhancer of the integrin–talin interaction is PI4,5P2, which induces an ∼10-fold increase of the binding affinity (Martel et al., 2001). Regulated production of PI4,5P2 at FAs, via Src-mediated phosphorylation of PIPKIγ661, may be key for modulation of the integrin–talin interaction, and subsequently, integrin activity and binding affinity for the ECM. This would provide a mechanism by which migrating cells can spatially and temporally control adhesion to the ECM differently at the leading or trailing edge.
Materials and methods
Constructs
Human talin1 head region (1–435) was cloned from HEK293T cells by RT-PCR using the OneStep RT-PCR kit (QIAGEN) and was subcloned into pET42. The COOH-terminal truncated or site-directed mutagenesis for the PIPKIγ661 mutants or talin head was performed using PCR-primer overlap extension with mutagenic primers. PIPKIγ661 mutants were then subcloned into pcDNA3.1, and talin head mutants were subcloned into pET42. His-tagged mouse β1-integrin tail was made following the published method (Pfaff et al., 1998), and was subcloned into pET28. The COOH terminus of PIPKIγ635 (439–635), PIPKIγ661 (439–661), or the Y to F mutants of PIPKIγ661 were PCR amplified from the corresponding pcDNA3.1 constructs and subcloned into pET28. PIPKIα/γ439–635 or PIPKIα/γ439–661 were constructed by fusing residues 1–444 of PIPKIα and residues 439–635 of PIPKIγ635 (PIPKIα/γ635) or 439–661 of PIPKIγ661 (PIPKIα/γ661) as described in Ling et al. (2002). PIPKIα/γ636–661 was constructed by fusion of the COOH-terminal 26 residues of PIPKIγ661 (PIPKIα/γ636–661) to the COOH terminus of PIPKIα as described by Ling et al. (2002). All mutants were confirmed by DNA sequence analysis. FAK, FAKY397F, and kinase-dead FAK constructs were gifts from Dr. Patricia J. Keely (University of Wisconsin, Madison, Madison, WI). The c-Src construct was provided by Dr. Alan C. Rapraeger (University of Wisconsin, Madison, Madison, WI).
Cell cultures and transfection
HEK293T cells, NRK cells, and A431 cells were cultured using DME supplemented with 10% FBS. FAK−/−, FAK+/+, SFY−/−, and Src+/+ cells, provided by Dr. Patricia J. Keely, were maintained in DME supplemented with 10% FBS. NRK cells were plated on glass coverslips and transfected using FuGENE™ 6 following the manufacturer's instructions. 0.5 × 106 HEK293T cells were transfected by calcium phosphate using 2 μg PIPKIγ expression vectors together with 4 μg empty pcDNA3 vector, 2 μg empty pcDNA3 vector plus 2 μg wild-type or mutant FAK, 2 μg empty pcDNA3 vector plus 2 μg c-Src, or 2 μg wild-type or mutant FAK plus 2 μg c-Src. SFY−/− and Src+/+ cells were plated on glass coverslips for 16 h and then used for immunofluorescence.
Antibodies
Anti-talin and anti-Flag (M2) antibodies were purchased from Sigma-Aldrich. Monoclonal mouse anti-PY (4G10), anti-Src (EC10), and anti-FAK (4.47) were obtained from Upstate Biotechnology. Anti-HA and anti-c-Myc antibodies were purchased from Covance. HRP-conjugated anti-GST antibody was purchased from Amersham Biosciences. Anti-His and HRP-conjugated anti-T7 antibodies were purchased from Invitrogen. Polyclonal PIPKIγ anti-serum was generated and purified as described previously (Ling et al., 2002). Secondary antibodies were obtained from Jackson ImmunoResearch Laboratories.
Protein expression and purification in Escherichia coli
pET28 or pET42 constructs were transformed in BL21(DE3) (Novagen). Proteins were expressed and purified using His resin following the manufacturer's instructions (Novagen).
In vitro Src kinase assay and phosphopeptide mapping
Primary chicken embryo cells were prepared as described previously (Reynolds et al., 1989). Exogenous c-SrcY527F was expressed using the RCAS B replication competent avian retroviral vector as described previously (Schaller et al., 1999). Cells were transfected with LipofectAMINE™ (Life Technologies) and LipofectAMINE™ Plus (Life Technologies) according to the manufacturer's instructions. Approximately 7–9 d after transfection, cells were lysed in RIPA buffer.
Overexpressed c-Src was immunoprecipitated from ∼1 mg chicken embryo cell lysates using EC10 and protein A–Sepharose (Amersham Biosciences) for 1 h at 4°C. Immune complexes were washed twice with RIPA buffer, twice with TBS, and twice with kinase reaction buffer (20 mM Pipes, pH 7.2, 5 mM MnCl2, and 5 mM MgCl2). The immune complexes were incubated with 4 μg recombinant substrates for various times at RT in kinase reaction buffer containing 35 μM ATP including 10 μCi γ[32P]ATP (6,000 Ci mmol−1; PerkinElmer), and were then terminated by adding sample buffer. Samples were resolved by SDS-PAGE, stained with SYPRO® Orange (Molecular Probes, Inc.), and analyzed after drying for fluorescence and by phosphorimaging, using a PhosphorImager® (Storm®; Molecular Dynamics).
4 μg recombinant substrates were tyrosine phosphorylated using c-SrcY527F in vitro as described above. The samples were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and visualized by autoradiography. The 32P-labeled substrate bands were excised and digested twice with 10 μg trypsin in 200 μl 0.05 M ammonium bicarbonate for 2 h each time at 37°C. The trypsinized membrane bands were oxidized on ice using 75 μl performic acid for 1 h and lyophilized as described by Boyle et al. (1991). Analysis by two-dimensional phosphopeptide mapping on thin-layer cellulose plates was performed using the Hunter thin-layer peptide mapping system (CBS Scientific) with methods described by Boyle et al. (1991). Electrophoretic resolution was performed in pH 1.9 buffer (2.2% formic acid, 7.8% acetic acid) for 30 min at 1,000 V. Then, ascending chromatography was performed for 3–4 h in phosphochromo buffer (37.5% _n_-butanol, 25% pyridine, and 7.5% acetic acid in water). The plates were dried and visualized by autoradiography at −70°C for 7 d with intensifying screens.
Immunoprecipitation and immunoblotting
Cells were harvested and lysed in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 5.0 mM NaF, 2 mM Na3VO4, 4 mM Na2P2O7, 1 mM EDTA, and 1 mM PMSF. 1–2-mg cell lysates were incubated with protein A–Sepharose and 5 μg anti-HA, anti-flag, anti-talin, or 10 μg purified anti-PIPKIγ antibody as indicated at 4°C overnight. The immunocomplexes were separated by SDS-PAGE, and analyzed as indicated.
Immunoblotting data were scanned, and the gray scale of each band was determined by NIH image 1.62 and then plotted using SigmaPlot 4.0 as described before (Ling et al., 2002).
Immunofluorescence and confocal microscopy
Immunofluorescence was performed as described previously (Ling et al., 2002). In brief, NRK cells 16 h after transfection, SFY−/−, or Src+/+ cells were washed with PBS at 37°C, fixed by 4% PFA at RT for 10 min, and permeabilized by 0.2% Triton X-100 in PBS at RT for 10 min. Then, cells were blocked by 3% BSA in PBS at RT for 30 min, incubated with the primary antibody for 1 h at 37°C, washed with 0.1% Triton X-100 in PBS, incubated with fluorescence-labeled secondary antibody at RT for 30 min, and then washed with 0.1% Triton X-100 in PBS. Cells were maintained and examined using a 60× Plan oil immersion lens on a confocal laser-scanning microscope (model MR-1000; Bio-Rad Laboratories) mounted transversely to an inverted microscope (Diaphot 200, Nikon; W.M. Keck Laboratory for Biological Imaging, Madison, WI). Images were processed as described previously (Ling et al., 2002) using Photoshop® 7.0.
Peptide synthesis and GST pull-down assay
Phosphorylated or nonphosphorylated peptides relevant to PIPKIγ661, named as PN (CDERSWVYSPLHYSAR), pY644 (CDERSWVpYSPLHYSAR), pY649 (CDERSWVYSPLHpYSAR), and pY644/649 (CDERSWVpYSPLHpYSAR), were synthesized and purified by HPLC at >95% purity (Invitrogen). Fluorescein-labeled tyrosine-phosphorylated or nonphosphorylated PIPKIγ661 peptides (labeled at the NH2 terminus) and peptides corresponding to β1-integrin tail, named as β1InPN (CMNAKWDTGENPIYKSA), β1InpT (CMNAKWDpTGENPIYKSA), β1InpY (CMNAKWDTGENPIpYKSA), and β1InpTpY (CMNAKWDpTGENPIpYKSA), were synthesized and purified by HPLC at >97% purity in the Peptide Synthesis Facility at the University of Wisconsin Biotechnology Center (Madison, WI). Purities, sequences, and correct phosphorylation of all synthesized peptides were confirmed by the Mass Spectrometry/Bioanalytical Facility at the University of Wisconsin (Madison, WI).
Purified GST-talin head proteins were incubated with PIPKIγ661 tails and/or β1-integrin tail, with or without synthesized PIPKIγ661 or β1-integrin peptides, together with glutathione Sepharose 4 Fast Flow (Amersham Biosciences) in 500 μl buffer B (PBS, 0.2% NP-40, and 2 mM DTT) for 4 h at 4°C. The beads were washed with 1 ml buffer B five times and analyzed by Western blot.
Fluorescence polarization assay
20 nM of each fluorescein-labeled PIPKIγ661 peptide was incubated without or with different titration of wild-type, K357Q, or R358Q mutated GST-talin head in PBS containing 0.1% BSA for 30 min at RT. Then the anisotropy value of each sample was determined using a photo counting spectroflurometer (SLM 8000C®; SLM Instruments) operating with an SLM 8100 hardware/software upgrade (Spectronic Instruments) at 25°C. Anisotropy value of peptide alone was abstracted from each of the relevant values of peptide bound to talin head. Data were analyzed and binding curves were plotted using SigmaPlot 4.0.
Acknowledgments
We thank Dr. Anna Huttenlocher and Dr. Patricia J. Keely at the University of Wisconsin, Madison (Madison, WI) for excellent discussions and reagents. We thank Martin G. Ensenberger for his technical help. We are grateful to Chateen Carbonara for her comments on the manuscript.
This work was supported by American Heart Association grant 133-EY51 to Kun Ling and by National Institutes of Health grant GM57549 to Richard A. Anderson and grant HL21644 to Deane F. Mosher. The authors declare that they have no competing financial interests.
Abbreviations used in this paper: FA, focal adhesion; FERM, band 4.1, ezrin, radixin, moesin homology; PI4,5P2, phosphatidylinositol 4,5-bisphosphate; PIPKIγ661, type Iγ phosphatidylinositol phosphate kinase 661; PTB, phosphotyrosine binding; pY, phosphotyrosine.
References
- Barsukov, I.L., A. Prescot, N. Bate, B.C. Patel, D.N. Floyd, N. Bhanji, C.R. Bagshaw, K. Letinic, G. Di Paolo, P. De Camilli, et al. 2003. PIP kinase type 1γ and β1-integrin cytoplasmic domain bind to the same region in the talin FERM domain. J. Biol. Chem. 278:31202–31209. [DOI] [PubMed] [Google Scholar]
- Boyle, W.J., P. van der Geer, and T. Hunter. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 201:110–149. [DOI] [PubMed] [Google Scholar]
- Burridge, K., and M. Chrzanowska-Wodnicka. 1996. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 12:463–518. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., and M.H. Ginsberg. 2003. Talin forges the links between integrins and actin. Nat. Cell Biol. 5:694–697. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., R. Zent, R. Grant, D.J. Rees, R.O. Hynes, and M.H. Ginsberg. 1999. The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274:28071–28074. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., B. Yan, J.M. de Pereda, B.G. Alvarez, Y. Fujioka, R.C. Liddington, and M.H. Ginsberg. 2002. The phosphotyrosine binding-like domain of talin activates integrins. J. Biol. Chem. 277:21749–21758. [DOI] [PubMed] [Google Scholar]
- Cary, L.A., and J.L. Guan. 1999. Focal adhesion kinase in integrin-mediated signaling. Front. Biosci. 4:D102–D113. [DOI] [PubMed] [Google Scholar]
- Di Paolo, G., L. Pellegrini, K. Letinic, G. Cestra, R. Zoncu, S. Voronov, S. Chang, J. Guo, M.R. Wenk, and P. De Camilli. 2002. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1γ by the FERM domain of talin. Nature. 420:85–89. [DOI] [PubMed] [Google Scholar]
- Felsenfeld, D.P., P.L. Schwartzberg, A. Venegas, R. Tse, and M.P. Sheetz. 1999. Selective regulation of integrin-cytoskeleton interactions by the tyrosine kinase Src. Nat. Cell Biol. 1:200–206. [DOI] [PubMed] [Google Scholar]
- Frame, M.C., V.J. Fincham, N.O. Carragher, and J.A. Wyke. 2002. v-Src's hold over actin and cell adhesions. Nat. Rev. Mol. Cell Biol. 3:233–245. [DOI] [PubMed] [Google Scholar]
- Garcia-Alvarez, B., J.M. de Pereda, D.A. Calderwood, T.S. Ulmer, D. Critchley, I.D. Campbell, M.H. Ginsberg, and R.C. Liddington. 2003. Structural determinants of integrin recognition by talin. Mol. Cell. 11:49–58. [DOI] [PubMed] [Google Scholar]
- Geiger, B., and A. Bershadsky. 2001. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13:584–592. [DOI] [PubMed] [Google Scholar]
- Geiger, B., A. Bershadsky, R. Pankov, and K.M. Yamada. 2001. Transmembrane crosstalk between the extracellular matrix–cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2:793–805. [DOI] [PubMed] [Google Scholar]
- Jiang, G., G. Giannone, D.R. Critchley, E. Fukumoto, and M.P. Sheetz. 2003. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature. 424:334–337. [DOI] [PubMed] [Google Scholar]
- Kellie, S., B. Patel, N.M. Wigglesworth, D.R. Critchley, and J.A. Wyke. 1986. The use of Rous sarcoma virus transformation mutants with differing tyrosine kinase activities to study the relationships between vinculin phosphorylation, pp60v-src location and adhesion plaque integrity. Exp. Cell Res. 165:216–228. [DOI] [PubMed] [Google Scholar]
- Liddington, R.C., and M.H. Ginsberg. 2002. Integrin activation takes shape. J. Cell Biol. 158:833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, K., R.L. Doughman, A.J. Firestone, M.W. Bunce, and R.A. Anderson. 2002. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 420:89–93. [DOI] [PubMed] [Google Scholar]
- Martel, V., C. Racaud-Sultan, S. Dupe, C. Marie, F. Paulhe, A. Galmiche, M.R. Block, and C. Albiges-Rizo. 2001. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J. Biol. Chem. 276:21217–21227. [DOI] [PubMed] [Google Scholar]
- McNamee, H.P., D.E. Ingber, and M.A. Schwartz. 1993. Adhesion to fibronectin stimulates inositol lipid synthesis and enhances PDGF-induced inositol lipid breakdown. J. Cell Biol. 121:673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panetti, T.S. 2002. Tyrosine phosphorylation of paxillin, FAK, and p130CAS: effects on cell spreading and migration. Front. Biosci. 7:d143–d150. [DOI] [PubMed] [Google Scholar]
- Parsons, J.T., K.H. Martin, J.K. Slack, J.M. Taylor, and S.A. Weed. 2000. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 19:5606–5613. [DOI] [PubMed] [Google Scholar]
- Pfaff, M., S. Liu, D.J. Erle, and M.H. Ginsberg. 1998. Integrin β cytoplasmic domains differentially bind to cytoskeletal proteins. J. Biol. Chem. 273:6104–6109. [DOI] [PubMed] [Google Scholar]
- Reynolds, A.B., S.B. Kanner, H.C. Wang, and J.T. Parsons. 1989. Stable association of activated pp60src with two tyrosine-phosphorylated cellular proteins. Mol. Cell. Biol. 9:3951–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, T., R. Jove, R. Fassler, and D.F. Mosher. 2001. Role of the cytoplasmic tyrosines of β1A integrins in transformation by v-src. Proc. Natl. Acad. Sci. USA. 98:3808–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry, S.K., and K. Burridge. 2000. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp. Cell Res. 261:25–36. [DOI] [PubMed] [Google Scholar]
- Schaller, M.D., J.D. Hildebrand, and J.T. Parsons. 1999. Complex formation with focal adhesion kinase: a mechanism to regulate activity and subcellular localization of Src kinases. Mol. Biol. Cell. 10:3489–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi, A.S., and J. Wehland. 2000. The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P 2 influences cytoskeletal protein activity at the plasma membrane. J. Cell Sci. 21:3685–3695. [DOI] [PubMed] [Google Scholar]
- Tadokoro, S., S.J. Shattil, K. Eto, V. Tai, R.C. Liddington, J.M. de Pereda, M.H. Ginsberg, and D.A. Calderwood. 2003. Talin binding to integrin β tails: a final common step in integrin activation. Science. 302:103–106. [DOI] [PubMed] [Google Scholar]
- Tapley, P., A. Horwitz, C. Buck, K. Duggan, and L. Rohrschneider. 1989. Integrins isolated from Rous sarcoma virus-transformed chicken embryo fibroblasts. Oncogene. 4:325–333. [PubMed] [Google Scholar]
- Ulmer, T.S., D.A. Calderwood, M.H. Ginsberg, and I.D. Campbell. 2003. Domain-specific interactions of talin with the membrane-proximal region of the integrin β3 subunit. Biochemistry. 42:8307–8312. [DOI] [PubMed] [Google Scholar]
- Volberg, T., L. Romer, E. Zamir, and B. Geiger. 2001. pp60c-src and related tyrosine kinases: a role in the assembly and reorganization of matrix adhesions. J. Cell Sci. 114:2279–2289. [DOI] [PubMed] [Google Scholar]
- Zamir, E., and B. Geiger. 2001. Molecular complexity and dynamics of cell-matrix adhesions. J. Cell Sci. 114:3577–3579. [DOI] [PubMed] [Google Scholar]