Two Roads Diverged: Interferon α/β– and Interleukin 12–mediated Pathways in Promoting T Cell Interferon γ Responses during Viral Infection (original) (raw)
Abstract
Viral infections induce CD8 T cell expansion and interferon (IFN)-γ production for defense, but the innate cytokines shaping these responses have not been identified. Although interleukin (IL)-12 has the potential to contribute, IL-12–dependent T cell IFN-γ has not been detected during viral infections. Moreover, certain viruses fail to induce IL-12, and elicit high levels of IFN-α/β to negatively regulate it. The endogenous factors promoting virus-induced T cell IFN-γ production were defined in studies evaluating CD8 T cell responses during lymphocytic choriomeningitis virus infections of mice. Two divergent supporting pathways were characterized. Under normal conditions of infections, the CD8 T cell IFN-γ response was dependent on endogenous IFN-α/β effects, but was IL-12 independent. In contrast, in the absence of IFN-α/β functions, an IL-12 response was revealed and substituted an alternative pathway to IFN-γ. IFN-α/β–mediated effects resulted in enhanced, but the alternative pathway also promoted, resistance to infection. These observations define uniquely important IFN-α/β–controlled pathways shaping T cell responses during viral infections, and demonstrate plasticity of immune responses in accessing divergent innate mechanisms to achieve similar ultimate goals.
Keywords: T cell, virus, interferon α/β, interleukin 12, interferon γ
Immune responses to various classes of infectious agents have many overlapping, but certain unique or uniquely dominant, characteristics (1). The elements appear to be in place to access and/or deliver mechanisms most effective in defense against the eliciting infectious organism. Innate cytokine responses can have roles in shaping downstream adaptive, as well as other innate, immune responses (2). The paradigm emerging from bacterial and parasite studies has IL-12 as the pivotal innate cytokine for promoting NK and T helper type 1 (Th1) cell IFN-γ responses (3–9). Roles for this cytokine in immune responses to viruses are less clear. Biologically active IL-12 is not induced during all viral infections (10–13). The cytokine is part of the innate immune response and stimulates NK cell IFN-γ production during murine cytomegalovirus (MCMV)1 (10, 11) and influenza virus (12), but not lymphocytic choriomeningitis virus (LCMV) (11), infections of mice. Moreover, IL-12–dependent T cell IFN-γ responses are not demonstrable in number of viral infections including those that do or do not induce detectable IL-12 (11, 12, 14).
In contrast to infections with other agents, many viruses elicit high levels of the innate cytokines, type 1 interferons (i.e., IFN-α/β) and adaptive CD8 T cell responses. During LCMV infections lacking detectable IL-12, early and dramatic elevations in IFN-α/β concentrations are induced on days 2 and 3 after infection (10, 11, 13, 15), and T cell immune responses characterized by profound CD8 T cell expansion and IFN-γ production are elicited on or after days 7–9 (16–22). Specificity of the CD8 T cell responses has been proven by direct visualization with binding of MHC class I tetramer molecules complexed to LCMV epitopes (23), and by stimulation of IFN-γ expression with LCMV peptides (23–25). The innate cytokines important for promoting these T cell responses have not been defined. IFN-α/β cytokines can contribute to a variety of immunoregulatory effects (26), and are reported to promote T cell IFN-γ production under certain culture conditions (27–29). Thus, they may be a class of innate cytokines uniquely regulating adaptive T cell responses to viral infections.
The studies reported here were undertaken to define roles for innate cytokines in supporting antiviral T cell responses. The experiments, carried out during LCMV infections of mice, demonstrate for the first time the major endogenous innate pathways to CD8 T cell IFN-γ during viral infections. They show that the IFN-α/β cytokines are dominant in promoting conditions for this T cell IFN-γ production. Moreover, they demonstrate that although T cell IFN-γ responses are IL-12 independent in the context of IFN-α/β induction and function, IL-12 revealed in the absence of IFN-α/β functions can substitute to promote IFN-γ production. This alternative pathway is beneficial but not sufficient for induction of optimal protection. Taken together, the data define unique factors and conditions regulating immune responses to viral infections. Furthermore, they result in the discovery of alternative innate cytokine pathways for promoting IFN-γ responses.
Materials and Methods
Mice.
Mice deficient in IL-12, as a result of targeted disruption of the IL-12p35 gene (IL-12p35 KO), were generated and bred at Genetics Institute, using the p35 SK+ vector (Stratagene Inc.), the embryonic stem cell system in 129/sv mice, and established procedures (30–32). Mice were backcrossed onto the C57BL/6 genetic background for two to four generations. Homozygous mutant (−/− or KO) and homozygous wild-type (WT) IL-12p35 gene mice were taken from littermates for use in these studies. The gene targeting vector eliminated exons 1–4 by replacement with the neomycin resistance gene (see Fig. 1 A). Disruption of the p35 gene was demonstrated by Southern blot analysis (see Fig. 1 B). Functional deletion of IL-12 was confirmed by absence of IL-12p70 heterodimers after challenge with the known stimulators lipopolysaccharide treatment and MCMV infection. For these studies, mice were injected at 0 h with 500 μg/kg LPS (3), and blood collected for serum sample preparation at 2.5 and 5 h (see Fig. 1 C), or infected with 5 × 104 PFU of Smith strain MCMV V70 salivary gland extract, and blood collected for serum sample preparation at 36 h (Fig. 1 D). Lack of IL-12 was shown to have biological consequences because the IL-12–dependent induction of natural killer cell IFN-γ production after MCMV infection was blocked in the IL-12p35 KO compared with WT mice (see Fig. 1 D).
Figure 1.

Development and testing of IL-12p35 KO mice. (A) Strategy for targeted disruption of the IL-12p35 locus is given. The IL-12p35 genomic structure is represented; closed boxes indicate location of the coding regions, which are numbered beneath, and white boxes indicate noncoding regions. The IL-12p35 targeting vector was constructed in the pBS SK+ vector and engineered by replacing a 5-kb HindIII-XbaI fragment containing exons 1–4 with a 1.6-kb neomycin resistance gene, represented as a striped box, under control of the PGK promoter. A 2.3-kb thymidine kinase cassette, represented as a stippled box, also under the control of the MC1 promoter, was inserted immediately adjacent to the 3′ end of the IL-12p35 genomic flanking sequence. A 1-kb Kpn1-XbaI fragment located downstream of exon 7, used as a probe for genomic Southern blot analysis, is indicated by a closed bar beneath. The size and location of fragments predicted for wild-type and mutant alleles expected during Southern analysis are indicated by double-headed arrows. The targeting construct was linearized at the pBS NotI site and electroporated into J1 embryonic stem cells grown and cultured under G418 and Gancyclovir selection. Restriction enzyme sites are indicated as follows: N, NeoI; H, HindIII; B, BamHI; X, XbaI; V, EcoRV; and K, KpnI. (B) Mouse genotyping using genomic Southern analysis. Genomic DNA was extracted from tail fragments of IL-12p35 +/+, IL-12p35 +/−, and IL-12p35 −/− mice. After BamHI/EcoRV restriction digestion, DNA was fractionated on 0.8% agarose gel, blotted onto membranes, and hybridized with 32P-labeled probe. (C) LPS induction of IL-12p70 expression. IL-12p35 +/+ and IL-12p35 −/− mice were injected with 500 μg/kg LPS. Blood was collected for serum sample preparation at 2.5 and 5 h after LPS injections. IL-12p70 was detected in an ELISA. Results are representative of two identical experiments containing six mice per group and are shown as means ± SEM. (D) MCMV induction of IL-12p70 and IFN-γ expression. IL-12p35 +/+ and IL-12p35 −/− mice were either uninfected or infected with 5 × 104 PFU of Smith strain MCMV for 36 h and serum was prepared. IL-12p70 was measured by antibody capture and use in a biological assay. IFN-γ was measured in an ELISA. Results represent experiments containing three mice per group and are shown as means ± SEM.
Mice deficient in IFN-α/β receptor function as a result of genetic mutation (IFN-α/βR KO) on the 129/Sv background (33, 34), originally obtained from B&K Universal Limited, were bred and maintained in strict isolation in the animal facility at Brown University. Age-matched WT 129 control (129 SvEvTacFBR) mice were purchased from Taconic Farms Inc. All mice used in experiments had the H-2b major histocompatibility complex and were handled in accordance with institutional guidelines for animal care and use.
Treatments.
The LCMV infections were established i.p. on day 0 with 2 × 104 PFU of Armstrong strain, clone E350 (11, 13, 35, 36). CD8 T cells were depleted in vivo by treatment i.p. with 0.5 mg of monoclonal anti–CD8 antibody 2.43, prepared from ascites, on day 5 after infection. Control treatment was with partially purified P3NS1 ascites containing 0.5 mg rat IgG (Sigma Chemical Co.). Sheep antimurine IFN-α/β IgG was injected (0.15 mg/mouse) to neutralize IFN-α/β with nonspecific sheep IgG used as control antibody (gifts of Ion Gresser, Center National de la Recherche Scientifique, Villejuif, France) (10, 13, 37, 38). The rat IgG monoclonal directed against the mouse IL-12 p40 chain, C17.8, was used to neutralize IL-12 function in vivo. Partially purified ascites preparations (1 mg) were injected to neutralize IL-12 with rat IgG added to P3NS1 ascites used as control antibody (10, 11). Injections were i.p. 14 h before, and day 4 of, LCMV infections. Splenic leukocytes were isolated from macerated whole spleens after passage through nylon mesh and osmotic lysis of erythrocytes by ammonium chloride treatment, with viable cell yields determined by trypan blue exclusion.
Cytokine Analyses.
Samples for quantitation of IFN-γ and IL-12 p70 were prepared as reported previously (11, 13, 17). Conditioned media (CM) were generated by incubating 107 splenic leukocytes/ml in 10% FBS-RPMI 1640 media for 24 h at 37°C before harvest of culture supernatants. Where indicated, CM samples were prepared by incubation of splenic leukocytes with or without 0.1 μg/ml LCMV peptides NP396-404 or GP33-41 in 48-well flat-bottom plates at concentrations of 2 × 106 cells per well in 0.4 ml of 10% FBS-RPMI supplemented with 50 U/ml human recombinant IL-2 (23, 25, 39). IFN-γ was measured by sandwich ELISA (11, 13, 17). Standard curves of mrIFN-γ (PharMingen) indicated detection limits of 10–40 pg/ml in serum, or 1–4 pg/106 cells in CM. IL-12 p70 was quantitated either in an ELISA (Genzyme Corp.) or in a biological assay (11, 13). Limit of detection for IL-12 p70 in the biological assay was 0.7 pg/ml serum. IL-2 was quantitated in ELISA using reagents from PharMingen according to the manufacturer's recommendations.
Flow Cytometric Analysis.
As per modification of published techniques (40), cells were stimulated on 24-well cluster plates previously coated overnight with 0.5 ml 10 μg/ml purified hamster anti–mouse CD3ε mAb 145-2C11 (PharMingen) in PBS. Plates were washed with PBS, and 2 × 106 cells in 2 ml 10% FBS-RPMI per well were incubated for a total of 6 h, with Brefeldin A (Sigma Chemical Co.) added the last 2 h. Alternatively, splenic leukocytes were stimulated with 0.1 μg/ml of the LCMV peptides NP396-404 or GP33-41 (23). Cells for these experiments were incubated in 48-well flat-bottom plates at concentrations of 2 × 106 cells per well in 0.4 ml of 10% FBS-RPMI supplemented with 50 U/ml human recombinant IL-2, at 37°C for 5 h total, with Brefeldin A added for the last 3 h. After stimulation, cells were washed in staining buffer (0.5% BSA and 0.006% NaN3 in PBS), incubated for 30 min at 4°C with biotinylated rat anti–CD4 mAb clone RM4-5 (PharMingen), washed twice with staining buffer, incubated 30 min at 4°C with streptavidin-PerCP (Becton Dickinson & Co.) and anti–CD8α FITC-conjugated rat mAb 53-6.7 (PharMingen), washed once with staining buffer, once with PBS, resuspended at 106 cells/ml, and fixed with an equal volume of 4% formaldehyde in PBS for 20 min at room temperature. For cytoplasmic staining, cells were washed once with PBS, once with staining buffer, treated with 150 μl of permeabilization buffer (1% saponin in staining buffer; Sigma Chemical Co.), resuspended in 25 μl permeabilization buffer containing 300 μg/ml rat IgG (Sigma Chemical Co.), incubated 10 min at room temperature, and incubated for 20 min at room temperature with anti–IFN-γ PE-conjugated rat mAb XMG1.2 (PharMingen). Specificity of IFN-γ staining was established by preincubation of anti–IFN-γ-PE with recombinant murine IFN-γ at 0.25 mg/test, before incubation with cells (cold block), or preincubation of cells with excess unconjugated XMG1.2 (cold competitor), and nonspecific staining assessed by use of control antibodies lacking specificities for murine determinants (PharMingen). Cells were washed twice with permeabilization buffer, once with staining buffer, and acquired immediately. H2Db tetramers, containing LCMV peptides NP396-404, GP33-41, and GP276-286, prepared, and conjugated with allophycocyanin as described, were used for surface staining along with anti–CD8 PE-conjugated rat mAb 52-6.7 (PharMingen) (23, 41). After incubation of 106 cells with tetramer for 1 h, cells were washed two times in 1× PBS supplemented with 2% FBS, fixed with 4% paraformaldehyde and washed one time. All samples were acquired at Brown University on a FACSCalibur® (Becton Dickinson & Co.), using CELLQUEST 3.0 software. More than 20,000 events were collected, with laser outputs of 15 mW at 488 or 635 nM.
Viral Plaque Assays.
Livers and spleens were frozen at −80°C, thawed, homogenized, and LCMV titers were measured by plaque formation on Vero cells as previously described (17, 35, 36).
Results
Requirements for Endogenous IL-12 Function.
To conclusively exclude a role for IL-12 in T cell responses to LCMV infection, mice were made deficient for the p35 subunit of IL-12 (IL-12p35 KO) by homologous recombination with a deleted gene construct, as described in Materials and Methods and Fig. 1. None of the day 8 splenic T cell responses to LCMV infection were significantly reduced in the IL-12p35 KO mice. Yield and flow cytometric analyses demonstrated that overall expansion of CD8 T cells was similar in both WT and IL-12p35 KO (Fig. 2 A). Furthermore, ELISA studies demonstrated that induction of IFN-γ expression, both in media conditioned with splenic leukocytes (CM) and in serum, was neither blocked nor significantly inhibited in the absence of IL-12 (Fig. 2 B). Because CD8 T cells are the predominant IFN-γ producers during LCMV infection, IFN-γ expression by CD8 T cells also was measured by flow cytometric analysis for cytoplasmic protein and shown to be unaffected by absence of IL-12. Upon stimulation ex vivo with immobilized anti–CD3, ∼60–70% of the cells induced to express cytoplasmic IFN-γ from both types of mice were CD8 T cells, and the proportions of IFN-γ–expressing CD8 T cells were 43.2% (± 8.0) and 43.5% (± 4.7) (means ± SEM) in WT and IL-12p35 KO mice, respectively (Fig. 2 C). Likewise, total numbers of CD8 T cells expressing IFN-γ, calculated based on CD8 T cell yields, were equivalent with 7.9 (± 2.2) and 9.6 (± 2.7) × 106 cells (Fig. 2 C). The endogenous responses were sufficient to mediate protection against infection because viral titers were below detectable levels in both infected WT and IL-12p35 KO mice by day 8 (data not shown). Thus, T cell proliferation and IFN-γ production occur in the complete absence of the biologically active IL-12 heterodimer during infections with this virus, and the immune responses induced under these conditions are protective.
Figure 2.

T cell responses in mice lacking endogenous sources of either IL-12 or IFN-α/β. Mice, WT and IL-12p35 KO 129/B6 (A–C) or WT and IFN-α/βR KO 129 (D–F), were either uninfected (open bars or symbols) or infected i.p. (closed bars or symbols) on day 0 with 2 × 104 PFU LCMV Armstrong strain. On day 8 after infection, blood and spleens were harvested and processed. Total numbers of CD8 T cell splenic leukocytes were calculated by multiplying the percentages of CD8 cells, determined by flow cytometry, with the total cell yields (A and D). IFN-γ levels were measured, in media conditioned with splenic leukocytes and in serum samples, by ELISA (B and E). For flow cytometric analysis of cytoplasmic IFN-γ expression, splenic leukocytes were stimulated for 6 h with anti–CD3, with Brefeldin A during the last 2 h. Cells were collected and stained for cell surface expression of CD8, CD4, and cytoplasmic expression of IFN-γ (C and F). Histogram plots were formed by gating on the CD8 population, and displaying CD8 T cell number versus IFN-γ fluorescence intensity. Numbers of CD8 T cells expressing IFN-γ were calculated by multiplying the percentage of CD8 T cells expressing IFN-γ by the number of CD8 T cells per spleen (× 106), and given as means ± SEM. Results are representative of two or more repetitive experiments with two to four mice per uninfected group and three to four mice per infected group. Data are shown as means ± SEM.
Requirements for Endogenous IFN-α/β Functions.
Experiments were carried out to characterize roles of IFN-α/β in regulating the T cell responses. Although of lower magnitude on the inbred genetic 129 background of these mice, inductions of CD8 T cell expansion and IFN-γ expression were observed and similar in both WT and IFN-α/βR KO mice (Fig. 2, D and E). IFN-γ levels, measured in samples from the IFN-α/βR KO mice, were equal to or greater than those from WT mice in CM and serum (Fig. 2 E). Interestingly, serum IFN-γ levels were enhanced significantly by more than threefold in samples obtained from IFN-α/βR KO, as compared with WT, mice. Studies, in IFN-α/βR KO mice depleted of endogenous CD8 T cells by antibody treatments, demonstrated that 66% of the enhanced serum IFN-γ was dependent on endogenous CD8 T cells. After anti–CD3 stimulation ex vivo, 50–60% of the IFN-γ expressing cell types from both IFN-α/βR KO and WT mice were CD8 T cells (data not shown), and similar proportions of the CD8 T cell subset from both types of mice were induced to express the cytokine (Fig. 2 F). As these T cell responses are intact, the data suggest that IFN-α/β deficiencies by themselves do not result in generalized T cell exhaustion or depletion. However, viral titers were increased in IFN-α/βR KO relative to WT mice (see below). Hence, the CD8 T cell expansion and IFN-γ production are not IFN-α/β dependent, but the conditions result in decreased resistance to infection.
Because total expansion and IFN-γ expression were intact, but levels of virus were increased, antigen specificity of the responding cells was tested. To enumerate CD8 cells bearing T cell receptors for LCMV antigens, splenic leukocytes were labeled with anti–CD8 and tetramers of Db MHC class I molecules containing LCMV peptides immunodominant for CD8 T cell responses (NP396-404 or GP33-41), as described in Materials and Methods. Flow cytometric analyses showed significant increases in percentages of CD8 T cells binding tetrameric Db NP396-404 or Db GP33-41 with cells from day 8 LCMV-infected, relative to uninfected, WT mice (Fig. 3, A and B). Samples from IFN-α/βR KO mice also had significant increases in proportions of tetrameric Db NP396-404– and Db GP33-41–binding CD8 T cells after infection (Fig. 3, A and B). T cell frequencies specific for tetramers with NP396-404 were higher in WT, whereas those with GP33-41 were higher in IFN-α/βR KO, populations. Total numbers of CD8 T cells binding the complexed tetramers were significantly increased after infection, relative to uninfected samples, in both WT and IFN-α/βR KO mice (Fig. 3 C). Moreover, because of increases in average splenic cell yields after infection, the total numbers of CD8 T cells specific for the tetrameric Db LCMV peptide complexes were higher in the IFN-α/βR KO than the WT mice, respectively, averaging 247 and 120 × 104 cells per spleen. As a result of their skewed proportional representation, however, the Db GP33-41 binding cells respectively comprised ∼75 and 20% of the populations, and appeared to account for a large proportion of the enhanced expansion of T cells recognizing viral epitopes in the IFN-α/βR KO mice. Thus, as in WT mice, expanding CD8 T cells in IFN-α/βR KO mice are specific for the virus. However, in the absence of IFN-α/β function, responses are differentially elicited to particular LCMV epitopes.
Figure 3.
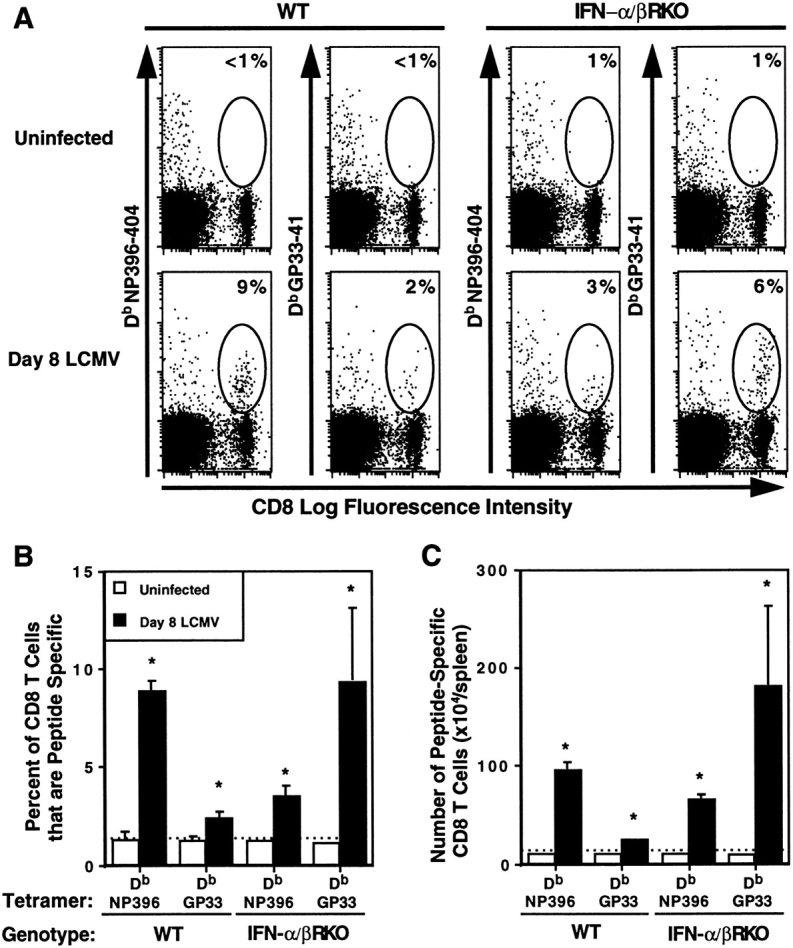
Specificity of CD8 T cell expansion after LCMV infection of IFN-α/βR KO mice. WT and IFN-α/βR KO mice were uninfected or were infected i.p. on day 0 with 2 × 104 PFU LCMV Armstrong strain, and spleens were harvested and processed on day 8. CD8 populations positive for binding Db tetramers complexed with LCMV peptides NP396-404 (NP396) or GP33-41 (GP33) were assessed by flow cytometric analysis. Dot plots (A) of CD8 versus tetramer fluorescence intensity are shown. Circles identify areas of CD8 T cells binding complexed tetramers. Numbers given in corners of dot plots indicate the percent of CD8 T cells that are tetramer positive for the individual sample. Averages of proportions (B) and numbers (C) of CD8 T cells that are tetramer positive for uninfected (open bars) and day 8 LCMV infected (closed bars) shown are for three mice per group, ± SEM. Results are representative of two repetitive experiments. *Infected samples are significantly different relative to uninfected samples, P ≤ 0.05, as determined by a two-tailed Student's t test. Broken lines across graphs in B and C signify basal staining of cells from uninfected mice.
To demonstrate specificity of functional CD8 T cells, IFN-γ expression responses to the LCMV NP396-404 and GP33-41 peptides were evaluated ex vivo. Cells from uninfected WT or IFN-α/βR KO mice were not stimulated by either of these peptides (data not shown). In contrast, on day 8 after LCMV infection, significant increases in CD8 T cells expressing cytoplasmic IFN-γ, relative to samples prepared in the absence of exogenous stimulation, were observed after peptide stimulation of either WT or IFN-α/βR KO splenic leukocytes (Fig. 4). Consistent with the Fig. 2 H experiments, proportions of total primed CD8 T cells responding to anti–CD3 stimulation for IFN-γ expression remained similar (Fig. 4 B). However, even though the total proportions of cells specific for binding tetramers with LCMV peptides (Fig. 3 B), and of cells expressing cytoplasmic IFN-γ after anti–CD3 stimulation (Fig. 4 B), were similar, lower proportions of cells from IFN-α/βR KO as compared with WT mice were stimulated in response to peptides (Fig. 4 B). Interestingly, despite dramatic increases in cells binding tetrameric Db GP33-41 (Fig. 3), the proportions and numbers of cells responding to this peptide for IFN-γ expression were similar or only marginally higher in IFN-α/βR KO compared with WT populations (Fig. 4). Thus, the reduced IFN-γ responses with cells from IFN-α/βR KO mice appeared to be a consequence of specifically binding populations failing to detectably respond to GP33-41 stimulation for IFN-γ expression. Evaluation of cytokine in CM demonstrated similar levels of population responsiveness to the LCMV peptides (Table I). These studies demonstrate induction of LCMV-specific CD8 T cell IFN-γ production responses in the absence of IFN-α/β functions. Taken together with the tetramer binding results, they provide evidence that a proportion of the CD8 T cells specifically expanding in response to viral epitopes are altered in their requirements for stimulation and/or magnitude of functional responses.
Figure 4.

LCMV epitope-specific stimulation of CD8 T cell IFN-γ expression in cells from IFN-α/βR KO mice. WT and IFN-α/βR KO mice were infected i.p. on day 0 with 2 × 104 PFU LCMV Armstrong strain. On day 8 after infection, spleens were harvested and processed. Splenic leukocytes were stimulated in the presence or absence of the immunodominant LCMV peptides NP396-404 or GP33-41, or anti–CD3, and labeled for flow cytometric analysis of cytoplasmic IFN-γ protein in CD8 T cells, as described in Materials and Methods. Dot plots (A) display CD8 versus IFN-γ fluorescence intensity, with CD8 T cells expressing IFN-γ circled. Numbers given in corners of dot plots are proportions of CD8 T cells expressing cytoplasmic IFN-γ for the individual samples. Averages of proportions (B) and numbers (C) of CD8 T cells positive for IFN-γ expression are shown for three mice per group, ± SEM. Results are representative of two repetitive experiments.
Table I.
IFN-α/β Effects on LCMV-specific IFN-γ Production on Day 8 after LCMV Infection
| Stimulation | IFN-γ Production (pg/106 cells) | |||
|---|---|---|---|---|
| WT | IFN-α/βR KO | |||
| Uninfected | Day 8 LCMV | Uninfected | Day 8 LCMV | |
| No peptide | <25 | 642.7 ± 164.3 | <25 | 461.6 ± 97.3 |
| NP396-404 | <25 | 18,778.1 ± 5,776.0 | <25 | 608.5 ± 102.8 |
| GP33-41 | <25 | 10,108.4 ± 4,909.5 | <25 | 4,437.4 ± 723.1 |
Cytokine Effects on Viral Burdens.
Kinetic studies of viral clearance during the experiments reported here, sampling on various days after LCMV infection, demonstrated that viral burdens in both IL-12p35 KO (data not shown) and WT mice were below detection by day 8 after infection. Extended studies of WT and IFN-α/βR KO mice showed that WT mice had viral burdens that peaked around day 4.5 at levels of 6.2 (± 0.2) and 5.0 (± 0.2) log PFU/g of spleen and liver, respectively, and were below detection by day 7 (Fig. 5). Similar kinetics of splenic and hepatic viral titers were observed in IFN-α/βR KO mice, but these peaked at the higher levels of 10.3 (± 0.2) and 10.5 (± 0.0) log PFU/g of respective tissue on day 4.5 after infection, and declined by day 7 (Fig. 5). Interestingly, decreases in splenic and liver viral loads were observed in the IFN-α/βR KO mice over time to 4.7 (± 0.2) and 4.2 (± 0.0) log PFU/g of tissue, respectively, on day 14 after infection, and below the limits of detection by day 28 after infection (Fig. 5). Thus, absence of an endogenous IL-12 response does not increase sensitivity to LCMV infection, and although absence of IFN-α/β functions does, viral clearance is eventually achieved.
Figure 5.

LCMV viral titers in IFN-α/βR KO as compared with WT mice. WT (○) and IFN-α/βR KO (•) mice were infected on day 0 with 2 × 104 PFU LCMV Armstrong strain. Spleens and livers were harvested on 1.5, 3, 4.5, 7, 8, 9.5, 11, 14, 21, 28, or 35 d after infection, for quantitation of LCMV titers in plaque assays. Data shown are means for three mice per group ± SEM, and the solid lines across the graphs represent the lower limits of detection.
Substitution of an IL-12 Response in the Absence of IFN-α/β.
Because IL-12 expression can be revealed during LCMV infection by neutralization of endogenous IFN-α/β functions (13), a released IL-12 induction may substitute in supporting T cell responses under these conditions. Measurements of IL-12 p70 in serum demonstrated that the factor was induced to detectable levels on days 1.5 and 3 of LCMV infection in IFN-α/βR KO, but not WT, mice (Fig. 6). Therefore, IL-12 effects on the T cell responses were examined in IFN-α/βR KO mice by treatments with antibodies neutralizing IL-12. Although anti–IL-12 treatments did not modify IFN-γ production responses as compared with control antibody treatments in WT mice, they significantly reduced IFN-γ production on day 8 in LCMV- infected IFN-α/βR KO mice; relative to cells isolated from control-treated IFN-α/βR KO mice, 70% decreases in CM spontaneous production of IFN-γ were observed (Fig. 7 A, left). Furthermore, anti–IL-12 treatment of IFN-α/βR KO mice, resulted in a >85% reduction in CM IFN-γ levels, as compared with anti–IL-12–treated WT mice (Fig. 7 A, left, striped bars); i.e., from 66.4 (± 16.0) to 7.8 (± 2.0) pg/106 cells. Consistent with the experiments shown in Fig. 2, B and E, only low levels of serum IFN-γ were detected in WT mice, but were increased by more than threefold in IFN-α/βR KO mice (Fig. 7 A, right). Given the contribution of CD8 T cells to serum IFN-γ (see above), the stimulation of CD8 T cell IFN-γ expression by LCMV epitopes (Fig. 4), and the significant increases in viral burdens (Fig. 5, day 8), elevated in vivo stimulation of the CD8 T cells by LCMV epitopes was likely to have contributed to higher serum IFN-γ levels in the IFN-α/βR KO mice. IL-12 also participated in this enhanced response because neutralization of the factor resulted in a 40% reduction in serum IFN-γ levels (Fig. 7 A, right). Thus, in the absence of endogenous IFN-α/β effects during a viral infection, IL-12 can be induced and substitute to provide conditions supporting IFN-γ responses.
Figure 6.

IL-12 p70 induction during LCMV infections of IFN-α/βR KO mice. Serum samples taken from WT (open bars) and IFN-α/βR KO (closed bars) mice that were either uninfected or infected for 1.5, 3, 4.5, 7, 8, or 9.5 d with 2 × 104 PFU LCMV Armstrong strain. A capture biological assay was used to measure IL-12 p70 levels. Values shown are means of two to three mice per group, ± SEM. Solid line across graph indicates limit of detection for the assay.
Figure 7.

T cell responses in mice lacking endogenous function of both IL-12 and IFN-α/β. Mice, WT and IFN-α/βR KO 129 (A) or WT and IL-12p35 KO 129/B6 (B), were infected i.p. on day 0 with 2 × 104 PFU LCMV Armstrong strain. The WT and IFN-α/βR KO 129 mice received either control or neutralizing C17.8 anti–IL-12p40 antibodies. The WT and IL-12p35 KO 129/B6 mice received either control or polyclonal neutralizing anti–IFN-α/β antibodies. All antibody treatments were given i.p. on days −1 and 4 relative to infection. On day 8 after infection, blood and spleens were harvested and processed. Levels of IFN-γ in splenic leukocyte CM and serum samples were measured by ELISA. Hatched bars represent samples from mice with antibody-mediated neutralization of cytokine functions. Data shown are means ± SEM. Significantly different P values were calculated by a two-tailed Student's t test comparing either anticytokine- to control-treated mutant mice (*,**) or anticytokine-treated mutant to wild-type mice (++), * P ≤ 0.05, ** P ≤ 0.01, ++ P ≤ 0.01. As a result of pooling results from two repetitive experiments, the values in A represent mean results with six mice per group. Those in B represent means of results from three mice per group.
To confirm and extend these studies, IFN-α/β effects were examined under the reciprocal conditions in mice lacking endogenous IL-12. For these experiments, WT and IL-12p35 KO mice were treated with antibodies neutralizing IFN-α/β or control antibodies. Cytokine neutralization of WT mice resulted in IFN-γ responses comparable with those in IFN-α/βR KO mice; i.e., IFN-γ production in CM was not blocked and was induced to elevated levels in serum (Fig. 7 B). Relative to control-treated mice, antibody-mediated neutralization of IFN-α/β in IL-12p35 KO mice significantly inhibited IFN-γ levels spontaneously produced in CM (Fig. 7 B, left). Moreover, in comparison with anti– IFN-α/β–treated WT mice, serum IFN-γ levels were reduced by >90% as a result of IFN-α/β neutralization in the IL-12p35 KO mice; i.e., whereas the anti–IFN-α/β– treated WT mice had levels of 1,454.2 (± 171.2) pg/ml in serum, the anti–IFN-α/β–treated IL-12p35 KO mice had only 110.9 (± 30.6) (Fig. 7 B, right, striped bars). Thus, in the absence of endogenous IL-12, IFN-α/β–mediated effects are primarily responsible for endogenous conditions promoting the IFN-γ responses to viral infections.
To evaluate the specificity of CD8 T cell responses for LCMV, under the conditions of anti–IL-12 treatment in IFN-α/βR KO mice, experiments were carried out examining peptide or anti–CD3 stimulation for cytoplasmic IFN-γ expression, specific stimulation by peptides for IFN-γ production in CM, and surface binding of tetramers complexed with peptides immunodominant for CD8 T cells. Overall, both the control and anti–IL-12 antibody treatments modestly blunted magnitudes of the ex vivo– detected specific responses. As a result, intracytoplasmic labeling was not sensitive enough to identify changes in the low proportions of CD8 T cells specifically responding to peptides with cytoplasmic IFN-γ expression (see Fig. 4). However, it was possible to demonstrate decreases, resulting from the blocking IL-12 function in IFN-α/βR KO mice, in the proportion of CD8 T cells primed for anti– CD3 stimulation of IFN-γ expression; i.e., 18.3% (± 2.2) to 12.8% (± 0.2). The anti–IL-12 treatments also resulted in significant 46 and 53% inhibitions of NP396-404– and GP33-41–stimulated IFN-γ production, respectively, in CM (Fig. 8). In contrast to the effects on IFN-γ responses, blocking of both cytokine pathways did not inhibit LCMV-induced CD8 T cell expansion; increased proportions and numbers of CD8 T cells specific for Db NP396-404 and Db GP33-41 were observed with or without IL-12 neutralization (data not shown). These studies demonstrate that the conditions of a revealed IL-12 pathway, in the absence of IFN-α/β–mediated functions, result in IFN-γ production promoted in a virus-specific manner by epitopes immunodominant for CD8 T cell responses. In contrast, the results indicate that specific CD8 T cell proliferation can occur in the absence of both IL-12 and IFN-α/β–mediated effects.
Figure 8.

LCMV-specific IFN-γ production in mice lacking endogenous IFN-α/β function. IFN-α/βR KO mice were infected i.p. on day 0 with 2 × 104 PFU LCMV Armstrong strain. They received either control (closed bars) or C17.8 anti– IL-12 (striped bars) antibodies, given i.p. on days −1 and 4 relative to infection. On day 8 after infection, spleens were harvested and processed to generate CM in the presence or absence of the immunodominant LCMV peptides NP396-404 (NP396) or GP33-41 (GP33). Levels of IFN-γ in samples were measured by ELISA. Data presented are means for three mice per group ± SEM. Significantly different (*) P values comparing anti–IL-12– to control-treated IFN-α/βR KO mice were calculated by a two-tailed Student's t test and were ≤0.05.
Antiviral Effect of Substituted IL-12 Response.
To assess the contribution of IL-12 to virus clearance in the absence of IFN-α/β function, viral titers were measured in LCMV-infected mice having had both factors blocked. Anti–IL-12 treatment of IFN-α/βR KO mice resulted in statistically significant (P < 0.01) increases in viral titers of almost 1 log by day 14 after infection; spleens and livers from anti–IL-12–treated IFN-α/βR KO had 5.8 (± 0.0) and 5.3 (± 0.1) log PFU/g, respectively, as compared with the 5.1 (± 0.1) and 4.4 (± 0.1) log PFU/g of tissue observed in control-treated IFN-α/βR KO mice (means of three mice per group ± SEM). These results show that the endogenous IL-12 response in IFN-α/βR KO mice promotes the antiviral state of the host, but cannot substitute for endogenous IFN-α/β in clearing virus expediently.
Discussion
These studies have characterized divergent innate pathways for promoting IFN-γ responses during viral infections, with induction of high level IFN-α/β resulting in conditions supporting one and acting to limit a potential alternative IL-12 pathway. The experiments demonstrate (a) an IFN-α/β pathway for IFN-γ induction, and (b) IL-12 independence of the CD8 T cell responses of expansion and IFN-γ production, during LCMV infections. Moreover, they show that in the absence of endogenous IFN-α/β– mediated functions, an IL-12 response is revealed and sufficient to support induction of an IFN-γ response, but not peak protection. Despite delayed clearance of viral burdens, the CD8 T cell expansion and IFN-γ responses elicited in the presence of the alternative IL-12 pathway are LCMV specific because the expanded cells bind MHC tetramer molecules complexed with the LCMV epitopes NP396-404 or GP33-41, and are stimulated to express IFN-γ by viral peptides. Although the substituted IL-12 acts to promote IFN-γ production, it does not appear to be required for expansion of virus-specific CD8 T cells. It is biologically significant, however, as viral titers increase if both pathways are blocked. Thus, major contributions to adaptive CD8 T cell responses are made by innate cytokines, predominantly IFN-α/β, but alternatively IL-12, during viral infection.
These innate cytokine immunoregulatory pathways can be contrasted to those characterized in response to nonviral intracellular pathogens. Similar to the Th1 responses defined under conditions of bacterial or parasitic stimuli (1, 3–9), LCMV infections induce IL-2 and IFN-γ production. However, in the other microbial infections, IL-12 is the pivotal cytokine promoting Th1 responses (4–6, 8, 42). The studies presented here conclusively demonstrate that, in the complete absence of endogenous biologically active IL-12 resulting from genetic mutation of the IL-12p35 subunit, LCMV-induced CD8 T cell expansion and IFN-γ expression proceed normally (Fig. 2, A–C). The lack of a role for IL-12 in induction of T cell IFN-γ expression confirms and extends an earlier report from this laboratory demonstrating that neutralization of endogenous IL-12 function by treatment with antibodies directed against the p40 chain does not inhibit LCMV induction of the T cell responses in IFN-α/β competent mice (11). Moreover, it is in agreement with the recent reports of lack of IL-12 effect on T cell IFN-γ responses under the conditions of treatments with antibody directed against the p40 chain during influenza virus infections (12) and genetic mutation of either the p35 or both the p35 and p40 molecules during mouse hepatitis virus infections (14). Thus, there are indications in a variety of viral infections that T cell IFN-γ responses are IL-12 independent.
In addition to showing the lack of importance for IL-12 in the presence of IFN-α/β functions, however, the studies demonstrate an IFN-α/β role in supporting T cell IFN-γ production during viral infections. They define the existence of this pathway for the first time and characterize the in vivo conditions under which it is important to the host. The results are consistent with reported enhancing effects of IFN-α/β for T cell IFN-γ production under certain specific conditions in culture (27–29, 43). However, the culture studies have been limited to examining the IFN-α roles for modest effects in association with IL-12 (28, 43), CD4 T cell subset IFN-γ responses (27, 43), and/or dramatic effects in association with the IFN-γ–inducing factor (IGIF), sometimes called IL-18 (29). Ongoing studies in our laboratory are evaluating a potential accessory role for IGIF under the conditions of viral infections. The results presented here contribute to defining the complete system by demonstrating that during viral infections IFN-α/β cytokines are dominant for T cell IFN-γ responses, mediate these effects in the absence of IL-12, and act on CD8 T cell subsets.
The experiments also identify a secondary IL-12 response absent in IFN-α/βR–competent but revealed in IFN-α/βR– deficient mice (Figs. 6–8). The lack of IL-12 appearance in the presence of IFN-α/β functions is consistent with the known negative regulation of IL-12 by the cytokines (13). The alternative IL-12 response is beneficial to the host because it can substitute in promoting IFN-γ production and facilitates clearance of virus. However, it is apparently sub-optimal because the conditions may fail to access the direct antiviral effects of IFN-α/β and result in delayed viral clearance. Thus, the responses elicited in the context of the IFN-α/β–mediated effects are clearly better for defense against this particular infectious agent, but the host can activate substitute defense mechanisms. Although the two divergent in vivo pathways to T cell IFN-γ are clearly established, it is not known if the IFN-α/β and IL-12 effects are mediated directly or indirectly, or at the priming as compared with the production phases. Experiments are underway examining these.
Remarkably, LCMV elicits particularly high circulating levels of IFN-α/β, and under these conditions does not induce IL-12 (10, 11, 13, 15). Immune responses to this infection may represent those on one end of a spectrum ranging from exclusive dependence on IFN-α/β, to codominant regulation by IFN-α/β and IL-12, to exclusive dependence on IL-12 for promoting IFN-γ production. Our hypothesis is that relative contributions would depend on presence or absence and magnitude of induction levels. MCMV and influenza infections induce more mixed responses with detectable IL-12 (10–12, 44), and certain bacterial and parasitic infections also may elicit both IL-12 and IFN-α/β expression (6, 45, 46). Other intracellular bacteria may preferentially elicit IL-12 responses (4). Direct comparison of infections indicates that LCMV induces up to threefold higher levels and sustains longer production periods of serum IFN-α/β relative to MCMV (Cousens and Biron, unpublished results). The levels achieved during LCMV infection are sufficient to mediate significant negative regulation of IL-12 (13). Thus, the picture emerging is that the immune system is equipped to induce both IFN-α/β and IL-12 simultaneously; either will lead to conditions promoting IFN-γ production, but conditions of high IFN-α/β expression make these cytokines dominant because they also are inhibiting the IL-12 response. As stated above, such conditions appear to be beneficial because they access direct antiviral functions and are particularly conducive for induction of protective responses. Moreover, as the dramatic T cell responses to LCMV render the host more sensitive to IL-12 toxicities (35, 36), they may additionally act to protect from detrimental immune responses.
Although effects of IFN-α/β on IFN-γ production are demonstrated, the results also indicate that total and virus-specific CD8 T cell expansions are IFN-α/β independent (Figs. 3 and 4), and that they occur even if both IFN-α/β and IL-12 functions are blocked (data not shown). Thus, other factors must be promoting CD8 T cell expansion during LCMV infections. At least one adaptive cytokine, IL-2, is apparently available to carry out this function. CM levels of IL-2 are similar with cells from IFN-α/βR KO, IL-12p35 KO, and WT mice, and only reduced by about half with cells from mice blocked in both IFN-α/β and IL-12 functions (data not shown). This cytokine is critical for CD8 T cell expansion, and as a result of supporting T cell proliferation, for peak T cell IFN-γ responses (17, 21). Thus, the innate cytokine IFN-α/β and/or IL-12 responses and the downstream consequences of these responses do not appear to be as important as IL-2 for T cell expansion. In this regard, it has been suggested that IFN-α/β may stimulate IL-15 production to promote proliferation of memory CD8 T cells during early viral infections (47, 48). Our results suggest that an IFN-α/β induction of IL-15 is not essential for virus-specific CD8 T cell proliferation during acute infections. However, it is interesting to note that specificities of the expanded T cells are somewhat skewed in the absence of IFN-α/β (Figs. 3 and 4). Thus, there are additional unidentified IFN-α/β effects contributing to selection of the T cell repertoire activated against infection.
The skewing of responses is observed at the level of relative proportions of CD8 T cells specifically binding tetrameric molecules complexed with NP396-404 or GP33-41 (Fig. 3). However, as the total numbers of cells binding one or the other are similar in WT and IFN-α/βR KO mice, the magnitude of the CD8 T cell proliferative responses is IFN-α/β function independent. Nevertheless, overall responses to peptide stimulation for CD8 T cell expression of cytoplasmic IFN-γ expression (Fig. 4), and peptide stimulation for IFN-γ production (Table I) are reduced. Moreover, the total proportions of CD8 T cells primed to specifically respond by expressing cytoplasmic IFN-γ after ex vivo stimulation with the LCMV peptides NP396-404 or GP33-41 account for 90–95% of those sensitized to anti– CD3 stimulation during infections of WT mice, but represent only 25–60% of those sensitized during infections of IFN-α/βR KO mice (Fig. 4). Thus, anti–CD3 reveals cells primed for T cell functions but failing to respond to the NP396-404 or GP33-41 peptides. The populations stimulated by anti–CD3, but not the tested LCMV peptides, could represent T cells having receptors (a) recognizing other LCMV epitopes, (b) nonspecifically activated, and/or (c) altered in magnitudes of functions and/or requirements for stimulation. There is evidence for the first two of these under other conditions. Although NP396-404 and GP33-41 represent the major immunodominant LCMV epitopes detected in MHC H-2b mice (23–25, 39, 49, 50), other minor epitopes have been identified (39). Despite prominence of the virus-specific T cell responses, “bystander” activation of memory phenotype T cells has been reported during LCMV infections (47). However, the last possibility seems most likely because CD8 T cells binding tetrameric Db GP33-41 are dramatically expanded. Further experiments are needed to conclusively distinguish between these possibilities. Nevertheless, the studies clearly document expansion of a large proportion of cells specific for LCMV epitopes and responding with IFN-γ production.
LCMV is a relatively noncytopathic virus. In the absence of T cell responses and/or under specific conditions of diminishing CTL responses, sometimes called “T cell exhaustion,” persistent LCMV infections can be established. Although detectable CTL function is not induced during LCMV infection in the absence of IFN-α/β (33; data not shown), our results indicate that the conditions are sufficient for resistance and eventual viral clearance (Fig. 8). Thus, they are in contrast to the suggestion of others that, in the absence of IFN-α/β–mediated regulation of viral replication, LCMV-induced T cell exhaustion results from an overwhelming viral burden (33, 34). Those investigators have based their hypothesis on the lack of CTL activity without enumerating CD8 T cell numbers. In our studies, the two are dissociated; i.e., CD8 T cell expansion and IFN-γ production occur in the absence of apparent virus-specific CTL function (data not shown). However, different isolates of LCMV vary for spontaneous induction, in immunocompetent mice, of T cell exhaustion as characterized by lack of CTL (51, 52) and persistent infection (51– 53), and it has been demonstrated that at least one of these conditions also results in the lack of virus-specific CD8 T cell expansion (54). Clearly, this is not the case under the conditions of infection in our studies. However, it is interesting to note that during chronic LCMV infections, the specificity of CD8 T cell responses can vary such that NP396-404–specific cells are deleted and functionally unresponsive GP33-41–specific cells are maintained (55), and that the skewing of specific T cells during infections of IFNα/βR KO mice is in this direction; i.e., reduced NP396-404 and increased GP33-41 specific cells (Fig. 3). Thus, parameters in addition to the absence of IFN-α/β functions must be required to extinguish defense and establish viral persistence, but protection mediated in the absence of IFN-α/β functions may be shifting in dependence towards T cell subsets sustained for longer periods of time during infections and extended antigen stimulation.
In summary, data presented here define unique divergent regulatory pathways promoting IFN-γ responses to viral infection, controlled by IFN-α/β or IL-12. They demonstrate that the strong and protective CD8 T cell responses of expansion and IFN-γ production are induced though IL-12–independent pathways during infections of immunocompetent hosts. Moreover, the studies show that in the absence of endogenous IFN-α/β, an IL-12 response can be revealed and substitute conditions to promote IFN-γ production. Although not resulting in induction of the most effective antiviral immune responses, the IL-12 substitution is beneficial. Thus, the results define uniquely IFN-α/β– controlled pathways for promoting peak defense during viral infections inducing these cytokines, and the plasticity of immune responses in accessing an alternative pathway to reach certain of the same goals.
Acknowledgments
The authors thank Drs. Ion Gresser, Phillip Scott, and Giorgio Trinchieri for their generous gifts of antibodies and hybridomas, Dr. Stan Wolf for helpful discussions, and Dr. Kaja Murali-Krishna for technical advice.
This work was supported in part by National Institutes of Health grants CA41268, AI42373, and NS21496. L.P. Cousens was supported in part by National Institutes of Health Environmental Science Training Grant T32-ES07272.
Abbreviations used in this paper
CM
conditioned media
LCMV
lymphocytic choriomeningitis virus
MCMV
murine cytomegalovirus
WT
wild-type
References
- 1.Biron CA, Gazzinelli RT. Effects of IL-12 on immune responses to microbial infections: a key mediator in regulating disease outcome. Curr Opin Immunol. 1995;7:485–496. doi: 10.1016/0952-7915(95)80093-x. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R, Janeway CA. Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 3.Heinzel FP, Rerko RM, Ling P, Hakimi J, Schoenhaut DS. Interleukin 12 is produced in vivo during endotoxemia and stimulates synthesis of gamma interferon. Infect Immunol. 1994;62:4244–4249. doi: 10.1128/iai.62.10.4244-4249.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh C-S, Macetionia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 5.Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, Romagnani S. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)–specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scharton-Kersten T, Afonso LCC, Wysocka M, Trinchieri G, Scott P. IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J Immunol. 1995;154:5320–5330. [PubMed] [Google Scholar]
- 7.Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+T cells. Annu Rev Immunol. 1994;12:635–673. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- 8.Sypek JP, Chung CL, Mayor SEH, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, Schaub RG. Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J Exp Med. 1993;177:1797–1802. doi: 10.1084/jem.177.6.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wysocka M, Kubin M, Vieira LQ, Ozmen L, Garotta G, Scott P, Trinchieri G. Interleukin-12 is required for interferon-γ production and lethality in lipopolysaccharide-induced shock in mice. Eur J Immunol. 1995;25:672–676. doi: 10.1002/eji.1830250307. [DOI] [PubMed] [Google Scholar]
- 10.Orange JS, Biron CA. Characterization of early IL-12, IFN-α/β, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol. 1996;156:4746–4756. [PubMed] [Google Scholar]
- 11.Orange JS, Biron CA. An absolute and restricted requirement for IL-12 in natural killer cell IFN-γ production and antiviral defense. J Immunol. 1996;156:1138–1142. [PubMed] [Google Scholar]
- 12.Monteiro JM, Harvey C, Trinchieri G. Role of interleukin-12 in primary influenza virus infection. J Virol. 1998;72:4825–4831. doi: 10.1128/jvi.72.6.4825-4831.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cousens LP, Orange JS, Su HC, Biron CA. Interferon-α/β inhibition of interleukin 12 and interferon-γ production in vitro and endogenously during viral infection. Proc Natl Acad Sci USA. 1997;94:634–639. doi: 10.1073/pnas.94.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schijns VECJ, Haagmans BL, Wierda CMH, Kruithof B, Heijnen IAFM, Alber G, Hornizek MC. Mice lacking IL-12 develop polarized Th1 cells during viral infection. J Immunol. 1998;160:3958–3964. [PubMed] [Google Scholar]
- 15.Welsh RM. Cytotoxic cells induced during lymphocytic choriomeningitis virus infection of mice. I. Characterization of natural killer cell induction. J Exp Med. 1978;148:163–181. doi: 10.1084/jem.148.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biron CA. Cytokines in the generation of immune responses to, and resolution of, virus infection. Curr Opin Immunol. 1994;6:530–538. doi: 10.1016/0952-7915(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 17.Cousens LP, Orange JS, Biron CA. Endogenous IL-2 contributes to T cell expansion and IFN-γ production during lymphocytic choriomeningitis virus infection. J Immunol. 1995;155:5690–5699. [PubMed] [Google Scholar]
- 18.Gessner A, Moskophidis D, Lehmann-Grube F. Enumeration of single IFN-γ producing cells in mice during viral and bacterial infection. J Immunol. 1989;142:1293–1298. [PubMed] [Google Scholar]
- 19.Kasaian MT, Biron CA. The activation of IL-2 transcription in L3T4+ and Lyt-2+lymphocytes during virus infection in vivo. J Immunol. 1989;142:1287–1292. [PubMed] [Google Scholar]
- 20.Moskophidis D, Cobbold SP, Waldmann H, Lehmann-Grube F. Mechanism of recovery from acute virus infection: treatment of lymphocytic choriomeningitis virus-infected mice with monoclonal antibodies reveals that Lyt-2+T lymphocytes mediate clearance of virus and regulate the antiviral antibody response. J Virol. 1987;61:1867–1874. doi: 10.1128/jvi.61.6.1867-1874.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su HC, Cousens LP, Fast LD, Slifka MK, Bunjiro RD, Ahmed R, Biron CA. CD4+ and CD8+T cell interactions in IFN-γ and IL-4 responses to viral infections: requirements for IL-2. J Immunol. 1998;160:5007–5017. [PubMed] [Google Scholar]
- 22.Zinkernagel RM, Doherty PC. The discovery of MHC restriction. Immunol Today. 1997;18:14–17. doi: 10.1016/s0167-5699(97)80008-4. [DOI] [PubMed] [Google Scholar]
- 23.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJD, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 24.Butz EA, Bevan MJ. Massive expansion of antigen-specific CD8+T cells during an acute virus infection. Immunity. 1998;8:167–175. doi: 10.1016/s1074-7613(00)80469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Most RG, Murali-Krishna K, Whitton JL, Oseroff C, Alexander J, Southwood S, Sidney J, Chesnut RW, Sette A, Ahmed R. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology. 1998;240:158–167. doi: 10.1006/viro.1997.8934. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed, R., and C.A. Biron. 1998. Immunity to viruses. In Fundamental Immunology. 4th ed. W.E. Paul, editor. Lippincott-Raven Publishers, New York. 1295–1334.
- 27.Brinkman V, Geiger T, Alkan S, Heusser CH. Interferon α increases the frequency of interferon γ–producing human CD4+T cells. J Exp Med. 1993;178:1655–1663. doi: 10.1084/jem.178.5.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manetti R, Annunziato F, Tomasevic L, Gianno V, Parronchi P, Romagnani S, Maggi E. Polyinosinic acid: polycytidylic acid promotes T helper type 1–specific immune responses by stimulating macrophage production of interferon-α and interleukin-12. Eur J Immunol. 1995;25:2656–2660. doi: 10.1002/eji.1830250938. [DOI] [PubMed] [Google Scholar]
- 29.Saraneva T, Matikainen S, Kurimoto M, Julkunen I. Influenza A virus–induced IFN-α/β and IL-12 synergistically enhance IFN-γ gene expression in human T cells. J Immunol. 1998;160:6032–6038. [PubMed] [Google Scholar]
- 30.Gossler A, Doetschman T, Korn R, Serfling E, Kemmler R. Transgenesis by means of blastocyst-derived embryonic stem cell lines. Proc Natl Acad Sci USA. 1986;83:9065–9069. doi: 10.1073/pnas.83.23.9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lufkin T, Dierich A, Le Meur M, Mark M, Chambon P. Disruption of the Hox-1.6 homeobox gene results in defects in a region corresponding to its rostal domain of expression. Cell. 1991;68:1105–1119. doi: 10.1016/0092-8674(91)90034-v. [DOI] [PubMed] [Google Scholar]
- 32.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo– derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 33.Muller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 34.van den Broek MF, Muller U, Huang S, Aguet M, Zinkernagel RM. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J Virol. 1995;69:4792–4796. doi: 10.1128/jvi.69.8.4792-4796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orange JS, Wolf SF, Biron CA. Effects of IL-12 on the response and susceptibility to experimental viral infections. J Immunol. 1994;152:1253–1264. [PubMed] [Google Scholar]
- 36.Orange JS, Salazar-Mather TP, Opal SM, Spencer RL, Miller AH, McEwen BS, Biron CA. Mechanism of interleukin 12-mediated toxicities during experimental viral infections: role of tumor necrosis factor and glucocorticoids. J Exp Med. 1995;181:901–914. doi: 10.1084/jem.181.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gresser I, Tovey MG, Maury C, Bandu M-T. Role of interferon in the pathogenesis of virus diseases in mice as demonstrated by the use of anti-interferon serum. J Exp Med. 1976;144:1316–1323. doi: 10.1084/jem.144.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moskophidis D, Battegay M, Bruendler M-A, Laine E, Gresser I, Zinkernagel RM. Resistance of lymphocytic choriomeningitis virus to alpha/beta interferon and to gamma interferon. J Virol. 1994;68:1951–1955. doi: 10.1128/jvi.68.3.1951-1955.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Most RG, Sette A, Oseroff C, Alexander J, Murali-Krishna K, Lau LL, Southwood S, Sidney J, Chesnut RW, Matloubian M, Ahmed R. Analysis of cytotoxic T cell responses to dominant and subdominant epitopes during acute and chronic lymphocytic choriomeningitis virus infection. J Immunol. 1996;157:5543–5554. [PubMed] [Google Scholar]
- 40.O'Garra, A., and K.M. Murphy. 1996. Role of cytokines in determining T cell function (mouse). In Weir's Handbook of Experimental Immunology. 5th ed. D.M. Weir, editor. Blackwell Science, Cambridge, MA. 226.1–226.10.
- 41.Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 42.Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, Gately M, Louis JA, Alber G. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania majorand mount a polarized Th2 cell response. Eur J Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- 43.Wenner CA, Guler ML, Macatonia SE, O'Garra A, Murphy KM. Roles of IFN-γ and IFN-α in IL-12– induced T helper cell-1 development. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 44.Grundy JE, Trapman J, Allan JE, Shellam GR, Melief CJM. Evidence for a protective role of interferon in resistance to murine cytomegalovirus and its control by non–H-2-linked genes. Infect Immun. 1982;37:143–150. doi: 10.1128/iai.37.1.143-150.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diefenbach A, Schindler H, Donhauser N, Lorenz E, Laskay T, MacMicking J, Röllinghoff M, Gresser I, Bogdan C. Type 1 interferon (IFN-α/β) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity. 1998;8:77–87. doi: 10.1016/s1074-7613(00)80460-4. [DOI] [PubMed] [Google Scholar]
- 46.Yaegashi Y, Nelson P, Sing A, Galanos C, Freudenberg MA. Interferon β, a cofactor in the interferon γ production induced by gram-negative bacteria in mice. J Exp Med. 1995;181:953–960. doi: 10.1084/jem.181.3.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tough DF, Burrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+T cells in vivo by IL-15. Immunity. 1998;8:591–599. doi: 10.1016/s1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- 49.Gairin JE, Mazarguil H, Hudrisier D, Oldstone MBA. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Dbmajor histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J Virol. 1995;69:2297–2305. doi: 10.1128/jvi.69.4.2297-2305.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yanagi Y, Tishon A, Lewicki H, Cubitt BA, Oldstone MBA. Diversity of T-cell receptors in virus-specific cytotoxic T lymphocytes recognizing three distinct epitopes restricted by a single major histocompatibility complex molecule. J Virol. 1992;66:2527–2531. doi: 10.1128/jvi.66.4.2527-2531.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahmed R, Simon RS, Matloubian M, Kolhekar SR, Southern PJ, Freedman DM. Genetic analysis of in vivo–selected viral variants causing chronic infection: importance of mutation in the L RNA segment of lymphocytic choriomeningitis virus. J Virol. 1988;62:3301–3308. doi: 10.1128/jvi.62.9.3301-3308.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral effector T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 53.Pfau CJ, Valenti JK, Pevear DC, Hunt KD. Lymphocytic choriomeningitis virus killer T cells are lethal only in weakly disseminated murine infections. J Exp Med. 1982;156:79–89. doi: 10.1084/jem.156.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliot T, Hengartner H, Zinkernagel R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I–peptide complexes. J Exp Med. 1998;187:1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]