B7-1 or B7-2 Is Required to Produce the Lymphoproliferative Phenotype in Mice Lacking Cytotoxic T Lymphocyte–associated Antigen 4 (CTLA-4) (original) (raw)
Abstract
The costimulatory molecules B7-1 and B7-2 regulate T lymphocyte activation by delivering activating signals through CD28 and inhibitory signals through cytotoxic T lymphocyte–associated antigen 4 (CTLA-4). The importance of CTLA-4–mediated inhibition was demonstrated by the uncontrolled T cell activation and lymphoproliferative disease that develops in CTLA-4–deficient (−/−) mice. To examine the role of B7 signaling in the activation of CTLA-4–deficient T cells, we bred CTLA-4−/− mice with mice lacking B7-1, B7-2, or both B7 molecules. The CTLA-4/B7-1−/− and the CTLA-4/B7-2−/− mice develop lymphoproliferation and enhanced T cell activation. Mice lacking CTLA-4, B7-1, and B7-2 have a normal life-span, and do not have lymphocytic infiltrates in any organs, or increased T cell activation. Therefore, the two B7 molecules have overlapping functions, since either B7-1 or B7-2 alone can cause the CTLA-4−/− phenotype. Elimination of both B7-1 and B7-2 from the CTLA-4– deficient mouse abrogates the lymphocyte activation and disease, and does not reveal evidence for additional stimulatory CD28 ligands. The CTLA-4−/− phenotype can be reproduced with anti-CD28 antibody in mice lacking CTLA-4, B7-1, and B7-2, but wild-type mice are unaffected by the same treatment. This suggests that the inhibitory function of CTLA-4 can overcome strong CD28-mediated signaling in vivo.
Keywords: cytotoxic T lymphocyte–associated antigen 4, B7, knockout mouse, costimulation, T lymphocyte
The importance of the B7-CD28/cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) pathway has been highlighted by studies showing that blockade of B7 costimulation can prolong allograft survival (1) and suppress autoimmunity (2). The two B7 ligands, B7-1 (CD80) and B7-2 (CD86), can both stimulate immune responses by binding to CD28 and downregulate responses by binding to CTLA-4. Because B7-2 is expressed constitutively and upregulated earlier than B7-1, it has been suggested that B7-2 participates in initiating an immune response, whereas B7-1 may be more important in sustaining or regulating ongoing responses. The upregulation of CTLA-4 on activated T cells parallels the kinetics of B7-1 expression. Together with the higher avidities of B7 ligands for CTLA-4 than CD28, this has raised the possibility that B7-1/CTLA-4 interactions predominate late to terminate immune responses. However, several reports suggest that B7-1 and B7-2 can provide similar signals (3, 4). Further understanding of the complexities in this pathway is required in order to optimize therapies that target this pathway.
To study the interactions between receptors and ligands in the B7-CD28/CTLA-4 pathway, we have generated and characterized mice lacking expression of one or more molecules in the pathway. The phenotype of mice lacking CTLA-4 (CTLA-4−/−) provides crucial evidence that CTLA-4 plays a negative regulatory role in T cell activation. These mice develop a fatal lymphoproliferative disease with lymphocytic infiltrates in multiple organs (5, 6). CTLA-4−/− T cells are highly activated in vivo and spontaneously proliferate in vitro without any added stimuli. Early treatment with CTLA4Ig blocks the T cell activation and lymphoproliferation, demonstrating that B7, presumably functioning by ligating CD28, is necessary for the development of the CTLA-4−/− phenotype (7). However, these studies do not address whether CD28 interacts only with B7-1 and B7-2, or if additional ligands for CD28/ CTLA-4 exist, as some have proposed (8, 9). The T cell activation observed in CTLA-4−/− mice and its dependence on B7 molecules provide a unique system for exploring interactions in this pathway.
Here we have used the CTLA-4−/− mouse as a tool to investigate interactions between CD28 and its ligands. We have generated and characterized three novel mouse strains, which are CTLA-4–deficient and also lack B7-1, B7-2, or both B7 molecules. If additional stimulatory CD28 ligands existed, then mice lacking CTLA-4, B7-1, and B7-2 would be expected to develop increased T cell activation. Alternatively, if there is not increased T cell activation in these mice, then this would suggest that in the absence of the two known B7 molecules, CD28 is no longer engaged. We found that mice lacking CTLA-4 and either B7-1 or B7-2 develop the lymphoproliferative phenotype observed in the CTLA-4−/− strain, but that mice deficient in CTLA-4 and B7-1 have a shorter life-span and show greater T cell activation in vivo than the CTLA-4/B7-2– deficient mice. In contrast, mice lacking CTLA-4, B7-1, and B7-2 (“triple knockout,” CTLA-4/B7-1/B7-2−/− TKO) show no evidence of T cell activation or lymphoproliferation, suggesting that there are no additional stimulatory ligands for CD28. We also show that administration of anti-CD28 antibody to CTLA-4/B7-1/B7-2−/− TKO mice can reproduce the phenotype of the CTLA-4−/− mouse, demonstrating the critical role of CD28 in activating CTLA-4−/− T cells.
Materials and Methods
Mice.
Mouse strains lacking CTLA-4 (6), B7-1 (10), B7-2, or both B7 molecules (11) have been described previously. Because CTLA-4–deficient mice die before reaching sexual maturity, the three CTLA-4/B7–deficient mice were generated by interbreeding CTLA-4 heterozygotes with B7-1–, B7-2–, or B7-1/B7-2-deficient mice. Mice heterozygous for both CTLA-4 and the B7 mutation were then intercrossed, and CTLA-4/B7–deficient mice were obtained. Because of the lethality of the CTLA-4/B7-1– and CTLA-4/B7-2–deficient phenotypes, these two strains were maintained by interbreeding CTLA-4+/−/B7−/− mice. The CTLA-4/B7-1/B7-2−/− TKO strain survives to adulthood, and was maintained by interbreeding these mice. All strains were genotyped by Southern blots, as described previously for CTLA-4 (6), B7-1 (10), and B7-2 (11). Genotypes were confirmed by flow cytometric analysis of splenocytes stimulated with LPS 20 μg/ml and dextran sulfate 10 μg/ml for expression of B7 molecules, and splenocytes stimulated with anti-CD3 (mAb 145-2C11) for expression of CTLA-4. All mice were generated on an inbred 129/ SvJae background. Brigham and Women's Hospital and Harvard Medical School are accredited by the American Association of Accreditation of Laboratory Animal Care (AALAC), and mice were cared for in accordance with institutional guidelines in a pathogen-free animal facility.
In Vivo Administration of Anti-CD28.
Wild-type and CTLA-4/B7-1/B7-2−/− TKO mice received either PV-1 mAb to CD28, provided by Dr. Vijay K. Kuchroo (Brigham and Women's Hospital), or hamster IgG (ICN Pharmaceuticals). 100 μg i.v. of purified antibody was administered on days 0, 7, and 14, and mice were analyzed between days 14 and 19.
Cell Preparation and Cultures.
Single cell suspensions from spleen and lymph nodes were prepared by dissociating tissue with sterile glass slides. Red blood cells were lysed by incubation in Tris-ammonium chloride for 5 min at 37°C. To assay proliferation, 2 × 105 cells/well were cultured in flat-bottomed 96-well plates in media as described previously (6). Cells were pulsed with 1 μCi [3H]thymidine for the last 8 h of the indicated day. Anti-CD3 stimulation used high titer supernatant prepared from the 145-2C11 hybridoma, obtained from the American Type Culture Collection.
Cell Surface Staining.
Spleen and lymph node cells were stained with fluorochrome-conjugated mAbs including anti– CTLA-4 (4F10), anti–B7-1 (1G10), anti–B7-2 (GL1), anti-CD62L (Mel 14), anti-CD69 (H1.2F3), anti-CD25 (7D4), and anti-CD3 (145-2C11). These antibodies were purchased from PharMingen. Stained cells were analyzed on a FACSCalibur® (Becton Dickinson).
Histology.
Tissue for light microscopy was fixed in 10% buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin using standard techniques.
Results
B7-1– or B7-2–mediated Signals Are Required to Produce Lymphoproliferation and Fatal Disease in the CTLA-4–deficient Mouse.
To assess the role of B7-1 and B7-2 in producing lymphoproliferative disease in mice lacking CTLA-4, we crossed the CTLA-4–deficient strain with mice lacking B7-1, B7-2, or both B7 molecules. Mice lacking CTLA-4 and B7-1 (CTLA-4/B7-1−/−) and mice lacking CTLA-4 and B7-2 (CTLA-4/B7-2−/−) develop the characteristic fatal phenotype of CTLA-4−/− mice. The survival of the CTLA-4/B7-1−/− mice (mean = 21.9 d; n = 8) was shorter than that of the CTLA-4/B7-2−/− mice (mean = 30.5 d; n = 8) (P = 0.02 by Wilcoxon signed rank test). The difference in survival between the CTLA-4/B7-1−/− and the CTLA-4−/− (mean = 17.5 d; n = 11) strains did not reach statistical significance (P > 0.05; Fig. 1). In contrast, CTLA-4/B7-1/B7-2−/− TKO mice lacking CTLA-4, B7-1, and B7-2 remain healthy and have a life-span and reproductive capacity comparable to wild-type mice.
Figure 1.

Either B7-1– or B7-2–mediated signaling is sufficient to produce fatal lymphoproliferative disease in mice lacking CTLA-4. Mice were observed daily, and survival was recorded. Survival of CTLA-4−/−, CTLA-4/B7-1−/−, and CTLA-4/B7-2−/− strains is shown. Each mouse is indicated by a different symbol, and mean life-span is shown as a horizontal bar. Wild-type and CTLA-4/B7-1/B7-2−/− TKO mice consistently live longer than 300 d.
Both the CTLA-4/B7-1−/− and CTLA-4/B7-2−/− strains develop splenomegaly, lymphadenopathy, and multiorgan lymphocytic infiltrates with tissue damage, similar to the CTLA-4−/− mice. The CTLA-4/B7-1−/− mice typically develop these pathologic changes earlier than the CTLA-4/B7-2−/− mice. In contrast, CTLA-4/B7-1/B7-2−/− TKO mice develop neither splenomegaly nor lymphadenopathy. Detailed histologic examination of CTLA-4/ B7-1/B7-2−/− TKO mice ranging in age from 2 to 8 mo reveals the complete absence of lymphocytic infiltrates (Fig. 2). These results indicate that either B7-1 or B7-2 can provide the necessary activating signal for the lymphoproliferative phenotype in CTLA-4−/− mice, but that the absence of both B7 molecules abrogates the phenotype.
Figure 2.
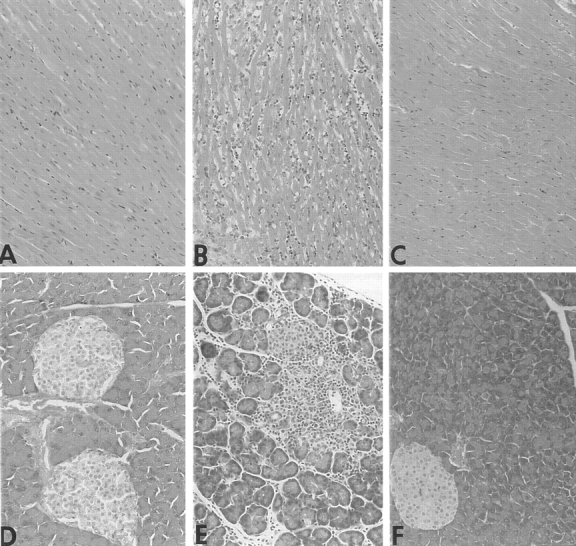
Removal of B7-1– and B7-2–mediated signaling prevents the lymphocytic infiltration and necrosis of organs observed in CTLA-4−/− mice. Heart (A, B, and C) and pancreas (D, E, and F) are shown (original magnification: ×50). CTLA-4−/− mice (B and E), as well as CTLA-4/B7-1−/− and CTLA-4/B7-2−/− mice (not shown), killed at 2 wk of age show severe pancreatitis and myocarditis, in contrast to wild-type littermates (A and D). CTLA-4/B7-1/B7-2−/− TKO mice killed at 2 mo show no evidence of inflammatory infiltrates (C and F).
In the Absence of CTLA-4, Extensive T Cell Activation Occurs Only in the Presence of B7-1 or B7-2.
To assess the degree of T cell activation in these mouse strains in vivo, freshly isolated splenocytes were stained for activation markers and analyzed by flow cytometry. T cells from CTLA-4−/−, CTLA-4/B7-1−/−, and CTLA-4/B7-2−/− mice show significant increases in the percentage that are positive for the activation marker CD69 (Fig. 3) and CD62L-low (data not shown) compared with wild-type mice. In four animals of each strain analyzed at 2 wk, there was a mean of 51% CD69+ T cells in CTLA-4−/−, 48% in CTLA-4/B7-1−/−, and 44% in CTLA-4/B7-2−/− mice. The same trend was observed when staining for CD62L, but the differences did not reach statistical significance. In contrast, <10% of CTLA-4/B7-1/B7-2−/− TKO cells are activated, as assessed by staining for CD69 (Fig. 3) and CD62L (data not shown). None of the CTLA-4/B7-1/ B7-2−/− TKO mice analyzed show significant T cell activation. Both the CTLA-4/B7-1/B7-2−/− TKO and B7-1/ B7-2−/− strains have consistently fewer activated T cells than wild-type mice.
Figure 3.

In the absence of CTLA-4, in vivo T cell activation requires B7-1 or B7-2. Splenocytes were isolated from 2-wk-old mice and double stained with PE–anti-CD3 and FITC– anti-CD69 for analysis by flow cytometry. The percentage shown next to each dot plot indicates the proportion of CD3+ cells that are CD69+. Results are representative of five mice of each strain.
We have previously reported that unfractionated splenocytes from CTLA-4−/− mice proliferate spontaneously in vitro in the absence of exogenous stimulation (7). This assay serves as another measure of unopposed T cell activation in CTLA-4−/− mice. Proliferation in both CTLA-4/ B7-1−/− and CTLA-4/B7-2−/− strains was significantly higher than in wild-type mice (Fig. 4 A). As might be expected from other measures of T cell activation in the two strains, CTLA-4/B7-1−/− cells proliferate more than those from age-matched CTLA-4/B7-2−/− cells. In contrast, CTLA-4/B7-1/B7-2−/− TKO splenocytes do not proliferate spontaneously, suggesting that no CD28 signaling occurs in the absence of B7-1 and B7-2.
Figure 4.


Proliferation of CTLA-4−/− T cells requires B7-1– or B7-2– mediated costimulation. (A) Proliferative responses of unstimulated splenocytes from 2-wk-old mice of the indicated strain at 24 h are shown. Data are representative of three experiments, and the mean of two mice of each strain are shown. (B) Proliferative responses of anti-CD3-stimulated splenocytes from 2-mo-old mice at 48 h are shown. Data are representative of four experiments. The mean of two mice of each strain are shown. Proliferation at all time points was assayed in triplicate, with SD < 15% of the mean.
We also assessed the in vitro proliferative response of CTLA-4/B7-1/B7-2−/− TKO T cells to anti-CD3 antibody stimulation. The response of CTLA-4/B7-1/B7-2−/− TKO splenocytes is markedly reduced compared with wild-type mice (Fig. 4 B). This deficit is indistinguishable from that observed with the B7-1/B7-2−/− mice. Splenocytes from CTLA-4−/−, CTLA-4/B7-1−/−, and CTLA-4/ B7-2−/− mice which are already activated in vivo showed a proliferative response to anti-CD3 stimulation that was highly variable between experiments, presumably because of varying numbers of in vivo–activated T cells, which undergo activation-induced cell death upon restimulation in vitro.
In Vivo Administration of Anti-CD28 to CTLA-4/B7-1/ B7-2−/− TKO Mice Activates T Cells and Produces Fatal Lymphoproliferative Disease.
The results showing that T cell activation in CTLA-4−/− mice requires B7-1 or B7-2 suggest that B7–CD28 interactions are critical for such activation. If CD28 signaling is sufficient to cause T cell activation in the absence of CTLA-4, then ligating CD28 artificially using anti-CD28 antibody should induce disease in CTLA-4/ B7-1/B7-2−/− TKO mice. To test this, we administered anti-CD28 mAb (PV-1) to adult mice for 2 wk. Mice were analyzed 14–19 d after initial treatment with anti-CD28 because they exhibited features typical of the CTLA-4−/− lymphoproliferative disease. At this time, they developed splenomegaly and lymphadenopathy, with greater than sixfold increases in cell numbers compared with anti-CD28– treated wild-type mice (data not shown). CTLA-4/B7-1/ B7-2−/− TKO mice treated with anti-CD28 also developed lymphocytic infiltrates and necrosis, in an organ distribution and severity comparable to the CTLA-4−/− mice (data not shown). No lymphoid or other organ involvement was observed in CTLA-4/B7-1/B7-2−/− TKO mice receiving control IgG, or wild-type mice receiving anti-CD28 or control IgG. Anti-CD28 administration also markedly increased the fraction of activated T cells in the CTLA-4/B7-1/ B7-2−/− TKO mice compared with wild-type, as determined by CD69 (Fig. 5) and CD62L expression (data not shown). Thus, signaling through CD28 in the CTLA-4/ B7-1/B7-2−/− TKO strain can reproduce the lymphoproliferative disease typical of CTLA-4−/− mice. The lack of effect of anti-CD28 antibody in wild-type mice in vivo is also noteworthy, demonstrating that engagement of CTLA-4 by endogenous levels of B7 can inhibit the proliferation of T cells receiving a strong positive signal through CD28.
Figure 5.

Administration of anti-CD28 antibody to CTLA-4/B7-1/ B7-2−/− TKO mice activates T cells. CTLA-4/B7-1/B7-2−/− TKO and wild-type mice received either anti-CD28 or control hamster IgG. At days 14–19, freshly isolated splenocytes were stained for CD3 and the activation marker CD69, and analyzed by flow cytometry. The percentage shown next to each dot plot indicates the proportion of CD3+ cells that are CD69+. Data are representative of at least three mice of each strain.
Discussion
The dual specificities of B7-1 and B7-2 for CD28 and CTLA-4 have made it challenging to elucidate physiological interactions in the B7-CD28/CTLA-4 pathway. The uncontrolled T cell activation seen in the CTLA-4−/− mice provides a valuable experimental system for dissecting in vivo functions of B7-1 and B7-2, and for searching for additional members of this family of costimulators. To do this, we have generated mouse strains lacking CTLA-4 and either B7-1, B7-2, or both B7 molecules, and analyzed them for evidence of T cell activation in vivo and in vitro. The phenotypes of the CTLA-4/B7-1−/− and CTLA-4/ B7-2−/− strains demonstrate overlapping roles for B7-1 and B7-2 in CD28-mediated signaling, as the presence of either B7-1 or B7-2 is sufficient to produce the CTLA-4−/− phenotype. However, the CTLA-4/B7-1−/− mice have shorter survival and greater T cell activation than age-matched CTLA-4/B7-2−/− mice. These differences may reflect earlier expression and higher cell surface levels of B7-2 compared with B7-1. Since either B7 molecule is capable of producing lymphoproliferation and T cell activation in the CTLA-4−/− mice, it seems unlikely that B7-1 and B7-2 produce fundamentally different signals through CD28.
The striking absence of T cell activation in the CTLA-4/ B7-1/B7-2−/− TKO strain demonstrates that B7-1 or B7-2 is required to produce the CTLA-4−/− lymphoproliferative disease. The absence of CD28-mediated costimulation in the CTLA-4/B7-1/B7-2−/− TKO strain is demonstrated by the (a) lack of lymphoproliferation in vivo, as evidenced by the normal life-span and absence of splenomegaly, lymphadenopathy, and lymphocytic infiltrates; (b) naive phenotype of T cells in vivo; (c) lack of spontaneous proliferation of splenocytes in vitro; and (d) impaired proliferative response to CD3 stimulation in vitro. Similar results have been obtained by treating CTLA-4−/− mice with CTLA4Ig. Our results extend these studies and show that B7-1 or B7-2 is capable of activating CTLA-4−/− T cells.
This requirement for B7-1 or B7-2 in activating CTLA-4−/− T cells is most consistent with a role of unopposed signaling by CD28, the only known activating T cell receptor for B7 molecules. We have formally established this by showing that anti-CD28 antibody can reproduce disease in CTLA-4/B7-1/B7-2−/− TKO mice. Interestingly, the same anti-CD28 antibody has no detectable effect on T cell activation in wild-type mice in vivo. These findings demonstrate the critical balance between CD28-mediated T cell activation and CTLA-4–mediated downregulation, and suggest that the inhibitory function of CTLA-4 can overcome strong CD28-mediated signals in vivo. These results may explain why many studies show no or inconsistent in vivo effects of anti-CD28 antibodies, which are known to be strong agonists when added to T cells in the absence of APCs in vitro.
We have not found evidence for additional stimulatory CD28 ligands in the CTLA-4/B7-1/B7-2−/− TKO mice. This strain provides an exquisitely sensitive means for examining whether additional stimulatory CD28 counterreceptors exist, since in the absence of CTLA-4, even the effects of weak CD28-mediated signaling would be apparent. The profound deficits observed in the CTLA-4/B7-1/ B7-2−/− TKO mice would suggest that if additional B7 ligands exist, they either do not stimulate T cell activation through CD28, or they are expressed in a restricted microenvironment. Such microenvironments might include organs such as the brain or kidney, which exhibit no inflammatory changes in CTLA-4−/− mice.
The CTLA-4/B7-1/B7-2−/− TKO strain has a phenotype that appears to be comparable to the B7-1/B7-2−/− strain both in vivo and in vitro. Therefore, in the absence of B7-1 and B7-2, the presence of CTLA-4 has no detectable downregulatory effect. Because T cells from the CTLA-4/B7-1/B7-2−/− TKO strain are mostly naive, unlike cells from CTLA-4−/− mice, CTLA-4/B7-1/B7-2−/− TKO mice also can serve as a useful tool to study the function of CTLA-4 during the initial activation of T cells.
Acknowledgments
We thank Abul Abbas for careful review of the manuscript, Vijay K. Kuchroo for the gift of anti-CD28 antibody, Frank Borriello and Elizabeth A. Tivol for initiating mouse breeding, and Sumi Scott for technical assistance.
This work was supported by National Institutes of Health grants K11 AI01212 to D.A. Mandelbrot, AI09709 to A.J. McAdam, and RO1 AI38310, RO1 AI40614, and PO1 AI35297 to A.H. Sharpe.
References
- 1.Turka LA, Linsley PS, Lin H, Brady W, Leiden JM, Wei RQ, Gibson ML, Zheng XG, Myrdal S, Gordon D, et al. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc Natl Acad Sci USA. 1992;89:11102–11105. doi: 10.1073/pnas.89.22.11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. 1994;265:1225–1227. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- 3.Lanier LL, O'Fallon S, Somoza C, Phillips JH, Linsley PS, Okumura K, Ito D, Azuma M. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol. 1995;154:97–105. [PubMed] [Google Scholar]
- 4.Schweitzer AN, Borriello F, Wong RC, Abbas AK, Sharpe AH. Role of costimulators in T cell differentiation: studies using antigen-presenting cells lacking expression of CD80 or CD86. J Immunol. 1997;158:2713–2722. [PubMed] [Google Scholar]
- 5.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 6.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 7.Tivol EA, Boyd SD, McKeon S, Borriello F, Nickerson P, Strom TB, Sharpe AH. CTLA4Ig prevents lymphoproliferation and fatal multiorgan tissue destruction in CTLA-4-deficient mice. J Immunol. 1997;158:5091–5094. [PubMed] [Google Scholar]
- 8.Boussiotis VA, Freeman GJ, Gribben JG, Daley J, Gray G, Nadler LM. Activated human B lymphocytes express three CTLA-4 counterreceptors that costimulate T-cell activation. Proc Natl Acad Sci USA. 1993;90:11059–11063. doi: 10.1073/pnas.90.23.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murakami M, Takahashi Y, Isashi Y, Kon S, Jia WY, Inobe M, Abe R, Uede T. Identification and characterization of an alternative cytotoxic T lymphocyte-associated protein 4 binding molecule on B cells. Proc Natl Acad Sci USA. 1996;93:7838–7842. doi: 10.1073/pnas.93.15.7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeman GJ, Borriello F, Hodes RJ, Reiser H, Hathcock KS, Laszlo G, McKnight AJ, Kim J, Du L, Lombard DB, et al. Uncovering of functional alternative CTLA-4 counter-receptor in B7-deficient mice. Science. 1993;262:907–909. doi: 10.1126/science.7694362. [DOI] [PubMed] [Google Scholar]
- 11.Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, Strom TB, Simpson EM, Freeman GJ, Sharpe AH. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]